
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRed aqueous room-temperature phosphorescence modulated by anion–π and intermolecular electronic coupling interactions†
Fengbo
Liu
 a,
Hai
Yang
a,
Dongdong
Sun
a,
Fang
Gao
a,
Xiongzhi
Zhang
ab,
Zhiyong
Zhao
ab,
Xie
Han
ab and
Simin
Liu
*ab
a,
Hai
Yang
a,
Dongdong
Sun
a,
Fang
Gao
a,
Xiongzhi
Zhang
ab,
Zhiyong
Zhao
ab,
Xie
Han
ab and
Simin
Liu
*ab
aSchool of Chemistry and Chemical Engineering, Wuhan University of Science and Technology, Wuhan 430081, China. E-mail: liusimin@wust.edu.cn
bThe State Key Laboratory of Refractories and Metallurgy, Institute of Advanced Materials and Nanotechnology, Wuhan University of Science and Technology, Wuhan 430081, China
First published on 25th May 2022
Abstract
Aqueous room temperature phosphorescence (aRTP) from purely organic materials has been intriguing but challenging. In this article, we demonstrated that the red aRTP emission of 2Br–NDI, a water-soluble 4,9-dibromonaphthalene diimide derivative as a chloride salt, could be modulated by anion–π and intermolecular electronic coupling interactions in water. Specifically, the rarely reported stabilization of anion–π interactions in water between Cl− and the 2Br–NDI core was experimentally evidenced by an anion–π induced long-lived emission (λAnion–π) of 2Br–NDI, acting as a competitive decay pathway against the intrinsic red aRTP emission (λPhos) of 2Br–NDI. In the initial expectation of enhancing the aRTP of 2Br–NDI by inclusion complexation with macrocyclic cucurbit[n]urils (CB[n]s, n = 7, 8, 10), we surprisingly found that the exclusion complexation between CB[8] and 2Br–NDI unconventionally endowed the complex with the strongest and longest-lived aRTP due to the strong intermolecular electronic coupling between the nπ* orbit on the carbonyl rims of CB[8] and the ππ* orbit on 2Br–NDI in water. It is anticipated that these intriguing findings may inspire and expand the exploration of aqueous anion–π recognition and CB[n]-based aRTP materials.
Introduction
Red and near infrared aqueous room temperature phosphorescence (aRTP) from purely organic materials has been intriguing in bioimaging due to the large Stokes shift, better tissue penetration, lower background fluorescence and biotoxicity.1 In the last decade, research has flourished on various strategies to boost RTP emission from solid purely organic phosphors, including crystallization, “direct heavy-atom effect”, H- & J-aggregate, host–guest doping, carbon dots, polymerization, intermolecular electronic coupling, host–guest complexation, and charge transfer (CT) mediated intersystem crossing (ISC).2–8 However, aRTP from water-soluble materials remains challenging and rarely reported yet, due to the severe non-irradiative quenching of phosphors by water and oxygen.The inclusion of phosphors in host molecules, such as proteins,9 molecular capsules,10 cyclodextrins11,12 and cucurbit[n]urils (CB[n]s),13–15 provides a feasible method for the construction of RTP materials in solution by segregating and restricting phosphors in their cavities. In particular, CB[n]s have been attractive because of their rigid hydrophobic cavities, ISC-boosting carbonyl rims and moderate water solubility of their host–guest complexes. For examples, Liu and coworkers reported the complexation of bromophenyl-methyl-pyridinium derivatives with CB[7] and CB[8] for the construction of aRTP pseudorotaxanes, supramolecular pins and supramolecular polymers for targeted cell imaging.13,14 Ma and coworkers constructed a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 host–guest complex of CB[8] and a triazine derivative modified with two 4-(4-bromophenyl)pyridine branches.15
:2 host–guest complex of CB[8] and a triazine derivative modified with two 4-(4-bromophenyl)pyridine branches.15
Naphthalene diimide (NDI) derivatives have been widely used for the construction of various supramolecular architectures due to their rigid and electron-deficient aromatic structures.16,17 Substitution on the naphthalene core can realize wide-range emissions over the full rainbow.18 Therefore, NDI derivatives have been practical in semiconductors,19 anion sensing,20 metal–organic framework materials,21 and anion–π catalysts.22 Intriguingly, NDI derivatives are also potential red RTP phosphors due to their ISC-boosting carbonyl groups and low-lying 3(π–π*) states.23,24 For example, in 2018, red aRTP was achieved by George's group by restricting a water-soluble 4,9-dibromonaphthalene diimide derivative (2Br–NDI, I− salt) in water-soluble LAPONITE®.25 The hybrid revealed an aRTP lifetime of 347 μs at 613 nm under the ambient environment.
Inspired by the work from George's group and our previous research on supramolecular recognition between CB[n]s (n = 7, 8, 10) and non-brominated NDI in water,25,26 we herein investigated the effects of complexation with CB[n]s (n = 7, 8, 10) on the aRTP emission of 2Br–NDI (chloride salt), in the initial expectation of enhancing the aRTP of 2Br–NDI by restricting it in the cavities of CB[n]s. To begin with, we found that the intrinsic aRTP (λPhos) properties of 2Br–NDI was also affected by its supramolecular structures in water, transforming from non-emitting aggregates along the short axis to long-lived emitting (λAnion–π) anion–π recognition upon dilution with a critical concentration near 125 μM (Fig. 1). 1H NMR, ESI-MS, spectroscopies and density functional theory (DFT) simulation experimentally supported the synergistic stabilization of anion–π interactions in water by H-bonding and coulombic interaction between Cl− and the ammonium alkyl chains of 2Br–NDI, which has rarely been evidenced in halogen anion–π recognition due to the hydration effect by water.27–29 CB[n]s (n = 7, 8, 10) were used to interact with 2Br–NDI and modulate the aRTP emission of excited 2Br–NDI. Distinguished from the expected dumbbell-shaped complex of CB[7]2·(2Br–NDI) and inclusion complex of CB[10]·(2Br–NDI)2, the exclusion complex of CB[8]·(2Br–NDI) exhibited strong intermolecular electronic coupling interaction between the carbonyl rims of CB[8] and the 2Br–NDI core (Fig. 1), which has been an efficient strategy to promote RTP emission for solid purely organic materials30,31 but yet reported for aRTP. Intriguingly, the complexation with CB[8] unconventionally induced the strongest and longest-lived aRTP of 2Br–NDI (τPhos = 302 μs and ΦPhos = 7.37%, in N2) among cases researched. The anion–π induced long-lived emission (λAnion–π) was also cut off due to the competitive complexation of CB[8] with the ammonium alkyl chains of 2Br–NDI. Additionally, CB[8]·(2Br–NDI) exhibited more significant hypoxia responsiveness due to the exposure of 2Br–NDI and also left the cavity of CB[8] free of guest, which is inspiring for the construction of more functional aRTP materials in the future.
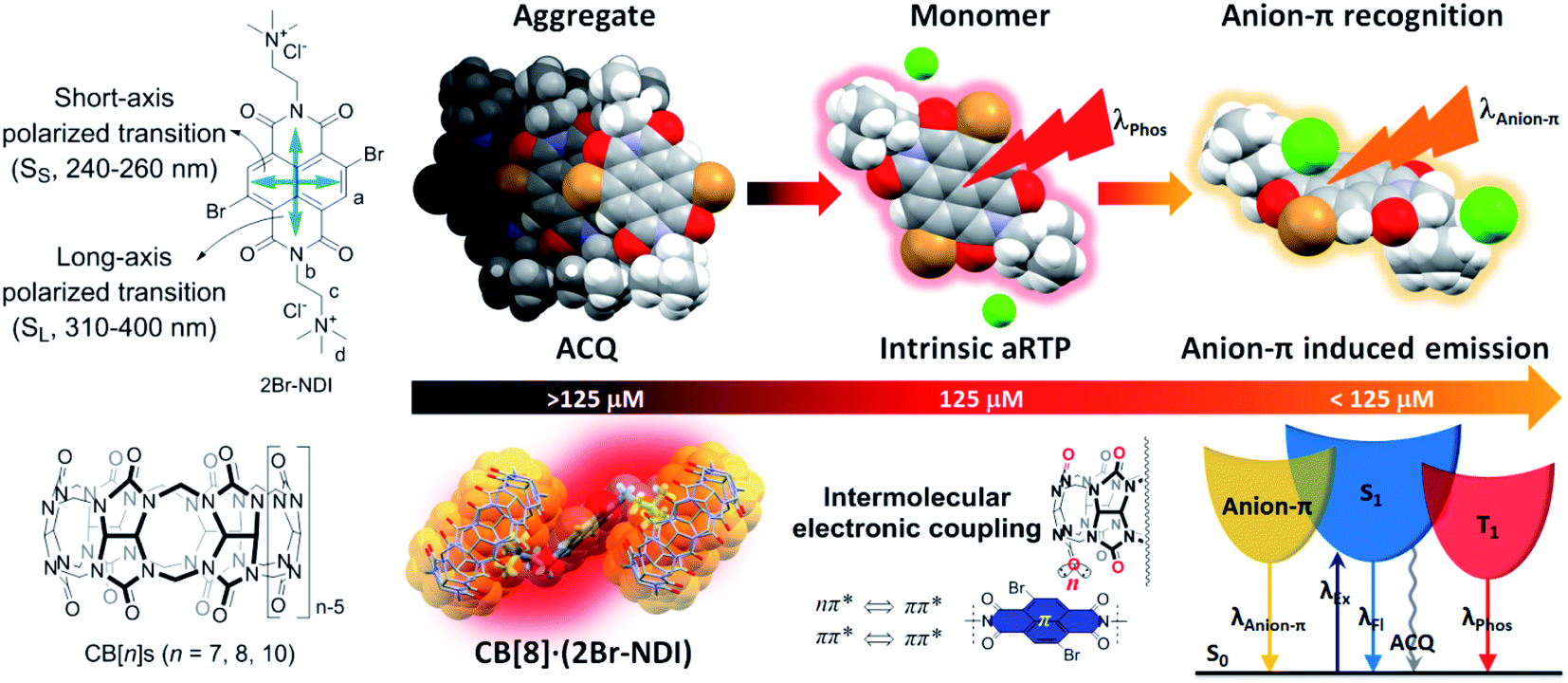 | ||
| Fig. 1 Structures of 2Br–NDI and CB[n]s and modulation of the aqueous room temperature phosphorescence of 2Br–NDI by anion–π and intermolecular electronic coupling interactions. | ||
Results and discussion
Anion–π modulated aRTP of 2Br–NDI
As shown in Fig. 2a and b, 2Br–NDI (330 μM) emitted fluorescence at 446 nm and phosphorescence in 600–800 nm, with peaks at 621.5 nm, 673.5 nm and 753.5 nm assigned to the vibrational energy levels of the 3(π–π*) state.25,32 Out of curiosity, the considerable aRTP in air and weak but detectable aRTP in O2 indicated moderate resistance of 2Br–NDI against quenching by water and O2. These remind us of the possible self-assembly of 2Br–NDI in water. | ||
| Fig. 2 (a) Steady (slitEx = 7.5 nm and slitEm = 15.0 nm) and (b) gated (slitEx = 4.0 nm, slitEm = 15.0 nm, τdelay = 60 μs, and τgate = 40 μs) emission of 2Br–NDI in air, and N2 and O2 ([2Br–NDI] = 0.33 mM and λEx = 385 nm). (c) Steady and (d) gated emission of 2Br–NDI in N2 upon dilution (λEx = 385 nm, slitEx = 6 nm, slitEm = 10 nm, τdelay = 60 μs, and τgate = 40 μs). (e) Concentration dependence of lifetime (λEx = 385 nm) at 583 nm vs. 621.5 nm of 2Br–NDI in N2. (f) DLS for 2Br–NDI upon dilution. | ||
To track the assembly behavior of 2Br–NDI in water, the concentration dependence (0.0039–8.0 mM) of steady and gated emission of 2Br–NDI was explored under N2 (Fig. 2c and d). It was surprisingly found that new long-lived emission at 583 nm showed up upon dilution to <125 μM. Both of the fluorescence and long-lived emission (583 nm & 621.5 nm) were enhanced at <125 μM and then decreased at >125 μM with concentration. The fluorescence hypsochromically shifted from 442.5 nm to 437 nm at <125 μM and then bathochromically shifted to 453 nm at >125 μM. The intensity at 583 nm and 621.5 nm were enhanced at <125 μM. However, upon further concentration, emission at 583 nm vanished along with the decreased emission at 621.5 nm. Therefore, transformation between two kinds of supramolecular states of 2Br–NDI in water could be speculated. Lifetime–concentration curves at 583 nm and λPhos = 621.5 nm (Fig. 2e and S4†) also unraveled transformation between two kinds of long-lived species with a critical concentration at 125 μM, showing monoexponential kinetics at lower concentrations while biexponential kinetics at higher concentrations.33
Dynamic light scattering (DLS) (Fig. 2f) was performed to verify the supramolecular states of 2Br–NDI at different concentrations. The results demonstrated the transformation from large-size (>100 nm) to small-size (<10 nm) assembly structures upon dilution. SEM (Fig. S5†) exhibited square lamella crystals after slow evaporation. The triclinic single crystal34 (Fig. S6, Tables S2–S7†) revealed layered stacking of 2Br–NDI with strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O···Br halogen bonding (3.0 Å) between adjacent coplanar 2Br–NDI, weak π–π overlaps (slipping angle = 32°) and weak inter-layer Br–π interaction (≈3.55 Å, the same as the sum of the van der Waals radius of Br and C). Considering the bathochromically shifted fluorescence at >125 μM and the LUMO/HOMO electron distribution of 2Br–NDI (Fig. S44b and e†), slipped aggregation along the short axis of 2Br–NDI with small π–π overlap could be responsible for the emission quenching of 2Br–NDI at high concentration. Meanwhile, the Cl− anion was positioned far away from the pyridinedione heterocycles, vanishing the anion–π induced long-lived emission (λAnion–π) at high concentration.
O···Br halogen bonding (3.0 Å) between adjacent coplanar 2Br–NDI, weak π–π overlaps (slipping angle = 32°) and weak inter-layer Br–π interaction (≈3.55 Å, the same as the sum of the van der Waals radius of Br and C). Considering the bathochromically shifted fluorescence at >125 μM and the LUMO/HOMO electron distribution of 2Br–NDI (Fig. S44b and e†), slipped aggregation along the short axis of 2Br–NDI with small π–π overlap could be responsible for the emission quenching of 2Br–NDI at high concentration. Meanwhile, the Cl− anion was positioned far away from the pyridinedione heterocycles, vanishing the anion–π induced long-lived emission (λAnion–π) at high concentration.
1H NMR (Fig. 3) of 2Br–NDI upon dilution from 8.0 mM to 3.9 μM revealed the switch from the aggregate state of 2Br–NDI into the anion–π recognition. Specifically, the resonance of aromatic protons (Ha) of 2Br–NDI underwent slight down-field shift upon decreasing the concentration from 8.0 mM to 125 μM, indicating the weakened aggregation of 2Br–NDI. At <125 μM, paired singlets at δ = 8.84 and δ = 8.09 were presented with a constant integral ratio of 1:1. This could be explained by the domination of anion–π interaction between Cl− and the electron-deficient 2Br–NDI core. The paired and up-field shifted singlets were formed due to the electron-shielding effect by one of the Cl− anions, which was preferentially bound to the pyridinedione heterocycle of 2Br–NDI as widely reported in DFT results for neutral core-disubstituted NDI derivatives.35,36 Additionally, chemical shift splitting and shifting of the ammonium alkyl chains (Hb, Hc & Hd) indicated the stabilization of the anion–π interaction by H-bonding and coulombic attraction, with down-field shift stemmed from Cl−-induced polarization while up-field shift from the shielding effect by the negative charge of Cl−.37 DFT simulation (Fig. S7†), with the independent gradient model (IGM) using Multiwfn software,38 supported that the anion–π interaction was synergistically stabilized by H-bonding and coulombic attraction between the ammonium alkyl chain and chloride anion with a binding energy of −37.30 kJ mol−1.39 The anion–π interaction was also evidenced by the presence of [2Br–NDI2+·Cl−]+ (m/z = 631.0) in the ESI-MS spectrum (Fig. S3†).
 | ||
| Fig. 3 1H NMR (600 MHz, D2O, and 298 K) of 2Br–NDI at (a) 23 mM, (b) 22 mM, (c) 21 mM, (d) 20 mM, (e) 2−1 mM, (f) 2−2 mM, (g) 2−3 mM, (h) 2−4 mM, (i) 2−5 mM, (j) 2−6 mM, (k) 2−7 mM and (l) 2−8 mM. | ||
To further evidence the existence of synergistically stabilized anion–π interaction in water and the anion–π induced long-lived λAnion–π emission of 2Br–NDI, excess sodium salts of various anions were added, respectively, into the aggregated 2Br–NDI solution. It was found that anions, such as F−, AcO−, HCO3− and HPO42−, could break the aggregation of 2Br–NDI in water and induced the appearance of 1:1 paired singlets in 1H NMR spectra (Fig. S9 and S10†) and long-lived λAnion–π emission from 550 nm to 600 nm (Fig. S11†). However, no characteristics for the anion–π interaction were observed upon addition of excess Cl− and Br− into the aggregated 2Br–NDI solution. The addition of excess I−, BF4− and PF4− resulted in the precipitation of 2Br–NDI.
In George's work,25 the emission at 580 nm was ascribed to the charge transfer emission driven by the anion–π interaction between I− and 2Br–NDI, but no long-lived emission was detected in gated emission of 1.0 mM 2Br–NDI under ambient conditions upon selective CT excitation. Therefore, the anion–π mediated long-lived emission was ruled out in their work. In our work, anion–π induced long-lived emission at 583 nm (λAnion–π) was observed at <125 μM in N2 (Fig. 2c–e) upon localized excitation (LE, λEx = 385 nm) of 2Br–NDI. To confirm the effect of the anion–π mediated CT process on this emission, steady and gated excitation spectra at 583 nm (Fig. S12†), and UV-Vis (Fig. S13†) and CT-excited emission spectra at λEx = 502 nm (Fig. S14†) of 125 μM 2Br–NDI were recorded. It revealed that no long-lived and oxygen-sensitive emission at 583 nm was recorded upon CT excitation, which was similar to the result in George's work for the I− salt. The temperature-dependent emission decay profile (Fig. S15†) from 0–60 °C for 125 μM 2Br–NDI showed that both λAnion–π and λPhos decreased as temperature increased due to the weakened anion–π interaction and enhanced non-irradiative decay of excited 2Br–NDI at high temperature.
Host–guest recognition between CB[n]s and 2Br–NDI
To further enhance the aRTP of 2Br–NDI, CB[n]s (n = 7, 8, 10) with different sizes of the rigid hydrophobic cavity were used to interact with 2Br–NDI in water, initially in expectation of reducing the molecular vibration of 2Br–NDI and protecting it from quenching by water and O2.As shown in Fig. S16,† the down-field shifted resonance of aromatic protons (Ha) and up-field shifted signals of ammonium alkyl chains (Hb, Hc, and Hd) suggested the dumbbell-shaped 2:1 complexation between CB[7] and 2Br–NDI.26,40 The binding underwent fast exchange on the 1H NMR time scale with a diffusion coefficient of 2.63 × 10−10 m2 s−1 (Fig. S17†).
1H NMR (Fig. S18 and S19†) demonstrated fast-exchanged kinetics for the complexation between CB[8] and 2Br–NDI, with a diffusion coefficient of 2.51 × 10−10 m2 s−1 (Fig. S20†). The down-field shifted signals of Ha, Hb and Hc and slightly up-field shifted Hd signal indicated that the 2Br–NDI core was outside but near the carbonyl portals of CB[8]. The doublet signal on CB[8] (δ = 5.83 and J = 15.2 Hz) was split into two broad signals with Δδ = 0.20 ppm (120 Hz), indicating asymmetrical surroundings on each carbonyl rim of CB[8]. ESI-MS (Fig. S21†) showed an exclusive m/z signal at 962.2 for the 1:1 stoichiometry between CB[8] and 2Br–NDI.
Monoclinic crystals for the CB[8]·(2Br–NDI) complex41 (Fig. 4 and Tables S8–S13†) showed that 2Br–NDI was externally bound to CB[8] in a linear polymer conformation. Crucially, the short distance of 2.859 Å between the carbonyl rims of CB[8] and the aromatic plane of 2Br–NDI indicated strong non-covalent interaction between them. Additionally, weak anion–π interaction was revealed with a distance of 3.658 Å between I− and 2Br–NDI (the sum of the van der Waals radius of iodide and carbon is 3.68 Å). CB[8] was gathered by the outer-surface H-bonding interaction (Fig. 4b). Inspiringly, the cavity of CB[8] is free of guest and occupied with six “high-energy” water molecules (Fig. 4c), suggesting that more fantastic and functional materials could be exploited based on this structure.
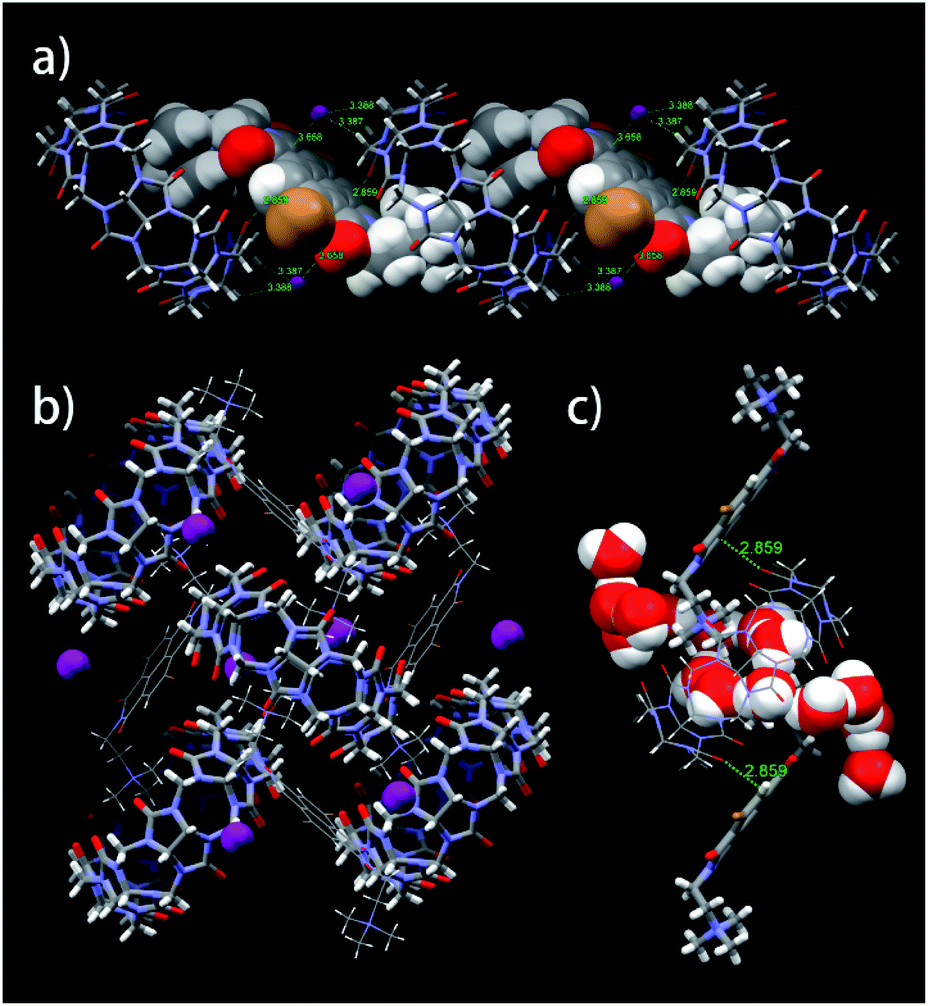 | ||
| Fig. 4 (a) Linear, (b) layered and (c) amplified views of single crystal of the CB[8]·(2Br–NDI) complex cultivated in the presence of KI. | ||
Similar to the non-brominated NDI,26 2Br–NDI exhibited slow-exchange binding with CB[10] in a host:guest ratio of 1:2 (Fig. S22 and S23†). The split and up-field shifted signals for aromatic protons Ha indicated two different surroundings of the 2Br–NDI partners in CB[10] and the integral ratio of Ha′:Ha′′ ≈ 1:3 in 1H NMR could be due to the slipping stacking of the 2Br–NDI homo-dimer along the central axis of CB[10] (conformation-2 in Fig. S24†). Signals for the methylene protons on 2Br–NDI were also split into two sets of multiplets. It could be concluded that Hb′ and Hb′′ were positioned near the cavity, while Hc′ and Hc′′ were positioned near the carbonyl rims of CB[10]. Meanwhile, the resonance of methyl protons was down-field shifted and split into two singlets, revealing their position near the carbonyl rims with different surroundings. For CB[10], the doublet at δ = 5.65 was split into two sets of doublets with coupling constants of 14.88 Hz and 14.82 Hz, respectively, indicating two different surroundings on CB[10] due to the asymmetric arrangement of the 2Br–NDI dimmer in CB[10].42 The DOSY spectrum (Fig. S25†) for CB[10]·(2Br–NDI)2 revealed a diffusion coefficient of 2.14 × 10−10 m2 s−1. ESI-MS (Fig. S26a†) showed a main peak at m/z = 713.6, corresponding to the 1:2 stoichiometry between CB[10] and 2Br–NDI. Interestingly, m/z at 963.2 (Fig. S26b†) supported the existence of [CB[10]·(2Br–NDI2+)2·(Cl−)]3+ and indicated the stabilization of Cl− by the CB[10]·(2Br–NDI)2 complex.
COSY (Fig. S27†) and ROESY (Fig. S28†) spectra were recorded to unravel the spatial correlation of the 2Br–NDI dimer in CB[10]. As shown in Fig. S27,† no scalar coupling was observed between Ha′ and Ha′′. As for the methylene groups, Hb′ and Hc′ were ascribed to the covalently linked methylene groups on two of the ammonium alkyl chains while Hb′′ and Hc′′ were ascribed to the covalently linked methylene groups on the other two chains. By contrast, weaker correlation intensity was observed for Hb′–Hc′′ and Hb′′–Hc′, indicating an intermolecular correlation between them. The ROESY spectrum (Fig. S28†) revealed intermolecular NOE effects for Hb′–Hb′′, Hc′–Hc′′ and interactions between methyl and methylene groups on 2Br–NDI partners. This suggested the proximity of neighboring ammonium alkyl groups. Positive cross peaks for Ha′–Ha′′, Hc′–Hc′′ and Hd′–Hd′′ indicated chemical exchange between the 2Br–NDI partners. Exchange spectroscopy (EXSY) (Fig. S29†) unravelled the slow exchange equilibrium with an apparent exchange equilibrium constant (Kapp) of 1/2.65, consistent with the 1H NMR integral (Fig. S22†). Therefore, the 2Br–NDI homo-dimer undergoes dynamic and slow conformation interconversion in CB[10].
CB[n]-modulated aRTP of 2Br–NDI
With these complexation behaviors in mind, the optical properties of 2Br–NDI in the presence of CB[n]s (n = 7, 8, 10) were researched. As shown in Fig. S30a,† complexing with CB[7] enhanced the absorption for the long-axis polarized transition of 2Br–NDI (SL, 310–400 nm),43 suggesting the segregation of 2Br–NDI in water. By contrast, upon addition of CB[8] (Fig. S30b†), the absorption for long-axis polarized transition of 2Br–NDI was enhanced with a bathochromic shift by 6–8 nm, as a result of dispersion of 2Br–NDI by CB[8] and hyperchromic effect from the carbonyl rims of CB[8] as an auxochrome. As for CB[10]·(2Br–NDI)2 (Fig. S30c†), both the absorption for short- (SS, 240–260 nm) and long-axis polarized transition of 2Br–NDI were weakened with a bathochromic shift of up to 7 nm, indicating enhanced J-aggregate of 2Br–NDI along its long axis in CB[10].44 DLS (Fig. S30d†) revealed that complexation with CB[n]s (n = 7, 8, 10) inhibited the aggregation of 2Br–NDI in water.As shown in steady and gated emission spectra (Fig. S31–S34†), the addition of CB[n]s (n = 7, 8, 10) decreased the fluorescence, while enhancing the phosphorescence of 2Br–NDI in both air and N2. By contrast to the free 2Br–NDI and CB[7]2·(2Br–NDI) complex, much stronger phosphorescence was observed for the CB[8]·(2Br–NDI) and CB[10]·(2Br–NDI)2 complexes. However, aRTP intensity for all the complexes of 2Br–NDI with CB[n]s (n = 7, 8, 10) revealed concentration dependence similar to free 2Br–NDI and was finally quenched at high concentration (Fig. S33†).
Statistical data (Fig. 5) unraveled the synergistic effects of self-assembly and CB[n]-based host–guest complexation of 2Br–NDI on its long-lived emission at 583 and 621.5 nm. As shown in Fig. 5a and d, anion–π induced long-lived emission at 583 nm was suppressed upon addition of CB[n]s (n = 7, 8, 10), consistent with the weakened anion–π interaction between Cl− and 2Br–NDI. In particular, the intensity at 583 nm was almost negligible for CB[8]·(2Br–NDI) (Fig. S32e and f†). In combination with the single crystal structure (Fig. 4a), it could be concluded that CB[8] acted as a strong competitor and inhibited the anion–π interaction between Cl− and 2Br–NDI in water by breaking the synergistic non-covalent interactions (H-bonding and coulombic attraction) between them. However, λAnion–π emission from the anion–π recognition was still observable for CB[7]2·(2Br–NDI) (Fig. S32c and d†) and CB[10]·(2Br–NDI)2 (Fig. S32g and h†) complexes, due to the exposure of the 2Br–NDI core for CB[7]2·(2Br–NDI) and the proximity of Cl− to the 2Br–NDI core for CB[10]·(2Br–NDI)2. aRTP emission at 621.5 nm in air (Fig. 5b and e) and N2 (Fig. 5c and f) showed that CB[8]·(2Br–NDI) featured more obvious hypoxia responsiveness than CB[10]·(2Br–NDI)2 due to the exposure of the 2Br–NDI core in the former complex. At concentrations ≤250 μM and in a N2 environment, CB[8]·(2Br–NDI) exhibited superior aRTP intensity at 621.5 nm to CB[7]2·(2Br–NDI) and CB[10]·(2Br–NDI)2, highlighting the significance of intermolecular interaction between the 2Br–NDI core and carbonyl rims of CB[8] in water. A higher concentration weakened the aRTP of CB[8]·(2Br–NDI) to slightly less than that of CB[10]·(2Br–NDI)2. Therefore, CB[10]·(2Br–NDI)2 possessed a higher binding constant than CB[8]·(2Br–NDI) and could resist more against the ACQ effect of 2Br–NDI at high concentrations. On the other hand, CB[10]·(2Br–NDI)2 exhibited excellent O2 resistance for efficient aRTP emission, even in an O2 environment (Fig. S34†).
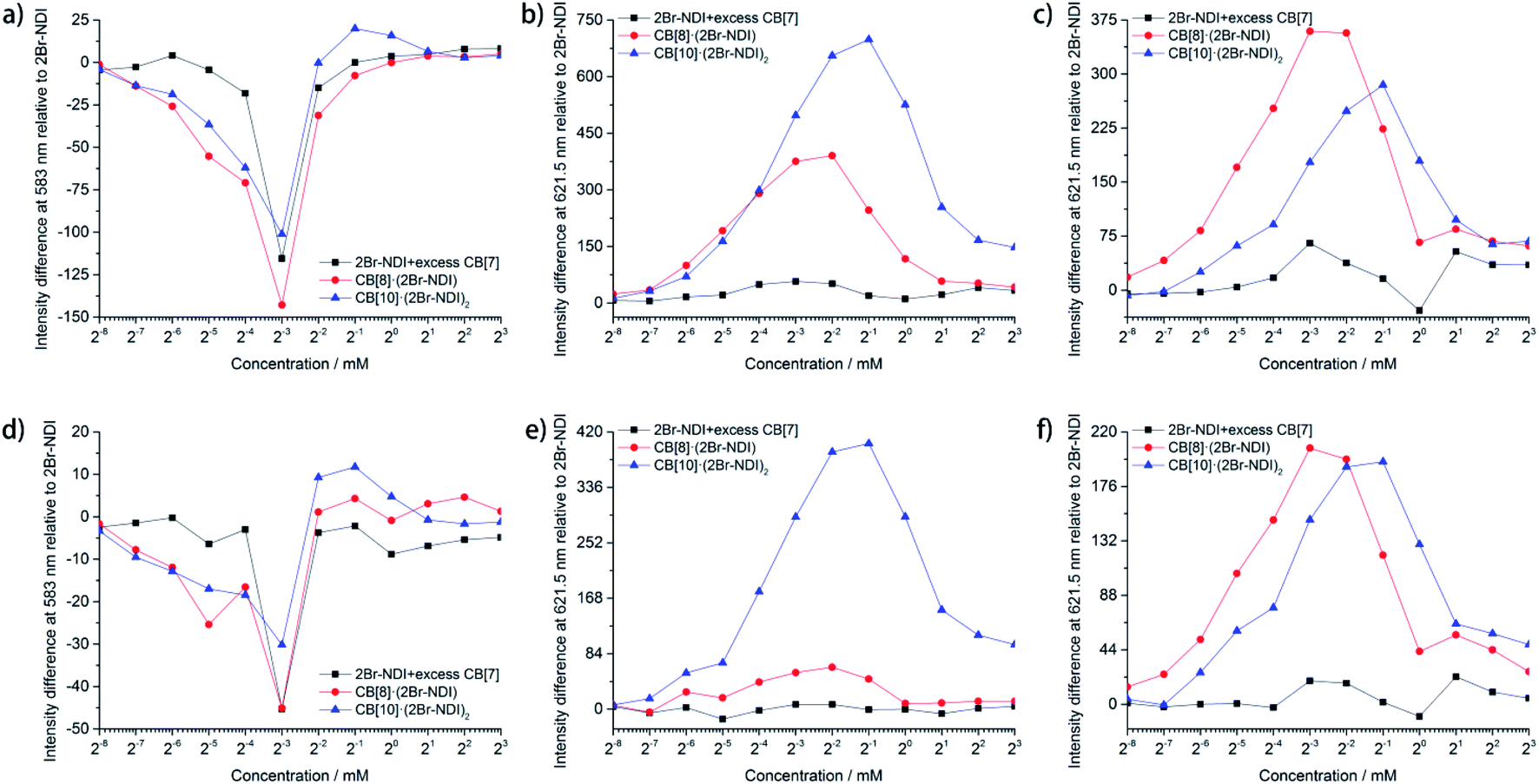 | ||
| Fig. 5 Statistical data for: the intensity difference of the steady (a) and gated (d) emission of 2Br–NDI at 583 nm in N2 upon complexation with CB[n]s (n = 7, 8, 10) at various concentrations of 2Br–NDI; the intensity difference of the steady (b and c) and gated (e and f) emission of 2Br–NDI at 621.5 nm in air (b and e) and N2 (c and f) upon complexation with CB[n]s (n = 7, 8, 10) at various concentrations of 2Br–NDI (measurements in air: slitEx = 3.5 nm and slitEm = 12.5 nm. Measurements in N2: slitEx = 6.0 nm, slitEm = 10 nm, τdelay = 0.06 ms, and τgate = 0.04 ms). | ||
Time-resolved phosphorescence measurements (Fig. 6a and b) revealed that complexation with CB[n]s (n = 7, 8, 10) prolonged the phosphorescent lifetime of 2Br–NDI at 621.5 nm. In particular, CB[10]·(2Br–NDI)2 exhibited the longest aRTP lifetime of up to 129 μs in air while CB[8]·(2Br–NDI) outstood with a longer phosphorescent lifetime of up to 302 μs in N2. Quantum yield determination (Fig. 6c, S35–S42†) in N2 showed that the phosphorescent quantum yield of 2Br–NDI increased up to 2.66% and 7.37% upon complexation with CB[10] and CB[8], respectively. These results were consistent with the results that CB[10]·(2Br–NDI)2 exhibits excellent resistance against O2 quenching, while CB[8]·(2Br–NDI) shows more efficient ISC to the triplet. The red aRTP emission of 2Br–NDI could be lightened up and observed by the naked eye upon complexation with CB[8] and CB[10] (Fig. 6d). However, negligible luminescent quantum yields were recorded for CB[7]2·(2Br–NDI).
 | ||
| Fig. 6 Phosphorescence lifetime of 2Br–NDI in the absence and presence of CB[n]s (n = 7, 8, 10) in (a) air and (b) N2 at λEm = 621.5 nm (λEx = 385 nm). (c) Fluorescence and phosphorescence quantum yields of 2Br–NDI in the absence and presence of CB[n]s (n = 7, 8, 10) in N2. (d) Photograph of 125 μM 2Br–NDI (from left to right) in the absence and presence of excess CB[7], 1.0 eq. CB[8] and 0.5 eq. CB[10] after N2 bubbling for 10 min. Gated emission–excitation spectra of 2Br–NDI in the (e) absence and presence of (f) excess CB[7], (g) 1.0 eq. CB[8], and (h) 0.5 eq. CB[10] in N2. | ||
To find out the synergistic effects of anion–π interaction and host–guest interaction with CB[n]s (n = 7, 8, 10) on the transition pathways of excited 2Br–NDI, steady and gated emission–excitation spectra (Fig. S43† and 6e–h) were recorded in N2. As shown in Fig. S43a† and 6e, upon long-axis polarized transition (S0 → SL, λEx = 310–400 nm), 2Br–NDI exhibited fluorescence (SL → S0, λEm = 400–450 nm) along with the anion–π induced long-lived emission (λAnion–π = 540–600 nm) and the intrinsic aRTP emission (T1 → S0, λPhos = 600–700 nm) of 2Br–NDI. The excitation of short-axis polarized transition (SS) by λEx = 240–260 nm gave mainly emission at λAnion–π and λPhos of 2Br–NDI, indicating a competitive relationship between emissions at λAnion–π and λPhos of 2Br–NDI.
Complexation with CB[n]s (n = 7, 8, 10) led to the bathochromic shift of the excitation wavelength for red aRTP emission. For CB[7]2·(2Br–NDI) (Fig. S43b† and 6f), a strong (Em and Ex) signal for the short-lived 1CT emission (500–680 nm and 440–560 nm) was observed due to the exposure of the 2Br–NDI core to Cl−. The excitation of short-axis polarized transition (λEx = 240–260 nm) also arouse intense emission from the short-lived 1CT state, indicating promoted formation of the 1CT state upon short-axis polarized transition for the dumbbell-shaped CB[7]2·(2Br–NDI) complex. As for CB[8]·(2Br–NDI) (Fig. S43c† and 6g) and CB[10]·(2Br–NDI)2 (Fig. S43d† and 6h), the anion–π interaction between Cl− and 2Br–NDI was inhibited due to the steric hindrance and competitive complexation of 2Br–NDI by CB[8] and CB[10]. As a result, both of the prompt 1CT and long-lived λAnion–π emissions were cut off and the long-lived emission was narrowed to mainly form the aRTP emission of 2Br–NDI. Interestingly, for CB[10]·(2Br–NDI)2, negligible emission was produced upon excitation of short-axis polarized transition (SS).
Theoretical calculations and cyclic voltammograms
DFT (Fig. 7 and S44†) and time-dependent DFT (TDDFT, Tables S14–S19†), based on the single crystal of CB[8]·(2Br–NDI), were carried out to have a deeper insight into the intermolecular interaction between CB[8] and 2Br–NDI and its effect on the optical properties of 2Br–NDI. As shown in the electrostatic potential (ESP) maps (Fig. 7a and b), CB[8] exhibited electron-rich carbonyl rims, while 2Br–NDI showed an electron-deficient core. This complementary donor–acceptor structure lays a foundation for the intermolecular electronic coupling interaction between CB[8] and 2Br–NDI. IGM analysis (Fig. 7c) demonstrated that the intermolecular electronic coupling interaction between CB[8] and 2Br–NDI was further locked by the characteristic ion–dipole interaction between the carbonyl rims of CB[8] and the cationic ammonium alkyl chains of 2Br–NDI, making it possible to realize intermolecular electronic coupling interaction even in water. Orbitals analyses for CB[8]·(2Br–NDI) revealed that the LUMO orbit of the complex was located on the 2Br–NDI core (Fig. 7d), while the HOMO orbit was located on the carbonyl rims of CB[8] (Fig. 7e). Additionally, the HOMO level of CB[8] was higher than the LUMO level of 2Br–NDI (ΔE = 2.27 eV, Fig. S44†), theoretically indicating thermal electron transfer from CB[8] to 2Br–NDI.36 According to perturbation theory,6,45 the rate for the ISC process is determined by both the spin–orbit coupling matrix element (SOCME, 〈1Ψ|ĤSOC|3Ψ〉) and the energy gap (ΔEST) between singlet and triplet states.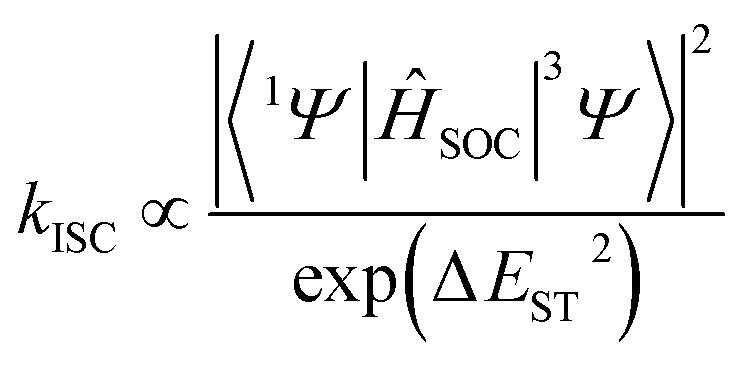
 | ||
| Fig. 7 Electrostatic potential of (a) CB[8] and (b) 2Br–NDI; (c) intermolecular interactions between CB[8] and 2Br–NDI; (d) LUMO and (e) HOMO energy levels of CB[8]·(2Br–NDI); singlet and triplet energy levels of (f) 2Br–NDI and (g) CB[8]·(2Br–NDI). | ||
Complexation with CB[8] generally reduced the SOCME of 2Br–NDI (Table S16†) between Sn and Tn. However, the strong intermolecular electronic coupling interaction between CB[8] and 2Br–NDI gathered energy levels of both high-lying singlets and triplets and opened up many more ISC pathways with ΔEST ≈ 0 (Fig. 7f and g and Tables S17 and S18†). Rates for ISC from high-lying singlets to T2 of 2Br–NDI were dramatically promoted (S5 → T2, S6 → T2, S8 → T2, S9 → T2, and S10 → T2 in Table S19†). Comprehensively, the almost eliminated ΔEST and the accelerated ISC from high-lying singlets to triplets ultimately strengthened the aRTP emission of CB[8]·(2Br–NDI).
Cyclic voltammograms were recorded to evidence the intermolecular electronic coupling interaction between CB[8] and 2Br–NDI in water. As shown in Fig. 8, complexation with CB[n]s (n = 7, 8, 10) induced a negative shift of the first reduction peak, while it induced a positive shift for the second reduction peak of 2Br–NDI. In particular, the CB[8]·(2Br–NDI) complex showed the most negative first reduction peak with a shift of up to −0.33 V related to that of free 2Br–NDI, supporting the strong intermolecular electronic coupling interaction between the carbonyl rims of CB[8] and the electron-deficient core of 2Br–NDI in water.
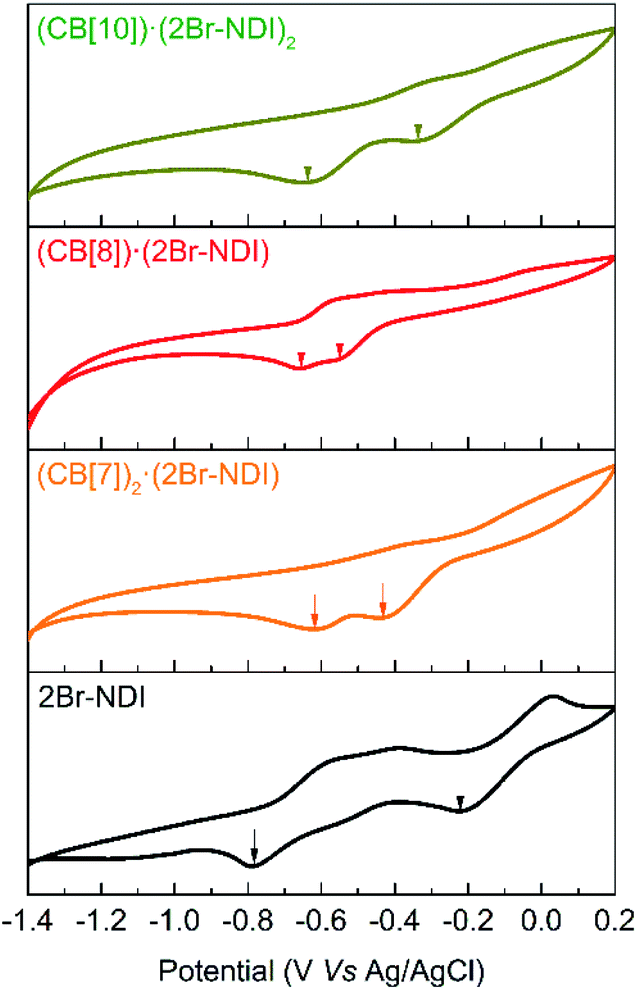 | ||
| Fig. 8 Cyclic voltammograms of 0.5 mM 2Br–NDI in the presence of excess CB[7], 1.0 eq. CB[8] and 0.5 eq. CB[10]. | ||
Conclusions
In conclusion, the aRTP emission of 2Br–NDI was tuned by its supramolecular structures in water. Significantly, the anion–π induced long-lived λAnion–π emission of 2Br–NDI in water was experimentally evidenced at low concentration. Relatively, high concentration quenched the aRTP emission of 2Br–NDI due to the slipped aggregation along the short axis. Complexation with CB[n]s (n = 7, 8, 10) weakened the anion–π interaction between Cl− and 2Br–NDI and modulated decay pathways of excited 2Br–NDI. Among them, the lightened-up aRTP of CB[10]·(2Br–NDI)2 with excellent O2 and ACQ resistance makes it potential in applications under ambient and extreme environments. Inspiringly, the exclusion complex between CB[8] and 2Br–NDI emitted the strongest red aRTP with obvious hypoxia responsiveness due to the strong intermolecular electronic coupling between the ISC-boosting carbonyl rims of CB[8] and the 2Br–NDI core, which provides a new strategy for designing novel aRTP materials based on exclusion complexation between CB[n]s family and electron-deficient RTP phosphors. What's more, in combination with the attractive research on CB[8]-based supramolecular materials,46,47 the cavity-free exclusion complex of CB[8]·(2Br–NDI) foreshadows more fantastic aRTP materials with stimuli-responsiveness in the future.Data availability
The crystalline data of two compounds in this article are available free of charge from the joint Cambridge Crystallographic Data Centre.Author contributions
F. L. and S. L. were responsible for the overall project concept. F. L. carried out experiments and manuscript writing. H. Y. helped with the observation by SEM. F. G. helped with the characterizations and analyses of 2D NMR. D. S., X. Z., Z. Z, X. H. and S. L. contributed to data analysis, results discussion and comments on the manuscript. S. L. as corresponding author was responsible for the funding acquisition and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21871216).Notes and references
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS.
- Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed.
- J. Yang, M. Fang and Z. Li, Acc. Mater. Res., 2021, 2, 644–654 CrossRef CAS.
- Z. Wu, J. Nitsch and T. B. Marder, Adv. Opt. Mater., 2021, 9, 2100411 CrossRef CAS.
- Q. Peng, H. Ma and Z. Shuai, Acc. Chem. Res., 2021, 54, 940–949 CrossRef CAS PubMed.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- S. Xu, R. Chen, C. Zheng and W. Huang, Adv. Mater., 2016, 28, 9920–9940 CrossRef CAS PubMed.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- M. L. Saviotti and W. C. Galley, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 4154–4158 CrossRef CAS PubMed.
- M. R. Ams, D. Ajami, S. L. Craig, J.-S. Yang and J. Rebek Jr, J. Am. Chem. Soc., 2009, 131, 13190–13191 CrossRef CAS PubMed.
- H. Chen, X. Ma, S. Wu and H. Tian, Angew. Chem., Int. Ed., 2014, 53, 14149–14152 CrossRef CAS PubMed.
- J.-J. Li, H.-Y. Zhang, Y. Zhang, W.-L. Zhou and Y. Liu, Adv. Opt. Mater., 2019, 7, 1900589 CrossRef CAS.
- H.-J. Yu, Q. Zhou, X. Dai, F.-F. Shen, Y.-M. Zhang, X. Xu and Y. Liu, J. Am. Chem. Soc., 2021, 143, 13887–13894 CrossRef CAS.
- X. K. Ma and Y. Liu, Acc. Chem. Res., 2021, 54, 3403–3414 CrossRef CAS PubMed.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
- M. Al Kobaisi, S. V. Bhosale, K. Latham, A. M. Raynor and S. V. Bhosale, Chem. Rev., 2016, 116, 11685–11796 CrossRef.
- S. V. Bhosale, M. Al Kobaisi, R. W. Jadhav, P. P. Morajkar, L. A. Jones and S. George, Chem. Soc. Rev., 2021, 50, 9845–9998 RSC.
- N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225–4237 RSC.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- V. A. Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef.
- M. Pan, X.-M. Lin, G.-B. Li and C.-Y. Su, Coord. Chem. Rev., 2011, 255, 1921–1936 CrossRef CAS.
- Y. Zhao, Y. Cotelle, L. Liu, J. López-Andarias, A.-B. Bornhof, M. Akamatsu, N. Sakai and S. Matile, Acc. Chem. Res., 2018, 51, 2255–2263 CrossRef CAS PubMed.
- A. Aster, C. Rumble, A.-B. Bornhof, H.-H. Huang, N. Sakai, T. Šolomek, S. Matile and E. Vauthey, Chem. Sci., 2021, 12, 4908–4915 RSC.
- S. Kuila, A. Ghorai, P. K. Samanta, R. B. K. Siram, S. K. Pati, K. S. Narayan and S. J. George, Chem.–Eur. J., 2019, 25, 16007–16011 CrossRef CAS.
- S. Kuila, K. V. Rao, S. Garain, P. K. Samanta, S. Das, S. K. Pati, M. Eswaramoorthy and S. J. George, Angew. Chem., Int. Ed., 2018, 57, 17115–17119 CrossRef CAS.
- X. Zhao, F. Liu, Z. Zhao, H. Karoui, D. Bardelang, O. Ouari and S. Liu, Org. Biomol. Chem., 2018, 16, 3809–3815 RSC.
- N. H. Evans and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11716–11754 CrossRef CAS.
- P. Gamez, T. J. Mooibroek, S. J. Teat and J. Reedijk, Acc. Chem. Res., 2007, 40, 435–444 CrossRef CAS.
- D.-X. Wang and M.-X. Wang, Acc. Chem. Res., 2020, 53, 1364–1380 CrossRef CAS.
- Z. Yang, Z. Mao, X. Zhang, D. Ou, Y. Mu, Y. Zhang, C. Zhao, S. Liu, Z. Chi, J. Xu, Y.-C. Wu, P.-Y. Lu, A. Lien and M. R. Bryce, Angew. Chem., Int. Ed., 2016, 55, 2181–2185 CrossRef CAS PubMed.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- F. Liao, J. Du, X. Nie, Z. Wu, H. Su, W. Huang, T. Wang, B. Chen, J. Jiang, X. Zhang and G. Zhang, Dyes Pigm., 2021, 193, 109505 CrossRef CAS.
- V. Martínez-Martínez, R. Sola Llano, S. Furukawa, Y. Takashima, I. López Arbeloa and S. Kitagawa, ChemPhysChem, 2014, 15, 2517–2521 CrossRef.
- Deposition number 2107788 contain the supplementary crystallographic data for this paper.†.
- R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533–538 CrossRef CAS.
- S. Guha, F. S. Goodson, L. J. Corson and S. Saha, J. Am. Chem. Soc., 2012, 134, 13679–13691 CrossRef CAS.
- R. Alberto, G. Bergamaschi, H. Braband, T. Fox and V. Amendola, Angew. Chem., Int. Ed., 2012, 51, 9772–9776 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS.
- Q. Song, F. Li, Z. Wang and X. Zhang, Chem. Sci., 2015, 6, 3342–3346 RSC.
- Deposition number 2100496 contain the supplementary crystallographic data for this paper.†.
- F. B. Liu, S. Chowdhury, R. Rosas, V. Monnier, L. Charles, H. Karoui, D. Gigmes, O. Ouari, F. Chevallier, C. Bucher, A. Kermagoret, S. M. Liu and D. Bardelang, Org. Lett., 2021, 23, 5283–5287 CrossRef CAS PubMed.
- J. Gawronski, M. Brzostowska, K. Kacprzak, H. Kolbon and P. Skowronek, Chirality, 2000, 12, 263–268 CrossRef CAS PubMed.
- H. Shao, T. Nguyen, N. C. Romano, D. A. Modarelli and J. R. Parquette, J. Am. Chem. Soc., 2009, 131, 16374–16376 CrossRef CAS PubMed.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS PubMed.
- E. Pazos, P. Novo, C. Peinador, A. E. Kaifer and M. D. Garcia, Angew. Chem., Int. Ed., 2019, 58, 403–416 CrossRef CAS PubMed.
- J. Liu, Y. Lan, Z. Yu, C. S. Tan, R. M. Parker, C. Abell and O. A. Scherman, Acc. Chem. Res., 2017, 50, 208–217 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of syntheses, ESI-MS, 1H NMR spectra, spectral characterization, crystalline data, SEM, cyclic voltammograms and theoretical calculations. CCDC 2100496 and 2107788. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d1sc06503c |
| This journal is © The Royal Society of Chemistry 2022 |