
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-catalyzed asymmetric trans-selective hydrogenation of 1,3-disubstituted isoquinolines†
Alexia N.
Kim
 a,
Aurapat
Ngamnithiporn
ab,
Michael D.
Bartberger
c and
Brian M.
Stoltz
*a
a,
Aurapat
Ngamnithiporn
ab,
Michael D.
Bartberger
c and
Brian M.
Stoltz
*a
aWarren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: stoltz@caltech.edu
bLaboratory of Medicinal Chemistry, Chulabhorn Research Institute, 54 Kamphaeng Phet 6 Road, Bangkok, 10210, Thailand
c1200 Pharma LLC, 844 East Green Street, Suite 204, Pasadena, CA 91101, USA
First published on 18th February 2022
Abstract
The development of the first asymmetric trans-selective hydrogenation of 1,3-disubstituted isoquinolines is reported. Utilizing [Ir(cod)Cl]2 and a commercially available chiral Josiphos ligand, a variety of differentially substituted isoquinolines are hydrogenated to produce enantioenriched trans-tetrahydroisoquinolines in good yield with high levels of enantioselectivity. Directing group studies demonstrate that the hydroxymethyl functionality at the C1 position is critical for hydrogenation to favor the trans-diastereomer. Preliminary mechanistic studies reveal that non-coordinating chlorinated solvents and halide additives are crucial to enable trans-selectivity.
Introduction
The asymmetric hydrogenation of heteroarenes has recently emerged as a powerful strategy to directly access enantioenriched, saturated heterocycles.1,2 While significant progress has been made in this field, controlling selectivity in the formation of multiple stereocenters in a single reaction remains challenging. Transition metal-catalyzed hydrogenation of arenes typically proceeds through initial dearomative reduction of the substrate and subsequent rapid hydrogenation, resulting in high cis-selectivity of product (Fig. 1A, path A).3,4 In contrast, accessing the trans-isomer requires a π-facial exchange of the arene to allow hydride delivery from the more sterically hindered face, rendering its synthesis significantly more difficult (Fig. 1A, path B). While several reports describe the trans-selective hydrogenation of arenes, most are limited in scope and not enantioselective.5,6 Considering the synthetic value of trans-substituted saturated heterocycles, a general method for an asymmetric trans-selective hydrogenation of heteroarenes would be a highly desirable and powerful strategy.7,8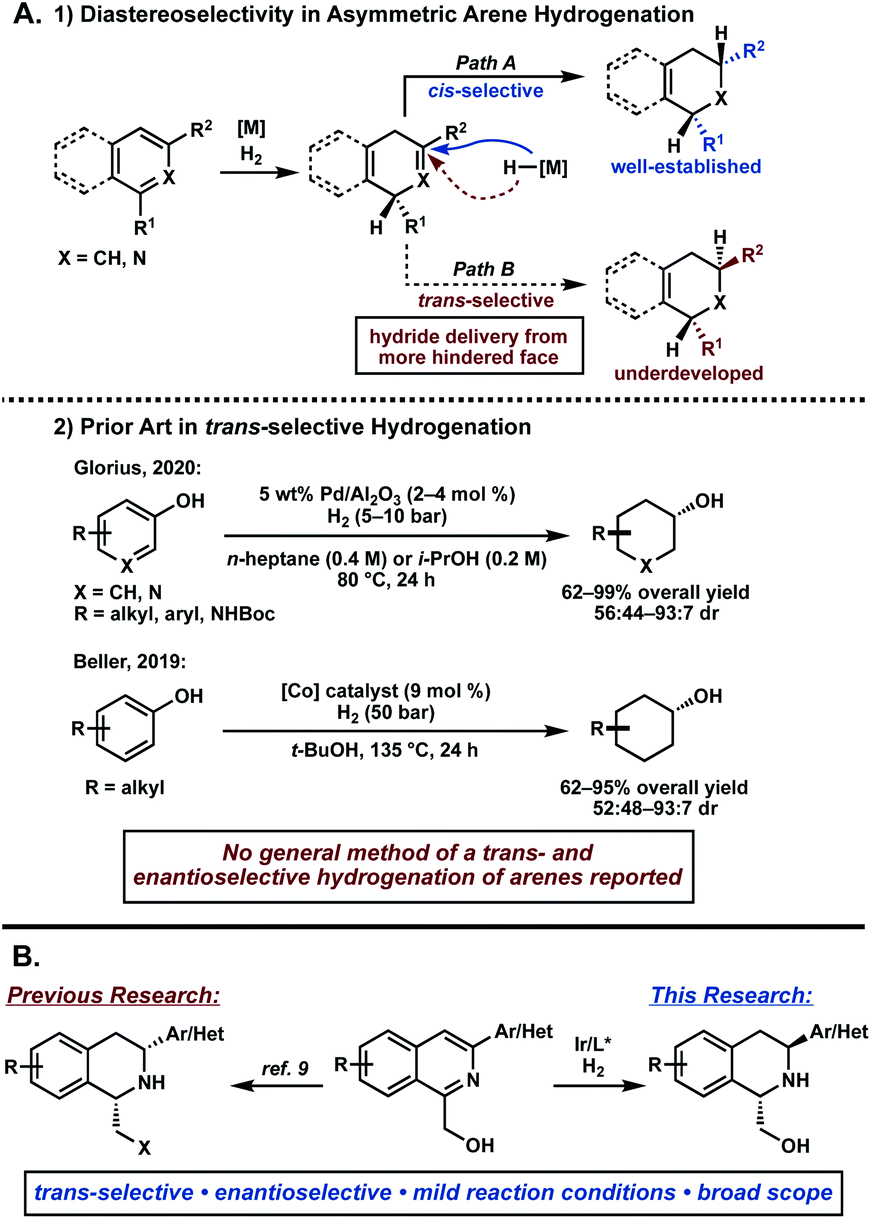 | ||
| Fig. 1 (A) Challenges in diastereoselectivity of trans-selective arene hydrogenation. (B) Our research on iridium-catalyzed asymmetric hydrogenation of 1,3-disubstituted isoquinolines. | ||
Recently, our group has reported the asymmetric hydrogenation of 1,3-disubstituted isoquinolines to access enantioenriched cis-1,2,3,4-tetrahydroisoquinolines (THIQs).9 This method enables the asymmetric hydrogenation of isoquinolines with Lewis basic functionalities, such as primary alcohols and heteroaryl-substituted isoquinolines, that significantly expanded the scope of the transformation compared to prior reports.10 During the course of this investigation, we also observed formation of the trans-THIQ under certain conditions in excellent enantioselectivity, albeit in small amounts. Herein, we disclose our efforts to develop the first examples of an asymmetric trans-selective hydrogenation of 1,3-disubstituted isoquinolines to access enantioenriched trans-THIQs (Fig. 1B).
Results and discussion
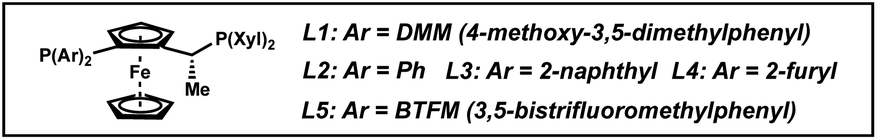
We began our studies with 1-(hydroxymethyl)-3-phenylisoquinoline (1a) as a model substrate. An initial experiment employing 1.25 mol% of [Ir(cod)Cl]2, 3 mol% of chiral Josiphos ligand L1, and 7.5 mol% of TBAI in THF delivered the THIQ product with high cis-selectivity (Table 1, entry 1).8 We observed that using CH2Cl2 as solvent gave higher levels of diastereoselectivity for trans-isomer 2a, with excellent enantioselectivity as well (97% ee, entry 2).11 Gratified by this result, we explored different additives and observed that smaller halides afforded higher levels of the desired diastereoselectivity, albeit with diminished conversions of 1a. Overall, TBABr provided the best combination of diastereo- and enantioselectivity (Table S1†). Other additives such as TBAPF6 previously investigated by Pfaltz and co-workers completely shut down the reaction, demonstrating that halide salts were crucial for this transformation (entry 5).12|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | X | Ligand | Solvent | Additive | % conv.b |
trans![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :cisb :cisb |
% ee of transc |
| a Performed on a 0.04 mmol scale. b Determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as standard. c Determined by chiral SFC analysis of Cbz-protected trans-product. | |||||||
| 1 | OH (1a) | L1 | THF | TBAI | >95 | 1:15.7 |
— |
| 2 | OH | L1 | CH2Cl2 | TBAI | >95 | 1:1.5 |
97 |
| 3 | OH | L1 | CH2Cl2 | TBABr | >95 | 2:1 |
93 |
| 4 | OH | L1 | CH2Cl2 | TBACl | 75 | 2.3:1 |
80 |
| 5 | OH | L1 | CH2Cl2 | TBAPF6 | <10 | — | — |
| 6 | OH | L2 | CH2Cl2 | TBABr | >95 | 1.8:1 |
94 |
| 7 | OH | L3 | CH2Cl2 | TBABr | 95 | 1.4:1 |
99 |
| 8 | OH | L4 | CH2Cl2 | TBABr | 45 | 1:2.3 |
35 |
| 9 | OH | L5 | CH2Cl2 | TBABr | 83 | 1:2.9 |
81 |
| 10 | OH | L1 | PhMe | TBABr | >95 | 1.2:1 |
91 |
| 11 | OH | L1 | EtOAc | TBABr | >95 | 1:1.1 |
89 |
| 12 | OH | L1 | CHCl3 | TBABr | 68 | 2.4:1 |
93 |
| 13 | OH | L1 | 1,2-DCE | TBABr | >95 | 2.4:1 |
92 |
| 14 | OMe (3a) | L1 | 1,2-DCE | TBABr | >95 | 1:17 |
N.D. |
| 15 | OBn (3b) | L1 | 1,2-DCE | TBABr | >95 | 1:>20 |
N.D. |
| 16 | OAc (3c) | L1 | 1,2-DCE | TBABr | 57 | 1:>20 |
N.D. |
| 17 | H (3d) | L1 | 1,2-DCE | TBABr | 92 | 1:>20 |
N.D. |
| 18 | NHBoc (3e) | L1 | 1,2-DCE | TBABr | >95 | 1.4:1 |
25 |
| 19 | NH2 (3f) | L1 | 1,2-DCE | TBABr | 0 | — | — |
|
|||||||
Seeking to improve the diastereoselectivity, we surveyed a variety of chiral ligand scaffolds and found the xyliphos ligand framework to be optimal (Table S2†). We observed that more electron-rich aryl groups on the chiral ligand provided the trans-product with higher selectivity, with the DMM-substituted phosphine L1 affording the highest diastereoselectivity of 2:1 trans:cis (entry 3 vs. entries 6–9). In contrast, more electron-withdrawing aryl groups such as L5 favored the formation of the cis-product (entry 9).
Having identified L1 as the optimal ligand, we briefly investigated different solvents. We observed that non-coordinating chlorinated solvents such as chloroform and 1,2-dichloroethane delivered product 2a with the highest trans-selectivity (entries 12–13), while non-chlorinated solvents toluene and ethyl acetate gave nearly a 1:1 diastereomeric ratio (entries 10–11).13
We also explored different directing groups at the C1 position to probe their effects on the diastereoselectivity of this reaction. Isoquinolines bearing functionalities such as a methyl ether, benzyl ether or acetate substituent (3a–c) solely provided the cis-THIQ, suggesting that they are not functioning as directing groups in the reaction (Table 1, entries 14–16). Indeed, the hydrogenation of isoquinoline 3d that lacks any potential directing group also afforded only the cis-diastereomer of product (entry 17). While Boc-protected amine 3e provided the product in 1.4:1 dr favoring the trans-THIQ, basic amine functionalities such as primary amine 3f gave trace product, potentially due to catalyst deactivation (entry 19). Nevertheless, the investigation of different directing groups demonstrates that the hydroxyl functionality serves as the best directing group to selectively access the trans-diastereomer by enabling π-facial exchange of the substrate and facilitating hydride delivery from the more sterically hindered face via a directed hydrogenation.
With optimized reaction conditions identified, we explored the substrate scope of this transformation (Table 2). Due to the inseparable nature of the cis- and trans-diastereomers of the hydrogenated products, the crude reaction mixture was subsequently treated with 1,1′-carbonyldiimidazole (CDI) to afford the oxazolidinone-fused THIQs that were easily separable by column chromatography. From 5a, the relative and absolute stereochemistry of the trans-THIQ product was confirmed by X-ray crystallography.14
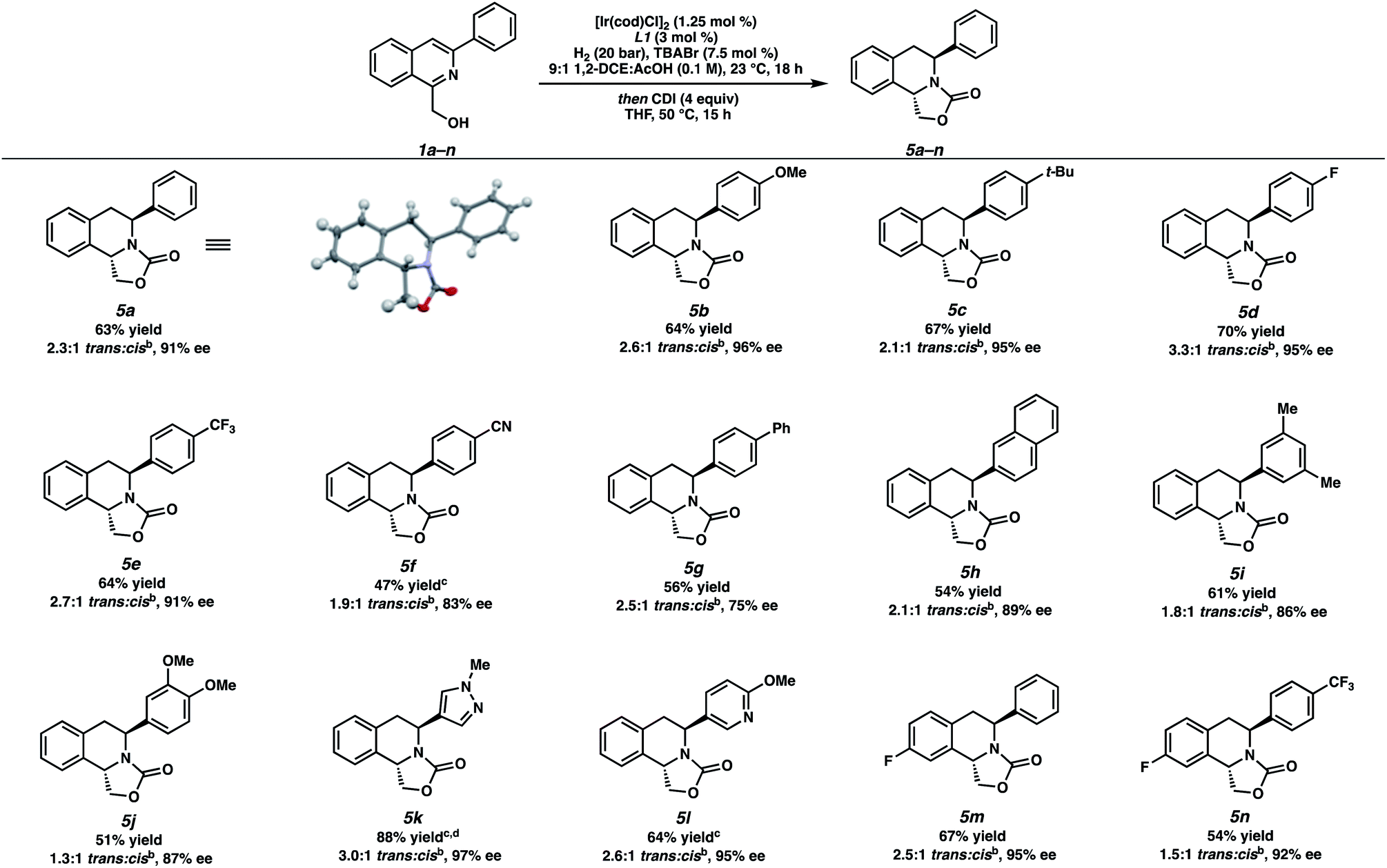
Gratifyingly, a wide variety of aryl substituents at the 3-position of the isoquinoline were well tolerated, selectively yielding the trans-product in moderate to excellent ee.15 Substitution at the para-position of the 3-aryl ring delivered hydrogenated products 5b–5g in high selectivities, ranging from electron-rich substrates 5b–5c to more electron-withdrawing substrates 5d–5f. Sterically encumbered substrates such as 3-naphthyl, 3-xylyl isoquinolines also afforded products 5h–5i in good isolated yields, with slightly diminished enantioselectivity. Furthermore, the nitrile functional group in 5f and naphthyl substituent of 5h were not reduced in this process, highlighting the chemoselectivity of this transformation.
Additionally, we were pleased to observe that heteroaryl-substituted isoquinolines were well tolerated at 60 °C and 60 bar H2 to produce trans-THIQs 5k,l in high enantioselectivities (97% and 94% ee, respectively), and with no erosion of diastereoselectivity. Finally, different electronics of the isoquinoline carbocycle such as fluorinated isoquinolines 1m–n were hydrogenated to afford electron-poor THIQs 5m–n in high selectivities under our standard conditions.
Having demonstrated that this transformation is general for a wide range of 1,3-disubstituted isoquinolines, we sought to derivatize the oxazolidinone-fused THIQs (Scheme 1). We were pleased to find that the oxazolidinone functional group could be efficiently removed with Ba(OH)2·8H2O to afford THIQ 2a in 81% yield.16,17 Alternatively, reduction with DIBAL afforded N-methyl THIQ 6a in 73% yield, providing a facile access of our hydrogenated products to N-methyl protected THIQs.
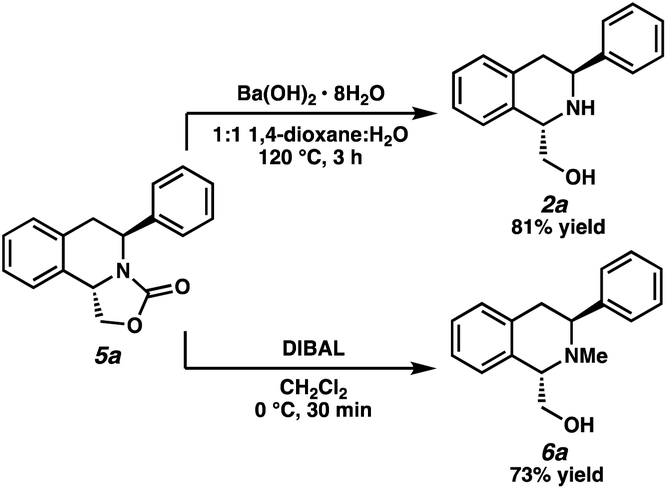 | ||
| Scheme 1 Synthetic derivatizations of product 5a. | ||
To elucidate the factors controlling the trans-selectivity in this transformation, several control experiments were conducted to probe the reaction mechanism (Scheme 2). Substituting TBABr for TBAI as the additive gave a 1:1.2 dr favoring the cis-product, with high enantioselectivities exhibited for both products (Scheme 2A). This suggests that the bromide ligand facilitates π-facial exchange of the substrate over iodide to afford higher levels of the trans-diastereomer.18,19 Replacing 1,2-DCE solvent for THF also delivered similar results, indicating that ethereal solvents inhibit the formation of trans-2a through stronger coordination with iridium (Scheme 2B).20 Overall, the combination of non-coordinating, chlorinated solvents and smaller halides are crucial in governing the observed trans-selectivity.
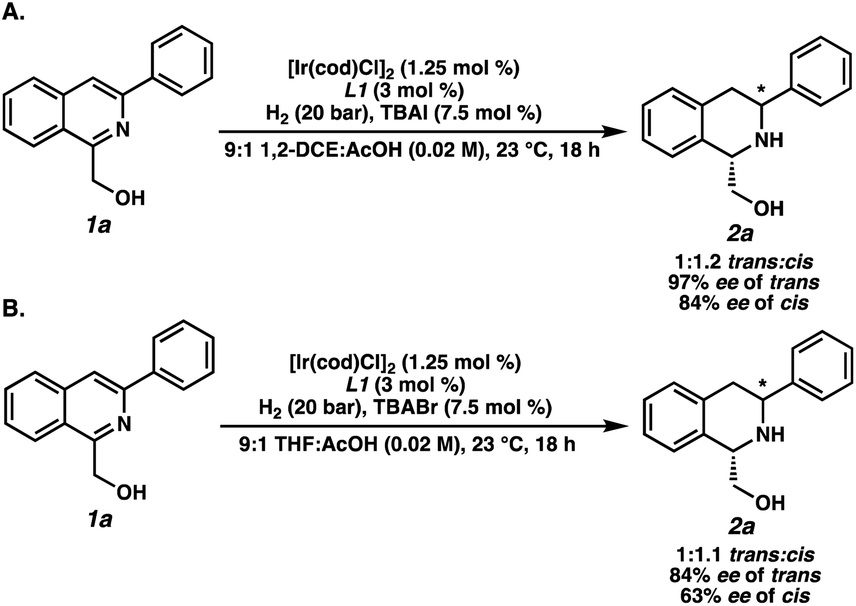 | ||
| Scheme 2 Control experiments of the asymmetric trans-selective hydrogenation by using (A) TBAI instead of TBABr and (B) THF instead of 1,2-DCE. | ||
Deuterium experiments were also conducted to determine the degree of deuteration of our hydrogenated products (Scheme 3). Interestingly, the combination of both D2 and CD3COOD delivered deuterium at the C1-, C3-, and C4-positions of the THIQ, as well as at the methylene carbon of the hydroxymethyl functional group (Scheme 3A). We attribute this exocyclic deuteration to a competitive β-hydride elimination pathway that is operative in situ under our trans-hydrogenation conditions (Scheme 3B).21,22 However, this is likely not a critical pathway toward the trans-product, as deuterium incorporation in the corresponding cis-isomer (cis-2a) is also observed. Exchanging either D2 or CD3COOD for their protic counterparts demonstrated deuteride delivery primarily from the gas at the C1- and C3-positions of 2a, yet the acid also enables reduction of the isoquinoline ring. This suggests a proton-hydride exchange occurring between the acid and the iridium hydride species for hydrogenation.19,23 Further investigations of the mechanism24 and other applications of this technology are currently underway and will be reported in due course.
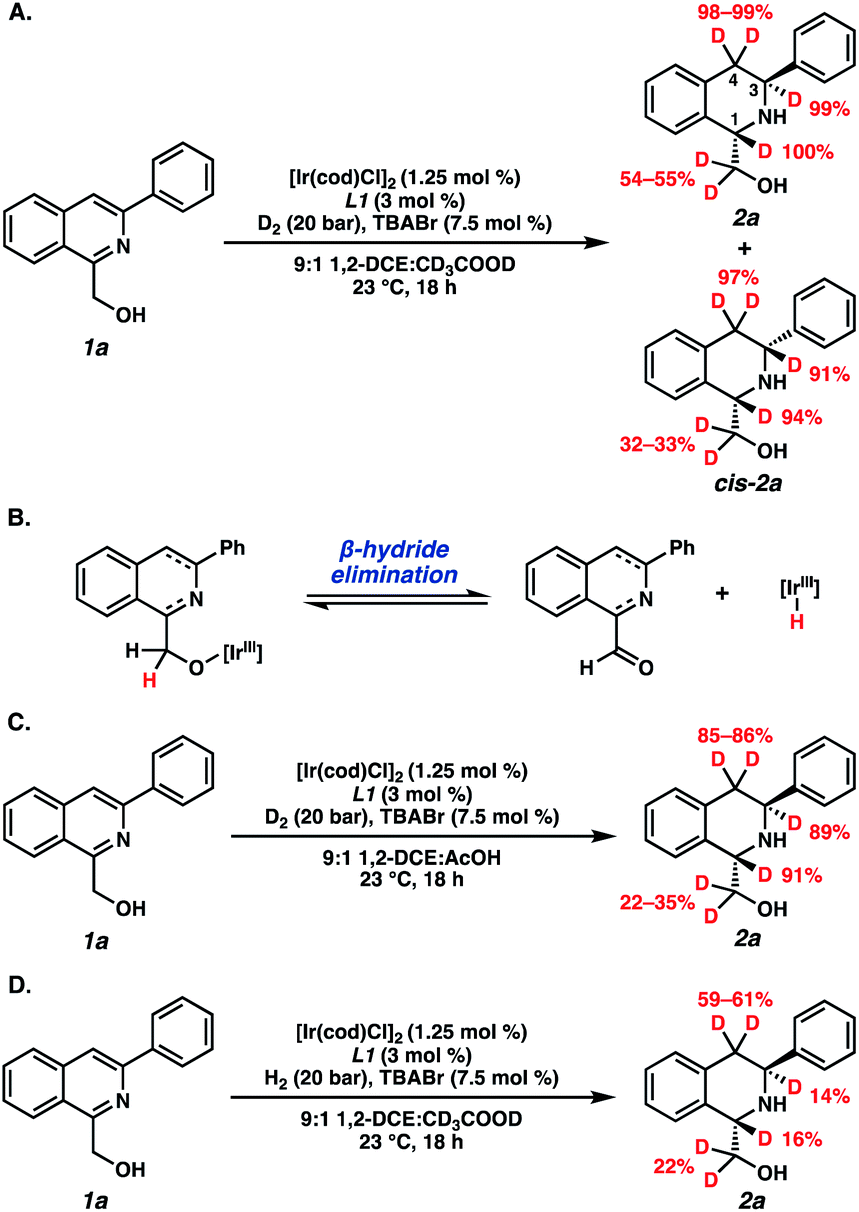 | ||
| Scheme 3 Deuterium experiments of (A) D2 and CD3COOD, (B) observed β-hydride elimination pathway, (C) D2 and protic AcOH, and (D) H2 and CD3COOD. | ||
Conclusions
In conclusion, we have developed an asymmetric trans-selective hydrogenation reaction of 1,3-disubstituted isoquinolines for the syntheses of enantioenriched trans-THIQs. Key to this enantio- and diastereoselective reaction is the hydroxymethyl directing group at the C1-position which enables π-facial exchange of the substrate and facilitates hydride delivery from the sterically more hindered face. This method tolerates a wide range of electronics, Lewis basic functionalities, and substitution at the C1, C3, and C8-positions of the isoquinoline core, representing one of the first examples of an asymmetric hydrogenation technology to selectively access the trans-diastereomer of hydrogenated aromatic compounds.Data availability
Data for this work, including optimization tables, general experimental procedures, characterization data for all new compounds and X-ray data are provided in the ESI.†Author contributions
B. M. S. conceived and directed the project. A. N. K. and A. N. designed, performed, and analyzed the synthetic chemistry experiments. M. D. B. assisted with VCD studies of THIQ 2a. A. N. K., A. N., M. D. B., and B. M. S. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NIH-NIGMS (R01GM127972A) and Caltech for financial support. A. N. thanks the Royal Thai Government Scholarship Program. We thank Dr David VanderVelde for NMR expertise, Dr Scott Virgil for instrumentation, Dr Mona Shahgholi for mass spectroscopy, and Dr Michael Takase and Larry Henling for assistance with X-ray analysis.Notes and references
- For recent reviews of asymmetric hydrogenation of heteroarenes, see: (a) D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed; (b) Y.-G. Zhou, Acc. Chem. Res., 2007, 40, 1357–1366 CrossRef CAS PubMed; (c) D. Zhao and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 9616–9618 CrossRef CAS PubMed; (d) A. N. Kim and B. M. Stoltz, ACS Catal., 2020, 10, 13834–13851 CrossRef CAS PubMed; (e) Y.-E. Luo, Y.-M. He and Q.-H. Fan, Chem. Rec., 2016, 16, 2697–2711 CrossRef PubMed; (f) N. Fleury-Brégeot, V. de la Fuente, S. Castillón and C. Claver, ChemCatChem, 2010, 2, 1346–1371 CrossRef.
- For recent reviews of asymmetric C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond hydrogenation, see:
(a) A. Cabré, X. Verdaguer and A. Riera, Chem. Rev., 2022, 122, 269–339 CrossRef PubMed;
(b) R. Abed Ali Abdine, G. Hedouin, F. Colobert and J. Wencel-Delord, ACS Catal., 2021, 11, 215–247 CrossRef;
(c) R. Molina Betancourt, P.-G. Echeverria, T. Ayad, P. Phansavath and V. Ratovelomanana-Vidal, Synthesis, 2021, 53, 30–50 CrossRef CAS;
(d) J. Barrios-Rivera, Y. Xu, M. Wills and V. K. Vyas, Org. Chem. Front., 2020, 7, 3312–3342 RSC;
(e) Y. Liu, X. Yue, C. Luo, L. Zhang and M. Lei, Energy Environ. Mater., 2019, 2, 292–312 CrossRef CAS.
N bond hydrogenation, see:
(a) A. Cabré, X. Verdaguer and A. Riera, Chem. Rev., 2022, 122, 269–339 CrossRef PubMed;
(b) R. Abed Ali Abdine, G. Hedouin, F. Colobert and J. Wencel-Delord, ACS Catal., 2021, 11, 215–247 CrossRef;
(c) R. Molina Betancourt, P.-G. Echeverria, T. Ayad, P. Phansavath and V. Ratovelomanana-Vidal, Synthesis, 2021, 53, 30–50 CrossRef CAS;
(d) J. Barrios-Rivera, Y. Xu, M. Wills and V. K. Vyas, Org. Chem. Front., 2020, 7, 3312–3342 RSC;
(e) Y. Liu, X. Yue, C. Luo, L. Zhang and M. Lei, Energy Environ. Mater., 2019, 2, 292–312 CrossRef CAS. - M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10460–10476 CrossRef CAS PubMed.
- C. Gelis, A. Heusler, Z. Nairoukh and F. Glorius, Chem.–Eur. J., 2020, 26, 14090–14094 CrossRef CAS PubMed.
- For examples of trans-selective arene hydrogenation, see: (a) M. Wollenburg, A. Heusler, K. Bergander and F. Glorius, ACS Catal., 2020, 10, 11365–11370 CrossRef CAS PubMed; (b) K. Murugesan, T. Senthamarai, A. S. Alshammari, R. M. Altamimi, C. Kreyenschulte, M.-M. Pohl, H. Lund, R. V. Jagadeesh and M. Beller, ACS Catal., 2019, 9, 8581–8591 CrossRef CAS; (c) H. Li, Y. Wang, Z. Lai and K.-W. Huang, ACS Catal., 2017, 7, 4446–4450 CrossRef CAS; (d) A. Tungler and E. Szabados, Org. Process Res. Dev., 2016, 20, 1246–1251 CrossRef CAS; (e) T. Maegawa, A. Akashi, K. Yaguchi, Y. Iwasaki, M. Shigetsura, Y. Monguchi and H. Sajiki, Chem.–Eur. J., 2009, 15, 6953–6963 CrossRef CAS PubMed; (f) S. Jansat, D. Picurelli, K. Pelzer, K. Philippot, M. Gómez, G. Muller, P. Lecante and B. Chaudret, New J. Chem., 2006, 30, 115–122 RSC.
- For an example of a sequential enantioselective hydrogenation to access the trans-diastereomer of product, see: M. P. Wiesenfeldt, D. Moock, D. Paul and F. Glorius, Chem. Sci., 2021, 12, 5611–5615 RSC.
- (a) H. Matsuda, K. Ninomiya, H. Shimoda, N. Nishida and M. Yoshikawa, Heterocycles, 2001, 55, 2043–2050 CrossRef; (b) S. J. Rochfort, L. Towerzey, A. Carroll, G. King, A. Michael, G. Pierens, T. Rali, J. Redburn, J. Whitmore and R. J. Quinn, J. Nat. Prod., 2005, 68, 1080–1082 CrossRef CAS PubMed.
- For recent total syntheses of trans-THIQ natural products, see: (a) S. Uenishi, R. Kakigi, K. Hideshima, A. Miyawaki, J. Matsuoka, T. Ogata, K. Tomioka and Y. Yamamoto, Tetrahedron, 2021, 90, 132165–132176 CrossRef CAS; (b) J. Kayhan, M. J. Wanner, S. Ingemann, J. H. van Maarseveen and H. Hiemstra, Eur. J. Org. Chem., 2016, 3705–3708 CrossRef CAS.
- (a) A. N. Kim, A. Ngamnithiporn, E. R. Welin, M. T. Daiger, C. U. Grünanger, M. D. Bartberger, S. C. Virgil and B. M. Stoltz, ACS Catal., 2020, 10, 3241–3248 CrossRef CAS PubMed; (b) E. R. Welin, A. Ngamnithiporn, M. Klatte, G. Lapointe, G. M. Pototschnig, M. S. J. McDermott, D. Conklin, C. D. Gilmore, P. M. Tadross, C. K. Haley, K. Negoro, E. Glibstrup, C. Grünanger, K. M. Allan, S. C. Virgil, D. J. Slamon and B. M. Stoltz, Science, 2019, 363, 270–275 CrossRef CAS PubMed.
- For recent reports of Ir-catalyzed asymmetric hydrogenation of 1,3-disubstituted isoquinolines, see: (a) M.-W. Chen, Y. Ji, J. Wang, Q.-A. Chen, L. Shi and Y.-G. Zhou, Org. Lett., 2017, 19, 4988–4991 CrossRef CAS PubMed; (b) J. Wen, R. Tan, S. Liu, Q. Zhao and X. Zhang, Chem. Sci., 2016, 7, 3047–3051 RSC; (c) Y. Kita, K. Yamaji, K. Higashida, K. Sathaiah, A. Iimuro and K. Mashima, Chem.–Eur. J., 2015, 21, 1915–1927 CrossRef CAS PubMed.
- The absolute configuration of the trans-diastereomer was assigned as (S,S) by VCD, see ESI 43.†.
- S. P. Smidt, N. Zimmermann, M. Studer and A. Pfaltz, Chem.–Eur. J., 2004, 10, 4685–4693 CrossRef CAS PubMed.
- Attempts to further improve the diastereoselectivity were unsuccessful, see Tables S1 and S2.†.
- The absolute stereochemistry for all other hydrogenated products were assigned by analogy to 5a.
- Attempts to prepare 3-alkyl substituted isoquinolines were unsuccessful.
- C.-Y. Liu, V. Angamuthu, W.-C. Chen and D.-R. Hou, Org. Lett., 2020, 22, 2246–2250 CrossRef CAS PubMed.
- Cbz-protected 2a was obtained in 91% ee, ruling out any racemization occurring in the reaction.
- K. Fagnou and M. Lautens, Angew. Chem., Int. Ed., 2002, 41, 26–47 CrossRef CAS.
- R. Dorta, D. Broggini, R. Stoop, H. Rüegger, F. Spindler and A. Togni, Chem.–Eur. J., 2004, 10, 267–278 CrossRef CAS PubMed.
- R. Díaz-Torres and S. Alvarez, Dalton Trans., 2011, 40, 10742–10750 RSC.
- (a) M. V. Jiménez, J. Fernández-Tornos, F. J. Modrego, J. J. Pérez-Torrente and L. A. Oro, Chem.–Eur. J., 2015, 21, 17877–17889 CrossRef PubMed; (b) O. Blum and D. Milstein, J. Am. Chem. Soc., 1995, 117, 4582–4594 CrossRef CAS.
- The same deuterium experiment under the cis-selective hydrogenation conditions demonstrates no deuteration at the methylene carbon of 1a, ruling out an alternative tautomerization mechanism.
- (a) H. Liu, W.-H. Wang, H. Xiong, A. Nijamudheen, M. Z. Ertem, M. Wang and L. Duan, Inorg. Chem., 2021, 60, 3410–3417 CrossRef CAS PubMed; (b) S. R. Klei, J. T. Golden, T. D. Tilley and R. G. Bergman, J. Am. Chem. Soc., 2002, 124, 2092–2093 CrossRef CAS PubMed.
- A proposed catalytic cycle is described in the ESI, see ESI 32.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2120281. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06729j |
| This journal is © The Royal Society of Chemistry 2022 |