
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free allylic C–H nitrogenation, oxygenation, and carbonation of alkenes by thianthrenation†
Ming-Shang
Liu
,
Hai-Wu
Du
and
Wei
Shu
 *
*
Shenzhen Grubbs Institute, Department of Chemistry, Guangdong Provincial Key Laboratory of Catalysis, Southern University of Science and Technology, Shenzhen 518055, Guangdong, P. R. China. E-mail: shuw@sustech.edu.cn
First published on 20th December 2021
Abstract
Selective functionalization of allylic C–H bonds into other chemical bonds is among the most straightforward and attractive, yet challenging transformations. Herein, a transition-metal-free protocol for direct allylic C–H nitrogenation, oxygenation, and carbonation of alkenes by thianthrenation was developed. This operationally simple protocol allows for the unified allylic C–H amination, esterification, etherification, and arylation of vinyl thianthrenium salts. Notably, the reaction furnishes multialkyl substituted allylic amines, ammonium salts, sulfonyl amides, esters, and ethers in good yields. The reaction proceeds under mild conditions with excellent functional group tolerance and could be applied to late-stage allylation of natural products, drug molecules and peptides with excellent chemoselectivity.
Introduction
Methods for direct allylic C–H functionalizations are among the most attractive transformations to streamline organic synthesis as they maximize the step- and atom-economy to generate stereodefined allylic species amenable to further chemical transformations, thus minimizing the cost and waste.1,2 Traditional allylic C–H functionalization reactions generally require the catalysis of transition-metals such as Pd, Cu, and Ir, which involves the formation of an allyl–metal complex via C–H activation followed by an intra- or intermolecular nucleometallation (Scheme 1a).3,4 Recently, the radical cleavage of an allylic C–H bond via hydrogen atom transfer is also developed to generate carbon-centered radical intermediates, which could be involved in following radical processes or transition-metal catalysis.3m,5 These two strategies proved to be powerful for organic synthesis and extensively investigated. The stereochemical outcomes heavily rely on the properties of transition-metals and anchoring ligands and are mostly dominated by thermodynamic control, leading to the formation of C–H functionalization products with more stable E-selectivity.3,6,7 On the contrast, the realization of Z-selective allylic C–H functionalizations is more challenging and remains elusive.8 On the other hand, organothianthrenium salts could be easily prepared from arenes and alkenes by thianthrenation using stoichiometric thianthrene S-oxide or phenoxathiine 10-oxide as the mediators, which could serve as the precursors for both cross-coupling reactions and radical processes.9 This two-step strategy creates potential chemical space for manipulating the C–H bond of arenes and alkenes. Recently, the Ritter group reported seminal work on selective C–H functionalizations of arenes via thianthrenation, providing access to diverse chemical bonds from aryl C–H bonds (Scheme 1b).10 The Wang group developed the Pd-catalyzed site-selective C–H borylation and arylation of arenes by employing the same strategy (Scheme 1b).11 Recently, the Shi group developed the utilization of alkyl thianthrenium salts for C–B and C–C bond formation.12 The Wickens group reported the electrochemical synthesis of 1,2-disubstituted thianthrenium salts from alkenes and thianthrene, which were used as the key intermediates to produce aziridines by C–N formation.13 In 2020, the Ritter group developed elegant examples of C–H functionalizations of alkenes via the isolated vinyl thianthrenium salts, leading to alkylation, alkynylation, arylation, halogenation, and trifluorosulfinylation of alkenes (Scheme 1b).14 Despite the promising progress, all the above-mentioned C–H functionalization reactions are restricted to ipso-functionalizations of arenes or alkenes via thianthrenation by replacing the C–S bond in the thianthrenium salts. Yet, no example of C–H functionalization at the allylic position of alkenes was reported. Herein, we report the metal-free selective allylic C–H functionalizations of alkenes via thianthrenation (Scheme 1c). The mild conditions allow for the allylic C–H nitrogenation, oxygenation, and carbonation of alkenes at room temperature by the formation of C–O, C–C, and C–N bonds, affording diverse esterifications, thioesterifications, etherifications, aminations, and arylations of allylic C–H bonds.15 Notably, the metal-free C–H functionalizations deliver allylic esters, ethers, amines, ammonium salts, and amides with preferred Z-selectivity.16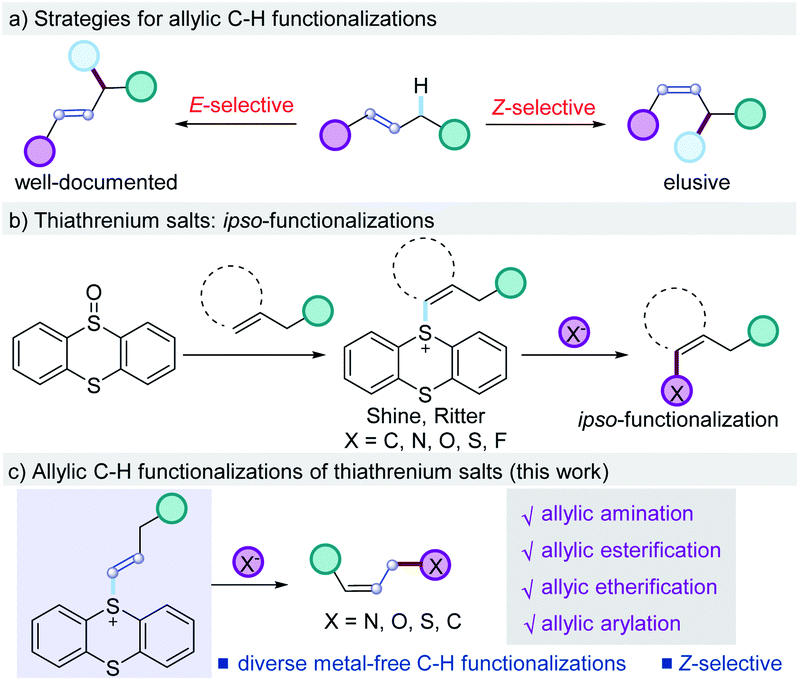 | ||
| Scheme 1 Impetus for metal-free allylic C–H functionalizations using thianthrenium salts. | ||
Results and discussion
We started to investigate the reaction of cyclohexylvinylthianthrenium salt 1a with benzoic acid 2a. Interestingly, translocation of the alkene by allylic C–H functionalization was observed, affording 1,1,2-trialkyl substituted alkene 3a instead of ipso-vinyl substitution of vinylthianthrenium. After evaluation of a variety of reaction parameters, we defined the use of potassium carbonate (1.0 equiv.) as the base in DCM (0.1 M) at room temperature as standard conditions, delivering the desired product 3a in 91% isolated yield (Table 1, entry 1). The use of other bases could also mediate the reaction. Cesium carbonate and potassium phosphate tribasic delivered 3a in 85% and 72% yields, respectively (Table 1, entries 2 and 3). Lithium carbonate led to no formation of 3a (Table 1, entry 4). The reaction proceeded in most tested solvents, including polar and nonpolar solvents, furnishing the desired product 3a in 47–88% yields (Table 1, entries 5–9).17|
|
||
|---|---|---|
| Entries | Variations as shown | Yield of 3a |
| a The reaction was conducted using 1a (0.1 mmol) and 2a (0.12 mmol) at room temperature for 24 h. Yield was determined by 1H NMR of the crude mixture using mesitylene as the internal standard. b Isolated yield after flash chromatography. | ||
| 1 | None | 92% (91%)b |
| 2 | Cs2CO3 as base | 85% |
| 3 | K3PO4 as base | 72% |
| 4 | Li2CO3 as base | N.D. |
| 5 | DCE as solvent | 88% |
| 6 | CH3CN as solvent | 82% |
| 7 | Acetone as solvent | 69% |
| 8 | THF as solvent | 47% |
| 9 | Toluene as solvent | 81% |
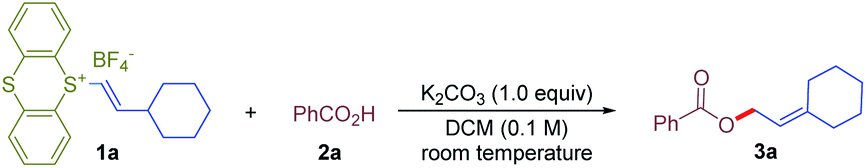
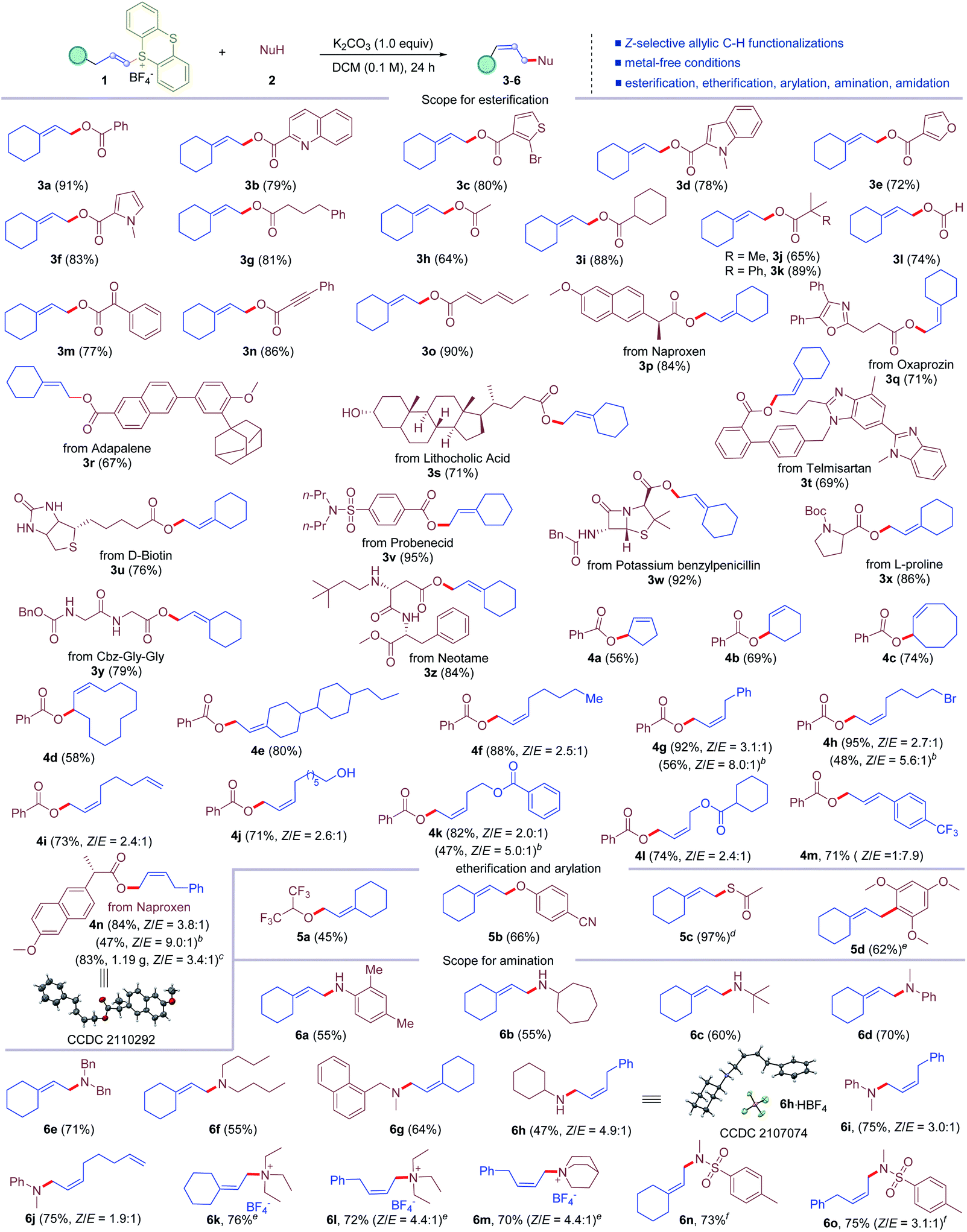
With the optimized conditions established, we turned to evaluate the scope of this reaction. It is found that the reaction conditions tolerate a variety of vinylthianthrenium salts and different nucleophiles with broad functional group and substitution pattern compatibility (Table 2). First, the scope of allylic C–H esterification was examined. A surprisingly wide range of carboxylic acids were tolerated (3a–3o). Aromatic and heteroaromatic carboxylic acids, such as quinoline carboxylic acid, thiophene carboxylic acid, indole carboxylic acid, furan carboxylic acid, and pyrrole carboxylic acid, could be involved in the reaction to deliver the allylic C–H esterification products (3b–3f) in 72–83% yields. Aliphatic acids, including α-linear, α-branched and α-tertiary carboxylic acids, are good substrates for this metal-free C–H functionalization process, giving corresponding esters (3g–3k) in 64–89% yields. Formic acid could form allylic formic ester 3l in 74% yield. Benzoylformic acid could form corresponding ester 3m in 77% yield via allylic C–H functionalization. Propiolic acid was tolerated to give corresponding allylic ester 3n in 86% yield. Conjugated dienoic acid was converted to 3o in 90% yield, leaving the conjugated diene intact. Moreover, this protocol was applicable to late-stage functionalization of complex molecules. Naproxen was transformed into corresponding allylic ester 3p in 84% yield without affecting the stereogenic center. Oxaprozin, adapalene, lithocholic acid, telmisartan, D-biotin, and probenecid were all good substrates for this allylic C–H esterification reaction, furnishing corresponding esters (3q–3v) in 67–95% yields. Potassium benzylpenicillin was compatible in the reaction, delivering the esterification product 3w in 92% yield in high chemoselectivity, without the formation of the N-allylation product. Notably, the reaction tolerated a wide range of natural and unnatural amino acids and peptides. N-Boc protected L-proline was successfully converted to allylic ester 3x in 86% yield. Peptides, such as CBz-Gly-Gly and neotame underwent allylic C–H esterification selectively to leave free amide and amine unreactive, affording 3y and 3z in 79% and 84% yields, respectively. Next, the scope of vinylthianthrenium salts was investigated. Five, six, and eight-membered cyclic alkenes could be involved to undergo allylic C–H oxygenation with benzoic acid to furnish cyclic allylic esters (4a–4c) in 56–74% yields. Cyclododecene was converted to corresponding allylic ester 4d in 58% yield with exclusive Z-selectivity. A mixture of isomers of alkene-derived thianthrenium salt delivered a single isomer of the corresponding allylic C–H oxygenation product 4e in 80% yield. It is noteworthy that 1-substituted alkene based thianthrenium salts were converted to allylic esterification products smoothly. Surprisingly, the reaction delivered 1,2-substituted alkenes favoring Z-selectivity. Alkenes with pendant bromides, alkenes, alcohols, and esters were all compatible in the reaction, leading to the esterification of allylic C–H bonds in 56–95% yields with 2.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–3.1:1 ratios of Z-selectivity (4f–4l). Additionally, 3-phenyl-1-propene derived thianthrenium salt furnished styrene type allylic ester 4m in 71% yield with E/Z = 7.9:1. Moreover, the reaction could be easily scaled up. The reaction on a 4.0 mmol scale afforded 1.19 g of 4n in 83% yield. Notably, the Z-selectivity could be further improved up to 9.0:1 (4g, 4h, 4k, and 4n) using pentamethyldiethylenetriamine (PMDTA) as the base, probably due to the rigid configuration with a more sterically hindered base. The configuration of the major product was confirmed unambiguously by X-ray diffraction of 4n. Next, the application of allylic C–H functionalization to other nucleophiles was examined. Allylic C–H etherification was successful using both alcohols and phenols as the nucleophile, furnishing alkyl and phenyl allylic ethers (5a and 5b) in 45% and 66% yields. Thioesterification of the allylic C–H bond was achieved in 97% yield (5c) using potassium thioacetate. Allylic C–H arylation was also accomplished in 62% yield (5d) with trimethoxybenzene as the nucleophile. Moreover, allylic C–H amination was also demonstrated. Primary anilines and aliphatic amines were all well tolerated, delivering allylic secondary amines (6a–6c) in 55–60% yields. Secondary amines with different substitution patterns were all good substrates for this reaction, giving diverse allylic tertiary amines (6d–6g) in 55–71% yields. Monosubstituted alkene based thianthrenium salts were converted to Z-selective 1,2-disubstituted allylic amines in 47–75% yields with 1.9:1–4.9:1 ratios (6h–6j). The configuration of the major isomer of allylic amines was further confirmed by the X-ray diffraction of the salt of 6h. Impressively, tertiary amines were also compatible in the allylic C–H amination reaction to afford allylic trialkyl ammonium salts in 70–76% yields (6k–6m). When 4-phenyl-1-butene derived thianthrenium salt was exposed to the reaction conditions with triethyl amine and quinuclidine, the desired allylic ammonium salts were obtained in 72% and 70% yields (6l and 6m), favoring Z-selectivity of 4.4:1. Notably, allylic C–H sulfonyl amidation of vinyl thianthrenium salts was also successful, affording corresponding allylic sulfonyl amides in 73% and 75% yields (6n and 6o), respectively.
:1–3.1:1 ratios of Z-selectivity (4f–4l). Additionally, 3-phenyl-1-propene derived thianthrenium salt furnished styrene type allylic ester 4m in 71% yield with E/Z = 7.9:1. Moreover, the reaction could be easily scaled up. The reaction on a 4.0 mmol scale afforded 1.19 g of 4n in 83% yield. Notably, the Z-selectivity could be further improved up to 9.0:1 (4g, 4h, 4k, and 4n) using pentamethyldiethylenetriamine (PMDTA) as the base, probably due to the rigid configuration with a more sterically hindered base. The configuration of the major product was confirmed unambiguously by X-ray diffraction of 4n. Next, the application of allylic C–H functionalization to other nucleophiles was examined. Allylic C–H etherification was successful using both alcohols and phenols as the nucleophile, furnishing alkyl and phenyl allylic ethers (5a and 5b) in 45% and 66% yields. Thioesterification of the allylic C–H bond was achieved in 97% yield (5c) using potassium thioacetate. Allylic C–H arylation was also accomplished in 62% yield (5d) with trimethoxybenzene as the nucleophile. Moreover, allylic C–H amination was also demonstrated. Primary anilines and aliphatic amines were all well tolerated, delivering allylic secondary amines (6a–6c) in 55–60% yields. Secondary amines with different substitution patterns were all good substrates for this reaction, giving diverse allylic tertiary amines (6d–6g) in 55–71% yields. Monosubstituted alkene based thianthrenium salts were converted to Z-selective 1,2-disubstituted allylic amines in 47–75% yields with 1.9:1–4.9:1 ratios (6h–6j). The configuration of the major isomer of allylic amines was further confirmed by the X-ray diffraction of the salt of 6h. Impressively, tertiary amines were also compatible in the allylic C–H amination reaction to afford allylic trialkyl ammonium salts in 70–76% yields (6k–6m). When 4-phenyl-1-butene derived thianthrenium salt was exposed to the reaction conditions with triethyl amine and quinuclidine, the desired allylic ammonium salts were obtained in 72% and 70% yields (6l and 6m), favoring Z-selectivity of 4.4:1. Notably, allylic C–H sulfonyl amidation of vinyl thianthrenium salts was also successful, affording corresponding allylic sulfonyl amides in 73% and 75% yields (6n and 6o), respectively.
| a The reaction was conducted on a 0.2 mmol scale. Standard conditions, see Table 1 for details. b PMDTA (pentamethyldiethylenetriamine, 1.0 equiv.) was used as the base with a nucleophile (2.0 equiv.) in DCE (0.05 M). c 4.0 mmol scale reaction. d Potassium thioacetate and H2O (0.2 μL) were used. e The reaction was conducted using amine (2.0 equiv.) and H2O (0.2 μL). f KOH (1.0 equiv.) was used as the base. |
|---|
|
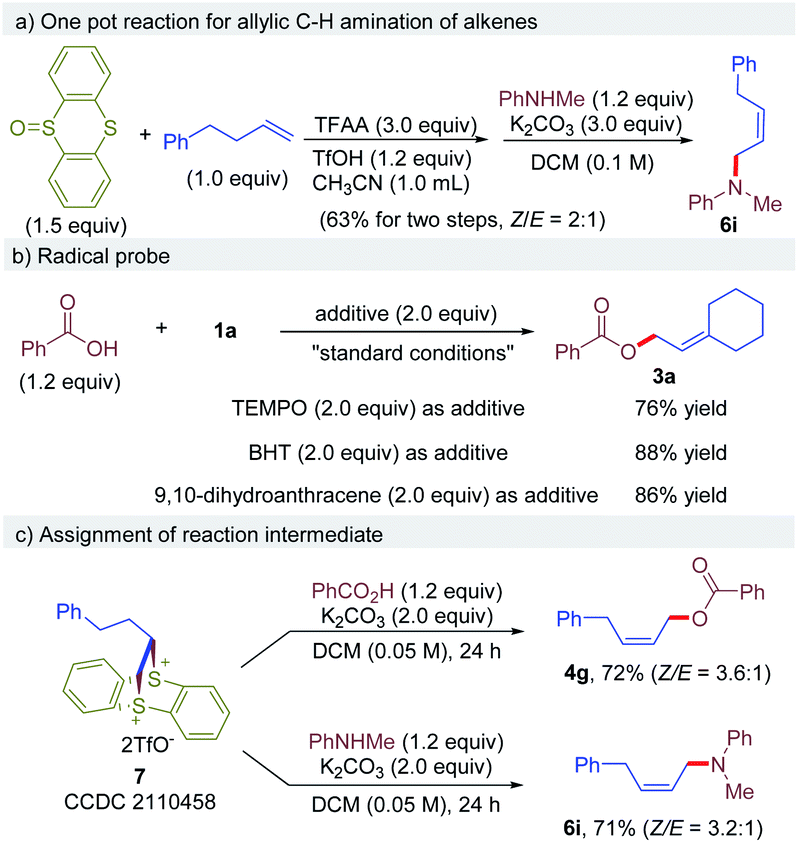
Next, a one-pot operation from an alkene and thianthreneoxide, followed by a nucleophile was demonstrated to prove the practicality of this reaction (Scheme 2a). The one pot reaction of 4-phenyl-1-butene with thianthrene S-oxide, followed by N-methylaniline afforded the desired allylic amine 6i in 63% yield without any workup or intermediate purification, which is comparable to previous results of step-wise procedure. Next, the reaction of 1a with 2a was conducted in the presence of a radical scavenger under otherwise identical to standard conditions (Scheme 2b). It is shown that the reaction proceeded smoothly in the presence of TEMPO, BHT or 9,10-dihydroanthracene, affording the desired product 3a without affecting the efficacy. These results exclude the involvement of radical intermediates in this reaction. To further probe the mechanism of the reaction, a dithianthrenium salt 7 was subjected to the reaction with benzoic acid or N-methylaniline, and corresponding allylic C–H esterification product 4g and amination product 6i were obtained in 72% and 71% yield, respectively (Scheme 2c). The yield and stereoselectivity are comparable to the results obtained using corresponding vinylthianthrenium. These results indicate that dithianthrenium salt could serve as the reactive intermediate for this reaction.
 | ||
| Scheme 2 One-pot synthesis and control experiments. | ||
Based on the literature and the experimental results, a plausible mechanism is described in Scheme 3. First, intramolecular attack of sulfur on the alkene moiety of vinylthianthrenium salt could deliver the dithianthrenium salt M1, which could further undergo site-selective ring-opening by intermolecular attack by a nucleophile to give an alkylthianthrenium salt intermediate M2. In the presence of a base, M2 would undergo a syn-elimination viaTS1 to give the final allylic C–H functionalization product in favor of Z-selectivity.
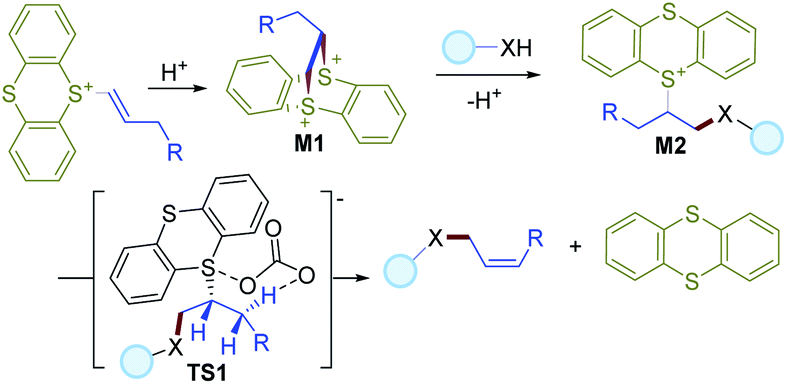 | ||
| Scheme 3 Proposed mechanism for the reaction. | ||
Conclusions
In summary, a unified transition-metal-free protocol for diverse functionalizations of allylic C–H bonds of alkenes by thianthrenation under mild conditions has been demonstrated for the first time. Notably, the reaction features Z-selectivity to afford multi-alkyl substituted allylic esters, thioesters, ethers, primary, secondary, tertiary amines, amides, and arenes in good yields without incorporation of any transition-metals. The one-pot procedure proved efficient to access direct allylic C–H functionalizations of alkenes. The reaction tolerates a wide range of O-, N-nucleophiles with excellent functional group tolerance, and could be applied to late-stage functionalizations of natural products, amino acids, and drug-like molecules with excellent chemoselectivity.Data availability
Experimental data has been provided as ESI.†Author contributions
M. S. L. discovered the reactions. W. S. and M. S. L. designed the experiments. M. S. L. and H. W. D performed and analysed the experiments. W. S. and M. S. L. wrote the manuscript. All authors discussed the experimental results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We sincerely acknowledge NSFC (21971101, 21801126, and 22171127), Thousand Talents Program for Young Scholars, Guangdong Basic and Applied Basic Research Foundation (2019A1515011976), Department of Education of Guangdong Province (2021KTSCX06), The Stable Support Plan Program of Shenzhen Natural Science Fund (No. 20200925152608001), the Pearl River Talent Recruitment Program (2019QN01Y261), and Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002) for financial support. We acknowledge the assistance of SUSTech Core Research Facilities. We thank Dr Xiaoyong Chang (SUSTech) for X-ray crystallographic analysis (CCDC 2110292, 2107074, and 2110458†) and Bi-Hong Chen (SUSTech) for reproducing the results of 3g, 4f, 6a and 6i.Notes and references
- (a) J. Eames and M. Watkinson, Angew. Chem., Int. Ed., 2001, 40, 3567–3571 CrossRef CAS; (b) W. Adam and O. Krebs, Chem. Rev., 2003, 103, 4131–4146 CrossRef CAS PubMed; (c) T. A. Ramirez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 931–942 RSC; (d) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173–6214 RSC; (e) L. Bayeh and U. K. Tambar, ACS Catal., 2017, 7, 8533–8543 CrossRef CAS PubMed; (f) A. M. Kazerouni, Q. A. McKoy and S. B. Blakey, Chem. Commun., 2020, 56, 13287–13300 RSC; (g) P.-S. Wang and L.-Z. Gong, Acc. Chem. Res., 2020, 53, 2841–2854 CrossRef CAS PubMed.
- G. Dyker, in Handbook of C-H Transformations, Wiley-VCH, Weinheim, 2005 Search PubMed.
- (a) R. C. Larock, T. R. Hightower, L. A. Hasvold and K. P. Peterson, J. Org. Chem., 1996, 61, 3584–3585 CrossRef CAS PubMed; (b) J. Franzen and J.-E. Backvall, J. Am. Chem. Soc., 2003, 125, 6056–6057 CrossRef CAS PubMed; (c) M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346–1347 CrossRef CAS PubMed; (d) K. J. Fraunhoffer and M. C. White, J. Am. Chem. Soc., 2007, 129, 7274–7276 CrossRef CAS PubMed; (e) G. Liu, G. Yin and L. Wu, Angew. Chem., Int. Ed., 2008, 47, 4733–4736 CrossRef CAS PubMed; (f) S. A. Reed and M. C. White, J. Am. Chem. Soc., 2008, 130, 3316–3318 CrossRef CAS PubMed; (g) S. Lin, C.-X. Song, G.-X. Cai, W.-H. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2008, 130, 12901–12903 CrossRef CAS PubMed; (h) A. J. Young and M. C. White, J. Am. Chem. Soc., 2008, 130, 14090–14091 CrossRef CAS PubMed; (i) N. A. Vermeulen, J. H. Delcamp and M. C. White, J. Am. Chem. Soc., 2010, 132, 11323–11328 CrossRef CAS PubMed; (j) A. N. Campbell, P. B. White, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 15116–15119 CrossRef CAS PubMed; (k) G.-W. Wang, A.-X. Zhou, S.-X. Li and S.-D. Yang, Org. Lett., 2014, 16, 3118–3121 CrossRef CAS PubMed; (l) B. L. Tran, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 17292–17301 CrossRef CAS PubMed; (m) X. Yi and X. Hu, Chem. Sci., 2021, 12, 1901–1906 RSC; (n) A. T. Parsons and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9120–9123 CrossRef CAS PubMed; (o) J. Xu, Y. Fu, D.-F. Luo, Y.-Y. Jiang, B. Xiao, Z.-J. Liu, T.-J. Gong and L. Liu, J. Am. Chem. Soc., 2011, 133, 15300–15303 CrossRef CAS PubMed; (p) X. Wang, Y. Ye, S. Zhang, J. Feng, Y. Xu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 16410–16413 CrossRef CAS PubMed.
- (a) T. Knecht, S. Mondal, J.-H. Ye, M. Das and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 7117–7121 CrossRef CAS PubMed; (b) H. Lei and T. Rovis, J. Am. Chem. Soc., 2019, 141, 2268–2273 CrossRef CAS PubMed; (c) Q. Li and Z.-X. Yu, J. Am. Chem. Soc., 2010, 132, 4542–4543 CrossRef CAS PubMed; (d) M. Sekine, L. Ilies and E. Nakamura, Org. Lett., 2013, 15, 714–717 CrossRef CAS PubMed.
- (a) X. Zhang, M. Wang, P. Li and L. Wang, Chem. Commun., 2014, 50, 8006–8009 RSC; (b) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74–77 CrossRef CAS PubMed; (c) R. Zhou, H. Liu, H. Tao, X. Yu and J. Wu, Chem. Sci., 2017, 8, 4654–4659 RSC; (d) L. Huang and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 10333–10337 CrossRef CAS PubMed; (e) J. L. Schwarz, F. Schafers, A. Tlahuext-Aca, L. Luckemeier and F. Glorius, J. Am. Chem. Soc., 2018, 140, 12705–12709 CrossRef CAS PubMed; (f) J. Li, Z. Zhang, L. Wu, W. Zhang, P. Chen, Z. Lin and G. Liu, Nature, 2019, 574, 516–521 CrossRef CAS PubMed; (g) C. Huang, R.-N. Ci, J. Qiao, X.-Z. Wang, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 11779–11783 CrossRef CAS PubMed.
- (a) K. B. Sharpless, T. Hori, L. K. Truesdale and C. O. Dietrich, J. Am. Chem. Soc., 1976, 98, 269–271 CrossRef CAS; (b) H. Yamanaka, J.-i. Matsuo, A. Kawana and T. Mukaiyama, Chem. Lett., 2003, 32, 626–627 CrossRef CAS; (c) H. Bao and U. K. Tambar, J. Am. Chem. Soc., 2012, 134, 18495–18498 CrossRef CAS PubMed; (d) J. A. Souto, D. Zian and K. Muñiz, J. Am. Chem. Soc., 2012, 134, 7242–7245 CrossRef CAS PubMed; (e) J. Trenner, C. Depken, T. Weber and A. Breder, Angew. Chem., Int. Ed., 2013, 52, 8952–8956 CrossRef CAS PubMed; (f) Z. Zhang, H. Du, J. Xu and P. Li, Chem. Commun., 2016, 52, 11547–11550 RSC.
- (a) L. Bayeh, P. Q. Le and U. K. Tambar, Nature, 2017, 547, 196–200 CrossRef CAS PubMed; (b) W. Liu, S. Z. Ali, S. E. Ammann and M. C. White, J. Am. Chem. Soc., 2018, 140, 10658–10662 CrossRef CAS PubMed; (c) H. Lei and T. Rovis, Nat. Chem., 2020, 12, 725–731 CrossRef CAS PubMed.
- (a) H.-C. Lin, P.-P. Xie, Z.-Y. Dai, S.-Q. Zhang, P.-S. Wang, Y.-G. Chen, T.-C. Wang, X. Hong and L.-Z. Gong, J. Am. Chem. Soc., 2019, 141, 5824–5834 CrossRef CAS PubMed; (b) T.-C. Wang, P.-S. Wang and L.-Z. Gong, Sci. China: Chem., 2020, 63, 454–459 CrossRef CAS; (c) Z. Y. Dai, P. S. Wang and L. Z. Gong, Chem. Commun., 2021, 57, 6748–6751 RSC.
- (a) J. J. Silber and H. J. Shine, J. Org. Chem., 1971, 36, 2923–2926 CrossRef; (b) D. Q. Qian, H. J. Shine, I. Y. Guzman-Jimenez, J. H. Thurston and K. H. Whitmire, J. Org. Chem., 2002, 67, 4030–4039 CrossRef CAS PubMed.
- (a) F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed; (b) P. S. Engl, A. P. Haring, F. Berger, G. Berger, A. Perez-Bitrian and T. Ritter, J. Am. Chem. Soc., 2019, 141, 13346–13351 CrossRef CAS PubMed; (c) R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161–16166 CrossRef CAS PubMed; (d) J. Li, J. Chen, R. Sang, W. S. Ham, M. B. Plutschack, F. Berger, S. Chabbra, A. Schnegg, C. Genicot and T. Ritter, Nat. Chem., 2020, 12, 56–62 CrossRef CAS PubMed; (e) E. M. Alvarez, T. Karl, F. Berger, L. Torkowski and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 13609–13613 CrossRef CAS PubMed; (f) H. Jia, A. P. Haring, F. Berger, L. Zhang and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7623–7628 CrossRef CAS PubMed; (g) B. Lansbergen, P. Granatino and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7909–7914 CrossRef CAS PubMed; (h) F. Juliá, Q. Shao, M. Duan, M. B. Plutschack, F. Berger, J. Meteos, C. Lu, X.-S. Xue, K. N. Houk and T. Ritter, J. Am. Chem. Soc., 2021, 143, 16041–16054 CrossRef PubMed.
- (a) J. Wu, Z. Wang, X.-Y. Chen, Y. Wu, D. Wang, Q. Peng and P. Wang, Sci. China: Chem., 2020, 63, 336–340 CrossRef CAS; (b) X.-Y. Chen, Y.-H. Huang, J. Zhou and P. Wang, Chin. J. Chem., 2020, 38, 1269–1272 CrossRef CAS; (c) X.-Y. Chen, X.-X. Nie, Y. Wu and P. Wang, Chem. Commun., 2020, 56, 5058–5061 RSC.
- (a) C. Chen, Z.-J. Wang, H. Lu, Y. Zhao and Z. Shi, Nat. Commun., 2021, 12, 4526 CrossRef CAS PubMed; (b) C. Chen, M. Wang, H. Lu, B. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 21756–21760 CrossRef CAS PubMed.
- D. E. Holst, D. J. Wang, M. J. Kim, I. A. Guzei and Z. K. Wickens, Nature, 2021, 596, 74–79 CrossRef CAS PubMed.
- (a) J. Chen, J. Li, M. B. Plutschack, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2020, 59, 5616–5620 CrossRef CAS PubMed; (b) F. Juliá, J. Yan, F. Paulus and T. Ritter, J. Am. Chem. Soc., 2021, 143, 12992–12998 CrossRef PubMed.
- Q. Cheng, J. Chen, S. Lin and T. Ritter, J. Am. Chem. Soc., 2020, 142, 17287–17293 CrossRef CAS PubMed.
- M.-S. Liu, H.-W. Du and W. Shu, ChemRxiv, 2021 DOI:10.33774/chemrxiv-2021-2w0jx.
- For more details on the condition optimization, see ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2110292, 2107074 and 2110458. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06577g |
| This journal is © The Royal Society of Chemistry 2022 |