
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetic trapping of a cobalt(II) metallocage using a carbazole-containing expanded carbaporphyrinoid ligand†
Weinan
Zhou
ab,
Tridib
Sarma
 c,
Yonghuan
Su
b,
Chuanhu
Lei
*b and
Jonathan L.
Sessler
*d
c,
Yonghuan
Su
b,
Chuanhu
Lei
*b and
Jonathan L.
Sessler
*d
aSchool of Materials Science and Engineering, Shanghai University, Shanghai 200444, China
bCenter for Supramolecular Chemistry and Catalysis and Department of Chemistry, College of Science, Shanghai University, Shanghai 200444, China. E-mail: chlei@shu.edu.cn
cDepartment of Chemistry, Cotton University, Guwahati 781001, Assam, India
dDepartment of Chemistry, The University of Texas at Austin, 105 East 24th Street, Stop A5300, Austin, Texas 78712-1224, USA. E-mail: sessler@cm.utexas.edu
First published on 20th December 2021
Abstract
The meso-unsubstituted expanded porphyrinoid 3, incorporating two carbazole moieties, acts as an effective ligand for Co(II) and permits the isolation and X-ray diffraction-based characterization of a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 metal-to-ligand metallocage complex that converts spontaneously to the constituent 2:1 metal-to-ligand metalloring species in chloroform solution. The discrete metalloring is formed directly when the Co(II) complex is crystallized from supersaturated solutions, whereas crystallization from more dilute solutions favors the metallocage. Studies with two other test cations, Pd(II) and Zn(II), revealed exclusive formation of the monomeric metalloring complexes with no evidence of higher order species being formed. Structural, electrochemical and UV-vis-NIR absorption spectral studies provide support for the conclusion that the Pd(II) complex is less distorted and more effectively conjugated than its Co(II) and Zn(II) congeners, an inference further supported by TD-DFT calculations. The findings reported here underscore how expanded porphyrins can support coordination modes, including bimetallic complexes and self-assembled cage structures, that are not necessarily easy to access using more traditional ligand systems.
:3 metal-to-ligand metallocage complex that converts spontaneously to the constituent 2:1 metal-to-ligand metalloring species in chloroform solution. The discrete metalloring is formed directly when the Co(II) complex is crystallized from supersaturated solutions, whereas crystallization from more dilute solutions favors the metallocage. Studies with two other test cations, Pd(II) and Zn(II), revealed exclusive formation of the monomeric metalloring complexes with no evidence of higher order species being formed. Structural, electrochemical and UV-vis-NIR absorption spectral studies provide support for the conclusion that the Pd(II) complex is less distorted and more effectively conjugated than its Co(II) and Zn(II) congeners, an inference further supported by TD-DFT calculations. The findings reported here underscore how expanded porphyrins can support coordination modes, including bimetallic complexes and self-assembled cage structures, that are not necessarily easy to access using more traditional ligand systems.
Introduction
Over the past two decades considerable efforts have been devoted to exploring the metal cation coordination chemistry of expanded porphyrins.1–3 This has led to advances that are not easily recapitulated in the case of other ligand systems. For instance, metalation of expanded porphyrins has been used to trigger changes in electronic structure, including the conversion between nonaromatic and aromatic forms.4–6 In addition, expanded porphyrins have been used for metal ion recognition and sensing;7–11 they have also been incorporated into stimuli responsive molecular machines.12 Other metallated expanded porphyrins have been investigated as catalysts for organic synthesis13,14 and as functional photoacoustic imaging agents.15 In 2014, Lash and coworkers described a so-called adj-dicarbaporphyrin that stabilises an unusual tris-palladium sandwich complex.16 However, as a general rule, and in contrast to what is true for porphyrins per se,17–19 the use of expanded porphyrins to self-assemble multi-metallated arrays is all but unexplored. Moreover, to our knowledge no expanded porphyrin has been used to trap a structurally characterized metallocage as an inherently unstable kinetic product. Here, we report that Co(II) complexation of a meso-unsubstituted expanded porphyrinoid incorporating two carbazole moieties (3)20 can, under appropriately chosen conditions, produce a 6:3 metal-to-ligand hexa-Co(II) metallocage ({3·2Co}3) comprising three bis-metallated expanded porphyrin subunits. This metallocage converts spontaneously in CHCl3 solution to the corresponding single component bis-Co(II) complex (3·2Co), a thermodynamically favored product that is readily obtained by independent synthesis. Discrete bis-metallated metalloring species (3·2M) are obtained exclusively in our hands when the Co(II) source, Co(OAc)2, is replaced by M(OAc)2, where M = Pd(II) or Zn(II) (Scheme 1). The present study demonstrates how the judicious interplay between expanded porphyrin ligand design and coordination chemistry can be used to access metal-containing ensembles that lie off equilibrium with regard to their more thermodynamically stable monomeric forms.
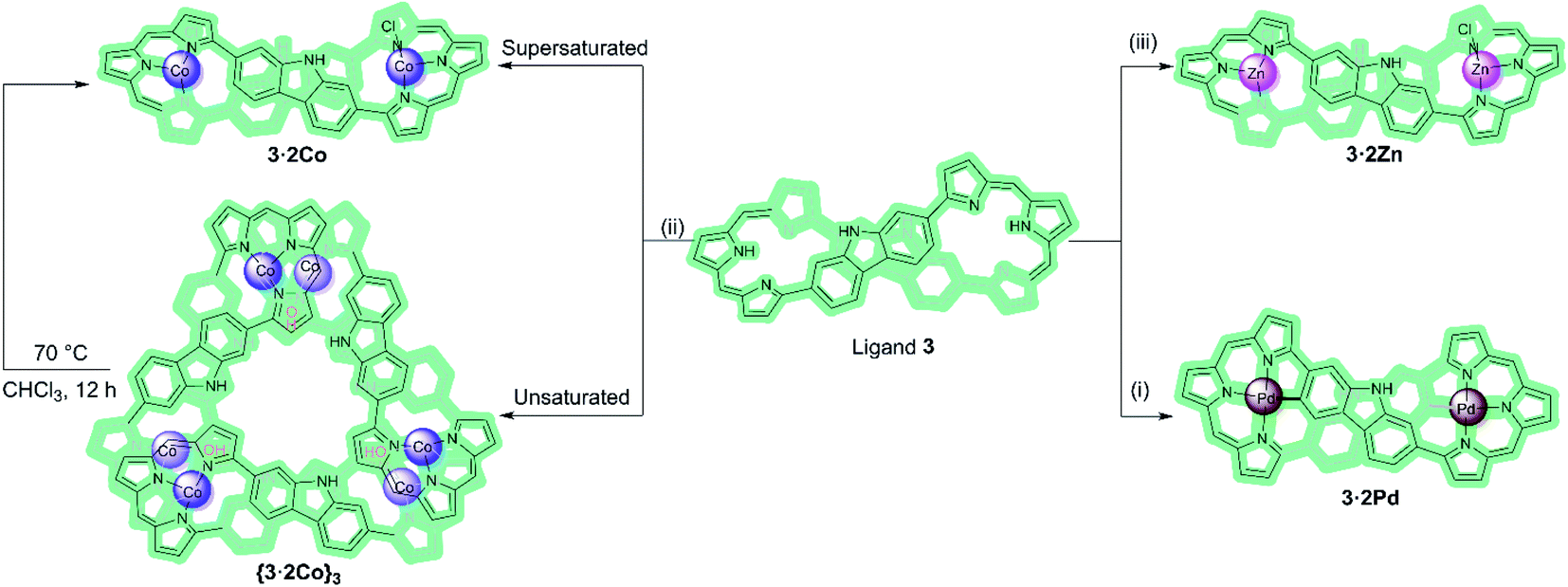 | ||
| Scheme 1 Metalation of ligand 3 with Pa(II), Co(II), and Zn(II) ions. Reagents and conditions: (i), (ii), and (iii) M(OAc)2, NaOAc, CHCl3/MeOH, RT. β-Pyrrolic alkyl groups are omitted for clarity. | ||
The present study relies on the use of a bis-carbazole expanded porphyrin (ligand 3). Carbazole-containing expanded porphyrins may be viewed as being a class of expanded carbaporphyrins,21,22 systems with one or more carbon donors incorporated into the central core.23–29 These unique macrocycles exhibit a diverse range of properties, including unusual aromaticity and chirality features,30–32 as well as serving as potential ligands.33–36 Among the various expanded carbaporphyrinoids reported to date, those with two embedded polyaromatic hydrocarbons or related heterocycles (PAHs) are still relatively rare and their metalation chemistry has not been extensively explored.37,38 Recently, we reported the synthesis of the meso-free expanded carbaporphyrinoid 3. In its as-prepared free-base form, 3 proved to be a flexible macrocycle that adopts figure-of-eight conformations that provide two tripyrrane-like pockets. We considered it likely that this system would prove useful as a ligand. What was less apparent was whether it would support chemistry that extended beyond the first coordination sphere. The present study was undertaken in an effort to test this possibility.
Results and discussion
Macrocycle 3 was prepared as detailed previously.20 Initially, we examined Pd(II) metalation (Scheme 1). Insertion of Pd(II) ions into ligand 3 was performed by treating with 10 molar equiv. of both palladium acetate and sodium acetate in CHCl3/MeOH (4:1) at room temperature for 8 h. After purification over neutral alumina followed by recrystallization from CH2Cl2/MeOH, the bis-Pd(II) complex (3·2Pd) was isolated in 68% yield. A MALDI-TOF mass spectrometric analysis revealed mass peaks at m/z = 1314.3255 ([M]+); calcd for C76H76N8Pd2: 1314.4267 (Fig. S10†).
Diffraction grade single crystals of 3·2Pd were obtained via the slow diffusion of methanol into a dichloromethane solution of the complex. An X-ray diffraction analysis revealed a solid-state structure possessing Ci molecular symmetry and a twisted figure-of-eight shape analogous to the free ligand 3 (Fig. 1). The two Pd(II) ions are each bound to three pyrrolic nitrogen atoms and one carbazole carbon atom. This leads to a slightly distorted square-planar NNNC coordination geometry with a τ4 (ref. 39) value = 0.17 reminiscent of what was seen in a previously reported core modified octaphyrin Pd(II) complex.29 Two independent structures are seen in the asymmetric unit, but they do not differ substantially (Fig. S15 and S16†). The distances between the palladium ions seen in these structures are 10.129 Å and 10.161 Å, respectively, whereas the Pd–N bond lengths vary from 1.959 to 2.095 Å and from 1.947 to 2.091 Å in these two structures, respectively. Likewise, Pd–C bond lengths of 2.037 and 2.041 Å, and 2.035 and 2.048 Å are found.
 | ||
| Fig. 1 Single crystal X-ray structures of 3·2Pd. (a) Top and (b) side views. Atom color key: carbon (light grey), nitrogen (light purple), palladium (maroon). Hydrogen atoms, β-pyrrolic alkyl groups and solvent molecules have been omitted for clarity. | ||
In contrast to what was seen for ligand 3, the 1H NMR spectrum of 3·2Pd recorded in CD2Cl2 is characterized by sharp signals at room temperature. Presumably, this reflects a system that is conformally rigid as the result of NNNC–Pd(II) coordination (Fig. S1†). Specific peak assignments were made on the basis of COSY and NOESY experiments (Fig. S4 and S5†). The chemical shifts of 3·2Pd are reminiscent of those seen for ligand 3 (Fig. 2). Of note are the absence of both pyrrolic NH signals, and the presence of two doublets and four singlets ascribed to the carbazole protons in the range of 5.97–7.95 ppm that further corroborated coordination occurs consistent with solid state structure. Bifurcated peaks corresponding to the four meso CH protons are also seen at 6.80 and 7.13 ppm that might reflect the Ci molecular symmetry of 3·2Pd and the chemical inequivalence of the meso CH protons.
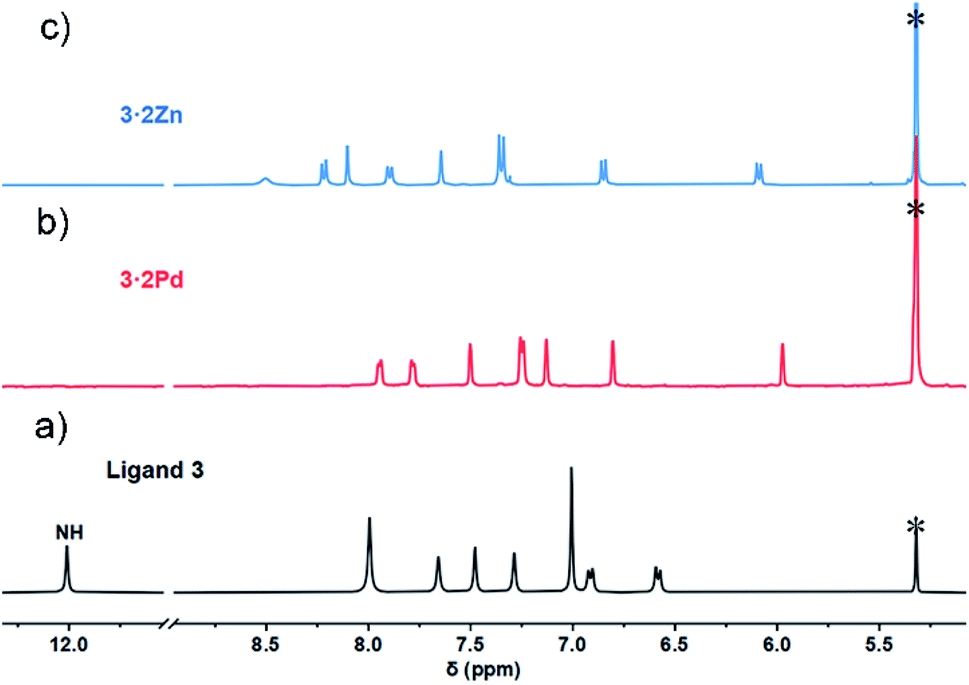 | ||
| Fig. 2 Comparative 1H NMR spectra (aromatic region) of ligand 3 and metal complexes. (a) Ligand 3 at −60 °C in CD2Cl2; (b) 3·2Pd at 25 °C in CD2Cl2; (c) 3·2Zn at −60 °C in CD2Cl2. Residual solvents were marked with asterisks. | ||
Co(II) metalation of ligand 3 was performed following the procedure used to effect Pd(II) insertion. Diffraction grade single crystals were then obtained by allowing n-hexane to diffuse slowly into a chloroform solution of 3·2Co. Structural analysis (Fig. 3a) revealed that under these conditions three units of [3·2Co–2Cl] and three hydroxy ligands trimerize to give a triangular-shaped metallocage that includes two water molecules and three chloride ions within its central cavity. This unique structure in the solid state may make this cage of interest in the context of chloride anion capture.40
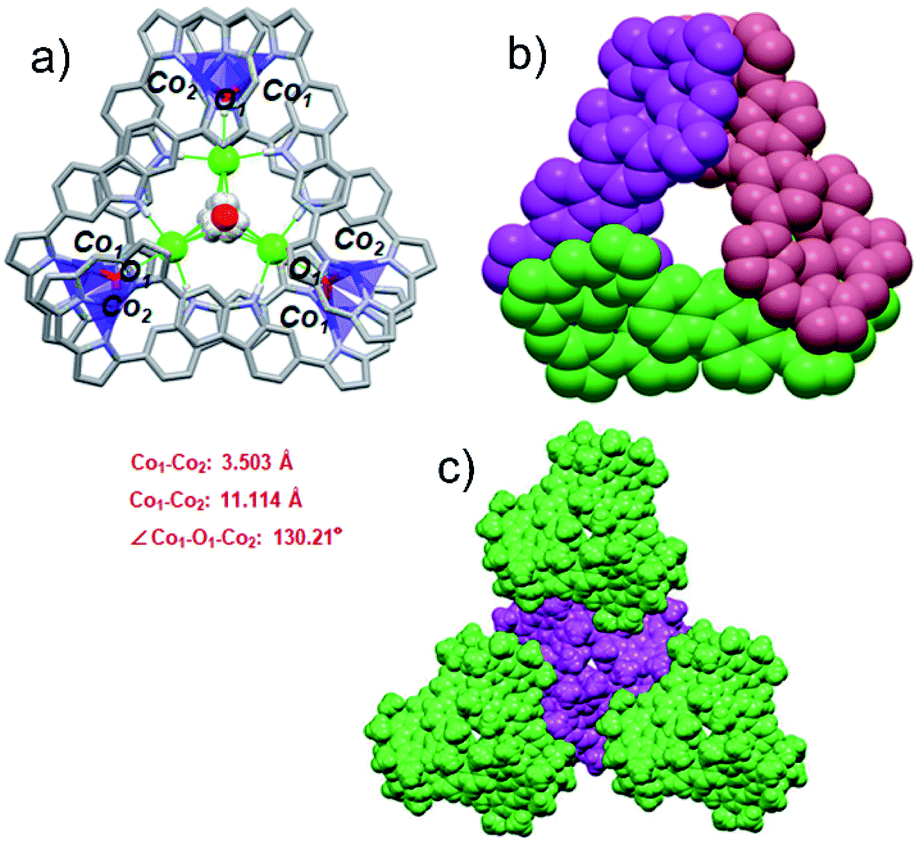 | ||
| Fig. 3 Single crystal X-ray structures of {3·2Co}3. (a) Top view of {3·2Co}3. Atom color key: hydrogen (white), carbon (light gray), nitrogen (light purple), cobalt (violet), chloride (green). Hydrogen atoms that are not involved in intermolecular interactions, β-pyrrolic alkyl groups and solvent molecules are omitted for clarity. (b) Top view in space-filling form showing individual 3·2Co subunits (each colored differently for clarity). (c) Top view of molecular packing arrangement showing individual {3·2Co}3 cages and the overall spiral arrangement (individual cages are colored differently for clarity). | ||
In the cage structure, referred to as {3·2Co}3, the three constituent 3·2Co monomers retain their figure-of-eight shape and are interconnected in a staggered manner. This results in overall C3 symmetry (Fig. 3b). In each monomer, the two Co ions are each bound to three tripyrrin-derived nitrogen atoms and share an axial hydroxy ligand with another monomer. Three axially coordinated oxygen atoms define a regular triangle defined by an O–O distance of 10.516 Å. The Co–O band lengths are 1.93(1) Å. Two water molecules are located above and below the mean plane (with respect to the three oxygen atoms) at distances of 1.617 Å and 1.719 Å, respectively. The two water molecules share three common chloride ions connected through O–H⋯Cl hydrogen bonds (av. OH⋯Cl: 2.581 Å). In addition, three chloride ions are directly bound to the carbazole NH protons (av. NH⋯Cl: 2.487 Å) and to axial OH moieties (av. OH⋯Cl: 2.339 Å) via an intermolecular hydrogen-bonding network. It is likely that both the chloride anions and water molecules help stabilize the cage structure. As shown in Fig. 3c, in the solid state each molecule cage unit ({3·2Co}3) is linked to three nearest neighbours via what are inferred to be weak intermolecular van der Waals interactions.
We next modified the crystallization conditions. Specifically, Co(OAc)2 (45 mg), NaOAc (15 mg), and ligand 3 (20 mg) in a mixture of CHCl3/MeOH (4:1; 20 mL) were allowed to react for 8 h at room temperature. After workup and purification, metallic rhombic-like crystals of 3·2Co were obtained by diffusion of n-hexane (liquid) into a supersaturated dichloromethane solution of the complex over the course of 3–5 days in a sealed vial at room temperature (Fig. S24a†). In contrast, when the concentration of the complex in the chloroform solution was kept low and slow vapor diffusion of hexane into a chloroform solution of the complex at room temperature was used to promote crystallization, brick-like crystal of {3·2Co}3 analogous to those generated originally were obtained after about three weeks (Fig. S24b†).
Crystallographic analysis (Fig. 4) of the crystals considered to consist of 3·2Co revealed a C2 symmetric figure-of-eight structure similar to that for the free ligand 320 and the 3·2Pd complex described above. Both Co ions are coordinated to three tripyrrane-like nitrogen atoms and an axial chloride. This results in a distorted tetrahedral coordination geometry with a τ4 value of 0.74. The distance between the Co centres is approximately 11.34 Å. The Co–N bond lengths vary from 1.967 to 2.027 Å, whereas the Co–Cl bond lengths range from 2.291 to 2.324 Å. The 1H NMR spectrum of 3·2Co revealed peaks at extremely low field (ca. 50 ppm) reflecting the paramagnetic effect of the coordinated Co(II) ions (Fig. S6†). Variable temperature magnetic susceptibility analyses further proved consistent with the two Co ions being in the +2, S = 3/2 high oxidation/spin state. A single-core model that did not account for possible intramolecular magnetic exchange interactions was used to fit the magnetic properties of 3·2Co; this gave values for ZJ and g of −0.033 and 2.57 cm−1, respectively (Fig. S35†). Unfortunately, the magnetic parameters of {3·2Co}3 could not be determined due to its instability; vide infra.
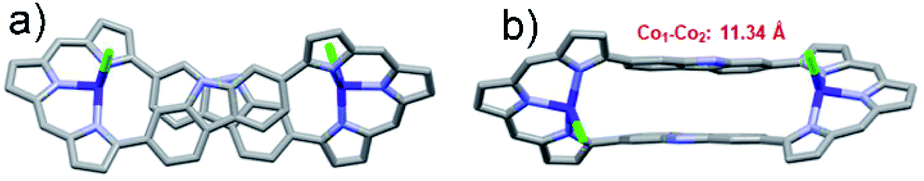 | ||
| Fig. 4 Single crystal X-ray structures of 3·2Co. (a) Top and (b) side views. Atom color key: carbon (light grey), nitrogen (light purple), cobalt (violet), chloride (green). Hydrogen atoms, β-pyrrolic alkyl groups and solvent molecules have been omitted for clarity. | ||
The two different crystalline species {3·2Co}3 and 3·2Co were analysed by scanning electron microscopy (SEM) (Fig. 5, S25 and S26†). These analyses revealed that crystals of {3·2Co}3 were prone to collapse or fracture even in the solid state. This provided a preliminary indication that this form might not be thermodynamically stable relative to the monomeric bis-Co(II) form, 3·2Co.
 | ||
| Fig. 5 SEM images of the crystalline entities: (a) and (b) 3·2Co; (c) and (d) {3·2Co}3. | ||
Consistent with the above inference, time-dependent UV-vis spectroscopic analyses revealed that the metallocage {3·2Co}3 decomposed gradually in CHCl3 solution at room temperature even when starting from pure crystals ({3·2Co}3) (Fig. S29†). We then confirmed that the metallocage {3·2Co}3 is transformed into the corresponding metalloring complex 3·2Co in CHCl3 solution at room temperature (Fig. S30†). Subjecting {3·2Co}3 to reflux in CHCl3 solution accelerated this transformation (Fig. S14 and S31†). HR-FT-ICR MS analyses revealed that the 2:1 complex produced as the result of this transformation bears a coordinated OH group on each Co(II) centre (Fig. S13†). We thus conclude that {3·2Co}3 lies off-equilibrium with respect to its constituent 3·2Co subunits. The relatively facile interconversion is ascribed to the lability of the coordination bonds in {3·2Co}3, which in other instances can allow for stimulus-induced structural rearrangements to give species of differing sizes and shapes.41 On the basis of the Gibbs free energy calculation, the corresponding ΔG value for going from the {3·2Co}3 cage to the 3·2Co macrocycle is −14.601 kcal mol−1, supporting the conclusion that the macrocycle is the thermodynamically favoured product (Fig. S42†).
In an effort to probe further the coordination chemistry of ligand 3, we explored its ability to stabilize complex(es) with Zn(II), a cation prone to form labile ligand–metal bonds. Metalation of 3 with zinc acetate, afforded the corresponding bis-metal complex, 3·2Zn, with no evidence of higher order species being detected via MALDI-TOF MS analysis (Fig. S12†). Single crystals of 3·2Zn were obtained through slow diffusion of methanol into a chloroform solution. The structure of 3·2Zn was confirmed by X-ray diffraction analysis and proved similar to that of 3·2Co (Scheme 1 and Fig. S19†).
The 1H NMR spectrum of 3·2Zn recorded in CD2Cl2 solution is characterized by a rather broad signal pattern in the aromatic region (Fig. S7†). This finding is consistent with a system that is conformationally flexible on the NMR time scale at ambient temperature. At −60 °C the 1H NMR spectrum becomes sharp with seven distinct set of protons, ascribed to the carbazole subunit, being observed in the range of 6.0–8.5 ppm region (Fig. 2). Bifurcated signal corresponding to the four meso CH protons are seen at 7.35 ppm. All signals were further assigned by COSY and NOESY experiments (Fig. S8 and S9†). The 1H NMR spectral analyses thus provide support for the conclusion, also drawn from the X-ray diffraction study above, that the basic structural features of ligand 3 are preserved upon Zn ion complexation.
The absorption spectra of ligand 3 and its corresponding metal complexes 3·2Pd, {3·2Co}3, 3·2Co, and 3·2Zn were measured in chloroform at room temperature. As can be seen from an inspection of Fig. 6 and Table 1, relative to ligand 3, the UV-vis-NIR absorption spectra of the metal complexes exhibit distinct bathochromic shifts, particularly in the NIR region; this is thought to reflect metal ion insertion into the cavity that serves to strengthen the extent of conjugation within the macrocycle.42 The absorption features of the metallocage {3·2Co}3 and metalloring 3·2Co were qualitatively similar, although differences are seen in the absorption tails, which extend out to ca. 1010 nm and ca. 960 nm in the case of {3·2Co}3 and 3·2Co, respectively.
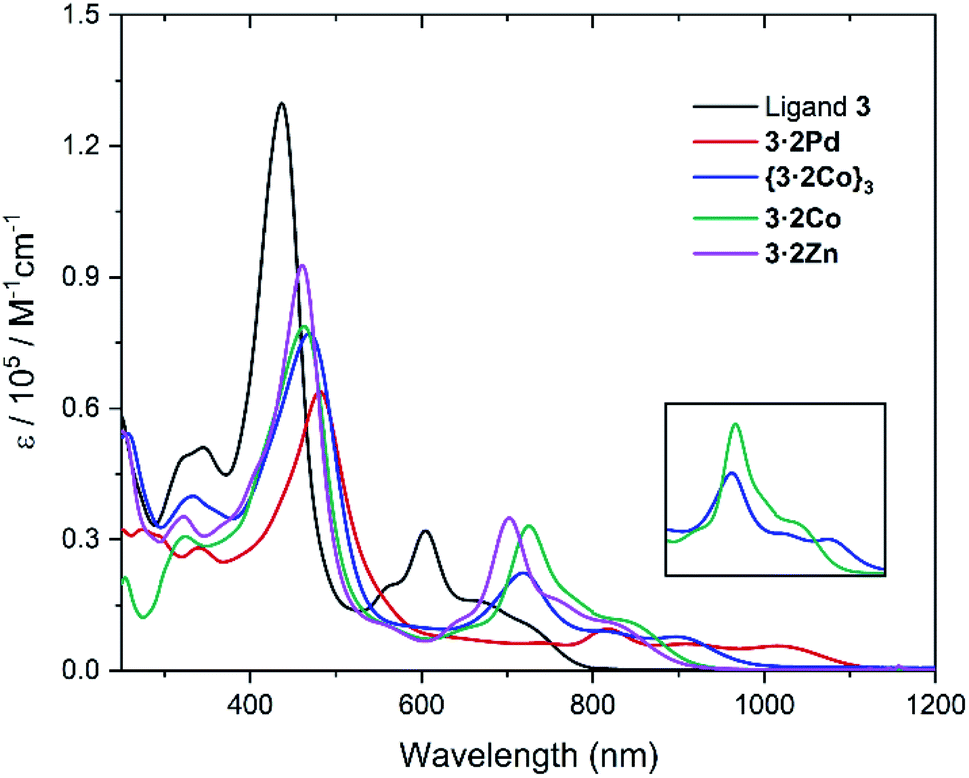 | ||
| Fig. 6 UV-vis-NIR absorption spectra of ligand 3, 3·2Pd, {3·2Co}3, 3·2Co, and 3·2Zn in CHCl3 at room temperature. (inset) Comparative of {3·2Co}3 and 3·2Co expanded region at 600–1000 nm. | ||
| Compound | λ [nm] (ε [×105 L mol−1 cm−1]) | E ox.3 | E ox.2 | E ox.1 | E red.1 | E red.2 | E red.3 | ΔEHLc [eV] | ΔEHLd [eV] |
|---|---|---|---|---|---|---|---|---|---|
| a UV-vis-NIR absorption spectra were recorded in CHCl3 at room temperature. b Cyclic voltammetry studies were conducted in CH2Cl2 containing 0.1 M n-Bu4NPF6 as the supporting electrolyte, Ag/Ag+ was used as the reference electrode, Pt wire was used as the counter electrode, and glassy carbon as the working electrode. Potentials were recorded vs. ferrocene/ferrocenium ion. Scan rates were 0.05 V s−1. These potentials were determined by differential pulse voltammetry. c Electrochemical HOMO–LUMO energy gap [eV] = Eox.1 − Ered.1. d HOMO–LUMO energy gap obtained by TD-DFT calculations (B3LYP/6-311G(d,p)). | |||||||||
| 3 | 436 (1.30), 605 (0.32) | — | 0.43 | 0.33 | −1.21 | −1.50 | — | 1.54 | 1.90 |
| 3·2Pd | 481 (0.64), 818 (0.10) | 0.84 | 0.34 | 0.19 | −1.07 | −1.13 | −1.71 | 1.26 | 1.67 |
| 3·2Co | 463 (0.79), 725 (0.33) | 1.13 | 0.58 | 0.49 | −0.85 | −1.15 | −1.65 | 1.34 | 1.88 |
| 3·2Zn | 461 (0.93), 702 (0.35) | 1.17 | 0.58 | 0.49 | −0.95 | −1.18 | — | 1.44 | 1.89 |
Particularly noteworthy are the low energy absorption maxima at 818 nm for Pd, 725 nm for Co, and 702 nm for Zn, respectively, which are approximately 100 nm to 200 nm bathochromically shifted as compared to the corresponding band in the case of ligand 3 (λmax = 605 nm). Also of interest is that, compared with the Zn and Co complexes (3·2Co and 3·2Zn), the absorption of the Pd complex (3·2Pd) is broader and is characterized by a tail that extends to ca. 1100 nm. This is taken as an indication that the π-conjugation is relatively enhanced in the case of the bis-Pd complex, an inference consistent with the solid state analyses, which revealed less twisting between the carbazole units and the tripyrranes in the case of 3·2Pd (av. 20.8°, Fig. S16†) as compared to 3·2Co (av. 35.23°, Fig. S17†) and 3·2Zn (av. 36.09°, Fig. S20†).
The redox properties of ligand 3 and the corresponding bis-metal complexes were also examined via cyclic voltammetry (CV) and differential pulse voltammetry (DPV) (Fig. S32–35†). The resulting redox potentials and electrochemically-derived HOMO–LUMO gaps (ΔEHL) are summarized in Table 1. With respect to 3, two irreversible oxidation waves at 0.33 and 0.43 V, along with one reversible reduction wave at −1.21 V and one irreversible wave at −1.50 V, are seen. Upon metal insertion, reduction becomes more facile. The extent of this anodic shift is smallest in the case of 3·2Pd. The first oxidation wave for 3·2Pd (0.19 V) also remains more negative than those for 3·2Zn and 3·2Co (both 0.49 V). This results in the smallest HOMO–LUMO gap (1.26 eV), a finding consistent with the low energy absorption features seen for this complex. (Note: the lifetime of metallocage {3·2Co}3 in solution proved too short to allow its analysis by electrochemical means.)
Time-dependent density functional theory (TD-DFT) calculations revealed that the frontier molecular orbitals (FMOs) of 3·2Co and 3·2Zn are similar and that these complexes possess analogous HOMO–LUMO energy gaps (Fig. S43 and S44†). In the case of 3·2Pd (Fig. S43†), the HOMO energy level is elevated as compared to the 3·2Zn and 3·2Co complexes. This leads to a slight decrease in the band gap (ΔE = 1.67 eV) for 3·2Pd as compared to 3·2Zn and 3·2Co (ΔE = 1.88–1.90 eV), in good agreement with the electrochemical HOMO–LUMO gaps (Table 1) and the relatively larger red shift in the absorption spectral features seen for 3·2Pd. Across the board TD-DFT studies revealed a good correspondence between the observed and predicted spectral features (Fig. S46–48† and Tables S3–S5†).
Conclusions
In summary, ligand 3 supports the formation of 2:1 metal-to-ligand complexes with Pd(II), Co(II), and Zn(II) (3·2Pd, 3·2Co, and 3·2Zn, respectively). Single-crystal X-ray diffraction structural analyses revealed distorted square-planar geometries about the metal centres in 3·2Pd, but tetrahedral coordination modes in the case of 3·2Co and 3·2Zn. These differences translate into a lower level of ligand distortion and better conjugation in the case of the Pd(II) complex, as inferred from spectroscopic and electrochemical analyses, as well as the TD-DFT calculations. In the case of Co(II), crystallization conditions (e.g., use of unsaturated solutions and antisolvent vapor diffusion) could be found that allowed for the kinetic trapping of a crystalline 6:3 metal-to-ligand metallocage ({3·2Co}3) containing three 3·2Co metallorings. This metallocage converts readily to the more thermodynamically favored 2:1 (3·2Co) metalloring form in CHCl3 solution. The present work thus highlights how expanded carbaporphyrinoids may be used to access structural forms, including self-assembled metal complexes, that are not necessarily accessible using simpler ligand systems.
Data availability
Crystallographic data for {3·2Co}3, 3·2Zn, 3·2Co, and 3·2Pd have been deposited at the CCDC under accession numbers 2105486–2105488 and 2105812 respectively, and can be obtained from Home – The Cambridge Crystallographic Data Centre (CCDC). Cyclic voltammetry, magnetic susceptibility measurement, NMR and MS spectra, DFT calculation details and Cartesian coordinates of calculated complexes supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization and supervision: JLS, TS, and CH; synthesis, characterization, NMR, spectroscopy, SEM, SQUID, CV and DPV studies: WN and YH; single crystal growing and data analysis: WN and CH; theoretical calculations: WN and XL; writing – original: WN; writing – review and editing: JLS, TS and CH. All authors proofread, commented on, and approved the final version of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for this work was sponsored by the National Natural Science Foundation of China (grant no. 22171179, 21901155, to C. L.), the Shanghai Rising-Star Program (no. 20QA1403700 to C. L.), Shanghai University (N.13-G210-21-234) and the Eastern Scholars program from the Shanghai Municipal Education Committee (to C. L.). We thank Prof. Yong Pei (Xiangtan University) for assistance in performing the quantum chemical calculations and Dr Lin Xiong (Xiangtan University) for providing computing resources and associated technical support. We thank Dr Zhaobo Hu (Nanjing University) for help in carrying out studies of the magnetic properties of 3·2Co. We also thank Mrs Lingling Li (Instrumental Analysis Center, Shanghai Jiao Tong University) for assistance with the single-crystal X-ray diffraction studies. J. L. S. acknowledges support from the Robert A. Welch Foundation (F-0018).Notes and references
- J. L. Sessler and E. Tomat, Acc. Chem. Res., 2007, 40, 371–379 CrossRef CAS.
- G. I. Vargas-Zúñiga and J. L. Sessler, Adv. Inorg. Chem., 2018, 71, 327–377 CrossRef.
- A. Alka, V. S. Shetti and M. Ravikanth, Coord. Chem. Rev., 2019, 401, 213063 CrossRef CAS.
- Y. Tanaka, S. Saito, S. Mori, N. Aratani, H. Shinokubo, N. Shibata, Y. Higuchi, Z. S. Yoon, K. S. Kim, S. B. Noh, J. K. Park, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 681–684 CrossRef CAS.
- T. Yoneda, Y. M. Sung, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 13169–13173 CrossRef CAS.
- S. L. Xue, D. Kuzuhara, N. Aratani and H. Yamada, Angew. Chem., Int. Ed., 2019, 58, 12524–12528 CrossRef CAS.
- M. Ishida, Y. Naruta and F. Tani, Angew. Chem., Int. Ed., 2010, 49, 91–94 CrossRef CAS.
- J.-I. Setsune, M. Kawama and T. Nishinaka, Tetrahedron Lett., 2011, 52, 1773–1777 CrossRef CAS.
- E. Ganapathi, W.-Z. Lee and M. Ravikanth, J. Org. Chem., 2014, 79, 9603–9612 CrossRef CAS.
- J. Wojaczyński, J. Maciołek and P. J. Chmielewski, Chem.–Asian J., 2017, 12, 643–647 CrossRef.
- G. Anguera, W.-Y. Cha, M. D. Moore, J. Lee, S. Guo, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 4028–4034 CrossRef CAS.
- B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS.
- Q. C. Chen, I. Saltsman, A. Kaushansky, Z. Y. Xiao, N. Fridman, X. Zhan and Z. Gross, Chem.–Eur. J., 2018, 24, 17255–17261 CrossRef CAS.
- H. Y. Chen, L. L. Wang, S. Xu, X. H. Liu, Q. He, L. J. Song and H. B. Ji, ACS Catal., 2021, 11, 6810–6815 CrossRef CAS.
- K. Shimomura, H. Kai, Y. Nakamura, Y. Hong, S. Mori, K. Miki, K. Ohe, Y. Notsuka, Y. Yamaoka, M. Ishida, D. Kim and H. Furuta, J. Am. Chem. Soc., 2020, 142, 4429–4437 CrossRef CAS.
- D. I. AbuSalim, G. M. Ferrence and T. D. Lash, J. Am. Chem. Soc., 2014, 136, 6763–6772 CrossRef CAS.
- T. Nakamura, H. Ube and M. Shionoya, Angew. Chem., Int. Ed., 2013, 52, 12096–12100 CrossRef CAS.
- F. J. Rizzuto, D. M. Wood, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2017, 139, 11008–11011 CrossRef CAS.
- E. G. Percástegui and V. Jancik, Coord. Chem. Rev., 2020, 407, 213165 CrossRef.
- W. Zhou, M. Hao, T. Lu, Z. Duan, T. Sarma, J. L. Sessler and C. Lei, Chem.–Eur. J., 2021, 27, 16173–16180 CrossRef CAS.
- T. D. Lash, Chem. Rev., 2017, 117, 2313–2446 CrossRef CAS.
- B. Szyszko and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2020, 59, 16874–16901 CrossRef CAS.
- H. Gopee, X. F. Kong, Z. Q. He, I. Chambrier, D. L. Hughes, G. J. Tizzard, S. J. Coles and A. N. Cammidge, J. Org. Chem., 2013, 78, 9505–9511 CrossRef CAS.
- M. Das, B. Adinarayana, M. Murugavel, S. Nayak and A. Srinivasan, Org. Lett., 2019, 21, 2867–2871 CrossRef CAS.
- M. Das, S. Chitranshi, M. Murugavel, B. Adinarayana, C. H. Suresh and A. Srinivasan, Chem. Commun., 2020, 56, 3551–3554 RSC.
- S. Chitranshi, M. Das, B. Adinarayana, W.-Y. Cha, D. Kim and A. Srinivasan, Org. Lett., 2020, 22, 1081–1085 CrossRef CAS.
- T. Sulfikarali, J. Ajay, C. H. Suresh, P. V. Bijina and S. Gokulnath, J. Org. Chem., 2020, 85, 8021–8028 CrossRef CAS.
- B. Szyszko, P. Rymut, M. Matviyishyn, A. Bialonska and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2020, 59, 20137–20146 CrossRef CAS.
- L. Liu, Z. Hu, F. Zhang, Y. Liu, L. Xu, M. Zhou, T. Tanaka, A. Osuka and J. Song, Nat. Commun., 2020, 11, 6206 CrossRef CAS.
- M. Stępień, L. Latos-Grażyński, N. Sprutta, P. Chwalisz and L. Szterenberg, Angew. Chem., Int. Ed., 2007, 46, 7869–7873 CrossRef.
- B. Szyszko, M. Przewoźnik, M. J. Białek, A. Białońska, P. J. Chmielewski, J. Cichos and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2018, 57, 4030–4034 CrossRef CAS.
- B. Szyszko, P. J. Chmielewski, M. Przewoźnik, M. J. Białek, K. Kupietz, A. Białońska and L. Latos-Grażyński, J. Am. Chem. Soc., 2019, 141, 6060–6072 CrossRef CAS.
- T. Chatterjee and M. Ravikanth, Coord. Chem. Rev., 2020, 402, 213480 CrossRef.
- W. Stawski, M. Kijewska and M. Pawlicki, Chem.–Asian J., 2020, 15, 8–20 CrossRef CAS.
- J.-I. Setsune, M. Toda, T. Yoshida, K. Imamura and K. Watanabe, Chem.–Eur. J., 2015, 21, 12715–12727 CrossRef CAS.
- A. Kalaiselvan, S. Dhamija, C. Aswathi, A. K. De and S. Gokulnath, Chem. Commun., 2021, 57, 11485–11488 RSC.
- X. S Ke, Y. Hong, P. Y. Tu, Q. He, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2017, 139, 15232–15238 CrossRef.
- X. S. Ke, Y. Hong, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2018, 140, 7579–7586 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159–161 CAS.
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- H. Mori and A. Osuka, Chem.–Eur. J., 2015, 21, 7007–7011 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2105486–2105488 and 2105812. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06514a |
| This journal is © The Royal Society of Chemistry 2022 |