
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium pincer complex-catalyzed heterocycle compatible alkoxycarbonylation of alkyl iodides: substrate keeps the catalyst active†
Han-Jun
Ai
 a,
Yang
Yuan
a and
Xiao-Feng
Wu
*ab
a,
Yang
Yuan
a and
Xiao-Feng
Wu
*ab
aLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: xiao-feng.Wu@catalysis.de
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Science, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
First published on 7th February 2022
Abstract
The electron pair of the heteroatom in heterocycles will coordinate with metal catalysts and decrease or even inhibit their catalytic activity consequently. In this work, a pincer ruthenium-catalyzed heterocycle compatible alkoxycarbonylation of alkyl iodides has been developed. Benefitting from the pincer ligand, a variety of heterocycles, such as thiophenes, morpholine, unprotected indoles, pyrrole, pyridine, pyrimidine, furan, thiazole, pyrazole, benzothiadiazole, and triazole, are compatible here.
Since the pioneering work on the catalytic alkoxycarbonylation of unactivated alkyl halides reported by Heck and Breslow in 1963,1 this transformation has attracted a great deal of interest due to its modularity and the direct employment of CO as a cheap and abundant C1 feedstock.2 However, compared with aryl halides, the development of alkoxycarbonylation of alkyl halides has been much more gradual.2,3 This situation is due to both the slow oxidative addition of C(sp3)–X bonds to the metal center and the easy β-hydride elimination of the alkyl-metal intermediate, particularly in the presence of carbon monoxide.4 Several catalytic systems for this process have been successfully developed in recent years (Scheme 1A), such as pure radical-based systems,5 palladium-based systems,6hν/palladium-based systems,7 rhodium-based systems,8 copper-based systems,9 and other metal carbonyl complex-based systems.10 Very recently, Neumann, Skrydstrup, and co-workers reported a nickel pincer-mediated alkoxycarbonylation for complete carbon isotope replacement, and this approach provided a procedure for generating carbon-labeled versions of potential simple carboxylate prodrug derivatives (Scheme 1B).11 Besides their advantages, in these cases the heterocycles, particularly those containing multiple N atoms or NH groups, are hardly compatible, which is considered as a remaining challenge. We attribute this to the Lewis-basic atoms in heterocyclic motifs being particularly detrimental to catalyst activity and potentially quenching the radical intermediates.12 Indeed, the development of heterocycle compatible catalytic systems remains an exciting task in the field of alkoxycarbonylation.
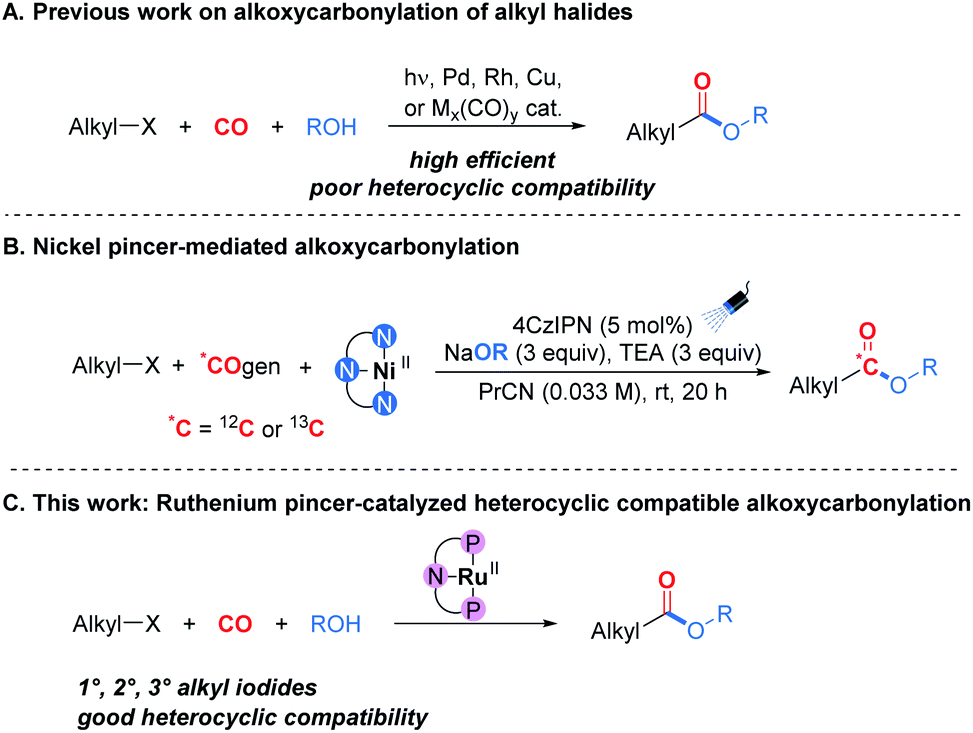 | ||
| Scheme 1 Approaches to alkoxycarbonylation of alkyl halides. | ||
On the other hand, heterocycles constitute important structural components of biologically active compounds and are ubiquitous in agrochemical and pharmaceutical industries.13 In a recent survey, 88% of small molecule drugs approved by the FDA between 2015 and June 2020 were found to contain at least one N-heterocycle.14 Specifically, heterocyclic subunits can modify the solubility, lipophilicity, polarity and hydrogen bonding ability of biologically active agents, thereby optimizing the corresponding ADME/Tox (absorption, distribution, metabolism, excretion, and toxicity) properties of drugs or drug candidates.15 Under this premise, the pursuit of new synthetic methods with good heterocycle compatibility is a worthwhile endeavor.
Herein we report a heterocycle compatible catalytic system for alkoxycarbonylation of alkyl iodides. With a ruthenium pincer complex as the catalyst, the tight coordination of the pincer ligand can effectively prevent the ruthenium from deactivation by heterocycle coordination (Scheme 1C). To the best of our knowledge, this is the first example of a ruthenium pincer complex-catalyzed carbonylation reaction.16 This new catalytic system might lead to novel synthetic routes toward heterocyclic carbonyl-containing compounds.
Pincer complexes of ruthenium are among the most effective catalysts for hydrogen transfer reactions between alcohols and unsaturated compounds.17 We initially used it to attempt the carbonylative coupling of acetophenone with iodobutane, as shown in eqn (1). Although we did not get the desired product I, the ester II could be obtained in 22% yield. By literature survey, we found there was no example showing that alkyl halides could be activated by ruthenium in previous reports on carbonylation reactions.3,16,18 We thus envisioned that the ruthenium pincer complex played a key role in this transformation.11,19
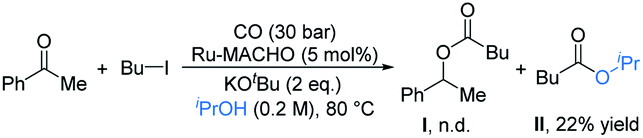 | (1) |
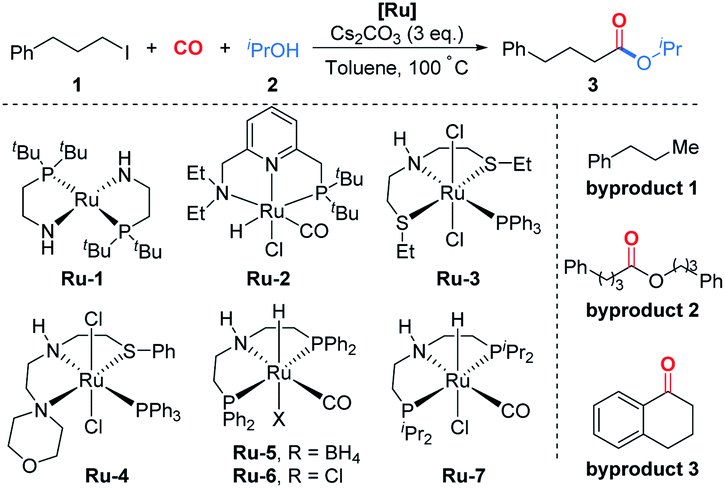
With this discovery in mind, we started the investigation of this ruthenium-catalyzed alkoxycarbonylation of alkyl halides by examining the reaction of (3-iodopropyl)benzene (1) with isopropanol (2) at 100 °C under a CO atmosphere (10 bar) in the presence of a catalytic amount of various readily available ruthenium pincer complexes (Table 1). Ordinary ruthenium catalysts, when used alone or in combination with monodentate/bidentate ligands, barely allow the reaction to proceed (Table 1, entries 1–3; for more details, see the ESI†). The improved yield of the desired product 3 was obtained when utilizing Milstein's catalyst Ru-220 (Table 1, entry 4). SNS or NNS-type pincer/Ru complexes Ru-3,4 afforded moderate conversions and poor selectivity (Table 1, entries 5 and 6). Meanwhile, Ru-MACHO-BH (Ru-5) and Ru-MACHO (Ru-6)21 were applied in the reaction; however, the selectivity obtained was unsatisfactory (Table 1, entries 7 and 8). Encouragingly, a significant improvement in the yield of 3 was achieved when employing Ru-7 as the catalyst precursor (Table 1, entry 9). The reaction could also be successfully performed under low CO pressure (1 bar), although with slightly lower yield (Table 1, entry 10). When using Ru-7, we were able to decrease the catalyst loading to 2.5 mol% without affecting the yield (Table 1, entries 11 and 12). Finally, after a judicious choice of the reaction temperature, optimal reaction conditions were constituted, delivering 3 in 86% yield (Table 1, entry 13). In most of these reactions, the formation of byproduct 1–3 was observed. As shown in eqn (2), when we removed isopropanol from the reaction, byproduct 2 which was produced by carbonylative homocoupling of the alkyl halide could be obtained in 71% yield.22 However, the reduced conversion and the absence of byproduct 3 implied that the alcohol plays more than a nucleophile role in this reaction. It is important to mention that the addition of water had no effect on the yield of byproduct 2. Concerning the effects from bases, organic bases, such as NEt3 and DBU, were tested, but no desired ester could be detected. Inorganic bases, including K2CO3 and K3PO4, were also tested, but very low yield of the ester was obtained. Notably, comparable yield of ester 3 can be obtained when LiOtBu was used as the base.
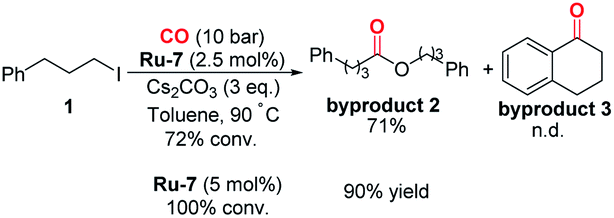 | (2) |
|
|
|||
|---|---|---|---|
| Entry | [Ru] | Conv.b (%) | Yieldb (%) |
| a Reaction conditions: 1 (0.2 mmol), 2 (0.6 mmol), [Ru] (5 mol%), Cs2CO3 (0.6 mmol), toluene (0.5 mL), CO (10 bar), 100 °C, 12 h. b Determined by GC with hexadecane as the internal standard. c CO (1 bar), N2 (9 bar). d [Ru] (2.5 mol%) was used. e [Ru] (1 mol%) was used. f 90 °C, average yield of two independent reactions. | |||
| 1 | Ru(acac)3 | 60 | 15 |
| 2 | RuH(Cl)(CO)(PPh3)3 | 21 | 11 |
| 3 | Ru-1 | 100 | 24 |
| 4 | Ru-2 | 93 | 41 |
| 5 | Ru-3 | 53 | 4 |
| 6 | Ru-4 | 49 | 5 |
| 7 | Ru-5 | 100 | 32 |
| 8 | Ru-6 | 100 | 38 |
| 9 | Ru-7 | 100 | 81 |
| 10 | Ru-7 | 100 | 63c |
| 11 | Ru-7 | 100 | 82d |
| 12 | Ru-7 | 100 | 72e |
| 13 | Ru-7 | 100 | 86d,f |
We next turned our attention to study the scope and the limitation of this transformation, as shown in Fig. 1. At the first stage, a variety of alcohols containing different functional groups and structural blocks were tested. In general, moderate to excellent yields were obtained under the standard conditions. For primary alcohols, the length of the carbon chain did not affect the good yield (4–7). The reaction tolerated the presence of ethers (8, 9), thioether (10), alkene (11), chlorine (12), trimethylsilyl (13), and amide (21). Benzyl alcohols and secondary alcohols were afterwards tested in this system and successfully transformed into the corresponding esters in good yields (14–17). With the further increase of the steric hindrance, tertiary alcohols hardly provided the desired products (18, 19). Phenol was also employed as the substrate in our attempt, and not surprisingly, phenyl 3-phenylpropyl ether (SN reaction product) was isolated as the main product (20).23 Interestingly, ethylene glycol could be converted to diester 22 in 83% yield, and no monocarbonylation product was detected, even though the alcohol was three equivalents. This suggests an interaction between the alcohol and the catalytic center, resulting in a higher rate of intramolecular reaction than intermolecular reaction. Subsequently, the excellent heterocycle compatibility of the method is nicely illustrated by the fact that thiophenes (23, 27), morpholine (24), unprotected indoles (25, 26), pyrrole (27), pyridine (28), pyrimidine (29), furan (30), thiazole (31), pyrazole (32), benzothiadiazole (33), and triazole (34) were perfectly tolerated under our protocol. The broad synthetic applicability of the reaction was also reflected in the successful alkoxycarbonylation of various primary iodides (35–44), secondary iodides (45–47), and even sterically hindered tertiary iodides (48–50).
 | ||
| Fig. 1 Scope of Ru pincer complex-catalyzed alkoxycarbonylation. Reactions run with 0.2 mmol of alkyl iodide and 3 equiv. of alcohol. Yield of the isolated product. aTogether with a 68% yield of the SN reaction product (phenyl 3-phenylpropyl ether). bEthylene glycol (3 equiv.) was used. cReduced yield of the isolated product because of the volatility of the product. | ||
In particular, the secondary iodides generated the corresponding esters in near quantitative yields. We also evaluated a substrate containing the C(sp2)–I bond to probe the chemoselectivity of our process (41). No trace of arylate was detected in the crude mixture by GC-MS, hence illustrating the good chemoselectivity of this catalytic system and offering opportunities for further structure modification. While this new methodology allows for the formation of a wide range of heterocycle-containing esters, some limitations still remain in terms of substrate scope. Bromoalkanes and chloroalkanes cannot be successfully converted under these conditions, even with the addition of equivalent amounts of NaI.
The alkoxycarbonylation could be applied to late-stage modification of a range of drugs and natural products, as shown in Fig. 2. trans-Sobrerol, a mucolytic, was successfully transformed, while the tertiary C–OH group was retained (51). A weak androgen, epiandrosterone, which is widely recognized to inhibit the pentose phosphate pathway and to decrease intracellular NADPH levels, provided 52 in 93% yield. Derivatives of estrone, cholesterol, and vitamin E also delivered the corresponding esters 53–55 in moderate to good yields. Common alcohol natural products, such as crotonyl alcohol, piperonyl alcohol, (−)-perillyl alcohol, (−)-borneol, (−)-menthol, and nerol, were tested as well and applicable to the reaction (56–61), which illustrated the utility of this method.
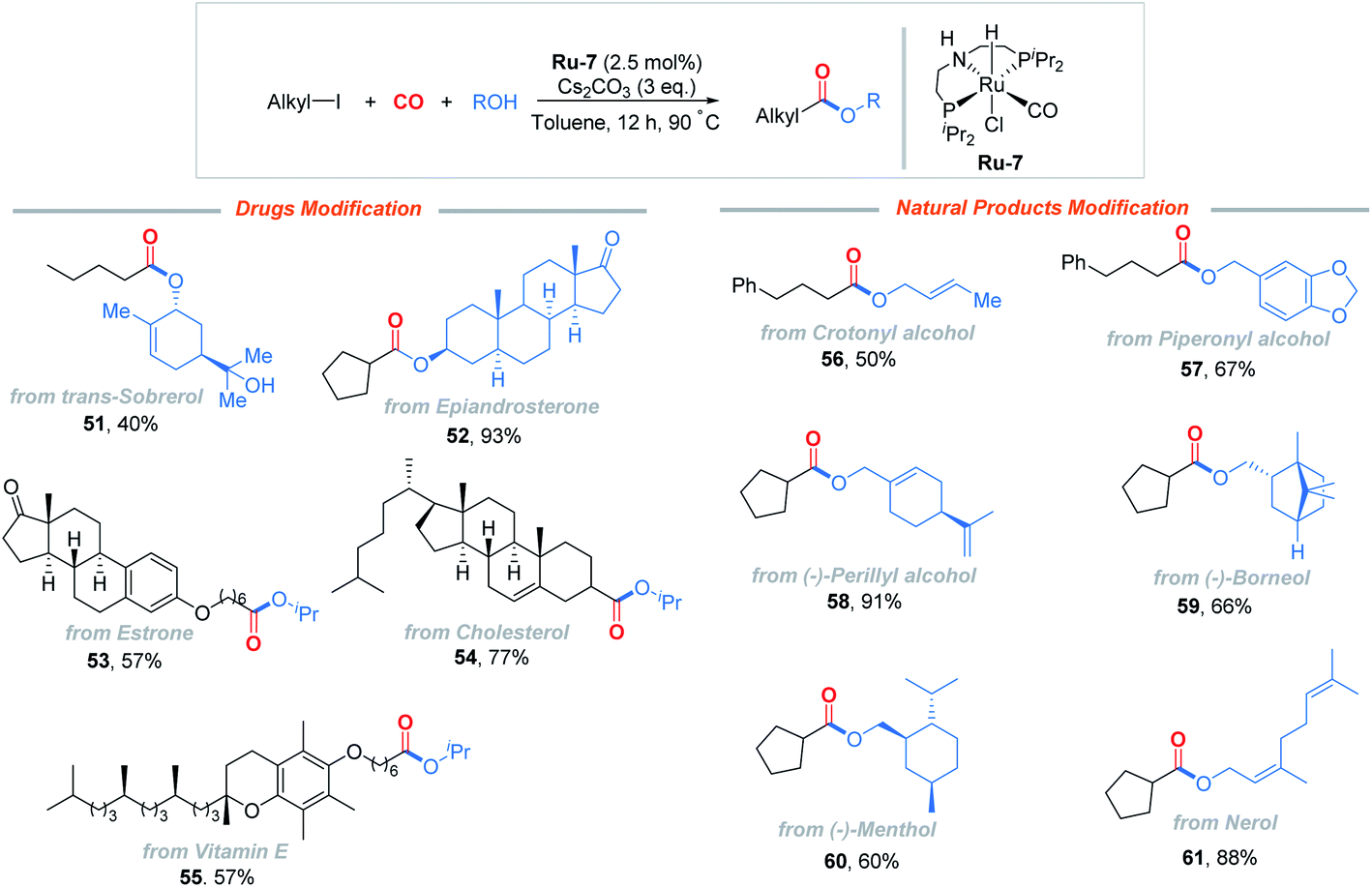 | ||
| Fig. 2 Modification of drugs and natural products. Reactions run with 0.2 mmol of alkyl iodide and 3 equiv. of alcohol. Yield of the isolated product. | ||
To gain more mechanistic insight into the reaction pathway, several experiments were conducted (Scheme 2). Under the standard conditions, the addition of TEMPO (radical capture agent) to the reaction led to the termination of the target reaction; meanwhile, the intermediate was captured (62) in 91% isolated yield (Scheme 2A, middle). In the control experiment, only limited conversion and no 62 was observed in the absence of the pincer catalyst (Scheme 2A, top), thus suggesting that the pincer/Ru activates the alkyl iodides to radicals. To ensure the radical pathway, we subsequently conducted radical inhibition experiments with BHT (butylated hydroxytoluene) as the radical inhibitor (Scheme 2B) and radical clock experiments (Scheme 2C). The model reaction was gradually suppressed with the addition of BHT. Furthermore, (iodomethyl)cyclopropane and 6-iodohex-1-ene under our optimized reaction conditions provided the corresponding ring-opening expansion product 64 and the cyclization product 65, respectively, with high selectivity.24
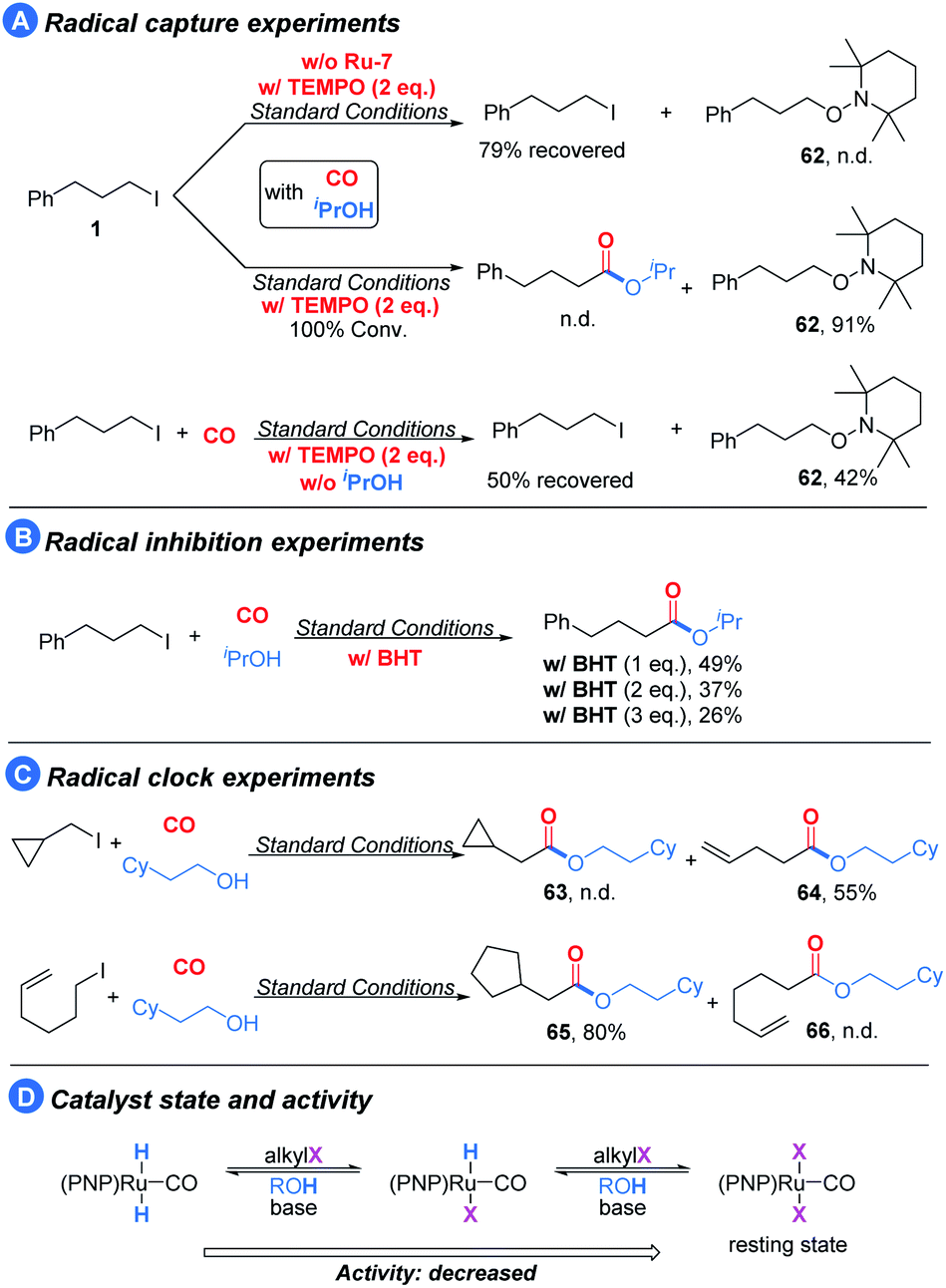 | ||
| Scheme 2 Mechanism studies. | ||
Based on the above results, we believe that the reaction involves a radical intermediate. In addition to this, as noted earlier, the alcohol appears to interact with the catalytic center and plays a role in promoting the activation of the alkyl halide. To probe this hypothesis, we removed the isopropanol from the reaction and utilized TEMPO to capture the radical intermediate (Scheme 2A, below). Compared with the reaction in the middle of Scheme 2A, the conversion and the yield of 62 significantly decreased in the absence of isopropanol. We explained that the (PNP)Ru(CO)X2 type complex is the catalyst resting state, and the alcohol may help it to return to the active state by hydrodehalogenation (Scheme 2D).25 Moreover, we could observe acetone during the optimization process, and when we subjected isopropanol alone to our optimized conditions, 57% yield of acetone could be detected,26 which suggests that (PNP)Ru(CO)HX can also undergo hydrodehalogenation to form (PNP)Ru(CO)H2.
Based on the above results and previous reports,16–18 a plausible mechanism is proposed (Scheme 3). Initially, the active 16 electron ruthenium complex A will be formed under the assistance of the base. Through a SET process, alkyl iodide will be activated and a 17 electron ruthenium complex B will be formed together with the corresponding alkyl radical which will immediately react with B to give 18 electron ruthenium complex C. The acylruthenium complex D will be produced after a CO insertion step. The possibility that the acylruthenium complex D might also be produced from complex B and the in situ formed acyl radical cannot be excluded. After X ligand exchange, ruthenium complex E will be formed which will provide the final ester product after a reductive elimination step and regenerate the active ruthenium catalyst A to finish the catalyst cycle. Alternatively, the direct nucleophilic attack at the acyl carbonyl of complex D by alcohol to give the ester product and complex F is also possible. Then complex F will be transformed into complex A under the assistance of the base.
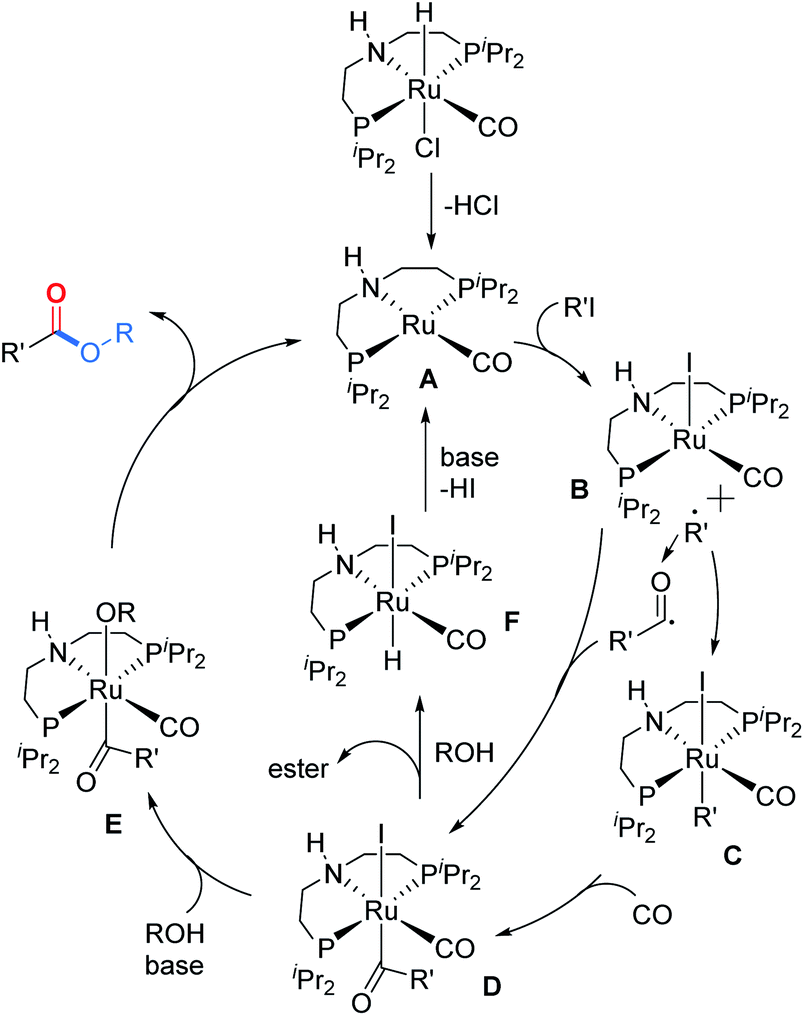 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a pincer ruthenium-catalyzed heterocycle compatible alkoxycarbonylation of alkyl iodides. By utilizing the tight coordination of the pincer ligand to effectively prevent the activity reduction of Ru, a variety of heterocycles are compatible. Remarkably, our approach also represents the first example of a pincer ruthenium-catalyzed carbonylation reaction. The excellent heterocycle compatibility, wide substrate scope, and generality of the protocol suggest that it can be a powerful tool for preparing heterocyclic carbonyl-containing compounds.Author contributions
XFW directed this project and revised the manuscript. HJA performed all the experiments and prepared the draft and ESI.† YY prepared a part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from K. C. Wong Education Foundation (GJTD-2020-08).Notes and references
- R. F. Heck and D. S. Breslow, J. Am. Chem. Soc., 1963, 85, 2779–2782 CrossRef CAS.
- For selected comprehensive reviews and books, see: (a) G. Kiss, Chem. Rev., 2001, 101, 3435–3456 CrossRef CAS PubMed; (b) B. Gabriele, Carbon Monoxide in Organic Synthesis: Carbonylation Chemistry, Wiley-VCH: Weinheim, Germany, 2021 CrossRef; (c) C. F. J. Barnard, Organometallics, 2008, 27, 5402–5422 CrossRef CAS; (d) L.-J. Cheng and N. P. Mankad, Chem. Soc. Rev., 2020, 49, 8036–8064 RSC; (e) S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2014, 47, 1563–1574 CrossRef CAS PubMed; (f) J.-B. Peng, F.-P. Wu and X.-F. Wu, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed; (g) D.-M. Yan, C. M. Crudden, J.-R. Chen and W.-J. Xiao, ACS Catal., 2019, 9, 6467–6483 CrossRef CAS; (h) K. László, in Modern carbonylation methods, John Wiley & Sons, 2008, pp. 1–367 Search PubMed.
- For selected reviews on carbonylation of alkyl halides, see: (a) L.-J. Cheng and N. P. Mankad, Acc. Chem. Res., 2021, 54, 2261–2274 CrossRef CAS PubMed; (b) A. Frisch and C. M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS PubMed; (c) L. Wu, X. Fang, Q. Liu, R. Jackstell, M. Beller and X.-F. Wu, ACS Catal., 2014, 4, 2977–2989 CrossRef CAS; (d) M. R. Kwiatkowski and E. J. Alexanian, Acc. Chem. Res., 2019, 52, 1134–1144 CrossRef CAS PubMed.
- (a) J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books: Sausalito, CA, 2010; pp. 301–320 Search PubMed; (b) A. C. Bissember, A. Levina and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 14232–14237 CrossRef CAS PubMed.
- (a) I. Ryu, Chem. Soc. Rev., 2001, 30, 16–25 RSC; (b) O. Itsenko and B. Långström, Org. Lett., 2005, 7, 4661–4664 CrossRef CAS PubMed.
- B. T. Sargent and E. J Alexanian, J. Am. Chem. Soc., 2016, 138, 7520–7523 CrossRef CAS PubMed.
- (a) T. Fukuyama, S. Nishitani, T. Inouye, K. Morimoto and I. Ryu, Org. Lett., 2006, 8, 1383–1386 CrossRef CAS PubMed; (b) A. Fusano, S. Sumino, S. Nishitani, T. Inouye, K. Morimoto, T. Fukuyama and I. Ryu, Chem.–Eur. J., 2012, 18, 9415–9422 CrossRef CAS PubMed; (c) G. M. Torres, Y. Liu and B. A. Arndtsen, Science, 2020, 368, 318–323 CrossRef CAS PubMed.
- (a) H.-J. Ai, H. Wang, C.-L. Li and X.-F. Wu, ACS Catal., 2020, 10, 5147–5152 CrossRef CAS; (b) X. Qi, R. Zhou, H.-J. Ai and X.-F. Wu, J. Catal., 2020, 381, 215–221 CrossRef CAS.
- (a) Y. Chen, L. Su and H. Gong, Org. Lett., 2019, 21, 4689–4693 CrossRef CAS PubMed; (b) Y. Li and X.-F. Wu, Chem. Commun., 2018, 1, 39 CrossRef.
- The only reported ruthenium-catalyzed alkoxycarbonylation reaction also requires a 200 W high-pressure mercury lamp (a) T. Kondo, Y. Tsuji and Y. Watanabe, Tetrahedron Lett., 1988, 29, 3833–3836 CrossRef CAS; (b) T. Kondo, Y. Sone, Y. Tsuji and Y. Watanabe, J. Organomet. Chem., 1994, 473, 163–173 CrossRef CAS; (c) H. Urata, H. Maekawa, S. Takahashi and T. Fuchikami, J. Org. Chem., 1991, 56, 4320–4322 CrossRef CAS.
- S. J. Ton, K. T. Neumann, P. Nørby and T. Skrydstrup, J. Am. Chem. Soc., 2021, 143, 17816–17824 CrossRef CAS PubMed.
- (a) S. Ge and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 12837–12841 CrossRef CAS PubMed; (b) M. A. Düfert, K. L. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 12877–12885 CrossRef PubMed; (c) V. F. Slagt, A. H. M. de Vries, J. G. de Vries and R. M. Kellogg, Org. Process Res. Dev., 2010, 14, 30–47 CrossRef CAS; (d) D. K. Whelligan, D. W. Thomson, D. Taylor and S. Hoelder, J. Org. Chem., 2010, 75, 11–15 CrossRef CAS PubMed; (e) V. M. Kassel, C. M. Hanneman, C. P. Delaney and S. E. Denmark, J. Am. Chem. Soc., 2021, 143, 13845–13853 CrossRef CAS PubMed.
- (a) C. Lamberth, Pest Manage. Sci., 2013, 69, 1106–1114 CrossRef CAS PubMed; (b) M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC; (c) A. Gomtsyan, Chem. Heterocycl. Compd., 2012, 48, 7–10 CrossRef CAS; (d) B. Eftekhari-Sis, M. Zirak and A. Akbari, Chem. Rev., 2013, 113, 2958–3043 CrossRef CAS PubMed; (e) M. A. P. Martins, C. P. Frizzo, D. N. Moreira, L. Buriol and P. Machado, Chem. Rev., 2009, 109, 4140–4182 CrossRef CAS PubMed.
- P. Bhutani, G. Joshi, N. Raja, N. Bachhav, P. K. Rajanna, H. Bhutani, A. T. Paul and R. Kumar, J. Med. Chem., 2021, 64, 2339–2381 CrossRef CAS PubMed.
- (a) D. Dalvie, P. Kang, C.-M. Loi, L. Goulet and S. Nair, Influence of Heteroaromatic Rings on ADME Properties of Drugs. Metabolism, Pharmacokinetics and Toxicity of Functional Groups: Impact of Chemical Building Blocks on ADMET, The Royal Society of Chemistry, 2010, 7, 328–369 Search PubMed; (b) J. Jampilek, Molecules, 2019, 24, 3839 CrossRef CAS PubMed.
- The only reported ruthenium-catalyzed alkoxycarbonylation reaction also requires a 200 W high-pressure mercury lamp, see ref. 10a,b. For other interesting approaches to Ru-catalyzed carbonylations, see: (a) X.-F. Wu and H. Neumann, ChemCatChem, 2012, 4, 447–458 CrossRef CAS; (b) J.-X. Xu, F. Zhao, Y. Yuan and X.-F. Wu, Org. Lett., 2020, 22, 2756–2760 CrossRef CAS PubMed; (c) A. Tlili, J. Schranck, J. Pospech, H. Neumann and M. Beller, Angew. Chem. Int. Ed., 2013, 52, 6293–6297 CrossRef CAS PubMed; (d) K. Shibata, N. Hasegawa, Y. Fukumoto and N. Chatani, ChemCatChem, 2012, 4, 1733–1736 CrossRef CAS; (e) S. Inoue, H. Shiota, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2009, 131, 6898–6899 CrossRef CAS PubMed; (f) N. Hasegawa, V. Charra, S. Inoue, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 8070–8073 CrossRef CAS PubMed; (g) K. Inamoto, J. Kadokawa and Y. Kondo, Org. Lett., 2013, 15, 3962–3965 CrossRef CAS PubMed.
- (a) C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed; (b) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (c) C. Bruneau and P. H. Dixneuf, Ruthenium in Catalysis, Topics in Organometallic Chemistry series, Springer, 2014 Search PubMed; (d) C. Bruneau and P. H. Dixneuf, Ruthenium catalysts in fine chemistry, Topics in organometallic chemistry, Springer, 2004 CrossRef.
- Grubbs and co-workers reported a Ru-catalyzed reduction of alkyl halides, see: M. C. Haibach, B. M. Stoltz and R. H. Grubbs, Angew. Chem., Int. Ed., 2017, 56, 15123–15126 CrossRef CAS PubMed.
- For recent examples on Ni-pincer complex promoted carbonylations, see: (a) A. S. Donslund, S. S. Pedersen, C. Gaardbo, K. T. Neumann, L. Kingston, C. S. Elmore and T. Skrydstrup, Angew. Chem., Int. Ed., 2020, 59, 8099–8103 CrossRef CAS PubMed; (b) A. S. Donslund, K. T. Neumann, N. P. Corneliussen, E. K. Grove, D. Herbstritt, K. Daasbjerg and T. Skrydstrup, Chem.–Eur. J., 2019, 25, 9856–9860 CrossRef CAS PubMed; (c) T. L. Andersen, A. S. Donslund, K. T. Neumann and T. Skrydstrup, Angew. Chem., Int. Ed., 2018, 57, 800–804 CrossRef CAS PubMed; (d) K. T. Neumann, A. S. Donslund, T. L. Andersen, D. U. Nielsen and T. Skrydstrup, Chem.–Eur. J., 2018, 24, 14946–14949 CrossRef CAS PubMed; (e) S. S. Pedersen, A. S. Donslund, J. H. Mikkelsen, O. Bakholm, F. Papp, K. B. Jensen, M. B. F. Gustafsson and T. Skrydstrup, Chem.–Eur. J., 2021, 27, 7114–7123 CrossRef CAS PubMed.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T. Kobayashi, N. Sayo and T. Saito, Org. Process Res. Dev., 2012, 16, 166–171 CrossRef CAS.
- In the reaction, Cs2CO3 is transformed into CsHCO3 which will give Cs2CO3, H2O, and CO2 after thermal disproportionation. The produced H2O will react with RX to give ROH which then produces RCO2R after the carbonylation reaction. See ref. 8a.
- This situation can mainly be explained from two aspects: (i) in the presence of a base, haloalkanes and phenols are prone to a nucleophilic substitution reaction to produce ethers; (ii) carbonylation of haloalkanes usually proceeds via free radical intermediates, which tend to be quenched by phenols. For solution, see ref. 8a.
- C. Walling and A. Cioffari, J. Am. Chem. Soc., 1972, 94, 6059–6064 CrossRef CAS.
- The catalyst resting state of the pincer/Ru complex, (PNN)RuX2(CO), was also proposed by Grubbs and co-workers by unlocked 31P NMR spectroscopy. See ref. 18.
- See the ESI for details.
Footnote |
| † Electronic supplementary information (ESI) available: General comments, general procedure, analytic data, and NMR spectra. See DOI: 10.1039/d1sc06581e |
| This journal is © The Royal Society of Chemistry 2022 |