
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel tyrosine hyperoxidation enables selective peptide cleavage†
Shengping
Zhang
ab,
Luis M.
De Leon Rodriguez
 a,
Freda F.
Li
a,
Renjie
Huang
a,
Ivanhoe K. H.
Leung
ac,
Paul W. R.
Harris
*abc and
Margaret A.
Brimble
*abc
a,
Freda F.
Li
a,
Renjie
Huang
a,
Ivanhoe K. H.
Leung
ac,
Paul W. R.
Harris
*abc and
Margaret A.
Brimble
*abc
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds St, Auckland, 1010, New Zealand. E-mail: paul.harris@auckland.ac.nz; m.brimble@auckland.ac.nz
bSchool of Biological Sciences, The University of Auckland, 3A Symonds St, Auckland, 1010, New Zealand
cMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, 1142, New Zealand
First published on 11th February 2022
Abstract
A novel tyrosine hyperoxidation enabling selective peptide cleavage is reported. The scission of the N-terminal amide bond of tyrosine was achieved with Dess–Martin periodinane under mild conditions, generating a C-terminal peptide fragment bearing the unprecedented hyperoxidized tyrosine motif, 4,5,6,7-tetraoxo-1H-indole-2-carboxamide, along with an intact N-terminal peptide fragment. This reaction proceeds with high site-selectivity for tyrosine and exhibits broad substrate scope for various peptides, including those containing post-translational modifications. More importantly, this oxidative cleavage was successfully applied to enable sequencing of three naturally occurring cyclic peptides, including one depsipeptide and one lipopeptide. The linearized peptides generated from the cleavage reaction significantly simplify cyclic peptide sequencing by MS/MS, thus providing a robust tool to facilitate rapid sequence determination of diverse cyclic peptides containing tyrosine. Furthermore, the highly electrophilic nature of the hyperoxidized tyrosine unit disclosed in this work renders it an important electrophilic target for the selective bioconjugation or synthetic manipulation of peptides containing this unit.
Introduction
Tyrosine (Tyr), a proteinogenic amino acid bearing an electron rich phenol moiety in the side chain, is one of the primary targets for protein oxidation, which plays an essential role in cellular physiology and pathology.1,2 The oxidation of Tyr in vivo, induced by biologically relevant radicals, UV radiation or enzymes, is a complex chemical process generating a diverse range of oxidized products whose formation critically depend on the type of oxidant as well as the residue's environment.3–6 Some Tyr oxidation products, such as dopamine, dopamine quinone and 5,6-dihydroxyindole, are important biological intermediates in neurotransmission and eumelanin biosynthesis.7,8 Their formation involves a cascade of chemical transformations including oxidation, cyclization, decarboxylation and proteolysis.6 Nitration of the aromatic ring is another major Tyr oxidative modification in vivo. Generated by the reaction between a Tyr radical (TyrO˙) and a nitrogen dioxide radical (˙NO2), the 3-nitroTyr residue within proteins serves as a key modulator of protein conformation and function, thereby exerting regulatory effects on cell signalling and cell dysfunction.9–11 Moreover, 3,3′-diTyr, a product formed via dimerization of two TyrO˙ during Tyr oxidation, can result in protein cross-linking and aggregation and is regarded as a diagnostic marker for damaged proteins and their selective proteolysis under oxidative stress.12–14In addition to its biological significance, Tyr oxidation has also been established as a robust tool in bioengineering and chemical biology studies. Dopaquinone is a chemically malleable species generated in the production of melanin, the natural pigment in human skin.15,16 After its formation by a tyrosinase-mediated Tyr oxidation, dopaquinone can undergo three different transformation pathways: crosslinking, conjugate addition or oxidative cleavage. The dopaquinone induced crosslinking of peptides and proteins usually proceeds via formation of Tyr–Tyr,17 Tyr–Cys,18 and Tyr–Lys19 linkages and has been extensively applied in the development of moisture-resistant adhesives and self-assembling materials for both industrial and biomedical applications.20,21 Furthermore, the dopaquinone species can undergo Michael addition with secondary amines and anilines22,23 or strain-promoted cycloaddition with cycloalkynes24 to achieve selective protein conjugation. The oxidative cleavage of proteins at a Tyr site, however, has long been an underexplored area. Accounts relating to this transformation are exceedingly scarce. Long and Hedstrom reported the fragmentation of a Tyr-rich hemagglutinin tag when treated with mushroom tyrosinase, but structural characterization of the cleaved peptide fragments was elusive.25 Chemical reagents, such as N-bromosuccinimide,26 were reported to cleave peptides at the amide bond C-terminal to Tyr, Trp and His.27 Moreover, 2-iodosobenzoic acid (IBA)28 is a well-established Trp-selective protein cleaving reagent, but can also lead to undesirable protein fragments at the Tyr site to a varying extent.
Site-selective cleavage of peptide bonds is a valuable chemical modification that has a plethora of applications in proteomics,29 chemical biology30 and drug development.31 One of its main uses is protein/peptide sequencing, wherein the primary structure of an unknown substrate can be determined by enzymatic or chemical cleavage of peptide bonds at specific residues followed by subsequent amino acid analysis or liquid chromatography with tandem mass spectrometry (LC-MS/MS).32,33 A series of proteases targeting specific residues, such as trypsin, chymotrypsin and pepsin, have been widely used for this purpose.34 However, the recent growing interest in chemically modified peptides, particularly cyclic peptides35 as novel therapeutics, poses a significant challenge to protease-enabled peptide sequencing as many modified peptides exhibit resistance to proteases.36,37 The use of chemical reagents to cleave peptide bonds, on the other hand, provides an inherent advantage over proteases when applied to chemically modified peptides. Nevertheless, their application to the cleavage of proteins/peptides is limited by low site-selectivity,38,39 undesirable modifications40,41 (such as oxidation of Cys, Met, Tyr, and Trp) and the use of harsh reaction conditions.42 The development of novel methods with improved selectivity and efficiency is therefore highly desirable to facilitate robust sequencing of chemically modified peptides, such as cyclic peptides.
Herein we report the first example of the selective cleavage of N-terminal amide bonds of Tyr residues using Dess–Martin periodinane (DMP). This cleavage affords a modified C-terminal peptide fragment bearing an unprecedented 4,5,6,7-tetraoxo-1H-indole-2-carboxamide (TICA) moiety along with an intact N-terminal peptide fragment. This efficient transformation proceeds selectively under mild conditions (40 °C) and can be applied to a broad range of substrates, including a diverse set of cyclic peptides.
Results and discussion
Peptide fragmentation at a tyrosine residue
We recently reported the successful synthesis of the naturally-occurring cyclic peptide callyaerin A, which contains a rare (Z)-2,3-diaminoacrylamide motif and has potent anti-tubercular activity.43 In one of our synthetic approaches to prepare callyaerin A, the oxidation of the hydroxyl side chain of the Ser residue in the peptide to an aldehyde with DMP did not occur under typical oxidation conditions (1.1 to 5 equiv. of DMP, RT to 50 °C), but surprisingly fragmentation at the Ser site was observed when increasing the equivalents of DMP (10 equiv.). Further studies of this reaction using other peptide substrates revealed that this oxidative peptide cleavage occurred not only at the Ser site but also at the Tyr position. Given the rarity of Tyr-selective peptide cleavage in the literature,44 we decided to examine this DMP-induced cleavage using the pentapeptide Fmoc–Gly–Tyr–Phe–Val–Phe–OH (1). The Fmoc group was retained as the N-terminal protecting group as it enabled easy quantification of the N-terminal fragment by HPLC monitoring.Peptide 1 was treated with DMP in DMSO at 40 °C, and the reaction progress was monitored by HPLC-MS. Within 1 h, the starting material was consumed and a new compound 1a with a molecular ion at m/z 868.2 (Fig. 1) was identified. This species was assigned as the peptidyl dopaquinone intermediate 1a, in line with the reported 2-iodoxybenzoic acid (IBX)-induced tyrosine oxidation.45,46 As the reaction proceeded, peptide 1a disappeared and two new peaks, 2 and 3 were observed in the HPLC chromatogram (Fig. 1). The peak at 22.8 min was identified as Fmoc-Gly-OH (2), the N-Fmoc protected-terminal amino acid. This result indicated that peptide fragmentation was taking place at the N-terminal tyrosyl amide bond. Accordingly, we postulated that the newly formed species 3 (m/z 651.2, 17.8 min), was derived from the C-terminal peptide fragment. The cleavage reaction went to completion cleanly after 48 h, affording only 2 and 3. To our surprise, the finding of peptide cleavage has not been recorded in previous reported IBX-induced tyrosine oxidation protocols, which for those cases is probably attributed to the addition of exogenous reducing reagents46 and nucleophiles45 during the reaction process.
 | ||
| Fig. 1 HPLC-MS analysis of the reaction mixture of 1 and DMP (10 equiv.) in DMSO at 40 °C at t = 1 h (top), 24 h (middle) and 48 h (bottom). The UV-Vis detector was set at a wavelength of 254 nm. The asterisk-labeled peaks correspond to the DMP-related byproducts that were consistently detected in all the cleavage reactions. MS characterization of dopaquinone intermediate 1a ([M + H]+ calcd/found 868.4/868.2), the generated peptide N-terminal fragment 2 ([M + H]+ calcd/found 298.1/298.2) and the modified C-terminal fragment 3 ([M + H]+ calcd/found 651.2/651.2). | ||
The structure of 3 was next elucidated by high-resolution mass spectrometry (HRMS) and nuclear magnetic resonance (NMR) analyses. HRMS of 3 in H2O/MeCN (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) showed an adduct ion at m/z 651.2470 corresponding to the chemical formula of C32H35N4O11. The decreased number of hydrogens and the unchanged number of carbon and nitrogen atoms in 3, relative to the unmodified C-terminal fragment of 1 comprising the N-terminal tyrosine (H-Tyr–Phe–Val–Phe-OH (S1), C32H38N4O6, ESI†), indicated a higher degree of unsaturation in the former. Furthermore, the significant increase in the number of oxygens in 3 revealed extensive oxidation of the phenyl ring of tyrosine and was attributed to the presence of multiple ketones.
:1) showed an adduct ion at m/z 651.2470 corresponding to the chemical formula of C32H35N4O11. The decreased number of hydrogens and the unchanged number of carbon and nitrogen atoms in 3, relative to the unmodified C-terminal fragment of 1 comprising the N-terminal tyrosine (H-Tyr–Phe–Val–Phe-OH (S1), C32H38N4O6, ESI†), indicated a higher degree of unsaturation in the former. Furthermore, the significant increase in the number of oxygens in 3 revealed extensive oxidation of the phenyl ring of tyrosine and was attributed to the presence of multiple ketones.
A comparison of the 1H NMR spectrum of 3 and H-Tyr–Phe–Val–Phe-OH revealed a similar pattern of signals for Phe and Val. Hence, we concluded that these residues were intact in 3. However, the signals of the methylene, amino and aromatic protons of Tyr were not observed in the 1H NMR spectrum of 3, thereby indicating that extensive chemical modification of Tyr had taken place (Fig. S1†). The subsequent spectroscopic analysis focused on the interpretation of the signals corresponding to the modified N-terminal tyrosine. These signals have been highlighted in red in Fig. 2 to facilitate the discussion.
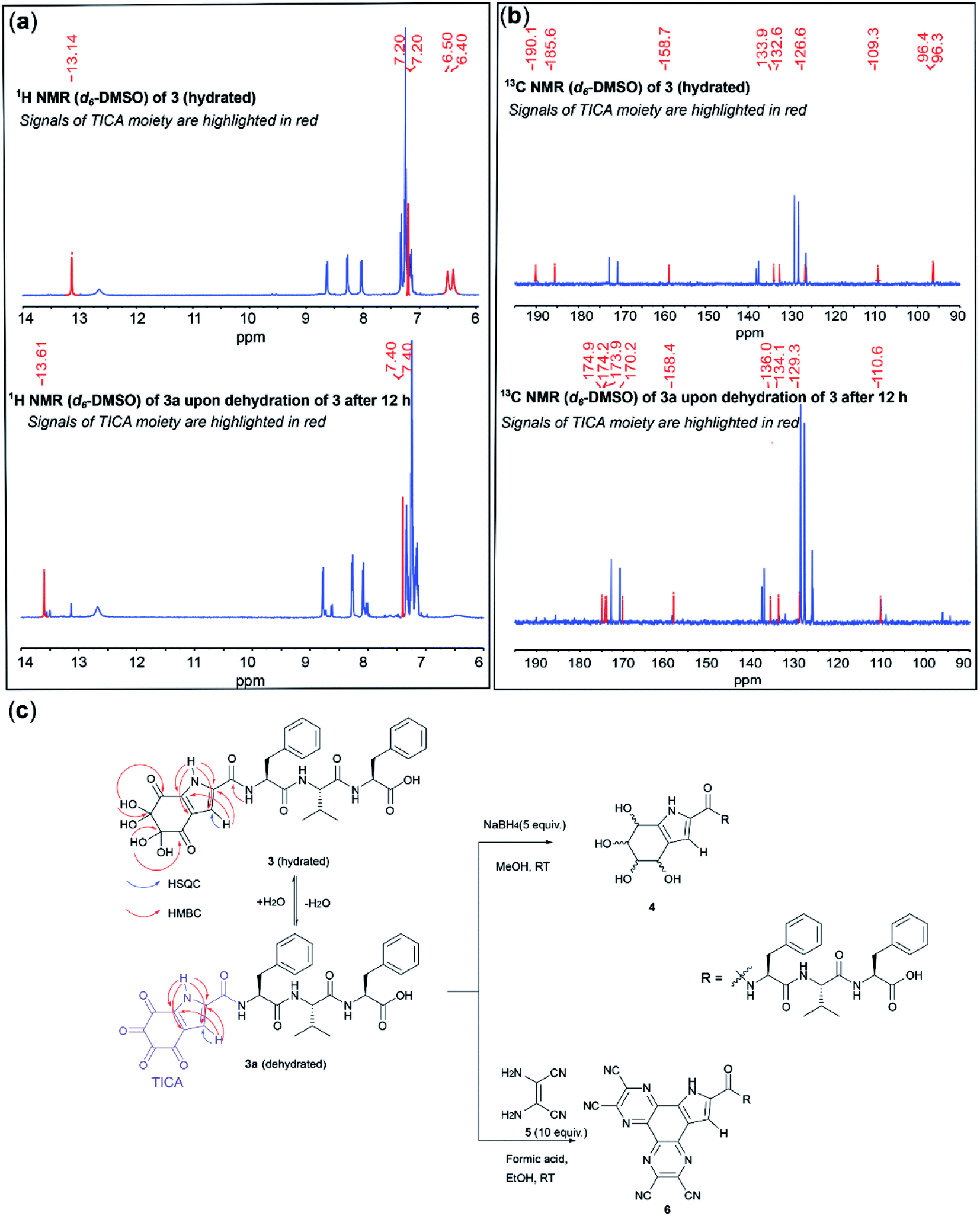 | ||
| Fig. 2 Structure determination of the C-terminal peptide fragment 3. (a) The stacked 1H-NMR (500 MHz, d6-DMSO) of 3 (top) and its dehydrated form 3a (bottom). (b) The stacked 13C-NMR (125 MHz, d6-DMSO) of 3 (top) and its dehydrated form 3a (bottom) (c) The proposed structure of 3 and 3a. Peptide 3 was reduced to a diastereomeric mixture 4 after treatment with NaBH4 or reacted with diaminomaleonitrile (5) to afford 6. | ||
NMR analysis of 3 in deuterated DMSO revealed a slow transition of 3 to another species 3a within 12 h (>80% conversion, Fig. 2a and c). The 1H NMR of 3 exhibited four signals attributed to the modified N-terminal moiety (Fig. 2a, top). One proton resonated significantly downfield at 13.14 ppm, suggesting its direct attachment to a nitrogen present within an aromatic system. A doublet at 7.20 ppm, which correlated to a carbon resonance at 109.3 ppm in the HSQC spectrum, was assigned to an aromatic proton. The two remaining signals at 6.40 and 6.50 ppm integrated to four protons and exhibited no correlations in the HSQC spectrum. In the 1H NMR spectrum of 3a, the NH and CH aromatic protons were found to be slightly deshielded at 13.61 and 7.40 ppm, respectively. However, the corresponding four-proton signals of the resonances of 3 at 6.40 and 6.50 ppm were not observed for 3a (Fig. 2a, bottom).
A comparison between the 13C NMR spectra of 3 and 3a revealed four resonances with a similar pattern (105–140 ppm) in the aromatic region, together with three and five carbons within the carbonyl region (160–190 ppm) for 3 and 3a, respectively. Additionally, two carbon signals resonating significantly more upfield (96.2 and 96.3 ppm) were observed for 3 (Fig. 2b). These carbons exhibited clear HMBC correlations to the four-proton signals at 6.40 and 6.50 ppm, suggesting the presence of two geminal-diol carbons in the structure of 3 (Fig. 2c). Thus, the disappearance of the two geminal-diol carbons of 3 concomitant with the appearance of two extra carbonyl signals (170 ppm) in the 13C NMR of 3a indicates the spontaneous dehydration of a dihydrate to a diketone.
Furthermore, the two carbonyl carbons of 3 were observed to resonate further downfield at 190.1 and 185.6 ppm in the 13C NMR spectrum (ca.170 ppm for 3a), and also exhibited HMBC correlations with the two gem-diol proton resonances, hence indicating that they are adjacent to the geminal-diol carbons (Fig. 2b). Both the HMBC spectra of 3 and 3a exhibited correlations from the NH aromatic proton to each of the four aromatic carbon resonances, and from the CH aromatic proton to two aromatic carbons, respectively. These NMR data allowed the assignment of a tri-substituted pyrrole ring within the highly oxidized moiety, reminiscent of the well-known tyrosine metabolite 5,6-dihydroxyindole-2-carboxylic acid (DHICA).47 Additionally, the carbonyl carbon at 158.7 ppm of 3 (158.4 ppm for 3a) appeared further upfield than other amide carbons in the peptide and exhibited an HMBC correlation with the neighbouring amide proton of Phe, indicating that the C-terminal amide of the tyrosine residue was attached to the pyrrole ring. Based on the above-mentioned NMR evidence, formation of an unprecedented 4,5,6,7-tetraoxo-1H-indole-2-carboxamide (TICA) moiety (derived from a hetero-annulation process during the cleavage) in 3a, which contains four consecutive ketones and forms dihydrate 3 when exposed to water, was proposed (Fig. 2c). To further prove the validity of the proposed structure, peptide 3 was subjected to NaBH4-mediated reduction. A diastereomeric mixture 4 (Fig. 2c) was obtained as expected and the 13C-NMR of this mixture exhibited a series of C(OH)H signals between 65–80 ppm with the concomitant disappearance of all the ketones and gem-diol carbon signals of 3, which confirmed the presence of the tetraketone system (Fig. S2 and S3†). Furthermore, treatment of 3 with diaminomaleonitrile (5) afforded a bis-conjugated peptide 6 and this result provided additional supporting evidence for the presence of highly electrophilic tetra ketone motif in 3a (Fig. 2c and S4–S7†).
A plausible mechanism for the DMP-mediated Tyr-specific cleavage reaction is shown in Scheme 1. The reaction commences with the known ortho-oxidation of Tyr to dopaquinone (1a) via intramolecular transfer of an oxygen atom from the iodine(V) intermediate 7 to the ortho-position of the phenol with concomitant reduction into a more stable iodine(III) o-quinolmonoketal 8.45,46 After tautomerization and reductive elimination of 2-iodobenzoic acid (10), the generated compound 1a then undergoes Michael addition with the adjacent amide nitrogen to give the indolium intermediate 12. The subsequent release of the N-acyl substituent, Fmoc-Gly, from indolium 12 leads to peptide cleavage at the N-terminal amide bond of tyrosine and the resulting dopachrome 13 undergoes tautomerization to afford the 5,6-dihydroxyl-indole species 15, similar to the formation of DHICA.47 Subsequent DMP-mediated ortho-oxidation of the two phenols in 15 could first occur at either site but hydroxylation at position 4 is shown as an example. Finally, ortho-oxidation of both phenols affords peptide fragment 3a bearing the TICA moiety. According to this mechanism, we anticipated that the DMP-mediated oxidation of the peptide containing a free Tyr at the N-terminus would lead to more efficient formation of the TICA moiety as the free amino group is more prone to participate in a Michael addition reaction than an amide functionality. Treatment of H-Tyr–Phe–Val–Phe-OH with DMP was therefore performed and the desired peptide 3a was rapidly generated within 30 min, thus corroborating the proposed pathway.
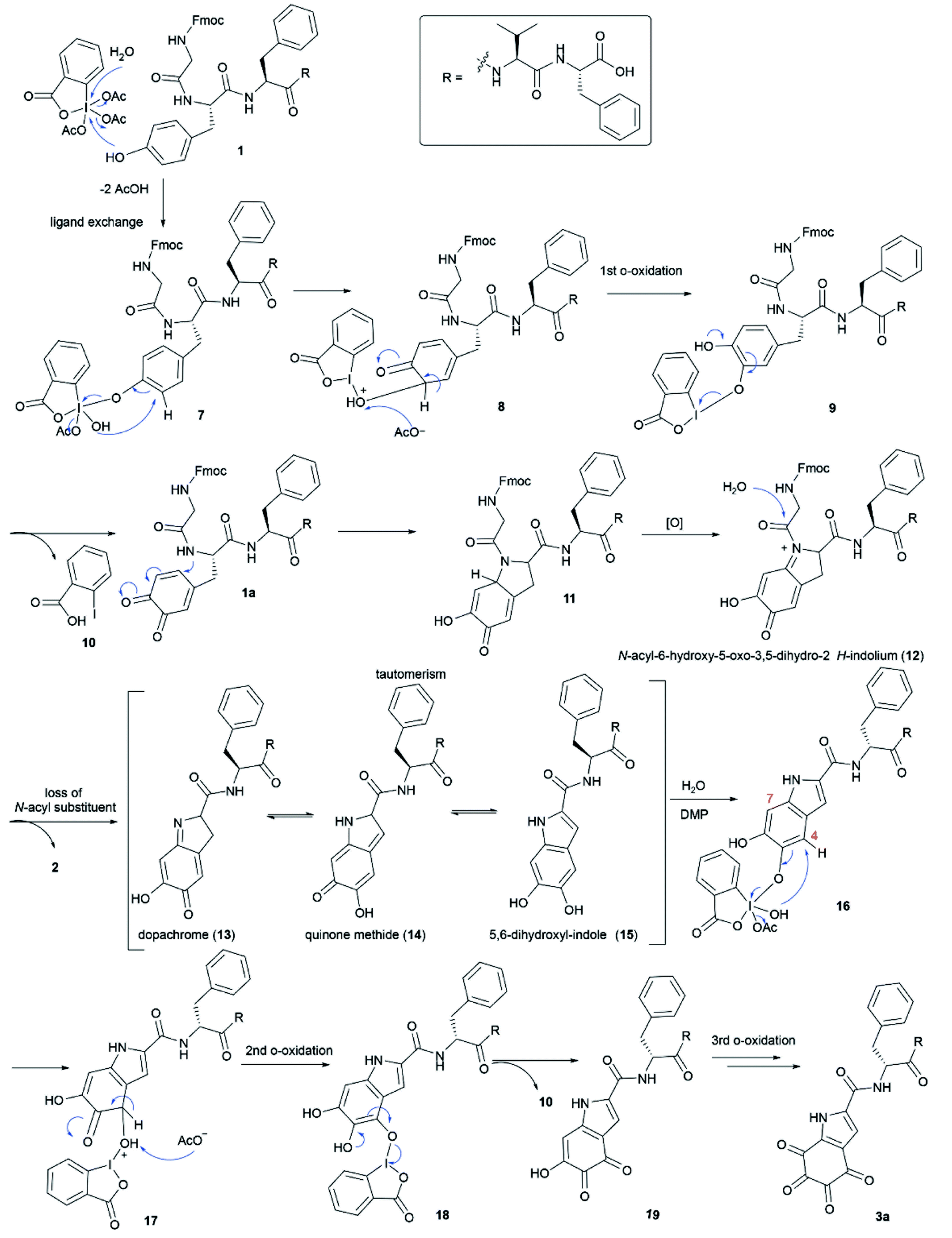 | ||
| Scheme 1 The proposed mechanism of the DMP-mediated peptide cleavage at the tyrosine residue. | ||
Selective cleavage at a tyrosine site over serine
As mentioned above, the DMP-mediated peptide cleavage also occurred at a Ser residue during our synthetic work towards callyaerin A. To further investigate the cleavage reaction at a Ser site, the peptide Fmoc-Pro–Ser–Phe–Pro–Ile-OH without a Tyr residue was first synthesized and cleaved with DMP in DMSO at 40 °C for 2 days. The resulting N-terminal peptide fragment was readily characterized as Fmoc-Pro-NH2 (20) (Fig. S8–S11†) and the C-terminal fragment was identified by HRMS and NMR as the oxalic acid mono amide peptide (see S2 in Fig. S12–S15†). A similar C-terminal peptide fragment was reported during the selective cleavage of Ser-containing peptides with Ru(VIII) (generated in situ from RuCl3 and NaIO4).48 Derived from this report one can posit that a mechanism for the DMP-mediated Ser-selective peptide rupture comprises (1) a Cα–Cβ oxidative scission at the Ser site, yielding an acylimine, (2) water addition to the acylimine, which results in a carbinolamide moiety, (3) oxidation of the carbinolamide to an oxalamide and (4) hydrolytic cleavage of the oxalamide to generate peptide amide and oxalic acid mono amide as peptide N- and C- terminal fragments, respectively (Scheme S1†). We then subjected the peptide Fmoc-Pro–Ser–Tyr–Pro–Ile-OH (21) to the DMP-mediated cleavage reaction conditions. After two-days, equivalent amounts of the N-terminal fragments cleaved at both the Ser (20) and Tyr (22) sites were identified by HPLC (Fig. 3, top, path b). This suggested that both cleavages proceeded at similar reaction rates in DMSO, precluding the option of a kinetically-controlled selective reaction. It has been reported that water could accelerate the Dess–Martin oxidation in various applications.49 It has also been indicated that the presence of water is required to facilitate hydrolysis of the tyrosine N-terminal amide bond (Scheme 1), while water is required not only during the hydrolytic cleavage of the oxalamide obtained during serine Cα–Cβ oxidative scission reaction but also in the formation of carbinolamide from the acylimine. Therefore, we envisaged that the addition of water might facilitate a differential effect on the reaction kinetics of these two cleavage reactions, thereby enabling selective control of the cleavage site. Aqueous phosphate buffer (DMSO/0.1 M phosphate buffer pH 7 (v/v): 2:1) was therefore added to the reaction mixture (Fig. 3, path a). Strikingly, the Tyr-selective peptide cleavage was complete within 2 h (Fig. 3, bottom) with a conversion to the Fmoc-Pro–Ser of 76% and no Ser cleavage side products were observed even after 12 h. Further optimization of the reaction temperature, equivalents of DMP and the ratio of DMSO/phosphate buffer did not lead to any further improvement of the conversion rate (Table S1†) nor any Ser cleavage derived peptides.
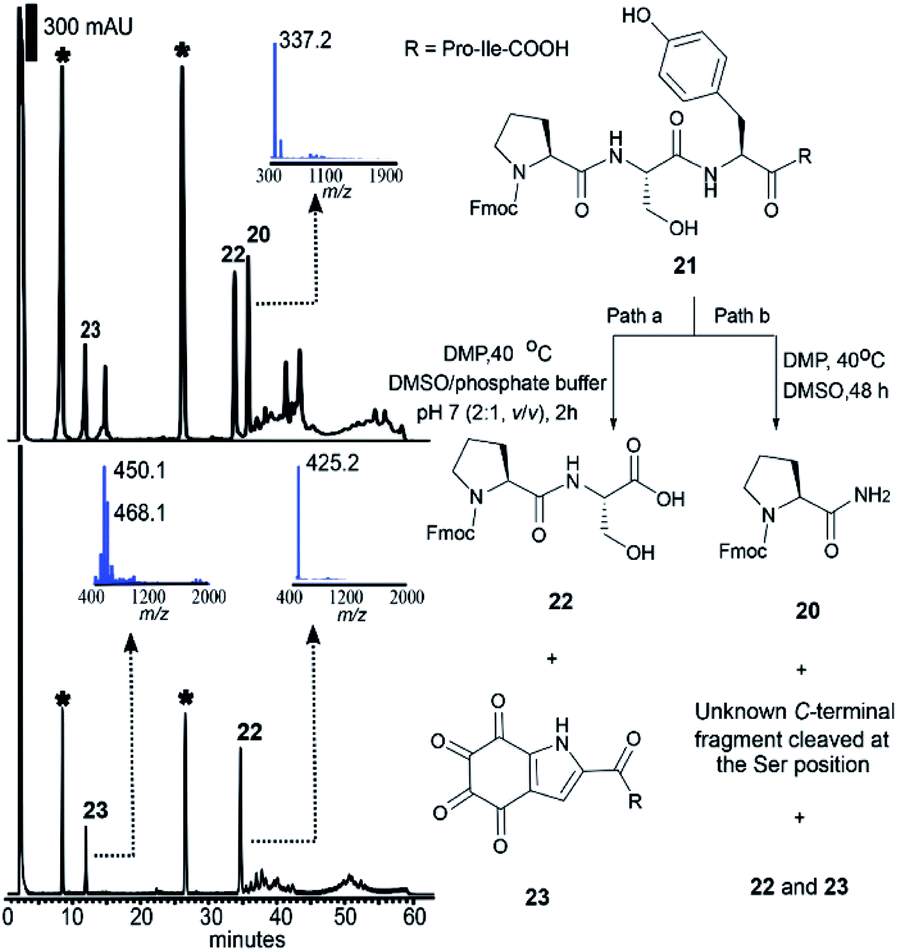 | ||
| Fig. 3 HPLC-MS analysis of the reaction mixture of 21 and DMP (10 equiv.) at 40 °C in DMSO after 48 h (top) and in DMSO/0.1 M phosphate buffer at pH 7 (2:1, v/v) after 2 h (bottom). The UV-Vis detector was set at a wavelength of 254 nm. The asterisk-labeled peaks correspond to the DMP-related byproducts that were consistently detected in all the cleavage reactions. MS characterization of the N-terminal fragment 22 ([M + H]+ calcd/found 425.2/425.2) and C-terminal fragment 23 ([M + H2O + H]+ calcd/found 450.2/450.1, [M + 2H2O + H]+ calcd/found 468.2/468.1) cleaved at the tyrosine site, as well as the N-terminal fragment 20 cleaved at the serine site ([M + H]+ calcd/found 337.2/337.2). | ||
Functional group tolerance
Next, we investigated the functional-group tolerance of this cleavage reaction using model pentapeptides Fmoc-Pro–Xaa–Tyr–Pro–Ile-OH (where Xaa represented different amino acids) (Table 1). Peptide substrates containing Leu (24a), Arg (24b), Asp (24c), Gln (24d), Thr (24e), Lys (24f) and His (24g) at Xaa position were all successfully cleaved at the tyrosine site, generating the corresponding N-terminal fragments (Fmoc–Pro–Xaa:Xaa = Leu, Arg, Asp, Gln, Thr, Lys and His) with conversions ranging from 76% to 88%. Cleavage of the Cys-containing peptide 24h using DMP resulted in the N-terminal fragment with a moderate conversion (46%) and the thiol group was oxidized to a sulfonate. The undesired modification of Cys is a common issue associated with many established protein cleavage protocols due to the strong nucleophilicity of the free thiol group and pre-capping of Cys is routinely performed to prevent the side reactions.50,51 Therefore, capping of the Cys sidechain of 24h with iodoacetamide before the cleavage was employed, which led to significant improvement of the cleavage conversion (24i, 91%). Importantly, for substrates containing electron-rich residues such as Met (24j) and Trp (24k), the N-terminal fragments (Fmoc-Pro–Met and Fmoc-Pro–Trp) were generated in high yield without oxidative modification of their sidechains. It is also noteworthy that unlike IBA, an iodine(III) oxidant, the DMP-mediated peptide cleavage demonstrated high site-selectivity for Tyr over Trp as no N-terminal fragment bearing the characteristic Trp-derived dioxindolylalanine28,39 motif was observed. Finally, cleavage of peptide 24l with a free N-terminal amino group did not lead to the expected peptide fragmentation. Instead, a cyclic peptide (see S3 Fig. S16–S21†) resulting from Michael addition of the α-amino group to the ortho-quinone intermediate was generated. Further attempts to prevent the peptide cyclization were made by lowering the pH of the reaction mixture (pH 2–6), conditions under which the N-terminal amino should be protonated, hence making it less nucleophilic. However, all attempts failed to prevent the formation of the undesired cyclic peptide side product, which indicates that it is necessary to protect the N-terminal amino group for the cleavage to occur (vide infra).| Entry | Substratea | Conversionb |
|---|---|---|
|
a Standard conditions: peptide (1 mg) was reacted with 10 equiv. of DMP in DMSO/phosphate buffer at pH 7 (2:1, v/v, 300 mL) at 40 °C for 2 h.
b Conversion to the N-terminal fragment was calculated from the HPLC peak area at 254 nm.
c The thiol group of cysteine was oxidized to a sulfonate.
d Peptide cleavage product was not detected.
e A disulfide bond forms between the two cysteine residues labelled with asterisk (*).
f Selective acetylation of the peptide N-terminus was performed before cleavage using the following conditions: (i) 50 equiv. of acetic anhydride, 30 equiv. of pyridine, DMSO, RT, 2 h; (ii) 2 M hydroxylamine buffer (pH = 6), 4 h, RT.
g Conversion to the linearized peptide was calculated from the HPLC peak area at 214 nm.
|
||
| 1 | Fmoc-Pro–Leu–Tyr–Pro–Ile-OH (24a) | 85% |
| 2 | Fmoc-Pro–Arg–Tyr–Pro–Ile-OH (24b) | 80% |
| 3 | Fmoc-Pro–Asp–Tyr–Pro–Ile-OH (24c) | 81% |
| 4 | Fmoc-Pro–Gln–Tyr–Pro–Ile-OH (24d) | 81% |
| 5 | Fmoc-Pro–Thr–Tyr–Pro-Ile-OH (24e) | 76% |
| 6 | Fmoc-Pro–Lys–Tyr–Pro–Ile-OH (24f) | 78% |
| 7 | Fmoc-Pro–His–Tyr–Pro–Ile-OH (24g) | 88% |
| 8 | Fmoc-Pro–Cys–Tyr–Pro–Ile-OH (24h) | 46%c |
| 9 | Fmoc-Pro–Cys(CH2CONH2)–Tyr–Pro–Ile-OH (24i) | 91% |
| 10 | Fmoc-Pro–Met–Tyr–Pro–Ile-OH (24j) | 87% |
| 11 | Fmoc-Pro–Trp–Tyr–Pro–Ile-OH (24k) | 85% |
| 12 | H-Leu–Val–Leu–Val–Tyr–Gly-OH (24l) | N.D.d |
| 13 | Fmoc-Gly–DTyr–DAsp–Phe–Gly-OH (24m) | 88% |
| 14 | Fmoc-Gly–(NMe)Ala–Tyr–Asn–Leu–Gly-OH (24n) | 83% |
| 15 | Fmoc-Gly–Tyrphos–Asp–Phe–Gly-OH (24o) | N.D.d |
| 16 | Fmoc-Cys*–Gly–Arg–Arg–Ala–Cys*–Tyr–Phe–Ala–Gly-OH (24p)e | 87% |
| 17 | H-Cys*–Tyr–Ile–Gln–Asn–Cys*–Pro–Leu–Gly-OH (24q)e,f | 49%g |
The applicability of this DMP-mediated cleavage for peptides comprising unnatural amino acids and post-translational modifications was also investigated. Substrates 24m and 24n, containing either D- or N-methyl amino acids, were both cleaved in high conversion after treatment with DMP (Table 1, entry 13 and 14). This result demonstrates a considerable advantage over conventional enzymatic digestion, where substrates bearing unnatural amino acids do not typically undergo enzymatic cleavage. However, no peptide fragmentation was observed in the reaction of peptide 24o bearing a phosphorylated Tyr residue. This is attributed to the masking of the phenol group, which prevents further reaction with DMP as shown in Scheme 1. In the case of 24p, which comprises an intramolecular disulfide bond, the N-terminal peptide fragment was successfully identified in the cleavage reaction with the disulfide bond remaining intact. Furthermore, the cleavage reaction was also applied to oxytocin (24q), a cyclic peptide containing a tyrosine residue within the disulfide bridge. After acetylation of the peptide N-terminus, the cleavage reaction afforded the expected linearized peptide bearing an N-terminal TICA moiety, which suggested that the position of the tyrosine residue relative to the disulfide ring does not influence the cleavage reaction. Therefore, this new method may provide additional information for assigning the location of disulfide bonds within polypeptides.
Substrate study of DMP-mediated peptide cleavage
The substrate scope of the present method was further investigated with longer and more complex peptide systems (Fig. 4). Peptide 25 featuring two tyrosine residues in the sequence was subjected to the DMP-mediated cleavage. However, the reaction of 25 with 10 equiv. of DMP was sluggish. The starting material was not totally consumed even after 12 h and a mixture of unknown side products were observed during reaction. An increased amount of DMP (40 equiv.) was required to bring the reaction to completion. Under these conditions, peptide cleavage at the two tyrosine sites proceeded smoothly, generating the expected three peptide fragments, the N-terminal fragment Fmoc-QQRLI-OH (25a), the middle fragment TICA-FLF-OH (25b) and the C-terminal fragment TICA-AILG-NH2 (25c) within 6 h (conversion to the N-terminal fragment 25a: 63%, Fig. 4, entry 1). The DMP cleavage method was next applied to the bioactive truncated relaxin-3 peptide 26.52 Similar to the case of 25, treatment of 26 with 10 equiv. of DMP led to a sluggish peptide cleavage. However, the use of an excess amount of DMP (40 equiv.) significantly improved the cleavage efficiency, producing the desired two peptide fragments 26a and 26b in a conversion of 83% after 3 h (Fig. 4, entry 2). It is important to note that in the reactions of 25 and 26 a precipitate formation was observed. This precipitate has previously been assigned to acetoxyiodinane oxide, a product derived from the partial hydrolysis of DMP, which in our case results from the slow reaction between long peptide substrates and DMP. Precipitation of the hydrolysed DMP intermediate competes with the peptide cleavage, thus requiring an increased amount of the peptide-cleaving reagent.49 | ||
| Fig. 4 The DMP-mediated peptide cleavage of peptide 25–29. Reaction conditions: (a) 40 equiv. of DMP, 40 °C, DMSO/phosphate buffer at pH 7 (2:1, v/v), 3–6 h; (b) (i) 50 equiv. of acetic anhydride, 30 equiv. of pyridine, DMSO, RT, 2 h; (ii) 2 M hydroxylamine buffer (pH = 6), 4 h, RT. | ||
The same conditions were then applied to the cleavage of human β-amyloid (1–19) (27). The Fmoc-protected peptide 27 was successfully cleaved by DMP at the Gly–Tyr site within 3 h and the resulting two fragments 27a and 27b were readily observed by LC/MS analysis with a high conversion rate of 89% (Fig. 4, entry 3).
Furthermore, DMP-mediated cleavage of a 30-mer truncated glucagon-like peptide 1 (28)53 containing multiple serine residues in the sequence was also successful after acetylation of the peptide N-terminus, producing the two fragments 28a and 28b (Fig. 4, entry 4).
Finally, a naturally occurring peptide, PlnJ (29) isolated from plantaricin JK, was selected to demonstrate the applicability of our method for non-synthetic peptides.54 In this case, acetylation of the N-terminus amino group is also performed to successfully achieve the selective cleavage reaction. The per-acetylated derivative of peptide 29 was subjected to the DMP-mediated cleavage for 3 h. Two major species were identified by LC/MS analysis of the reaction mixture, corresponding to the expected C- and N-terminal peptide fragments (29a and 29b) (Fig. 4, entry 5).
DMP-mediated peptide cleavage of naturally occurring cyclic peptides
Cyclic peptides are an important class of natural products used in drug discovery and biochemical research. Conventionally, structural determination of cyclic peptides relies on NMR spectroscopy, which requires relatively large quantities of material.55 Furthermore, MS/MS of cyclic peptides can give complex fragmentation patterns, rendering structural analysis impractical.56 We therefore postulated that DMP-mediated cleavage of cyclic peptides would facilitate peptide sequencing by linearizing the cyclic peptide at a tyrosine site. This hypothesis was tested with three cyclic peptides. Due to the high conformational constraint of cyclic peptides, we anticipated that the cleavage of cyclic peptides would be slow relative to their linear counterparts, thus an increased amount of DMP (40 equiv.) was employed. Dichotomin J (30) is a vasodilating cyclic peptide isolated from Stellaria dichotoma.57 Treatment of 30 with DMP for 3 h successfully cleaved the cyclic peptide, affording the corresponding linear peptide 30a (yield 28%) bearing the TICA moiety at the N-terminus ([M + 2H2O + H]+ calcd/found 759.3196/759.3211) (Fig. 5a). The MS/MS spectrum of 30a revealed a simplified fragmentation pattern (Fig. 5a) in comparison with its cyclic counterpart 30 (Fig. S22†). The N-terminal TICA moiety remained intact throughout the MS/MS experiment and most of the b ions (in red) were readily identified in the MS/MS spectrum, which facilitated the rapid determination of the peptide sequence.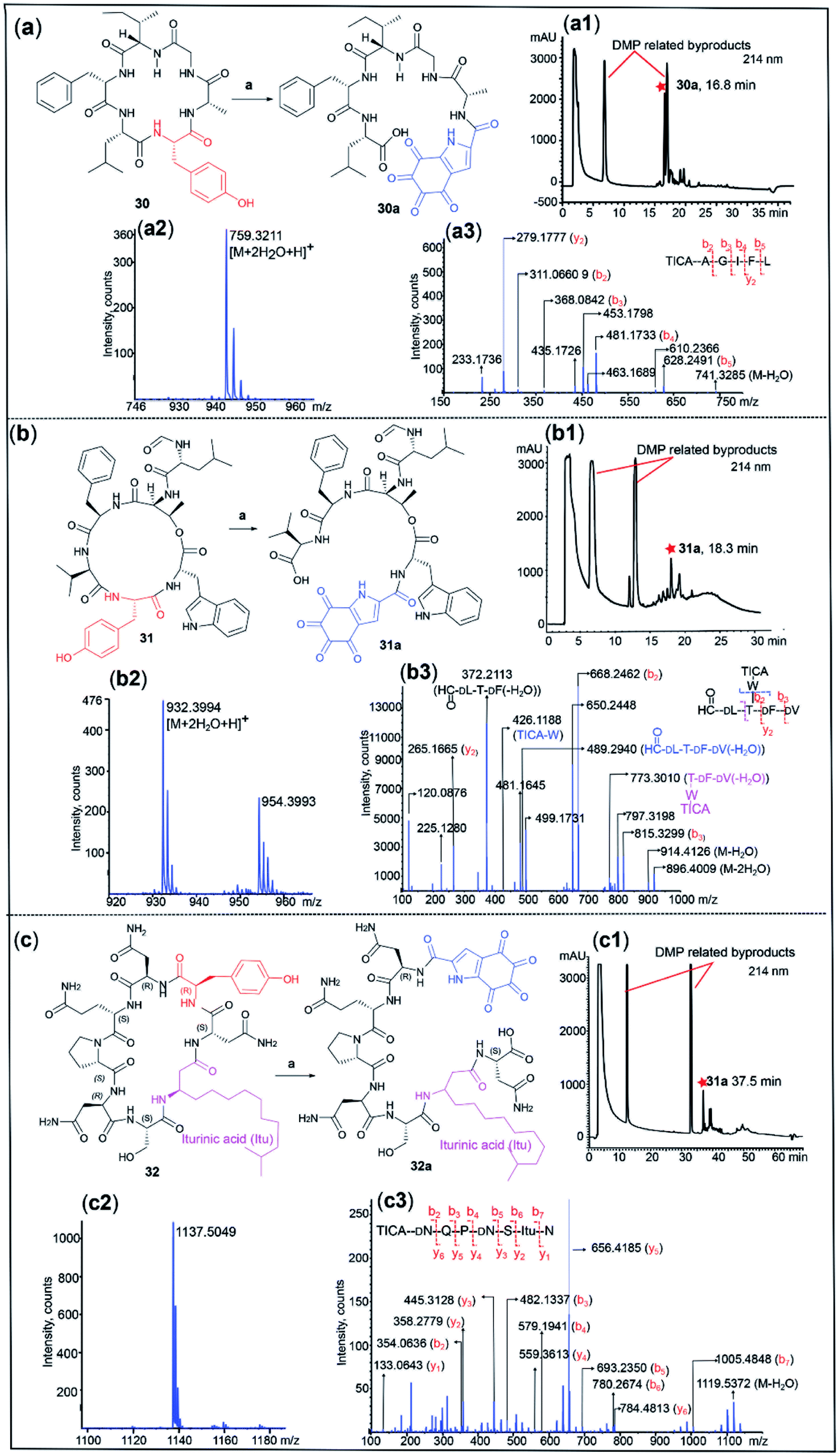 | ||
| Fig. 5 The DMP-mediated cleavage of cyclic peptides 30–32. (a) Cleavage of dichotomin J (30) at the tyrosine site: HPLC traces of the reaction between 30 and DMP (a1), MS characterization of 30a ([M + 2H2O + H]+ calcd/found 759.3196/759.3211) (a2) and the MSMS spectrum of 30a (a3); (b) cleavage of szentiamide (31) at the tyrosine site: HPLC traces of the reaction between 31 and DMP (b1), MS characterization of 31a ([M + 2H2O + H]+ calcd/found 932.3678/932.3994) (b2) and the MS/MS spectrum of 31a (b3); (c) cleavage of iturin a (32) at the tyrosine site: HPLC traces of the reaction between 32 and DMP (c1) and MS characterization of 32a ([M + 2H2O + H]+ calcd/found 1137.5059/1137.5049) (c2) and the MS/MS spectrum of 32a (c3). Reaction conditions: (a) 40 equiv. of DMP, 40 °C, DMSO/phosphate buffer at pH 7 (2:1, v/v), 3 h. | ||
The N-formylated cyclic depsipeptide szentiamide (31) from Xenorhabdus szentirmaii58 underwent DMP-mediated peptide cleavage at the tyrosine site within 3 h, generating the TICA-containing linear peptide 31a ([M + 2H2O + H]+ calcd/found 932.3678/932.3994) (Fig. 5b). Transformation of cyclic peptide 31 to linear 31a led to a significant reduction in the number of fragment ions generated in the MS/MS experiment, facilitating peptide sequencing (Fig. 5b and S23†). The MS/MS spectrum of 31a showed a series of b and y ions (in red), as well as several informative fragment ions. The fragment ion at 773.3010 represented the peptide C-terminal fragment after loss of formyl D-Leu and water (in pink) and the breakdown of the depsipeptide bond led to formation of the sidechain fragment ion TICA-Trp (m/z = 426.1188) and the tetrapeptide ion OHC-D-Leu–Thr–D-Phe–D-Val(-H2O) at 489.2940 (in blue). This result demonstrates the compatibility of our method with depsipeptide sequencing.
Finally, a commercially available sample of the antifungal cyclic lipopeptide iturin A59(32), incorporating a β-lipoamino acid (Fig. 5c), was also successfully linearized at the tyrosine site using DMP and the resulting major product was identified as the TICA-containing peptide 32a ([M + 2H2O + H]+ calcd/found 1137.5059/1137.5049). The MS/MS spectra of 32a showed all the expected b and y ions, enabling facile determination of the peptide sequence (Fig. 5c) while the MS/MS profile of the parent cyclic peptide 32 was more complex (Fig. S24†).
Conclusion
We herein report a novel tyrosine-selective peptide bond cleavage via DMP-mediated hyperoxidation of the tyrosine sidechain. This highly site-selective reaction was performed under mild conditions and was significantly faster than most of the existing peptide digestion methods. Extensive NMR and HRMS studies revealed that this cleavage reaction generates an oxidized C-terminal peptide fragment comprising an unprecedented TICA moiety, together with an unmodified N-terminal peptide fragment. High functional group tolerance to proteinogenic amino acids and several unnatural amino acids was demonstrated. The successful application of the current method to cleave a series of biorelevant oligopeptides with a protected N-terminus further demonstrated its broad substrate scope. Importantly, this method was also applicable to cleavage of cyclic peptides including depsipeptides and lipopeptides. The corresponding linear peptides generated from the cleavage contain an N-terminal TICA moiety, MS/MS analysis of which simplifies peptide sequencing of the corresponding cyclic peptides. These results together demonstrate that this novel DMP-mediated selective peptide cleavage at tyrosine can be used as a valuable complementary tool to enzymatic methods for the determination of the primary structure of a diverse range of peptides.Albeit outside the scope of this work, it is important to highlight that the presence of the four contiguous ketone functionalities in the TICA moiety appoint this moiety as a highly reactive electrophilic target for the selective bioconjugation or synthetic manipulation of TICA-peptides. Thus, protocols similar to aldehyde-mediated bioconjugation or the preparation of azine derivatives of interest in the development of organic electrodes or fluorescent materials can be proposed. The latter concept was demonstrated herein by the robust reaction of a model TICA-peptide with diaminomaleonitrile to afford a highly conjugated four-ring system at the peptide N-terminus.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
S. Z. and L. M. D. L. R. conceived and designed the experiments. S. Z. and F. F. L. performed the chemical synthesis. S. Z., L. M. D. L. R., F. F. L., R. H. and I. K. H. L. performed spectroscopic analysis. S. Z., L. M. D. L. R., F. F. L., P. W. R. H. and M. A. B. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for a PhD scholarship from the China Scholarship Council (S. Z.).Notes and references
- B. S. Berlett and E. R. Stadtman, J. Biol. Chem., 1997, 272, 20313–20316 CrossRef CAS PubMed.
- C. Houée-Lévin, K. Bobrowski, L. Horakova, B. Karademir, C. Schöneich, M. J. Davies and C. M. Spickett, Free Radical Res., 2015, 49, 347–373 CrossRef PubMed.
- G. Cohen, S. Yakushin and D. Dembiec-Cohen, Anal. Biochem., 1998, 263, 232–239 CrossRef CAS PubMed.
- S. Bartesaghi, J. Wenzel, M. Trujillo, M. López, J. Joseph, B. Kalyanaraman and R. Radi, Chem. Res. Toxicol., 2010, 23, 821–835 Search PubMed.
- M. J. Davies, J. Clin. Biochem. Nutr., 2010, 48, 8–19 Search PubMed.
- C. Giulivi, N. Traaseth and K. Davies, Amino Acids, 2003, 25, 227–232 CrossRef CAS PubMed.
- A. K. Chakraborty, J. T. Platt, K. K. Kim, B. Se Kwon, D. C. Bennett and J. M. Pawelek, Eur. J. Biochem., 1996, 236, 180–188 CrossRef CAS PubMed.
- D. Sulzer and L. Zecca, Neurotoxic. Res., 1999, 1, 181–195 CrossRef PubMed.
- S. N. Savvides, M. Scheiwein, C. C. Böhme, G. E. Arteel, P. A. Karplus, K. Becker and R. H. Schirmer, J. Biol. Chem., 2002, 277, 2779–2784 CrossRef CAS PubMed.
- S.-K. Kong, M. B. Yim, E. R. Stadtman and P. B. Chock, Proc. Natl. Acad. Sci., 1996, 93, 3377–3382 CrossRef CAS PubMed.
- L. MacMillan-Crow, J. Greendorfer, S. Vickers and J. Thompson, Arch. Biochem. Biophys., 2000, 377, 350–356 CrossRef CAS PubMed.
- C. Giulivi and K. J. Davies, J. Biol. Chem., 2001, 276, 24129–24136 CrossRef CAS PubMed.
- F. Leinisch, M. Mariotti, P. Hägglund and M. J. Davies, Free Radical Biol. Med., 2018, 126, 73–86 CrossRef CAS PubMed.
- T. DiMarco and C. Giulivi, Mass Spectrom. Rev., 2007, 26, 108–120 CrossRef CAS PubMed.
- J. J. Bruins, B. Albada and F. van Delft, Chem.–Eur. J., 2018, 24, 4749–4756 CrossRef CAS PubMed.
- J. P. Ortonne, Br. J. Dermatol., 2002, 146, 7–10 CrossRef CAS PubMed.
- L. A. Burzio and J. H. Waite, Biochemistry, 2000, 39, 11147–11153 CrossRef CAS PubMed.
- R. J. Martinie, P. I. Godakumbura, E. G. Porter, A. Divakaran, B. J. Burkhart, J. T. Wertz and D. E. Benson, Metallomics, 2012, 4, 1037–1042 CrossRef CAS PubMed.
- B. Liu, L. Burdine and T. Kodadek, J. Am. Chem. Soc., 2006, 128, 15228–15235 CrossRef CAS PubMed.
- M. Yu, J. Hwang and T. J. Deming, J. Am. Chem. Soc., 1999, 121, 5825–5826 CrossRef CAS.
- B. Sun, A. D. Ariawan, H. Warren, S. C. Goodchild, L. M. Ittner and A. D. Martin, J. Mater. Chem. B, 2020, 8, 3104–3112 RSC.
- A. M. Marmelstein, M. J. Lobba, C. S. Mogilevsky, J. C. Maza, D. D. Brauer and M. B. Francis, J. Am. Chem. Soc., 2020, 142, 5078–5086 CrossRef CAS PubMed.
- J. C. Maza, D. L. Bader, L. Xiao, A. M. Marmelstein, D. D. Brauer, A. M. ElSohly, M. J. Smith, S. W. Krska, C. A. Parish and M. B. Francis, J. Am. Chem. Soc., 2019, 141, 3885–3892 CrossRef CAS PubMed.
- J. J. Bruins, A. H. Westphal, B. Albada, K. Wagner, L. Bartels, H. Spits, W. J. van Berkel and F. L. van Delft, Bioconjugate Chem., 2017, 28, 1189–1193 CrossRef CAS PubMed.
- M. J. Long and L. Hedstrom, ChemBioChem, 2012, 13, 1818–1825 CrossRef CAS PubMed.
- G. L. Schmir, L. A. Cohen and B. Witkop, J. Am. Chem. Soc., 1959, 81, 2228–2233 CrossRef CAS.
- L. K. Ramachandran and B. Witkop, Methods Enzymol., 1967, 11, 283–299 CAS.
- A. Fontana, D. Dalzoppo, C. Grandi and M. Zambonin, in Methods in Protein Sequence Analysis, ed. M. Elzinga, Humana Press, Totowa, NJ, 1982, vol. 3, pp. 325–334 Search PubMed.
- R. L. Lundblad, Techniques in Protein Modification, CRC press, Boca Raton, Florida, 1994 Search PubMed.
- J. Prakash and J. J. Kodanko, Curr. Opin. Chem. Biol., 2013, 17, 197–203 CrossRef CAS PubMed.
- T. Y. Lee and J. Suh, Chem. Soc. Rev., 2009, 38, 1949–1957 RSC.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CrossRef CAS PubMed.
- B. T. Chait, Science, 2006, 314, 65–66 CrossRef CAS PubMed.
- L. Tsiatsiani and A. J. R. Heck, FEBS J., 2015, 282, 2612–2626 CrossRef CAS PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- P. K. Madala, J. D. A. Tyndall, T. Nall and D. P. Fairlie, Chem. Rev., 2010, 110, PR1–PR31 CrossRef CAS PubMed.
- V. Baeriswyl and C. Heinis, Protein Eng., Des. Sel., 2013, 26, 81–89 CrossRef CAS PubMed.
- H. E. Elashal and M. Raj, Chem. Commun., 2016, 52, 6304–6307 RSC.
- W. Mahoney, P. Smith and M. Hermodson, Biochemistry, 1981, 20, 443–448 CrossRef CAS PubMed.
- K. Tanabe, A. Taniguchi, T. Matsumoto, K. Oisaki, Y. Sohma and M. Kanai, Chem. Sci., 2014, 5, 2747–2753 RSC.
- Y. Seki, K. Tanabe, D. Sasaki, Y. Sohma, K. Oisaki and M. Kanai, Angew. Chem., 2014, 126, 6619–6623 CrossRef.
- R. Kaiser and L. Metzka, Anal. Biochem., 1999, 266, 1–8 CrossRef CAS PubMed.
- S. Zhang, L. M. De Leon Rodriguez, I. K. H. Leung, G. M. Cook, P. W. R. Harris and M. A. Brimble, Angew. Chem., Int. Ed., 2018, 57, 3631–3635 CrossRef CAS PubMed.
- S. Mahesh, K.-C. Tang and M. Raj, Molecules, 2018, 23, 2615 CrossRef PubMed.
- B. M. Bizzarri, C. Pieri, G. Botta, L. Arabuli, P. Mosesso, S. Cinelli, A. Schinoppi and R. Saladino, RSC Adv., 2015, 5, 60354–60364 RSC.
- R. Bernini, M. Barontini, F. Crisante, M. C. Ginnasi and R. Saladino, Tetrahedron Lett., 2009, 50, 6519–6521 CrossRef CAS.
- G. Prota, Melanins and Melanogenesis, Academic Press, San Diego, CA, 2012 Search PubMed.
- D. Ranganathan, N. K. Vaish and K. Shah, J. Am. Chem. Soc., 1994, 116, 6545–6557 CrossRef CAS.
- S. D. Meyer and S. L. Schreiber, J. Org. Chem., 1994, 59, 7549–7552 CrossRef CAS.
- A. Fontana, D. Dalzoppo, C. Grandi and M. Zambonin, Biochemistry, 1981, 20, 6997–7004 CrossRef CAS PubMed.
- E. Gross, in Methods Enzymol, Academic Press, 1967, vol. 11, pp. 238–255 Search PubMed.
- L. M. Haugaard-Kedström, F. Shabanpoor, M. A. Hossain, R. J. Clark, P. J. Ryan, D. J. Craik, A. L. Gundlach, J. D. Wade, R. A. D. Bathgate and K. J. Rosengren, J. Am. Chem. Soc., 2011, 133, 4965–4974 CrossRef PubMed.
- A. Evers, M. Bossart, S. Pfeiffer-Marek, R. Elvert, H. Schreuder, M. Kurz, S. Stengelin, M. Lorenz, A. Herling and A. Konkar, J. Med. Chem., 2018, 61, 5580–5593 CrossRef CAS PubMed.
- P. Rogne, C. Haugen, G. Fimland, J. Nissen-Meyer and P. E. Kristiansen, Peptides, 2009, 30, 1613–1621 CrossRef CAS PubMed.
- J. W.-H. Li and J. C. Vederas, Science, 2009, 325, 161–165 CrossRef PubMed.
- H. Mohimani, Y. L. Yang, W. T. Liu, P. W. Hsieh, P. C. Dorrestein and P. A. Pevzner, Proteomics, 2011, 11, 3642–3650 CrossRef CAS PubMed.
- H. Morita, T. Iizuka, C.-Y. Choo, K.-L. Chan, H. Itokawa and K. Takeya, J. Nat. Prod., 2005, 68, 1686–1688 CrossRef CAS PubMed.
- B. Ohlendorf, S. Simon, J. Wiese and J. F. Imhoff, Nat. Prod. Commun., 2011, 6, 1247–1250 CrossRef CAS PubMed.
- A. Isogai, S. Takayama, S. Murakoshi and A. Suzuki, Tetrahedron Lett., 1982, 23, 3065–3068 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06216f |
| This journal is © The Royal Society of Chemistry 2022 |