
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-induced transition metal and photosensitizer free decarbonylative addition of amino-arylaldehydes to ketones†
Yi
Wang‡
a,
Yatao
Lang‡
a,
Chao-Jun
Li
 b and
Huiying
Zeng
*a
b and
Huiying
Zeng
*a
aThe State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, 222 Tianshui Road, Lanzhou, 730000, P. R. China. E-mail: zenghy@lzu.edu.cn
bDepartment of Chemistry, FQRNT Centre for Green Chemistry and Catalysis, McGill University, 801 Sherbrooke St. West, Montreal, Quebec H3A 0B8, Canada
First published on 21st December 2021
Abstract
The decarbonylative-coupling reaction is generally promoted by transition metals (via organometallic complexes) or peroxides (via radical intermediates), often at high temperatures to facilitate the CO release. Herein, a visible-light-induced, transition metal and external photosensitizer free decarbonylative addition of benzaldehydes to ketones/aldehydes at room temperature is reported. Tertiary/secondary alcohols were obtained in moderate to excellent yields promoted by using CsF under mild conditions. The detailed mechanistic investigation showed that the reaction proceeded through photoexcitation–decarbonylation of the aldehyde to generate an aromatic anion, followed by its addition to ketones/aldehydes. The reaction mechanism was verified by the density functional theory (DFT) calculations.
Introduction
The cleavage and formation of the C–C bond have always played a key role in the reconstruction of organic compound skeletons, being the essence of organic chemistry.1 The C–C bond activation via the decarbonylative process has emerged as an important approach for such transformations.2 Meanwhile, aldehydes are relatively abundant and easily available chemicals, and have been widely used as precursors for a large number of decarbonylation reactions,3 such as the Tsuji–Wilkinson decarbonylation,4 decarbonylative coupling reactions,5 and retro-hydroformylation-type reactions.6 Among them, the decarbonylative coupling of aldehydes and unsaturated compounds by transition metal catalysis (via organometallic complexes, Scheme 1a)7 or with peroxides (via radical intermediates, Scheme 1b)8 has shown great potential in organic synthesis. For example, our group9 has developed a Ru-catalyzed olefination via decarbonylative addition of aldehydes and alkynes; Shi10 reported the Rh-catalyzed intramolecular cascade decarbonylative cyclization reaction of ortho-formyl alkylidenecyclopropanes (ACPs); Yang11 exploited the Fe-catalyzed decarbonylative alkylation–azidation cascade reaction of styrene derivatives to provide aliphatic azides via a one-pot procedure.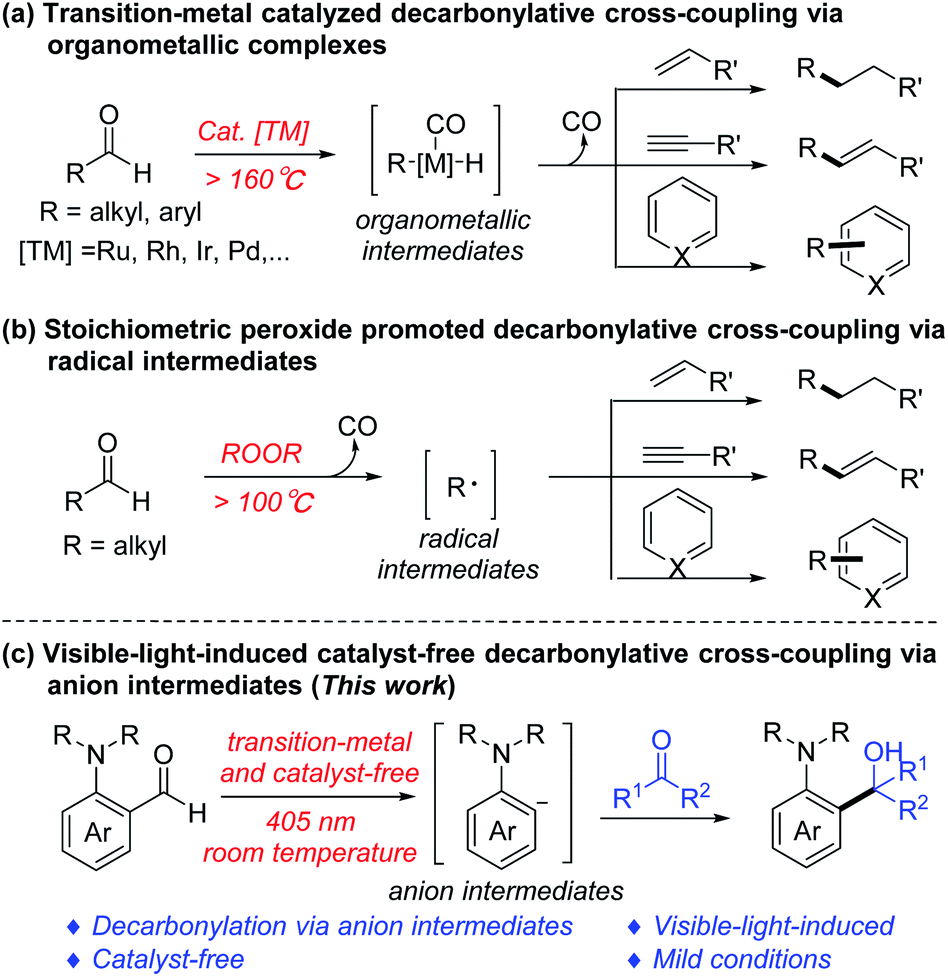 | ||
| Scheme 1 Decarbonylative cross-coupling reactions via different intermediates. | ||
However, these catalytic decarbonylative coupling reactions require transition metals and special ligands, and most of them require high temperature. Likewise, decarbonylative coupling reactions mediated by peroxides also necessitate high temperature to promote the homo-cleavage of peroxide, and most of them are only applicable to aliphatic aldehydes. Therefore, it is of great significance to develop more efficient decarbonylative coupling strategies that can overcome the above shortcomings in the absence of transition metal catalysts or peroxides under mild conditions.
More recently, our group has reported a series of photoinduced transition metal- and photosensitizer-free cross-coupling reactions,12 which showed potential green chemistry advantages. Herein, we disclose a visible-light-induced transition metal and external photosensitizer free decarbonylative nucleophilic addition of amino-benzaldehydes to ketones/aldehydes at room temperature via anion intermediates (Scheme 1c).
Results and discussion
2-(Diethylamino)benzaldehyde (1a) was selected as a model substrate in acetone (as both the reactant and solvent), and was irradiated with a 405 nm LED at room temperature for 24 h. To our delight, decarbonylative nucleophilic addition product 2-(2-(diethylamino)phenyl)propan-2-ol (3aa) was obtained with 70% yield (Table 1, entry 1). Encouraged by this result, different bases, both organic and inorganic bases commonly used in laboratories, were examined for this reaction (see details in Table S1†). Interestingly, the reaction could be satisfactorily performed by using CsF as a base, forming the product in 88% yield (entry 2). Considering that the cesium cation or fluoride anion might play an important role in this decarbonylative coupling reaction, different fluoride and cesium salts were investigated (entries 3–6), but they did not show further improvement. Increasing the amount of CsF to 2.0 equiv. or reducing to 0.5 equiv. gave almost the same yields (entries 7–8). Although using LEDs of other wavelengths (380 nm or 425 nm, entries 9–10) was less effective, illumination was necessary (entry 11). Finally, reducing the reaction time to 12 h gave 89% yield (entry 12). Further reducing the reaction time led to a lower yield (entry 13) (see details in Table S1 in the ESI†).|
|
||
|---|---|---|
| Entry | Variations from standard conditions | Yieldb/% |
| a Reaction conditions: 1a (0.1 mmol) and CsF (1.0 equiv.) in acetone 2a (1.0 mL) were irradiated with a LED (405 nm, 3 W × 2) for 24 h under an argon atmosphere. b Yields were determined by 1H NMR using nitromethane as an internal standard; isolated yields are shown in parentheses. | ||
| 1 | — | 70 |
| 2 | CsF (1.0 equiv.) | 88 |
| 3 | Cs2CO3 (1.0 equiv.) | 75 |
| 4 | CsBr (1.0 equiv.) | 76 |
| 5 | LiF (1.0 equiv.) | 68 |
| 6 | KF (1.0 equiv.) | 76 |
| 7 | CsF (2.0 equiv.) | 86 |
| 8 | CsF (0.5 equiv.) | 88 |
| 9 | CsF (0.5 equiv.), 380 nm | 83 |
| 10 | CsF (0.5 equiv.), 425 nm | 41 |
| 11 | In the dark | n.r. |
| 12 | CsF (0.5 equiv.), 12 h | 89(83) |
| 13 | CsF (0.5 equiv.), 8 h | 78 |
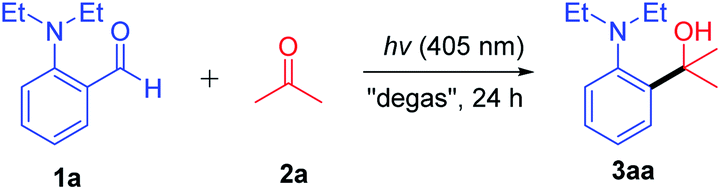
With the optimized conditions in hand, the scope of substrates was explored by introducing various functional groups into compound 1 (Table 2). Regardless of the alkyl-chain length of the N-substituent, the yield could be obtained in 63–80% (3aa–3ca). It is interesting to note that N-allyl substituted derivatives also successfully gave the product in moderate yields (3da–3ea). In addition, the cyclic amino substrate also had good compatibility to produce 82% yield of 3fa. With halogen (Cl and Br) atoms on the benzene ring of 1, the corresponding products (3ga and 3ha) were generated in excellent yields with the halogen atom intact. Substrates bearing strong electron-donating substituents, such as ethoxy (3ia) and ethylthio groups (3ja), also afforded the corresponding products in good to high yields. Likewise, both weak electron-donating groups (ethyl) and strong electron-withdrawing groups (trifluoromethyl) were also tolerated, producing the decarbonylative coupling products (3ka and 3la) in good yields. The 2,6-disubstituted substrate also successfully reacted with acetone, generating the desired product 3ma in 78% yield even if the allyl group is present at the ortho-position. Besides, the 2,5-disubstituted substrate also reacted successfully, generating product 3na in 70% yield.
| a Reaction conditions: 1 (0.1 mmol) and CsF (0.5 equiv.) in acetone (2a, 1.0 mL) were irradiated with a LED (405 nm, 3 W × 2) for 12 h at room temperature; isolated yields were given. |
|---|
|
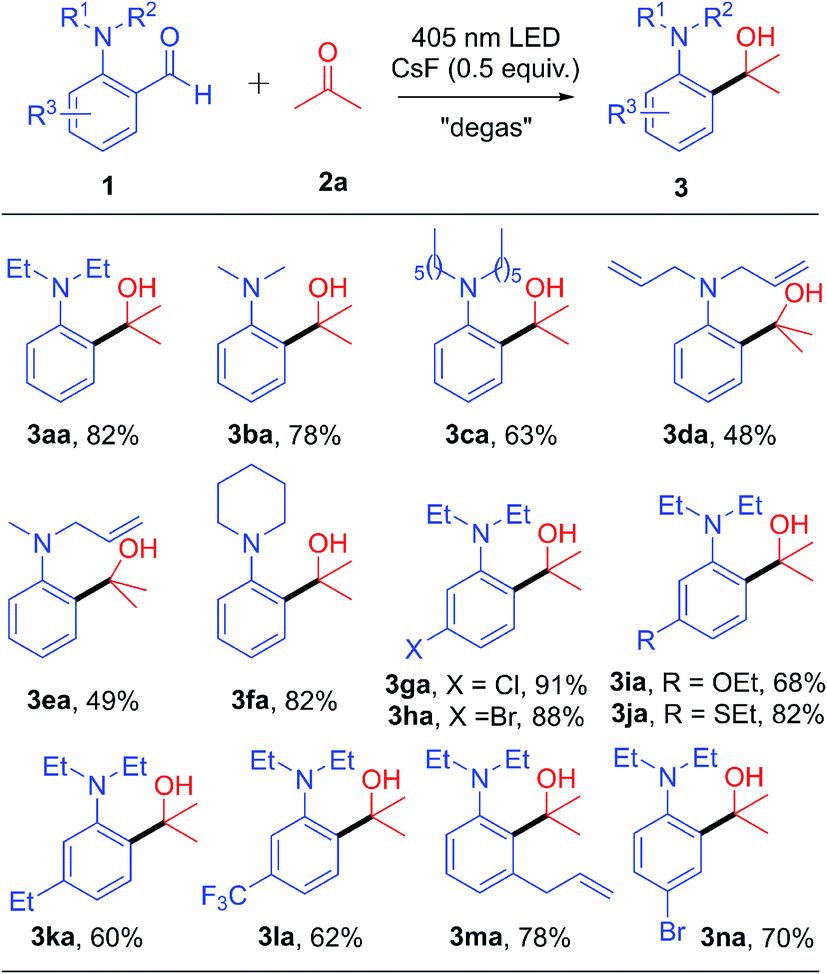
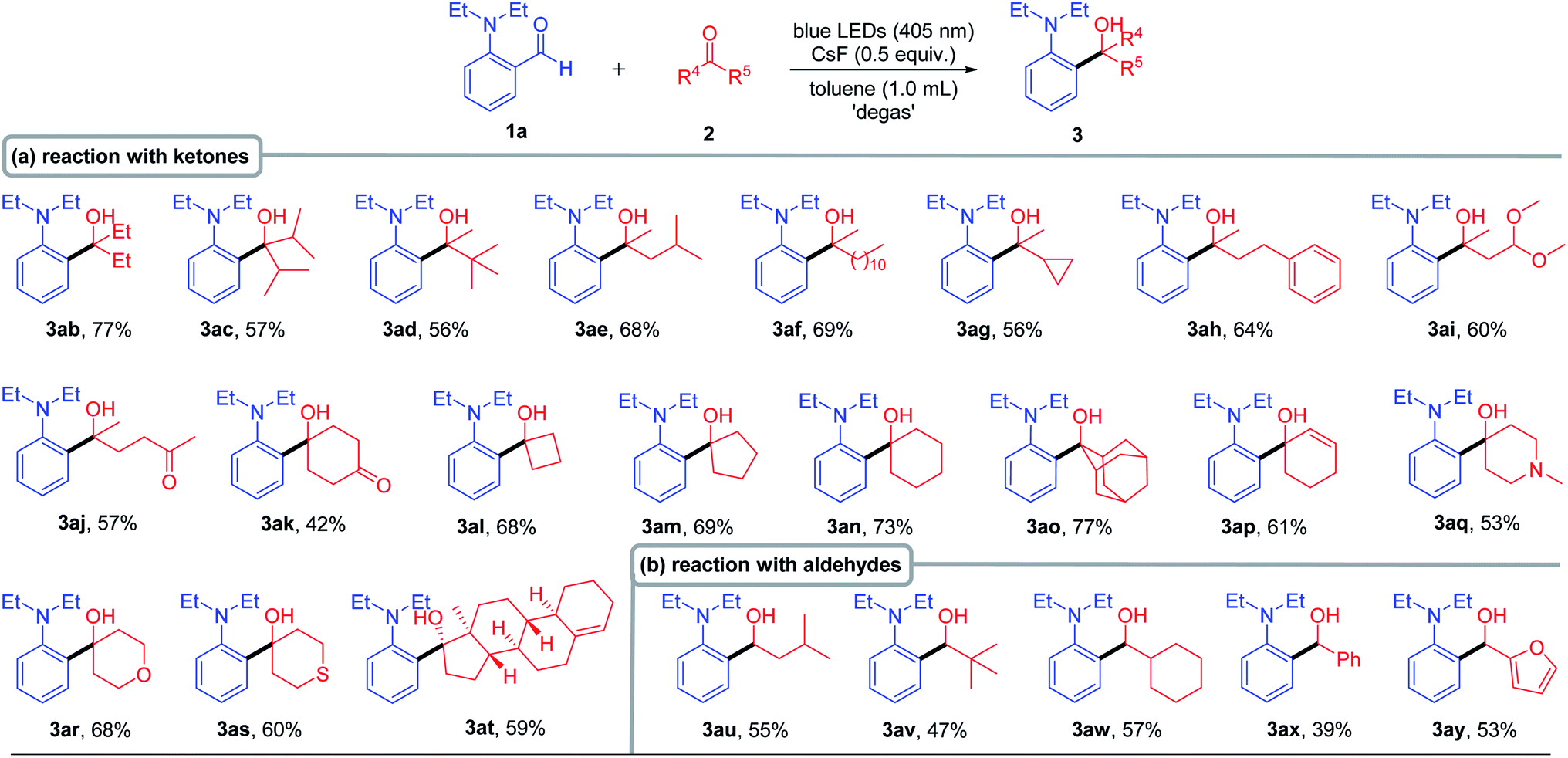
When we tried to expand the scope of ketones, some ketones were solid and expensive, which made the experiment progress sluggish. Therefore, we optimized the reaction solvent (including cyclohexane, THF, toluene, CH3CN and CH3OH) again to reduce the dosage of ketone 2b (please see the detail in Table S2 in the ESI†). We found that the corresponding cross-coupling product 3ab was obtained with the highest yield (85% NMR yield and 77% isolated yield) when toluene was used as the solvent. After obtaining the new reaction conditions, we set out to investigate the scope of ketone 2 (Table 3). For aliphatic ketones bearing a bulky alkyl group, such as diisopropylketone and tert-butyl methyl ketone, the corresponding products (3ac and 3ad) were obtained in good yields, indicating that neither the steric hindrance nor the length of the carbon chain had significant effect on this transformation, as both cyclopropyl and benzyl group substituted ketones gave the corresponding products in good yields (3ae–3ah). It is worth mentioning that the reaction has good compatibility with the ketal group (3ai). Moreover, only mono-cross-coupling products 3aj and 3ak were formed with moderate to good yields when 2,5-hexadione and 1,4-dioxocyclohexane were used as substrates, respectively. In addition, different cyclo-ketones provided satisfactory yields (3al–3ao), and even the sterically hindered adamantanone was an effective substrate (3ao, 77% yield). The reaction showed high chemo-selectivity when cyclohexenone was used as the substrate, only forming 1,2-addition product 3ap in 61% yield. Next, we evaluated the reactivity of the cyclo-ketone containing heteroatoms, including 1-methyl-4-piperidinone, tetrahydro-4-pyrone and 1-thiacyclohexan-4-one, all of which gave the corresponding products (3aq–3as) in good yields. Remarkably, the complex structure 4-estren-17-one (3at) could react smoothly, in spite of having a sterically hindered quaternary carbon center adjacent to the carbonyl group. Considering the excellent reaction generality of ketones, we then extended the reaction to aldehydes. The corresponding coupling products (3au–3ay) were successfully generated with medium to good yields for both aliphatic and aromatic aldehydes.
| a Reaction conditions: 0.1 mmol 1a, ketone or aldehyde 2 (8.0 equiv.) and CsF (0.5 equiv.) in toluene (1.0 mL) were irradiated with LEDs (405 nm, 3 W × 2) for 12 h at room temperature; isolated yields were given. |
|---|
|
To gain insight into the decarbonylative cross-coupling mechanism, we first performed UV-vis spectroscopy measurements (Fig. S3 in the ESI†). We found that 1a could be directly excited using a 405 nm LED, and the addition of ketone 2b or CsF did not affect the absorption wavelength of 1a, which showed no EDA complex formation. Besides, the Stern–Volmer quenching experiments illustrated that the excited state 1a* could not directly interact with ketone 2b because of its low quenching coefficient Ksv (Fig. S4 and S5 in ESI†). The above experimental results indicated that 1a may directly dissociate via its excited state. The isotope labelling experiments (Scheme 2) showed that the 13C labelled carbonyl group of 1a′ released 13CO (Scheme 2a), and the deuterated 4a′ was formed after adding D2O to the system (Scheme 2b), which indicated that there was a high probability of forming a phenyl anion intermediate after decarbonylation. It is well known that carbonyl compounds can be excited by light and generate a 1,2-diradical species through intersystem crossing.13 However, when deuterated 1b′ was used as the substrate, deuterium on the alkyl group was intact, without intramolecular [1,6]-D transfer or D/H exchange (Scheme 2c).14 Besides, no radical intermediates were detected when various radical trapping agents (TEMPO, BHT, isoprene and cyclohexene) were added, with only limited impact on the yields (Scheme 3a). Attempts to capture the possible radical intermediate with an intramolecular double bond did not give any cyclization product (Scheme 3b–d). Thus, this reaction might not generate the common 1,2-diradical intermediates, but went through a new dissociative process to afford a phenyl anion intermediate. The ortho-substituted alkylamine maybe important for this transformation. Thus, we investigated the effect of amino-substituted aryl aldehyde (Scheme 4). Without the alkyl substituent on the amino group 1o or with the monosubstituent on the amino group 1p or with the substituent on the para-position of 1q, the reaction could not proceed at all (Scheme 4a and b). In addition, when ketone 7 was used as the substrate, no cross-coupling product was detected, proving that the aldehyde hydrogen is essential for this transformation (Scheme 4c).
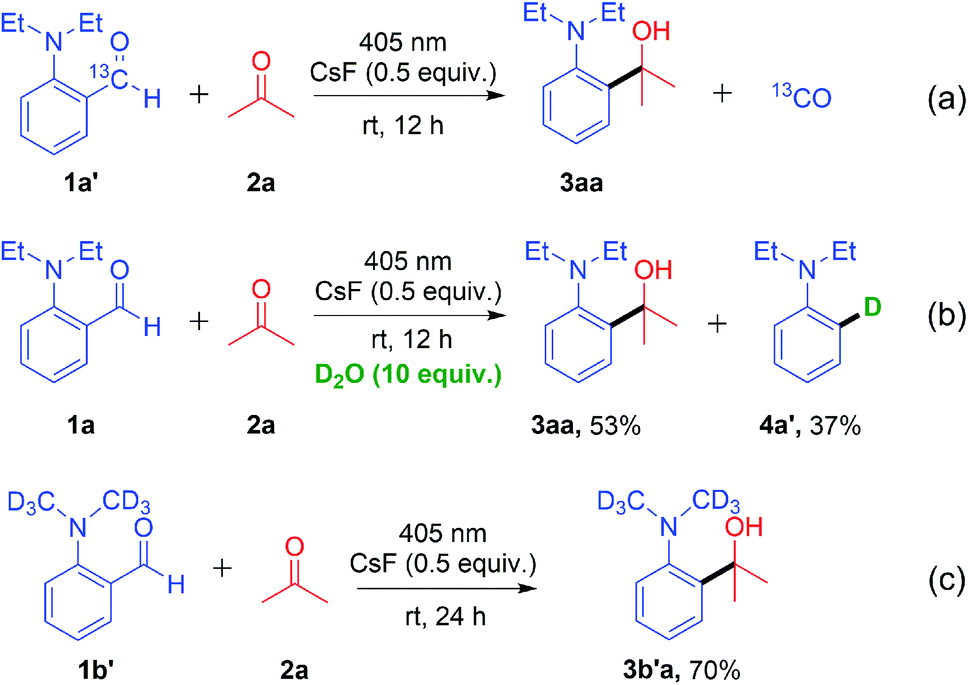 | ||
| Scheme 2 Isotope labelling experiments. | ||
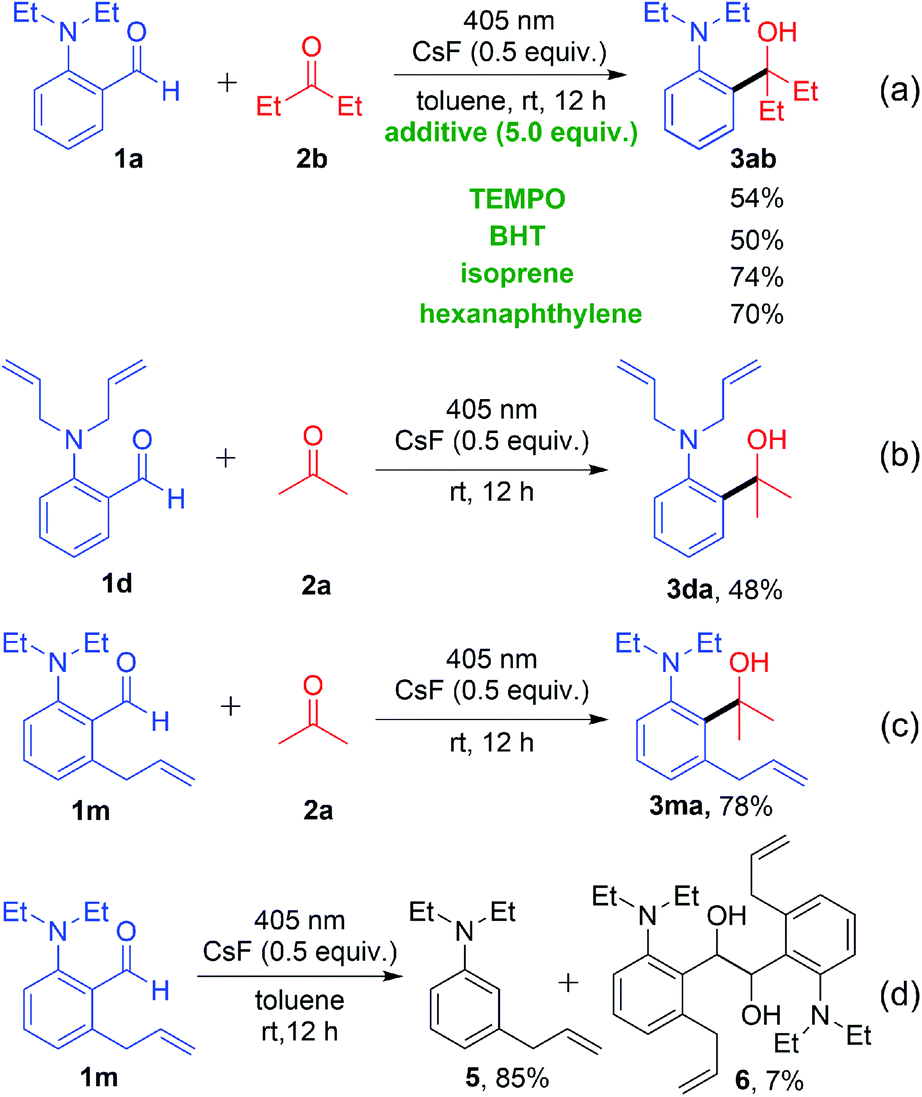 | ||
| Scheme 3 Radical capture experiments. | ||
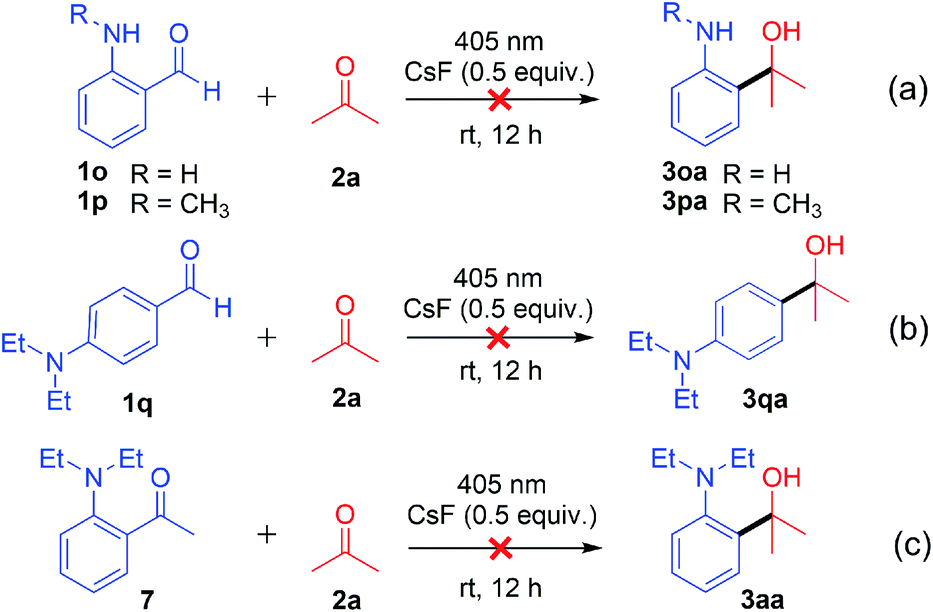 | ||
| Scheme 4 Investigating the effect of the amino-substituent on aryl aldehyde. | ||
To study the role of CsF, we carried out several control experiments (Scheme 5a and b). When only aldehyde 1a was added in toluene and irradiated with a 405 nm LED for 12 hours in the absence of CsF, four products 4a and 8–10 were generated (Scheme 5a). Among them, products 4a and 8 could be generated through phenyl anion intermediates, and products 9 and 10 could be generated through 1,2-diradical intermediates.15 When CsF was added to the reaction system, the production of 9 and 10 was inhibited, while the yields of 4a and 8 were improved. These experiments demonstrated that the addition of CsF could inhibit the formation of 1,2-diradical intermediates and promote the formation of phenyl anion intermediates. Similar results were obtained when aldehyde 1a and ketone 2b were irradiated with a 405 nm LED in toluene without CsF, obtaining only 45% yield of the cross-coupling product 3ab (Scheme 5b). However, adding CsF could significantly increase the yield of product 3ab to 88%, and inhibit the by-products 9 and 10.
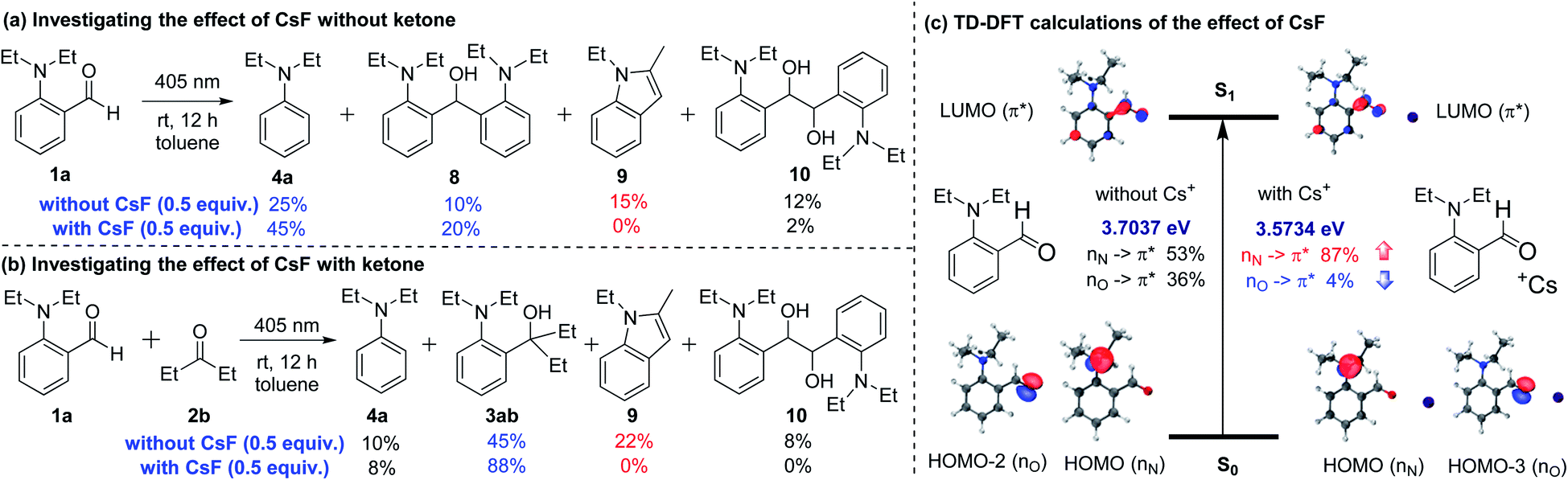 | ||
| Scheme 5 (a) Investigating the effect of CsF without ketone; (b) investigating the effect of CsF with ketone; (c) TD-DFT calculations of the effect of CsF. | ||
In order to further investigate the effect of ortho-substituted alkylamines and how CsF could inhibit the formation of 1,2-diradical intermediates, the time-dependent density functional theory (TD-DFT) calculations were performed at the CAM-B3LYP/6-311G(d,p)/IEFPCM (toluene) level (Scheme 5c). The calculation results showed that the first excited state S1 of 1a comprised two different orbital transitions. Among them, the local transition of lone pair electrons on the oxygen atom to the carbonyl π anti-bonding orbital (nO → π*) accounts for 36%, which was a common transition mode in carbonyl compounds and contributed to the generation of 1,2-diradical intermediates.13,16 Besides, a new charge-transfer transition of lone pair electrons on the nitrogen atom to the carbonyl π anti-bonding orbital (nN → π*) accounts for 53%, which may afford the phenyl anion intermediates. When the Cs ion was present in the system, the nO → π* transition dropped to 4%, and the nN → π* transition increased to 87%, possibly due to the coordination between the Cs ion and carbonyl n electrons. The DFT calculations strongly supported the results shown in Scheme 5a and b. Regarding the lack of reactivity of 2-aminobenzaldehyde (1n) and 4-(diethylamino)benzaldehyde (1o) under the standard conditions (Scheme 4a and b), the TD-DFT calculations showed that the S1 states of 1n and 1o were mainly composed of the nO, π* transition (1n, nO → π* accounts for 76%, Fig. S8 in the ESI;† and 1o, nO → π* accounts for 89%, Fig. S9 in the ESI†), rather than the nN, π* transition.
Based on the above experimental results, we proposed two possible mechanisms and the potential energy surfaces were calculated by the CAM-B3LYP/6-311G(d,p)/IEFPCM (toluene) level of theory (Fig. 1). Initially, aldehyde 1a was excited by light to reach its S1 state via the SET process (the lone pair electrons on the nitrogen atom to the carbonyl π anti-bonding orbital (nN → π*)). Then, the S1 state could undergo intramolecular [1,4]-hydrogen migration via singlet transition state TS1 to afford the singlet intermediate IM1 (path a). Alternatively, 1a in the S1 state may transform into the triplet state T1via intersystem crossing (ISC), which could also undergo the intramolecular [1,4]-hydrogen migration via triplet transition state 3TS1 to afford the triplet intermediate 3IM1 (path b). However, the results of kinetic isotope experiments showed that when the aldehyde hydrogen was replaced with deuterium, the reaction rate did not decline (please see Scheme S2 in the ESI†), which indicated that the hydrogen migration process was not the rate-limiting step. The results of calculations show that the triplet hydrogen migration process (path b) has a significant energy barrier (23.3 kcal mol−1), so the path a was more feasible. Then, IM1 could undergo the decarbonylative process via transition state TS2 to afford the phenyl anion intermediate IM2 with a 14.3 kcal mol−1 energy barrier. Finally, the intermediate IM2 could undergo nucleophilic addition to the ketone to form the target product (please see the complete energy surface in ESI, Fig. S10†).
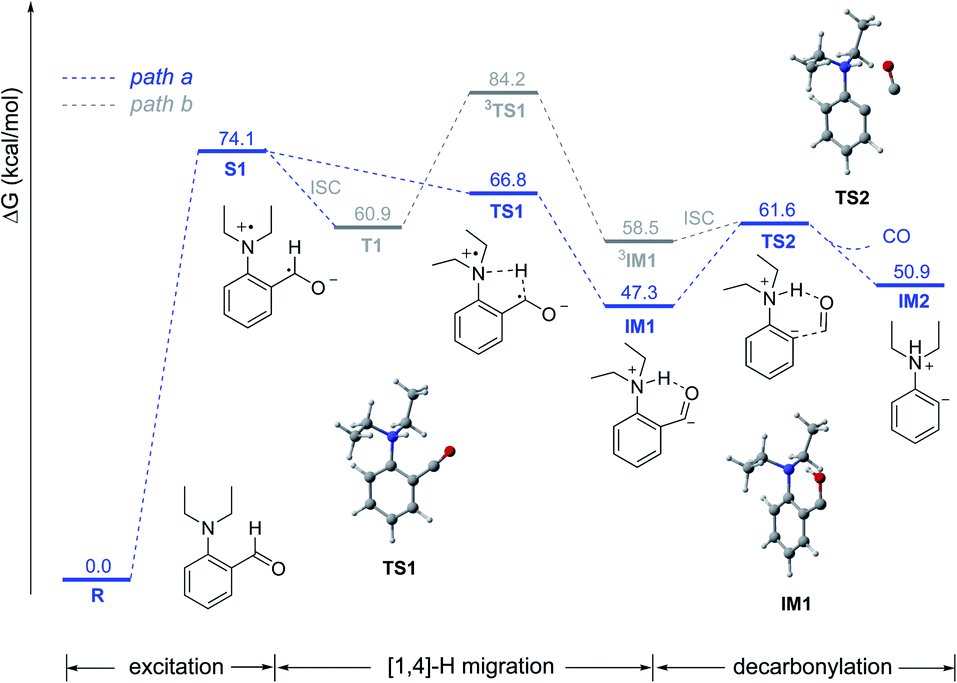 | ||
| Fig. 1 Potential energy surfaces. | ||
According to the above results, a possible mechanism is shown in Scheme 6. Aldehyde 1a is directly excited with a 405 nm LED, and the n electron on the nitrogen atom is transferred to the carbonyl π anti-bonding orbital (nN → π*) forming intermediate A. Then the generated nitrogen radical cation A abstracts the hydrogen atoms of aldehyde generating intermediate B and C, which further converts to aromatic anion D through the decarbonylative process. Then, the hydrogen bonding of intermediate E helps in the nucleophilic attack of anion D on the carbonyl of ketone via C–C bond formation and intramolecular proton transfer to generate the product 3aa.
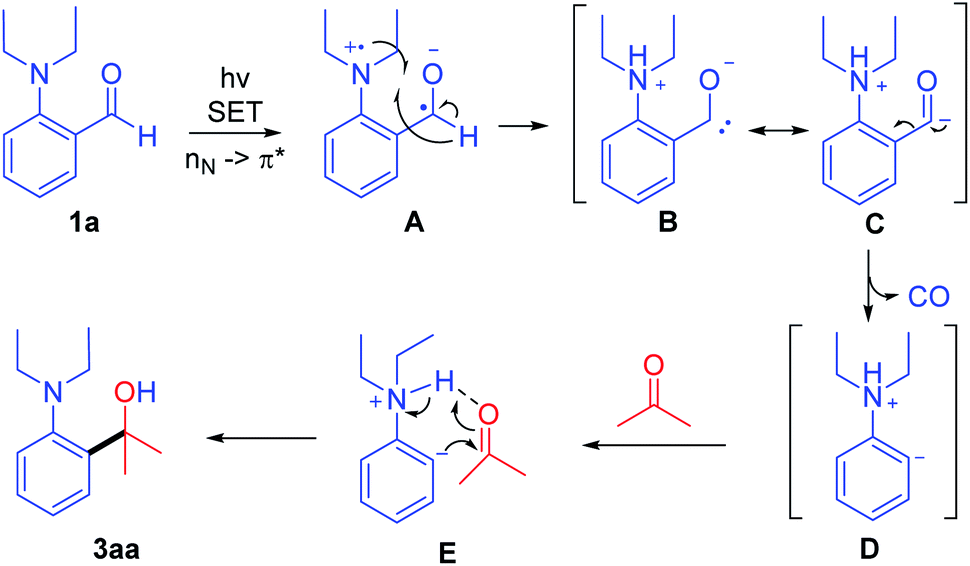 | ||
| Scheme 6 The plausible mechanism. | ||
Conclusions
In summary, a novel visible-light-induced decarbonylative cross-coupling of arylaldehydes with ketones/aldehydes under mild conditions was developed, realizing the activation of the C(sp2)–C(sp2) bond to release CO in the absence of transition-metal or external photosensitizer at room temperature. A series of tertiary/secondary benzyl alcohols were synthesized with good functional group tolerance in moderate to excellent yields under mild conditions. The detailed mechanistic investigation and DFT calculations illustrated that the reaction proceeded through photoexcitation and decarbonylation of aldehyde to generate an aromatic anion, followed by the nucleophilic addition to ketones/aldehydes. Furthermore, the key role of cesium fluoride was confirmed by control experiments and DFT calculations. This new mechanism provides an opportunity to develop new catalyst-free organic reactions.Data availability
All experimental and computational data is available in the ESI.†Author contributions
Y. W. performed the experiments with contributions from Y. L., Y. L. performed DFT calculations. H. Z. and C.-J. L. supervised the project. All the authors analyzed the data, discussed results and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSFC (21971093), the International Joint Research Centre for Green Catalysis and Synthesis (grant No. 2016B01017 and 18JR4RA003) and the 111 Project for support of our research. We also thank the Canada Research Chair (Tier I) foundation, the E.B. Eddy Endowment Fund, the CFI, NSERC, and FQRNT to C.-J. Li.Notes and references
- G. B. Dong, C–C Bond Activation, ed. G. Dong, Springer, Berlin, 2013, vol. 346, pp. 1–258 Search PubMed.
- A. Dermenci and G. Dong, Sci. China: Chem., 2013, 56, 685–701 CrossRef CAS.
- (a) W.-C. Yang, J.-G. Feng, L. Wu and Y.-Q. Zhang, Adv. Synth. Catal., 2019, 361, 1700–1709 CrossRef CAS; (b) H. Lu, T.-Y. Yu, P.-F. Xu and H. Wei, Chem. Rev., 2021, 121, 365–411 CrossRef CAS PubMed.
- (a) E. P. K. Olsen and R. Madsen, Chem.–Eur. J., 2012, 18, 16023–16029 CrossRef CAS PubMed; (b) J. Weatherly, K. Ding, S. Xu, R. Alotaibi, K. Paudel and E. W. Reinheimer, J. Org. Chem., 2017, 82, 4924–4929 CrossRef PubMed; (c) Y.-M. Pan, Y.-J. Ding, W.-H. Li, C.-Y. Li, Y. Li, H.-T. Tang and H.-S. Wang, Chem. Commun., 2018, 54, 8446–8449 RSC; (d) A. Voutchkova-Kostal, D. Ainembabazi, C. Reid, A. Chen, N. An and J. Kostal, J. Am. Chem. Soc., 2020, 142, 696–699 CrossRef PubMed.
- (a) C.-J. Li, L. Yang, T. Zeng, Q. Shuai and X. Guo, Chem. Commun., 2011, 47, 2161–2163 RSC; (b) L. Yang, L. Kang, F. Zhang and L.-T. Ding, RSC Adv., 2015, 5, 100452–100456 RSC; (c) M. Rueping, L. Guo, W. Srimontree, C. Zhu, B. Maity, X. Liu and L. Cavallo, Nat. Commun., 2019, 10, 1957 CrossRef PubMed.
- (a) V. M. Dong, S. K. Murphy, J.-W. Park and F. A. Cruz, Science, 2015, 347, 56 CrossRef PubMed; (b) S. Kusumoto, T. Tatsuki and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 8458–8461 CrossRef CAS PubMed; (c) V. M. Dong, X. Wu, F. A. Cruz and A. Lu, J. Am. Chem. Soc., 2018, 140, 10126–10130 CrossRef PubMed.
- (a) C.-J. Li, X. Guo and J. Wang, Org. Lett., 2010, 12, 3176–3178 CrossRef PubMed; (b) H. Yao, P. Sun, S. Gao, C. Yang, S. Guo and A. Lin, Org. Lett., 2016, 18, 6464–6467 CrossRef PubMed; (c) C.-J. Li, N. Moitessier, A. Tomberg, S. Kundu and F. Zhou, ACS Omega, 2018, 3, 3218–3227 CrossRef PubMed.
- (a) W.-Y. Li, Q.-Q. Wang and L. Yang, Org. Biomol. Chem., 2017, 15, 9987–9991 RSC; (b) X. Xu, Z. Luo, X. Han, Y. Fang, P. Liu, C. Feng and Z. Li, Org. Chem. Front., 2018, 5, 3299–3305 RSC.
- C.-J. Li, X. Guo and J. Wang, J. Am. Chem. Soc., 2009, 131, 15092–15093 CrossRef PubMed.
- M. Shi, X. Fan, R. Liu and Y. Wei, Org. Chem. Front., 2019, 6, 2667–2671 RSC.
- L. Yang, C.-S. Wu, R. Li and Q.-Q. Wang, Green Chem., 2019, 21, 269–274 RSC.
- (a) L. Li, W. Liu, H. Zeng, X. Mu, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 8328–8331 CrossRef CAS PubMed; (b) P. Liu, W. Liu and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 14315–14321 CrossRef CAS PubMed; (c) D. Cao, C. Yan, P. Zhou, H. Zeng and C.-J. Li, Chem. Commun., 2019, 55, 767–770 RSC; (d) H. Zeng, Q. Dou and C.-J. Li, Org. Lett., 2019, 21, 1301–1305 CrossRef CAS PubMed; (e) W. Liu, J. Li, C.-Y. Huang and C.-J. Li, Angew. Chem., Int. Ed., 2020, 59, 1786–1796 CrossRef PubMed; (f) Y. Lang, X. Peng, C.-J. Li and H. Zeng, Green Chem., 2020, 22, 6323–6327 RSC; (g) Q. Dou, C.-J. Li and H. Zeng, Chem. Sci., 2020, 11, 5740–5744 RSC; (h) D. Cao, Z. Chen, L. Lv, H. Zeng, Y. Peng and C.-J. Li, iScience, 2020, 23, 101419 CrossRef CAS PubMed; (i) Y. Lang, C.-J. Li and H. Zeng, Org. Chem. Front., 2021, 8, 3594–3613 RSC; (j) D. Cao, P. Pan, C.-J. Li and H. Zeng, Green Synthesis and Catalysis, 2021, 2, 303–306 CrossRef; (k) Q. Dou, Y. Lang, H. Zeng and C.-J. Li, Fundamental Research, 2021, 1, 742–746 CrossRef; (l) Q. Dou, L. Geng, B. Cheng, C.-J. Li and H. Zeng, Chem. Commun., 2021, 57, 8429–8432 RSC; (m) D. Cao, S. Xia, P. Pan, H. Zeng, C.-J. Li and Y. Peng, Green Chem., 2021, 23, 7539–7543 RSC.
- W. M. Nau, F. L. Cozens and J. C. Scaiano, J. Am. Chem. Soc., 1996, 118, 2275–2282 CrossRef CAS.
- I. D. Jurberg, B. Peng, E. Wostefeld, M. Wasserloos and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 1950–1953 CrossRef CAS PubMed.
- (a) J. Zhou, L. Li, M. Yan and W. Wei, Green Chem., 2019, 21, 5521 RSC; (b) Z. Qiu, H. D. M. Pham, J. Li, C.-C. Li, D. J. Castillo-Pazos, R. Z. Khaliullin and C.-J. Li, Chem. Sci., 2019, 10, 10937 RSC; (c) X. Yuan, X. Wu, S. Dong, G. Wu and J. Ye, Org. Biomol. Chem., 2016, 14, 7447 RSC; (d) W. Li, Y. Duan, M. Zhang, J. Cheng and C. Zhu, Chem. Commun., 2016, 52, 7596 RSC; (e) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828 CrossRef CAS PubMed.
- (a) I. K. Sideri, E. Voutyritsa and C. G. Kokotos, Org. Biomol. Chem., 2018, 16, 4596 RSC; (b) M. A. Theodoropoulou, N. F. Nikitas and C. G. Kokotos, Beilstein J. Org. Chem., 2020, 16, 833 CrossRef CAS PubMed; (c) N. F. Nikitas, P. L. Gkizis and C. G. Kokotos, Org. Biomol. Chem., 2021, 19, 5237 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06278f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |