
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn asymmetric 2,3-fluoranthene imide building block for regioregular semiconductors with aggregation-induced emission properties†
Xianglang
Sun
a,
Ming-Yun
Liao
b,
Xinyu
Yu
a,
Ying-Sheng
Wu
b,
Cheng
Zhong
c,
Chu-Chen
Chueh
 b,
Zhen
Li
c and
Zhong'an
Li
*a
b,
Zhen
Li
c and
Zhong'an
Li
*a
aKey Laboratory for Material Chemistry of Energy Conversion and Storage, Ministry of Education, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan, 430074, China. E-mail: lizha@hust.edu.cn
bDepartment of Chemical Engineering, National Taiwan University, Taipei, 10617, Taiwan
cSauvage Center for Molecular Sciences, Department of Chemistry, Wuhan University, Wuhan, 430072, China
First published on 6th January 2022
Abstract
For organic semiconductors, the development of electron-deficient building blocks has lagged far behind that of the electron-rich ones. Moreover, it remains a significant challenge to design organic molecules with efficient charge transport and strong solid-state emission simultaneously. Herein, we describe a facile synthetic route toward a new π-acceptor imide building block, namely 2,3-fluoranthene imide, based on which four regioregular small molecules (F1–F4) are synthesized by tuning the imide orientations and the central linkage bridges. All molecules exhibit attractive aggregation-induced emission (AIE) characteristics with strong far-red emission in the powder state, and F3 shows the highest photoluminescence quantum yield of 5.9%. F1 and F3 with a thiophene bridge present an obvious p-type characteristic, while for F3 with an outward imide orientation, the maximum hole mobility from a solution-processed field-effect transistor (FET) device reaches 0.026 cm2 V−1 s−1, being ∼104 times higher than the value of F1 with an inward imide orientation. By using a fluorinated thiophene bridge, the resulting F2 and F4 can be turned into n-type semiconductors, showing an electron mobility of ∼1.43 × 10−4 and ∼3.34 × 10−5 cm2 V−1 s−1, respectively. Our work not only demonstrates that asymmetric 2,3-fluoranthene imide is a promising building block for constructing organic materials with high carrier mobility and strong solid-state emission, but also highlights the importance of regioregular structures in the materials' properties.
Introduction
Organic semiconductors show great importance for next-generation flexible and printed optoelectronic devices as well as biomedicines.1–4 Among them, donor–acceptor (D–A) type π-conjugated systems have received the most interest because their optical and electronic properties can be conveniently and precisely adjusted by changing the D and A units.5–10 However, compared to the diverse D units, the development of A units is severely restricted by the synthetic challenges of reduced reactivity caused by the electron-withdrawing groups.11–14 Currently, arene imides such as perylene diimide (PDI) and naphthalene diimide (NDI) (Fig. 1a) are two of the most successful A units for organic optoelectronics due to their straightforward synthesis, deep lowest unoccupied molecular orbital (LUMO) level, facile material accessibility, etc.15–21 However, due to the strong π–π intermolecular interactions as a result of highly planar π-conjugated structures, they usually suffer from aggregation-caused quenching (ACQ) in the solid state that limits their further applications.22–25 Thus, it remains a challenge to design organic semiconductors with both high carrier mobility and strong solid-state emission, which thus warrants the exploration of new elementary building blocks.26–31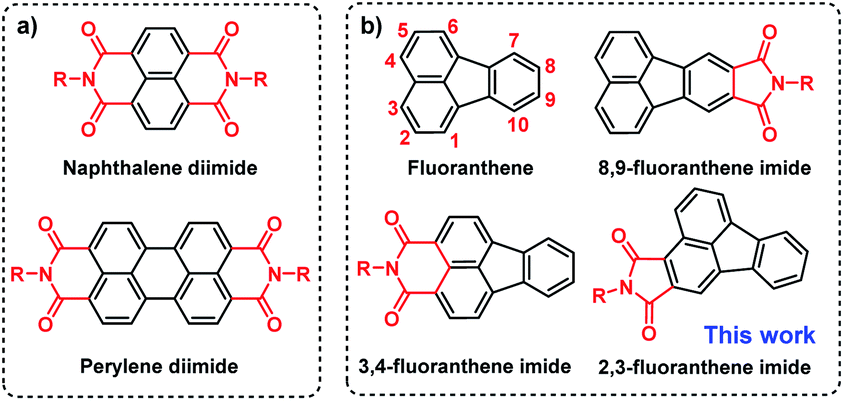 | ||
| Fig. 1 Structures of (a) perylene diimide (PDI) and naphthalene diimide (NDI) and (b) fluoranthene and fluoranthene-fused imide derivatives. | ||
Fluoranthene is one typical polycyclic aromatic hydrocarbon, which possesses a rigid, planarized structure that is beneficial for intramolecular charge transport via π–π stacking. Moreover, it can be regarded as a basic unit of fullerene, exhibiting electron-deficient characteristics due to the central five-membered ring.32–34 In this regard, the electron-deficient fluoranthene should be a suitable building block for imide-based semiconductors. Indeed, fluoranthene-fused imides have been successfully developed as non-fullerene acceptors for organic solar cells, the power conversion efficiency (PCE) of which can reach up to 3.12%.35–39 Nonetheless, the structural diversity and synthetic strategy of fluoranthene imides are still limited (Scheme S1†). At present, the imidization sites of fluoranthene are mainly restricted to the 3, 4-sites or 8, 9-sites (Fig. 1b), as limited by the synthetic challenges of functionalization at other sites.38,40–42 Besides, the applicability of fluoranthene derivatives including fluoranthene imides in organic field-effect transistors (OFETs) has rarely been explored so far.43,44
Recently, our group developed a facile synthetic route to functionalize fluoranthene at the 2,3-sites for the first time, that is, using dibenzofulvene as the diene to undergo a typical Diels–Alder reaction, rather than using acenaphthenequinone in most cases.45 The resulting 2,3-dicyanofluoranthene unit was shown as an efficient electron-deficient core to construct D–A type dopant-free hole transporting materials for perovskite solar cells.45,46 Therefore, it would be interesting to know whether this is also a feasible synthetic method to develop new fluoranthene imide building blocks. Herein, we successfully synthesize 2,3-fluoranthene imide through a simple one-pot reaction (Fig. 1). We find that the 4-site of 2,3-fluoranthene imide exhibits an enhanced reactivity compared to the 9-site, thus allowing us to synthesize two types of regioregular small molecules with different imide orientations (F1–F4, Fig. 2a). By regulating the imide orientation and the central linkage bridge, we show that the molecular configuration and packing of the derived molecules can be accordingly adjusted to not only realize high hole mobility and attractive aggregation-induced emission (AIE) properties in one molecule, but also enable the conversion from p-type to n-type charge transport characteristics.
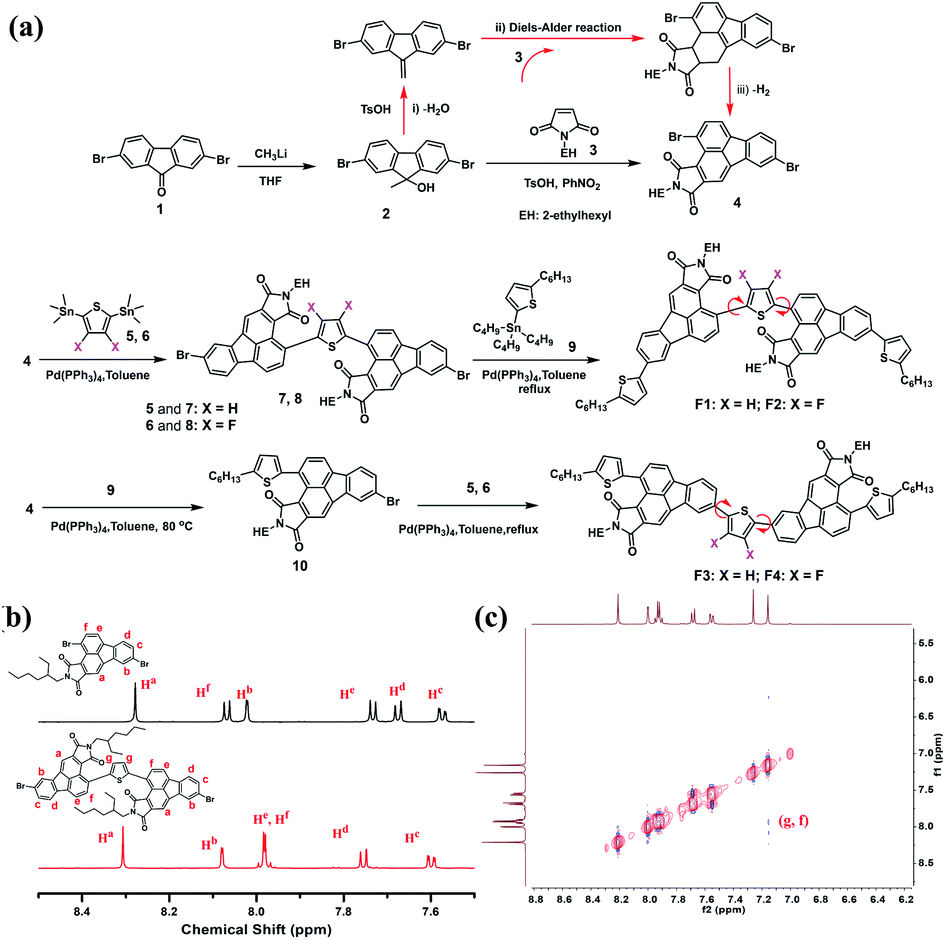 | ||
| Fig. 2 (a) Synthetic routes for regioregular F1–F4; (b) comparison of 1H NMR signals between intermediates 4 and 7; (c) 1H–1H NOESY spectra of 7. | ||
Results and discussion
Fig. 2a illustrates the synthetic routes for F1–F4. First, 2,7-dibromofluorenone (1) reacted with methyllithium to afford compound 2, which then underwent an optimized one-pot reaction to obtain a new 2,3-fluoranthene imide (4) with two bromide atoms substituted at the 4- and 9-sites. As shown, this one-pot reaction involves three consecutive steps; compound 2 first underwent dehydration with a small amount of p-toluenesulfonic acid (TsOH) as the catalyst (i), and then the resulting dibenzofulvene product carried out a Diels–Alder reaction with maleimide 3 at 160 °C (ii), followed by dehydrogenation (iii). We further found that the bromine substitutions at the 4- and 9-sites possess different reactivity for the Stille coupling reaction, and the 4-site shows a higher reactivity. By controlling the reaction temperature and the feeding ratio between compounds 4 and 5, we successfully obtained a new regioregular intermediate (7) with an inward imide orientation. The comparison of the 1H NMR spectra of 4 and 7 shows that the proton Hf in 7 has a visible shift to the high field, indicating that the reaction occurs at the 4-site (Fig. 2b). We also performed the 1H–1H NOESY measurements of 7 (Fig. 2c), and the correlation between Hg and Hf reveals that Hf is the proton adjacent to the thiophene group, further confirming the structure. Using the same method, the intermediate 8 was synthesized with two fluorine atoms on the thiophene bridge. Finally, F1 and F2 were obtained via another Stille coupling reaction with compound 9. Similarly, F3 and F4 with an outward imide orientation were yielded via two consecutive coupling reactions; however, the reaction conditions such as temperature need to be carefully optimized. The structure of the critical intermediate 10 was also confirmed by 1H NMR and 1H–1H NOESY spectra as shown in Fig. S1–S2.†Thermogravimetric analysis was used to study the thermal properties of F1–F4, and all exhibit high thermal stability with a 5% weight loss temperature over 410 °C (Fig. S3†). Differential scanning calorimetry (DSC) (Fig. S4†) shows that F3 exhibits the most intense melting/crystallization peaks at 275.4/238.0 °C, whereas F1 exhibits the weakest melting/crystallization peaks at 210.5/123.0 °C, indicating a higher crystallinity of F3 powder (Fig. S5†). Besides, for F2 and F4, the melting/crystallization peaks were shifted to 273.7/244.6 °C and 279.2/247.3 °C, respectively, mainly due to their structural difference induced by the imide direction and fluorination effect.
The absorption spectra of F1–F4 in dichloromethane (DCM) solutions and as thin films are shown in Fig. 3a and S6,† respectively, with relevant data listed in Table 1. The absorption profiles of F1 and F2 have a triple-band shape while F3 and F4 have a typical dual-band shape, indicating that the imide orientation has a clear impact on the molecular conjugation. The low-energy absorption band is ascribed to the intramolecular charge transfer (ICT) between the thiophene unit and the imide group, and fluorination leads to a blue-shifted ICT band, ∼10 nm. Furthermore, all molecules exhibit a red-shifted ICT absorption band in the solid state, ∼30 nm, compared to the solution ones, possibly attributed to the planarization of the conjugated backbone as a result of aggregation. To gain insight into the redox behaviours, cyclic voltammetry (CV) measurements were conducted (Fig. 3b). As listed in Table 1, F1 and F2 with an inward imide orientation both exhibit slightly deepened HOMO/LUMO levels compared to those with an outward imide orientation, while fluorination can further slightly decrease the HOMO/LUMO levels due to their enhanced electronegativity.
| λ sol (nm) | λ fil (nm) | E g (eV) | HOMOb (eV) | LUMOb (eV) | T d (°C) | T m (°C) | |
|---|---|---|---|---|---|---|---|
| a Optical energy gaps calculated according to the equation Eg = 1240/λonset eV. b Estimated values using the equations ELUMO/EHOMO = −(4.80 + Ered/Eox) eV. c The 5% weight loss temperature. d Melting point. | |||||||
| F1 | 341, 411, 503 | 344, 530 | 2.07 | −5.51 | −3.46 | 429.6 | 210.5 |
| F2 | 337, 420, 493 | 307, 454, 522 | 2.12 | −5.56 | −3.50 | 417.7 | 273.7 |
| F3 | 367, 505 | 376, 532 | 2.11 | −5.34 | −3.44 | 438.9 | 275.4 |
| F4 | 358, 492 | 369, 519 | 2.16 | −5.50 | −3.44 | 416.2 | 279.2 |
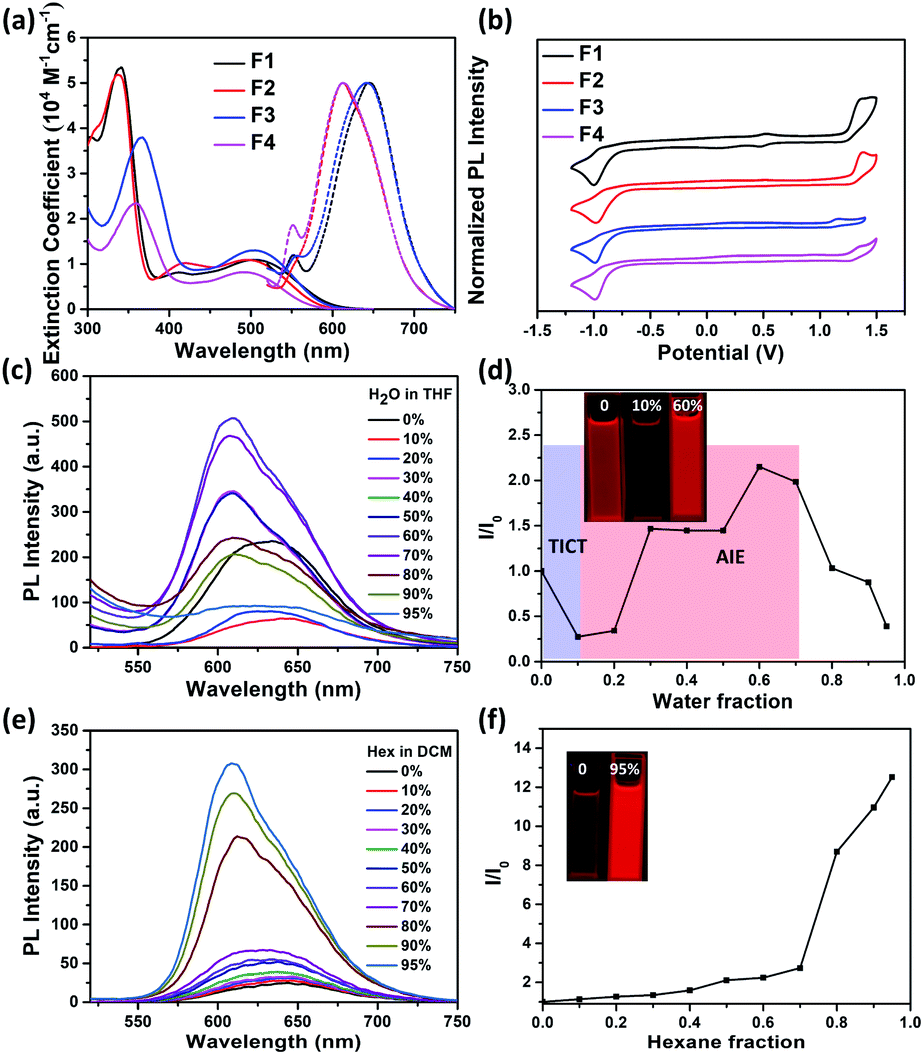 | ||
| Fig. 3 (a) UV-vis spectra (solid) and normalized emission spectra (dash) and (b) cyclic voltammograms of F1–F4 in DCM solutions; emission spectra of F3 in THF/water mixtures (c) and DCM/hexane mixtures (e); plots of (I/I0) vs. fw in THF/water mixtures (d) and DCM/hexane mixtures (f). Inset: images of F3 in solution mixtures under 365 nm ultraviolet light. | ||
We further studied their photoluminescence (PL) properties in different organic solvents (Fig. 3a and S7†), with relevant data listed in Table 2. Interestingly, all the PL spectra show an obvious solvatochromic effect; that is, the emission spectra are red-shifted and broadened upon increasing the solvent polarity, accompanied by a significantly decreased PL intensity. This phenomenon can be attributed to the well-known twisted intramolecular charge transfer (TICT) effect, for which the intramolecular rotation can bring the molecule from the locally excited (LE) state to the TICT state in polar solvents, leading to a pronounced charge separation between the D and A units to quench the fluorescence.47–50
| λ em (nm) | Φ PL | Φ PL | Φ PL | |||||
|---|---|---|---|---|---|---|---|---|
| Hexane | Toluene | THF | DCM | Powder | ||||
| a Absolute photoluminescence quantum yields tested in DCM solutions. b In DCM/hexane mixtures, and the hexane fraction for F1–F4 is 95%, 70%, 95% and 90%, respectively. c In solid powders. | ||||||||
| F1 | 610 | 634 | 643 | 645 | 650 | 2.7% | 5.0% | 3.8% |
| F2 | 577 | 594 | 605 | 613 | 642 | 3.0% | 7.0% | 3.9% |
| F3 | 598 | 615 | 629 | 641 | 640 | 0.3% | 4.3% | 5.9% |
| F4 | 590 | 608 | 613 | 620 | 653 | 2.1% | 3.8% | 3.8% |
Encouragingly, the formation of aggregates for F1–F4 can effectively suppress the negative TICT effect (Fig. 3c–d, S8–S10†). As an example, Fig. 3c–d show the change of the PL intensity of F3 as a function of water fraction (fw). When the fw increases from 0 to 10%, the PL intensity of F3 is decreased by 50%, accompanied by a red shift, indicating the occurring of a “dark” TICT state. However, when increasing the fw to 40%, an obvious AIE phenomenon is observed, realizing a ∼9-fold intensity enhancement at fw = 60%. This could be due to the restriction of intramolecular rotation between the D and A units due to the formation of aggregates.51–54 Note that adding more water further causes a decrease in emission due to sedimentation (Fig. S11†). The emission properties of F1–F4 in DCM/hexane mixtures were also investigated (Fig. 3e–f, S12–S14†), and upon increasing the apolar hexane fraction, their emission all was increased obviously with a gradual blue-shift, attributed to the synergetic effects from AIE and mitigated TICT. The AIE effect can be further confirmed by dynamic light scattering results (Fig. S15†) as well as enhanced absolute photoluminescence quantum yields (ΦPLs) in DCM/hexane mixtures (Table 2). The above results are indeed encouraging, because (i) most of the imide-based arenes such as PDI and NDI usually suffer from a serious ACQ effect due to their rigid and highly planar structure; (ii) the AIE properties for F1–F4 are achieved without needing the decoration of any AIE-active building blocks or large steric spacers that usually break the original π-conjugated structure.29,55–57
In the solid state, F1–F4 all show bright red emission (Fig. S16†) with ΦPLs (Table 2) estimated to be 3.8, 3.9, 5.9 and 3.8%, respectively. It is worth noting that both F2 and F4 exhibit an obvious red-shifted solid-state emission compared to those in solutions, while no clear change is observed for F1 and F3 (Fig. S17† and Table 2), ascribed to the amorphous state of former powders with tight intermolecular interactions confirmed by powder XRD patterns (Fig. S5†).
Theoretical computations were also performed by employing the density functional theory (DFT) method at the B3LYP/6-31G level to understand the effect of the regioregular structure, and the results are shown in Fig. S18.† All molecules show a twisted structure between the thiophene bridge and two fluoranthene imide units. In particular, F1 and F2 exhibit much larger dihedral angles than F3 and F4, which can be rationalized by the steric hindrance between the two inward imide units of the former. Possibly due to the larger steric hindrance, the two fluoranthene imides in F1 are located on the upper and lower sides of the thiophene unit, while they are located on the same sides for the others. In addition, fluorinated F2 and F4 both show reduced dihedral angles compared to the corresponding nonfluorinated analog, suggesting enhanced backbone planarity achieved by the non-covalent interactions due to the fluorine substitutions. F1–F4 also show similar energy level distributions, in which the HOMOs delocalize over the entire conjugated backbone, while the LUMOs mainly localize on the two fluoranthene imide units. Notably, the trend of calculated HOMO/LUMO levels is almost consistent with that obtained by CV measurements.
We further employed grazing-incidence wide angle X-ray scattering (GIWAXS) and atomic force microscope (AFM) measurements to investigate the impact of geometric differences on the crystalline properties and the film morphology. The films were prepared by spin-coating on a SiO2 dielectric layer and annealing at 180 °C. As shown in Fig. 4a–d, all films show a weak (010) π–π stacking peak along the in-plane direction, being beneficial to charge transport. Both F1 and F3 films further show a strong (001) diffraction peak at ∼0.35 Å along the out-of-plane direction, assigned to a highly ordered lamella packing. In addition, the F1 film possesses large crystalline domains with obvious grain boundaries and a large root mean square roughness (RMS) value of 4.7 nm, while F3 molecules assemble into uniform and compact nanosheets with a small RMS value of 1.7 nm. In principle, the discontinuous grain boundaries observed in F1 are not favorable for charge transport while the interconnected nanosheets for F3 are conducive for efficient charge transport.58,59 As a result, the regioregular configuration exerted by the imide orientation leads to a discrepancy in the molecular packing behavior and film morphology. On the other hand, both F2 and F4 films show a ring-like scattering feature with a relatively weak and broadened peak at 0.35 Å along the out-of-plane direction, suggesting a partially ordered lamellar packing but with anisotropy. Moreover, as shown in Fig. 4c–d, they also exhibit a fiber-like feature with large RMS values of 13.7 and 14.3 nm, respectively. Hence, there is a compromise existing between fluorination and regioisomerism for the molecule's crystallinity and aggregation in our case.
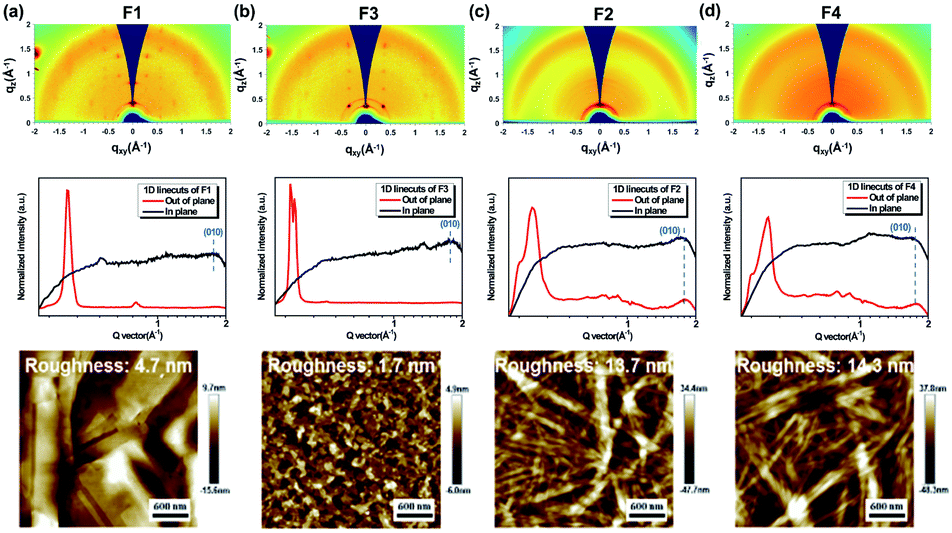 | ||
| Fig. 4 GIWAXS 2D patterns, 1D scattering profiles, and AFM images of films of (a) F1, (b) F3, (c) F2, and (d) F4. Note that all films are annealed at 180 °C. | ||
We finally examined the charge transport properties of F1–F4 by fabricating bottom-gate/top-contact (BG/TC) OFETs. The details of device fabrication and characterization are described in the Experimental section in the ESI. Fig. 5 shows the optimized transfer curves of OFET devices based on the films of F1–F4 annealed at 180 or 200 °C and the detailed performance is summarized in Table 3. The transfer curves based on these films annealed at other temperatures (180 or 250 °C) are presented in Fig. S19.† As seen, all devices show distinguishable FET characteristics, except for F1 (Fig. 5a). This can be attributed to its twisted molecular structure and unfavorable film morphology as discussed earlier. In contrast, F3, the regioisomer of F1, delivers a conspicuous p-type characteristic (Fig. 5b) and its corresponding output curves are presented in Fig. S20.† The maximum hole mobility (μh) value of a FET device based on the F3 film reaches 0.026 cm2 V−1 s−1, being ∼104 times greater than the value of F1. Note that there are very few reports on fluoranthene-based FETs (Table S1†), and the high μh value of F3 is comparable to that of most solution-processed small molecule semiconductors.60–62 The critical effect of regioregular structures on the charge transport properties closely correlates with their distinct molecular packing patterns and film morphology.63–66 Here, the outward imide units in F3 are more favorable for intermolecular packing than the inward ones in F1, thereby enabling a much higher μh value.
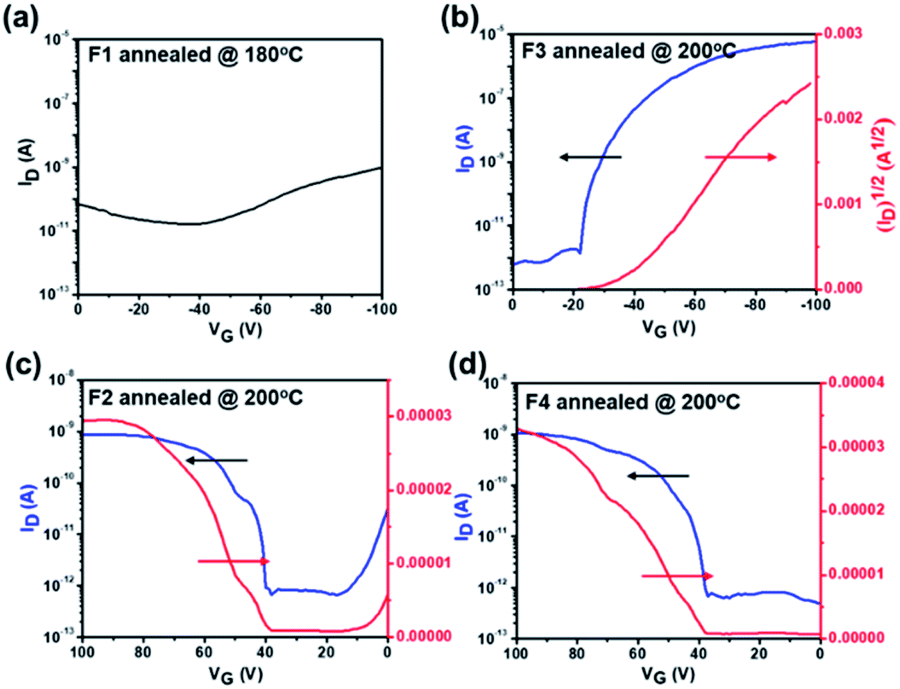 | ||
| Fig. 5 P-type transfer curves of FET devices based on (a) the F1 film annealed at 180 °C and (b) the F3 film annealed at 200 °C. N-type transfer curves of FET devices based on (c) the F2 film and (d) the F4 film annealed at 200 °C. | ||
| T anneal (°C) | μ (cm2 V−1 s−1) | V T (V) | I on /I off | |
|---|---|---|---|---|
| a Maximum values in brackets. | ||||
| p-type character | μ h | |||
| F1 | 180 | 3.03 × 10−6 | −46.41 | 5.8 × 101 |
| F3 | 180 | 1.88 × 10−3 | −30.88 | 8.3 × 106 |
| F3 | 200 | 2.41 × 10−2 (2.63 × 10−2)a | −38.96 | 1.8 × 108 |
| F3 | 250 | 2.21 × 10−2 | −50.87 | 1.7 × 108 |
| n-type character | μ e | |||
| F2 | 180 | 2.20 × 10−5 | 46.06 | 1.9 × 104 |
| F2 | 200 | 5.81 × 10−5 (1.43 × 10−4)a | 41.36 | 2.8 × 105 |
| F2 | 250 | 1.35 × 10−5 | 58.67 | 5.7 × 103 |
| F4 | 180 | 6.91 × 10−6 | 54.84 | 9.0 × 104 |
| F4 | 200 | 2.22 × 10−5 (3.34 × 10−5)a | 39.00 | 5.5 × 103 |
| F4 | 250 | 1.04 × 10−5 | 42.46 | 1.2 × 105 |
On the other hand, as presented in Fig. 5c–d, both F2 and F4 comprising a fluorinated thiophene bridge present n-type characteristics, and their maximum electron mobility (μe) values are ∼1.43 × 10−4 and ∼3.34 × 10−5 cm2 V−1 s−1, respectively. Clearly, the fluorination of the bridge thiophene unit changes the polarity of the compounds from p-type to n-type as a result of enhanced electronegativity. We notice that the low μe values might be attributed to their inferior crystal orientation. In addition, the LUMO level below −3.6 eV has been considered as the window of trap-free electron transport for n-type semiconductors.67 Thus, the relatively higher LUMO levels of both F2 (−3.50 eV) and F4 (−3.44 eV) could also be responsible for their poor electron transport. Nonetheless, this is the first demonstration of n-type FET devices based on fluoranthene molecules (Table S1†), manifesting the potential of asymmetric 2,3-fluoranthene imide units for new n-type semiconductors. Further device optimization like using Ag electrodes with a more compatible work function to F2 and F4 is in progress.
In addition, as presented in Table 3, the annealing temperature of the active layer has a germane correlation with the resultant performance. Due to the high melting points, F2–F4 films deliver the most optimized FET performance at high annealing temperature (200 °C) as a result of the facilitated molecular packing. Taking F3 as an example, the FET device based on the as-cast F3 film shows no distinguishable FET characteristic (Fig. S21†). However, owing to the low melting point, the optimized annealing temperature of the F1 film is restricted to 180 °C, and annealing the F1 film at 200 °C results in severe film degradation.
Conclusions
In conclusion, starting from the simple fluorenone, a new type of π-acceptor building block, namely 2,3-fluoranthene imide, is synthesized through a facile one-pot synthetic route. Unlike the fluoranthene imide derivatives reported to date, this new asymmetric building block shows a significant reactivity difference at the 4- and 9-substitution sites, thereby allowing us to successfully synthesize two types of regioisomers (F1–F4) with outward and inward imide orientations. We unveil that the distinct molecular geometry for these regioisomers results in different molecular crystallinity in the solid state. Due to the twisted configuration, all molecules exhibit an obvious twisted TICT decay pathway, leading to red-shifted and reduced emission in polar solvents. However, the negative TICT effect can be effectively suppressed by the attractive AIE process, and F3 shows the brightest red emission at 640 nm in the powder state with the highest ΦPL of 5.9%. The charge transporting abilities of these molecules were systematically studied by fabricating solution-processed FETs. Impressively, F3 delivers a conspicuous p-type character with a maximum μh value of 0.026 cm2 V−1 s−1, being ∼104 times greater than the value of F1, owing to its superior crystallinity and film morphology. Moreover, the fluorination on the central thiophene bridge effectively converts the polarity of the molecules from p-type to n-type, resulting in μe values of ∼1.43 × 10−4 and ∼3.34 × 10−5 cm2 V−1 s−1 for F2 and F4, respectively. Our results clearly suggest that 2,3-fluoranthene imide is a promising building block for high-performance organic semiconductors with AIE properties.Data availability
Data associated with this article, including synthetic details, materials characterization and transistor fabrication details are available in the ESI.†Author contributions
X. Sun and Z. A. Li conceived and designed the study. X. Sun and X. Yu performed the materials synthesis. X. Sun performed the materials characterization. M.-Y. Liao and Y.-S. Wu performed the morphology characterization and the transistor fabrication. C. Zhong performed the DFT calculations. X. Sun, M.-Y. Liao, C.-C. Chueh and Z. A. Li analyzed the obtained data. X. Sun wrote the original draft. Z. A. Li and C.-C. Chueh reviewed and edited the paper. C.-C. Chueh, Z. Li and Z. A. Li supervised the work. All authors contributed to the finalization of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. L. acknowledges the financial support from the Natural Science Foundation of China (No 21975085 and 22175067) and the Innovation and Talent Recruitment Base of New Energy Chemistry and Device (No B21003). X. S. acknowledges the financial support from the China Postdoctoral Science Foundation (2021M691111). C.-C. C acknowledges the financial support from the Ministry of Science and Technology of Taiwan (MOST 110-2634-F-002-043 and 110-2628-E-002-011-) and Top University Project of National Taiwan University (110L7836). The authors from HUST would also like to thank the Analytical and Testing Center at HUST for use of their facilities.Notes and references
- D. Li, W.-Y. Lai, Y.-Z. Zhang and W. Huang, Adv. Mater., 2018, 30, 1704738 CrossRef PubMed.
- C. Cui, D. H. Park and D. J. Ahn, Adv. Mater., 2020, 32, 2002213 CrossRef CAS PubMed.
- H. Xu, L. Yin, C. Liu, X. Sheng and N. Zhao, Adv. Mater., 2018, 30, 1800156 CrossRef PubMed.
- A. Facchetti, Mater. Today, 2007, 10, 28–37 CrossRef CAS.
- L. Duan, J. Qiao, Y. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137–1144 CrossRef CAS PubMed.
- T. Lei, J.-Y. Wang and J. Pei, Acc. Chem. Res., 2014, 47, 1117–1126 CrossRef CAS PubMed.
- Z. Liu, G. Zhang and D. Zhang, Acc. Chem. Res., 2018, 51, 1422–1432 CrossRef CAS PubMed.
- M. Kim, S. U. Ryu, S. A. Park, K. Choi, T. Kim, D. Chung and T. Park, Adv. Funct. Mater., 2020, 30, 1904545 CrossRef CAS.
- J. Zhang, W. Xu, P. Sheng, G. Zhao and D. Zhu, Acc. Chem. Res., 2017, 50, 1654–1662 CrossRef CAS PubMed.
- X. Liu, Y.-Z. Long, L. Liao, X. Duan and Z. Fan, ACS Nano, 2012, 6, 1888–1900 CrossRef CAS PubMed.
- J. Yang, Z. Zhao, S. Wang, Y. Guo and Y. Liu, Chem, 2018, 4, 2748–2785 CAS.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed.
- C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS PubMed.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS PubMed.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 15259–15278 CrossRef CAS PubMed.
- M. M. Ling, P. Erk, M. Gomez, M. Koenemann, J. Locklin and Z. Bao, Adv. Mater., 2007, 19, 1123–1127 CrossRef CAS.
- X. Zhan, Z. a. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS PubMed.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- Z. A. Li, C. C. Chueh and A. K. Y. Jen, Prog. Polym. Sci., 2019, 99, 101175 CrossRef CAS.
- J. Liu, H. Zhang, H. Dong, L. Meng, L. Jiang, L. Jiang, Y. Wang, J. Yu, Y. Sun, W. Hu and A. J. Heeger, Nat. Commun., 2015, 6, 10032 CrossRef CAS PubMed.
- F. Bu, R. Duan, Y. Xie, Y. Yi, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2015, 54, 14492–14497 CrossRef CAS PubMed.
- Z. Li and A. Qin, Natl. Sci. Rev., 2014, 1, 22–24 CrossRef.
- Y. Huang, J. Xing, Q. Gong, L.-C. Chen, G. Liu, C. Yao, Z. Wang, H.-L. Zhang, Z. Chen and Q. Zhang, Nat. Commun., 2019, 10, 169 CrossRef PubMed.
- H. Jiang and W. Hu, Angew. Chem., Int. Ed., 2020, 59, 1408–1428 CrossRef CAS PubMed.
- Z. Qin, H. Gao, H. Dong and W. Hu, Adv. Mater., 2021, 33, 2007149 CrossRef CAS PubMed.
- D. Liu, Q. Liao, Q. Peng, H. Gao, Q. Sun, J. De, C. Gao, Z. Miao, Z. Qin, J. Yang, H. Fu, Z. Shuai, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2021, 60, 20274–20279 CrossRef CAS PubMed.
- Z. Zhao, S. M. Gao, X. Y. Zheng, P. F. Zhang, W. T. Wu, R. T. K. Kwok, Y. Xiong, N. L. C. Leung, Y. C. Chen, X. K. Gao, J. W. Y. Lam and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1705609 CrossRef.
- A. Dadvand, A. G. Moiseev, K. Sawabe, W. H. Sun, B. Djukic, I. Chung, T. Takenobu, F. Rosei and D. F. Perepichka, Angew. Chem., Int. Ed., 2012, 51, 3837–3841 CrossRef CAS PubMed.
- A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed.
- M. Kyeong, J. Lee, K. Lee and S. Hong, ACS Appl. Mater. Interfaces, 2018, 10, 23254–23262 CrossRef CAS PubMed.
- X. Sun, F. Wu, C. Zhong, L. Zhu and Z. a. Li, Chem. Sci., 2019, 10, 6899–6907 RSC.
- A. Modelli, L. Mussoni and D. Fabbri, J. Phys. Chem. A, 2006, 110, 6482–6486 CrossRef CAS PubMed.
- L. Ding, C.-Y. Yang, Y.-Q. Zheng, J.-Y. Wang, J. Pei and Z. Su, Asian J. Org. Chem., 2017, 6, 1231–1234 CrossRef CAS.
- Y. Zhou, Y. Z. Dai, Y. Q. Zheng, X. Y. Wang, J. Y. Wang and J. Pei, Chem. Commun., 2013, 49, 5802–5804 RSC.
- S. Chatterjee, Y. Ie, T. Seo, T. Moriyama, G.-J. A. H. Wetzelaer, P. W. M. Blom and Y. Aso, NPG Asia Mater., 2018, 10, 1016–1028 CrossRef CAS.
- Y. Q. Zheng, Y. Z. Dai, Y. Zhou, J. Y. Wang and J. Pei, Chem. Commun., 2014, 50, 1591–1594 RSC.
- Y. Zhou, L. Ding, K. Shi, Y.-Z. Dai, N. Ai, J. Wang and J. Pei, Adv. Mater., 2012, 24, 957–961 CrossRef CAS PubMed.
- H. Ishikawa, K. Katayama, J.-i. Nishida, C. Kitamura and T. Kawase, Tetrahedron Lett., 2018, 59, 3782–3786 CrossRef CAS.
- L. Ding, C. Yang, Z. Su and J. Pei, Sci. China: Chem., 2015, 58, 364–369 CrossRef CAS.
- L. Ding, H. Z. Ying, Y. Zhou, T. Lei and J. Pei, Org. Lett., 2010, 12, 5522–5525 CrossRef CAS PubMed.
- Q. Yan, Y. Zhou, B.-B. Ni, Y. Ma, J. Wang, J. Pei and Y. Cao, J. Org. Chem., 2008, 73, 5328–5339 CrossRef CAS PubMed.
- Y. Kunugi, T. Kosuge and K. Okamoto, Electrochemistry, 2008, 76, 865–867 CrossRef CAS.
- X. Sun, Q. Xue, Z. Zhu, Q. Xiao, K. Jiang, H.-L. Yip, H. Yan and Z. a. Li, Chem. Sci., 2018, 9, 2698–2704 RSC.
- X. Yu, Z. Li, X. Sun, C. Zhong, Z. Zhu, Z. a. Li and A. K. Y. Jen, Nano Energy, 2021, 82, 105701 CrossRef CAS.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- J. Katuri, X. Ma, M. M. Stanton and S. Sánchez, Acc. Chem. Res., 2017, 50, 2–11 CrossRef CAS PubMed.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Peña-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CrossRef CAS.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef PubMed.
- J. Yang, Z. Chi, W. Zhu, B. Z. Tang and Z. Li, Sci. China: Chem., 2019, 62, 1090–1098 CrossRef CAS.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- Y. Li, Z. Cai, S. Liu, H. Zhang, S. T. H. Wong, J. W. Y. Lam, R. T. K. Kwok, J. Qian and B. Z. Tang, Nat. Commun., 2020, 11, 1255 CrossRef CAS PubMed.
- K. Katayama, I. Kawajiri, Y. Okano, J. I. Nishida and T. Kawase, Chempluschem, 2019, 84, 722–729 CrossRef CAS PubMed.
- D. Guo, L. Li, X. Q. Zhu, M. Heeney, J. Li, L. C. Dong, Q. S. Yu, Z. H. Gan, X. G. Gu and L. X. Tan, Sci. China: Chem., 2020, 63, 1198–1207 CrossRef CAS.
- L. Zong, Y. Xie, C. Wang, J. R. Li, Q. Li and Z. Li, Chem. Commun., 2016, 52, 11496–11499 RSC.
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- Y. Zhang, C. Kim, J. Lin and T. Q. Nguyen, Adv. Funct. Mater., 2012, 22, 97–105 CrossRef CAS.
- G. Zhang, Y. Zhao, B. Kang, S. Park, J. Ruan, H. Lu, L. Qiu, Y. Ding and K. Cho, Chem. Mater., 2019, 31, 2027–2035 CrossRef CAS.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2018, 30, 1801079 CrossRef PubMed.
- M. Chen, L. Yan, Y. Zhao, I. Murtaza, H. Meng and W. Huang, J. Mater. Chem. C, 2018, 6, 7416–7444 RSC.
- K. Takimiya, M. Nakano, H. Sugino and I. Osaka, Synth. Met., 2016, 217, 68–78 CrossRef CAS.
- L. H. Jimison, M. F. Toney, I. McCulloch, M. Heeney and A. Salleo, Adv. Mater., 2009, 21, 1568–1572 CrossRef CAS.
- T. Bjørnholm, D. R. Greve, N. Reitzel, T. Hassenkam, K. Kjaer, P. B. Howes, N. B. Larsen, J. Bøgelund, M. Jayaraman, P. C. Ewbank and R. D. McCullough, J. Am. Chem. Soc., 1998, 120, 7643–7644 CrossRef.
- D. H. Kim, Y. D. Park, Y. Jang, H. Yang, Y. H. Kim, J. I. Han, D. G. Moon, S. Park, T. Chang, C. Chang, M. Joo, C. Y. Ryu and K. Cho, Adv. Funct. Mater., 2005, 15, 77–82 CrossRef CAS.
- Y. Wang, H. Tatsumi, R. Otsuka, T. Mori and T. Michinobu, J. Mater. Chem. C, 2018, 6, 5865–5876 RSC.
- N. B. Kotadiya, A. Mondal, P. W. M. Blom, D. Andrienko and G.-J. A. H. Wetzelaer, Nat. Mater., 2019, 18, 1182–1186 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06807e |
| This journal is © The Royal Society of Chemistry 2022 |