
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented mid-infrared nonlinear optical materials achieved by crystal structure engineering, a case study of (KX)P2S6 (X = Sb, Bi, Ba)†
Vivian
Nguyen
a,
Bingheng
Ji
a,
Kui
Wu
 b,
Bingbing
Zhang
*b and
Jian
Wang
*a
b,
Bingbing
Zhang
*b and
Jian
Wang
*a
aDepartment of Chemistry and Biochemistry, Wichita State University, Wichita, Kansas 67260, USA. E-mail: jian.wang@wichita.edu
bCollege of Chemistry and Environmental Science, Hebei University, Key Laboratory of Analytical Science and Technology of Hebei Province, Baoding 071002, China
First published on 2nd February 2022
Abstract
Three acentric type-I phase-matchable infrared nonlinear optical materials KSbP2S6, KBiP2S6, and K2BaP2S6, showing excellent balance between the second harmonic generation coefficient, bandgap, and laser damage threshold, were synthesized via a high-temperature solid-state method. KSbP2S6 is isostructural to KBiP2S6, which both crystallize in the β-KSbP2Se6 structure type. K2BaP2S6 was discovered for the first time, which crystallizes in a new structure type. KSbP2S6 and KBiP2S6 exhibit close structural similarity to the parent compound, centrosymmetric Ba2P2S6. The [P2S6] motifs, isotypic to ethane, exist in Ba2P2S6, KSbP2S6, KBiP2S6, and K2BaP2S6. The mixed cations, K/Sb pair, K/Bi pair, and K/Ba pair, play a dual-role of aligning the [P2S6] structure motifs, contributing to a high SHG coefficient, as well as enlarging the bandgap. KSbP2S6, KBiP2S6, and K2BaP2S6 are direct bandgap semiconductors with a bandgap of 2.9(1) eV, 2.3(1) eV and 4.1(1) eV, respectively. KSbP2S6, KBiP2S6, and K2BaP2S6 exhibit a high second harmonic response of 2.2× AgGaS2, 1.8× AgGaS2, and 2.1× AgGaS2, respectively, coupled with a high laser damage threshold of 3× AgGaS2, 3× AgGaS2, and 8× AgGaS2, respectively. The DFT calculations also confirm that the large SHG coefficient mainly originates from [P2S6] anionic motifs.
Introduction
Middle infrared lasers have various important applications such as instrumental spectroscopy,1 optical sensing,2 optical imaging,3,4 and long-distance communications.5 One critical way to obtain middle infrared lasers is via the nonlinear harmonic process, where middle infrared nonlinear optical materials (MIR NLO) are the key components. Even after many years of intense research, state-of-the-art MIR NLO materials utilized to cover the spectrum range of 3–25 μm are still underdeveloped. The commercial materials such as ZnGeP2, AgGaS2, and AgGaSe2 are impeded from use in the range of 3–25 μm due to their intrinsic efficiency loss originating from double photon absorption (ZnGeP2), the low laser damage threshold (AgGaS2), and non-phase matchable behavior (AgGaSe2).6–9 A state-of-the-art MIR NLO should balance a large second harmonic generation coefficient (dij > AgGaS2), moderate birefringence Δn for phase matchability, a high laser damage threshold (LDT, many times > AgGaS2), and a large bandgap for a good transmission range (>3.5 eV), which is almost impossible for many systems. Many studies, including experimental and theoretical studies, all proved the existence of a “3.5 eV wall” for middle IR NLO.10–15 Now the major strategy to achieve a large bandgap is through the investigation of oxides, with good examples of Cs4V8O22,16 La3SnGa5O14,17 LiCd(IO3)3,18 Li2MTeO6 (M = Ti, Sn),19 and M2LiVO4 (M = Rb, Cs).20 Compared with oxygen, which has intrinsic vibration modes within the infrared spectrum range, sulfides constituted from heavier sulfur atoms can have better performance.21–24 For sulfide MIR NLO materials, they currently remain in a vacuum without a promising material to balance dij, Δn, and LDT with a bandgap breaking through the “3.5 eV wall”. A new research strategy is called for to break the “3.5 eV wall” with a good balance of dij, Δn, and LDT for MIR NLO.Mixed cations with a combination of two chemically different atoms may increase the structural complexity of the target system, which has a higher chance of forming NCS structures, moderate birefringence for phase-matching behavior, and interactions between cations and anionic groups for better NLO properties.21,25–30 Our research was firstly inspired by Ba2P2S6, which contains [P2S6] ethane-like motifs and crystallizes in a centrosymmetric structure.31 The K+ cation was selected to replace Ba2+ cations due to their comparable ionic sizes: 152 pm and 149 pm, respectively.32 Trivalent cations containing stereochemically active lone pair electrons (SCALP) such as Sb and Bi were also incorporated into the system. Many previous studies have demonstrated that SCALP play an important role in enhancing NLO properties.33–38 Herein, in this study, we verified that a new strategy of applying crystal structure engineering via mixed cations plays a dual-role in three emerging MIR NLO materials, KSbP2S6, KBiP2S6, and K2BaP2S6. The mixed cations, K/Sb, K/Bi and K/Ba, align the [P2S6] structure motifs, contributing to both a high SHG coefficient and enlarging the bandgap. The bandgap of K2BaP2S6 is 4.1(1) eV, which sets a new record for superior MIR NLO sulfide materials. KSbP2S6, KBiP2S6, and K2BaP2S6 are all type-I phase-matchable materials with very large bandgaps (2.9(1) eV, 2.3(1) eV, and 4.1(1) eV, respectively), high SHG coefficients (2.2× AgGaS2, 1.8× AgGaS2, and 2.1× AgGaS2, respectively) and large LDT (3× AgGaS2, 3× AgGaS2, and 8× AgGaS2, respectively). The structural chemistry between KSbP2S6, KBiP2S6, K2BaP2S6, and the parent centrosymmetric compound Ba2P2S6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31 is presented. The synthesis, crystal structure, electronic structure, linear and nonlinear optical properties, and DFT calculations of KSbP2S6, KBiP2S6, and K2BaP2S6 and the parent centrosymmetric compound Ba2P2S6 are discussed.
31 is presented. The synthesis, crystal structure, electronic structure, linear and nonlinear optical properties, and DFT calculations of KSbP2S6, KBiP2S6, and K2BaP2S6 and the parent centrosymmetric compound Ba2P2S6 are discussed.
Experimental
Synthesis
All starting materials were stored and used in an Ar-filled glove box. Starting materials were used as received without further handling: K metal (Alfa Aesar, 99.95%), P powder (Alfa Aesar, 99.5%), S powder (Alfa Aesar, 99.5%), Ba rod (Sigma-Aldrich 99.9%), Bi (Fisher, 99.99%), and Sb (Fisher, 99.5%). The P2S5 precursor was made via loading P and S at a molar ratio of 2:5 and heating them to 573 K in 10 hours and holding for 10 hours. Polycrystalline samples of K2BaP2S6, KSbP2S6, and KBiP2S6 were grown by a two-step solid-state reaction. First step: stoichiometric amounts of reactants (K2BaP2S6: K/Ba/P/S = 2:1:2:6, KSbP2S6: K/Sb/P/S = 1:1:2:6, KBiP2S6: K/Bi/P/S = 1:1:2:6) were loaded into silica quartz tubes and were flame-sealed under vacuum at 10−2 torr and placed into a programmable furnace. The sealed silica tubes were heated to 398 K in 10 h, held for 10 h, heated to 923 K in 10 h, held for 144 h, and cooled to RT over 48 h. Second step: the quartz tubes from the first step were opened in a glovebox and the products were manually ground using a mortar and pestle. The finely mixed powders were then loaded into a new carbonized quartz tube. The ampoules were sealed and heated again using an identical temperature profile to the first step. After the second step, the homogeneous powders appearing in various colors (K2BaP2S6: white transparent, KSbP2S6: yellow-green, KBiP2S6: red) were collected and checked by powder X-ray diffraction. The powder X-ray diffraction verified the single-phase nature of K2BaP2S6, KSbP2S6, and KBiP2S6 (Fig. S1–S3†).
Single crystal X-ray diffraction
X-ray diffraction data were collected at 298(2) K for K2BaP2S6 using a Bruker Kappa APEX II diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Data reduction and integration, together with global unit cell refinements, were performed in the APEX2 software.39 Multi-scan absorption corrections were employed.39 The structures were solved by direct methods and refined by full matrix least-squares methods on F2 using the SHELX package with anisotropic displacement parameters for all atoms.40 In the last refinement cycles, the atomic positions for the three compounds were standardized using the program structure TIDY.41,42 Details of the data collection and structure refinement are provided in Table 1. Atomic coordinates and selected bond distances are summarized in Tables S1 and S2.† During the structure refinement process, a residual peak separated from the K2 atom by 2.87(2) Å with an intensity of 5.22 e Å−3 was found in K2BaP2S6. The final refinement ended up with a high wR2 of 0.1458 ([I > 2σ(I)]) and G.O.F of 1.200. The twinning refinement and reproduced data collection with better crystal quality did not improve the refinement results. With defining the residual peak as K1 with adding constraints of occupancy of K1 + K2 = 100%, the final structure model ended up with a much lower wR2 of 0.0807 ([I > 2σ(I)]), wR2 of 0.0874 (all data), and G.O.F of 1.071. When solely refining the occupancy of K2, a value of 97% was obtained. The occupancy of interstitial K1 is around 13% from single crystal refinement results. Since K2BaP2S6 was revealed as a large bandgap semiconductor by theoretical simulation and experimental UV-vis results (vide infra), the charge-balanced formula (K+)2(Ba2+)(P4+)2(S2−)6 can be established by assigning a formal charge of 1+ to the K atoms, 2+ to the Ba atoms, 4+ to the P atoms due to the presence of homoatomic interactions, and 2− to the S atoms. Hence, the actual occupancy of interstitial K1 would be much lower than 10% to keep the charge balanced nature of K2BaP2S6. To simplify the discussion of K2BaP2S6, the interstitial K1 was not included. The crystallographic data such as atomic coordinate parameters and selected interatomic distances of K1 atoms are listed in Tables S1 and S2,† respectively. Crystallographic data for K2BaP2S6 have been deposited with the Cambridge Crystallographic Data Centre, CCDC, 12 Union Road, Cambridge CB21EZ, UK. Copies of the data can be obtained free of charge on quoting the depository number CCDC-2119928 (K2BaP2S6).| Empirical formula | K2BaP2S6 | ||
|---|---|---|---|
| a R 1 = ∑‖Fo| − |Fc‖/∑|Fo|; wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2, and w = 1/[σ2Fo2 + (A·P)2 + B·P], P = (Fo2 + 2Fc2)/3; A and B are weight coefficients. | |||
| Formula weight | 469.84 g mol−1 | Unit cell volume | 1211.8(2) Å3 |
| Temperature | 173(2) K | Z | 4 |
| Radiation, wavelength | Mo Kα, 0.71073 Å | Density (calc.) | 2.575 g cm−3 |
| Crystal system | Orthorhombic | Absorption coefficient | 5.203 mm−1 |
| Space group | Pna21 (no. 33) | ||
| Unit cell dimensions | a = 12.3592(13) Å | Final R indicesa [I > 2σ(I)] | R 1 = 0.0373 |
| wR2 = 0.0807 | |||
| b = 14.4324(14) Å | Final R indicesa [all data] | R 1 = 0.0561 | |
| wR2 = 0.0874 | |||
| c = 6.7939(7) Å | G.O.F | 1.071 | |
Powder X-ray diffraction
Data were collected at room temperature using a Rigaku Mini Flex II diffractometer with Cu Kα radiation (λ = 1.5406 Å).UV-vis measurements
Diffuse-reflectance spectra were recorded at room temperature using a PERSEE-T8DCS UV-vis spectrophotometer equipped with an integration sphere in the wavelength range of 230–850 nm. The reflectance data, R were recorded and converted to the Kubelka–Munk function, f(R) = (1 − R)2(2R)−1. The Tauc plots, (KM × E)2 and (KM × E)1/2, were applied to estimate direct and indirect bandgaps, respectively.Second harmonic measurements
Using the Kurtz and Perry method,43 powder SHG responses of title compounds were investigated using a Q-switch laser (2.09 μm, 3 Hz, 50 ns) with different particle sizes, including 38.5–54, 54–88, 88–105, 105–150, and 150–200 μm. The detailed process of sample preparation would be: the as-synthesized sample and the as-synthesized reference AgGaS2 sample were manually ground into particles within a glovebox first. The ground samples were sieved using standard brass sieves with various mesh sizes. The particles were sorted out and separated using standard brass sieves with various mesh sizes with the potential of a large range of particle size distribution. The LDTs of the title compounds were evaluated on powder samples (150–200 μm) with a pulsed YAG laser (1.06 μm, 10 ns, 10 Hz). The judgment criterion is as follows: with increasing laser energy, the color change of the powder sample is constantly observed using an optical microscope to determine the damage threshold. To adjust different laser beams, an optical concave lens is added to the laser path. The damaged spot is measured using the scale of the optical microscope.Theoretical calculations
The density of states (DOS), partial density of states (PDOS), band structure, crystal orbital Hamilton population (COHP), and electron localization function (ELF) of Ba2P2S6, KBiP2S6, and K2BaP2S6 are calculated using the tight binding-linear muffin tin orbitals-atomic sphere approximation (TB-LMTO-ASA) program.44,45 The Barth–Hedin exchange potential was employed for the LDA calculations.45 The radial scalar-relativistic Dirac equation was solved to obtain the partial waves. The basis set used contained K (4s), Ba (6s, 5d, 4f), Bi (6s, 5d, 6p), P (3s, 3p) and S (3s, 3p) orbitals, and was employed for a self-consistent calculation, with downfolded functions of K (4p, 3d), Ba (5f, 6p), Bi (6d, 5f), P (3d) and S (3d). The density of states and band structures were calculated after converging the total energy on a dense k-mesh of Ba2P2S6 (20 × 20 × 10 points with 1122 irreducible k-points), K2BaP2S6 (12 × 12 × 24 points with 1176 irreducible k-points), and KBiP2S6 (16 × 16 × 8 points with 1056 irreducible k-points). The optical properties were calculated based on ab initio calculations implemented in the CASTEP package through density functional theory (DFT).46 The generalized gradient approximation (GGA)47 was adopted, and the Perdew–Burke–Ernzerhof (PBE)48 function was chosen to calculate the exchange–correlation potential, with an energy cutoff of 650 eV. The numerical integration of the Brillouin zone was performed using Monkhorst–Pack k-point sampling. The k-point separation for each material was set as 0.04 Å−1. The geometry optimizations were applied prior to property calculations. Norm-conserving pseudopotentials were employed.Results and discussion
Crystal structure
KSbP2S6 is isostructural to KBiP2S6, which crystallizes in the β-KSbP2Se6 structure type.49 KSbP2S6 and KBiP2S6 form in the monoclinic space group P21 (no. 4), which were discovered by Manrique, V. et al. without investigating any properties.50 K2BaP2S6 crystallizes in the orthorhombic space group Pna21 (no. 33) with unit cell parameters of a = 12.018(1) Å, b = 23.762(2) Å, and c = 3.984(1) Å. The Wyckoff sequence of K2BaP2S6 is a11 with Pearson symbol oP44. Searching for isostructural compounds through the ICSD database did not return any results. Therefore, K2BaP2S6 crystallizes in a new structure type. The selected crystal data and structure refinement parameters for K2BaP2S6 are summarized in Table 1. The atomic coordinate parameters and selected interatomic distances are listed in Tables S1 and S2.†To discuss the structural chemistry between KSbP2S6, KBiP2S6, and K2BaP2S6, a centrosymmetric compound Ba2P2S631 must be discussed for a few reasons: (1) the fundamental building units of Ba2P2S6 are the [P2S6] motifs, which are the same fundamental building units in KSbP2S6, KBiP2S6, and K2BaP2S6; (2) there are close unit cell parameters between Ba2P2S6, KSbP2S6, and KBiP2S6 with very comparable unit cell volumes as shown in Table S3;† (3) the chemical formula of Ba2P2S6 can be presented by [Ba2+]2[P2S6]4−, where the charge of cations would be 4+. The same charge requirements remain in KSbP2S6, KBiP2S6, and K2BaP2S6 with representation as [K+Sb3+][P2S6]4−, [K+Bi3+][P2S6]4−, and [K2+Ba2+][P2S6]4− respectively; (4) Ba2P2S6 inspired our research interest in compounds containing [P2S6] motifs. Since KSbP2S6 is isostructural to KBiP2S6, only KBiP2S6 is selected for structural discussion. The structural comparisons between centrosymmetric Ba2P2S6, KSbP2S6, and K2BaP2S6 are summarized in Fig. 1 with emphasis on the [P2S6] motifs on the right.
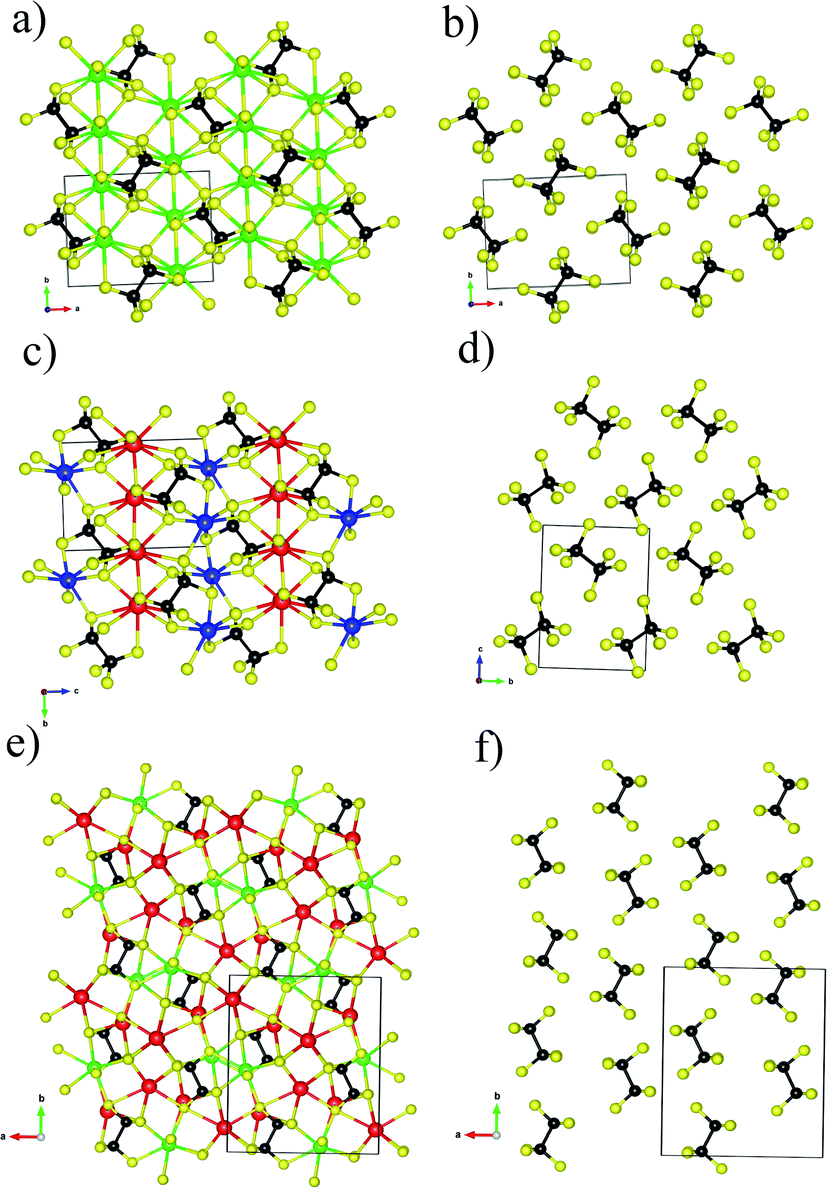 | ||
| Fig. 1 (a) Ball-stick structure model of Ba2P2S6 viewed along the [001] axis, (b) the arrangement of [P2S6] motifs within Ba2P2S6. (c) Ball-stick structure model of KBiP2S6 viewed along the [100] axis, (d) the arrangement of [P2S6] motifs within KBiP2S6. (e) Ball-stick structure model of K2BaP2S6 viewed along the [001] axis, (f) the arrangement of [P2S6] motifs within K2BaP2S6. K red color, Ba: green color, Bi: blue color, P: black color, S: yellow color. | ||
Ba2P2S6 crystallizes in the centrosymmetric P21/n (no. 14) space group with unit cell parameters of a = 6.7459(6) Å, b = 7.5871(6) Å, and c = 9.9983(9) Å, β = 91.204(3)°, which were redetermined in this work. The crystal structure of Ba2P2S6 is shown in Fig. 1a and b. The three-dimensional (3D) framework of Ba2P2S6 is constructed by isolated [P2S6] motifs interconnected by 2D [Ba2S6] slabs, where the center Ba atoms are surrounded by nine sulfur atoms. The Ba–S interactions within [BaS9] polyhedra fall into the range of 3.217–3.567 Å. The theoretical simulation also verified the ionic bonding nature of Ba–S interactions (vide infra). The crystal structure of KBiP2S6 shows high similarity to Ba2P2S6, as summarized in Table S3† and Fig. 1. KBiP2S6 crystallizes in the acentric P21 (no. 4) space group with unit cell parameters of a = 6.6200(6) Å, b = 7.4058(7) Å, and c = 9.9002(9) Å, β = 92.108(1) °.50 The unit cell volume of Ba2P2S6 and KBiP2S6 is 508.82 Å3 and 485.04 Å3, respectively. The 3D framework of KBiP2S6 is formed by isolated [P2S6] motifs linked by alternatively arranged 2D [KS5] slabs and 2D [BiS5] slabs, which are shown in Fig. 1c. The 2D [KS5] slabs are constructed by [KS9] polyhedra interlinked via sharing vertices and edges. The K–S interactions within [KS9] polyhedra are 3.191–3.592 Å, comparable to Ba–S interactions of 3.217–3.567 Å within Ba2P2S6, which are revealed as dominantly ionic bonds (vide infra). The 2D [BiS5] slabs are made by distorted [BiS7] polyhedra with Bi–S interactions of 2.706–3.210 Å. The Bi–S interactions of 2.71 Å show strong polarized ionic bonding characteristics (vide infra).
K2BaP2S6 exhibits a much more complex structure than Ba2P2S6 and KBiP2P6. K2BaP2S6 crystallizes in the orthorhombic space group Pna21 (no. 33) with unit cell parameters of a = 12.3592(13) Å, b = 14.4324(14) Å, and c = 6.7939(7) Å with unit cell volume of 1211.8(2) Å3, about two times higher than those of Ba2P2S6 (508.82 Å3) and KBiP2P6 (485.04 Å3). The 3D framework of K2BaP2S6 can be viewed as isolated [P2S6] motifs connected by 2D [K2BaS6] slabs, where the K/Ba ratio is two. The 2D [K2BaS6] slabs are made of [KS7] polyhedra, [KS8] polyhedra, and [BaS10] polyhedra. The K–S interactions in [KS7] and [KS8] fall into the range of 3.213(16)–3.679(5) Å, which is close to those of many potassium-sulfides such as K2FeGe3S8 (3.131–3.762 Å),51 K2CoGe3S8 (3.130–3.765 Å),51 KBiP2S6 (3.191–3.562 Å),50 K2MnP2S6 (3.226–3.600 Å),52 KSbP2S6 (3.206–3.592 Å).50 The 3.220(5) Å K–S interactions are revealed by theoretical calculations as weak ionic bonds (vide infra). The Ba–S interactions in [BaS10] polyhedra fall into the range of 3.226(3)–3.535(7) Å, which is close to those of many barium-sulfides such as Ba6(Cu2Mg)Ge4S16 (3.084–3.454 Å),53 Ba2P2S6 (3.217–3.567 Å),31 Ba2AlSbS5 (3.217–3.567 Å),54 BaAg2GeS4 (3.251–3.306 Å),55 BaCd2As2S6 (3.248–3.374 Å),56etc. The 3.24 Å ionic Ba–S interactions exhibit certain bond strengths as revealed by theoretical calculations (vide infra). The structural details of K2BaP2S6 are shown in Fig. 1e and f. The [P2S6] motifs are the common features existing in Ba2P2S6, KBiP2P6 and K2BaP2S6. The [P2S6] motif is isotypic to ethane, constructed by a P–P dimer with three sulfur atoms surrounding each P atom. The interatomic P–P bonds for Ba2P2S6, KBiP2P6 and K2BaP2S6 are 2.205 Å, 2.219 Å, and 2.223(4) Å, respectively, which are typical for homoatomic P–P bond distances such as BaCu5P3 (2.263 Å),57 La2Ba6Cu16P30 (2.071–2.428 Å),58 BaCu2P4 (2.183 and 2.289 Å),59 Ba8Cu16P30 (2.159–2.299 Å),60 La7Zn2P11 (2.207(7) Å),61 Ba2Cu3P4 (2.236 Å),62 La4Zn7P10 (2.151(8) Å),63etc. The P–S interatomic distances within Ba2P2S6 are 2.016–2.025 Å, and are slightly expanded into a wider range of distances to 1.962–2.057 Å and 1.958(17)–2.066(15) Å for KBiP2P6 and K2BaP2S6, respectively. Even though all [P2S6] motifs have identical geometry within Ba2P2S6, KBiP2P6, and K2BaP2S6, the slightly different interatomic distances coupled with various ∠S–P–S angles indicate that the degrees of distortion of the [P2S6] motifs are distinct. The strong covalent bonding nature of P–P interactions and P–S interactions together with complex interactions between cations and [P2S6] was uncovered by theoretical simulation (vide infra).
Another intriguing structural feature of Ba2P2S6, KBiP2P6 and K2BaP2S6 is the arrangement of [P2S6] motifs as shown in Fig. 1b, d and f. To discuss the alignment of [P2S6] motifs, we introduce the ∠P–P–P angles as a measure, where the three P atoms are coming from two neighbor [P2S6] motifs. The ∠P–P–P angles are summarized in Table 2. From Table 2, the ∠P–P–P angle significantly increases from Ba2P2S6 to K2BaP2S6 of 70.73(4)° and 89.4(3)° respectively. The ∠P–P–P angle in KBiP2S6 resides between Ba2P2S6 and K2BaP2S6 of 73.57(3)°. The ∠P–P–P angle is closer to 90°, which indicates that [P2S6] motifs are arranged more parallel to each other. As shown in Fig. 1f, the [P2S6] motifs within K2BaP2S6 are arranged nearly parallel. The later studies demonstrate that the alignment of [P2S6] motifs is crucial for the optical properties of K2BaP2S6 (vide infra).
| Ba2P2S6 | KbiP2S6 | K2BaP2S6 | |
|---|---|---|---|
| ∠P–P–P/° | 70.53(4) | 73.57(3) | 89.4(3) |
Electronic structures
To elucidate the electronic structures of Ba2P2S6, KBiP2S6, and K2BaP2S6, the calculations of band structure and density of states (DOS) were employed. The band structures are summarized in Fig. S4–S11.† The density of states of Ba2P2S6, KBiP2S6, and K2BaP2S6 is shown in Fig. 2. To validate the accuracy of theoretical simulations, two different methods, TB-LMTO-ASA and VASP, were employed to study the electronic structures of Ba2P2S6, KBiP2S6, and K2BaP2S6 (Fig. 2 and S4–S14†), which exhibited comparable results.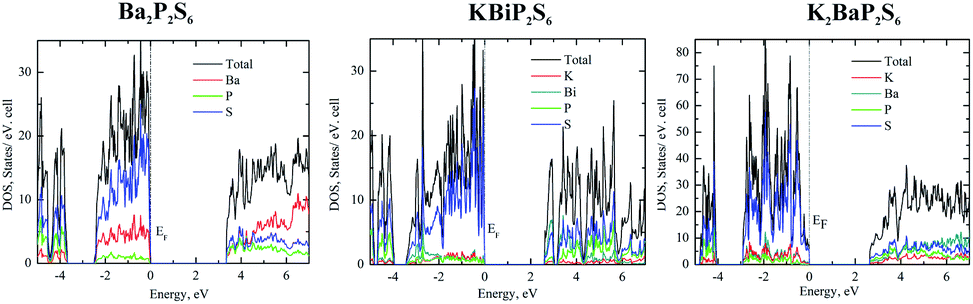 | ||
| Fig. 2 Left: density of states of Ba2P2S6, middle: density of states of KBiP2S6, right: density of states of K2BaP2S6. | ||
The charge-balanced formula (Ba2+)2(P4+)2(S2−)6, (K1+)(Bi3+)(P4+)2(S2−)6, and (K1+)2(Ba2+)(P4+)2(S2−)6 can be easily achieved via assigning a formal charge of 2+ to the Ba atoms, 1+ to the K atoms, 3+ to the Bi atoms, 4+ to the P atoms due to the presence of P–P homoatomic bonds, and 2− to the S atoms. The Bi3+ is confirmed by the presence of 6p2 lone pair electrons elucidated by ELF calculations (vide infra) and bond valence sum calculations.64–66 Bond valence sum calculations indicated that the BVS of K atoms, Ba atoms and Bi atoms is 1.1, 1.9, and 2.5, respectively, which suggests K atoms, Ba atoms and Bi atoms with oxidation states of +1, +2, and +3, respectively. The semiconductor nature of Ba2P2S6, KBiP2S6, and K2BaP2S6 was verified by DFT calculations and UV-vis measurements (Fig. S15–S19†). The band structure calculation indicated the direct semiconductor nature of Ba2P2S6, KBiP2S6, and K2BaP2S6 (Fig. S6, S7, S9 and S10†). As shown in Fig. 2 left, the calculated bandgap of Ba2P2S6 is 3.3 eV, which is lower than the measured value of 4.3(1) eV (Fig. S15 and S16†). In Ba2P2S6, the top of the valence band is dominantly contributed by S 3p orbitals and Ba 5s orbitals with some contributions from P 3p orbitals. The bottom of the conduction band is mainly contributed by S 3p orbitals and P 3p orbitals with certain contribution from Ba orbitals. The Ba orbitals significantly contribute a high energy interval of 5–7 eV as shown on the left of Fig. 2. We can anticipate that the Ba–S interactions are crucial for enlarging the bandgap of Ba2P2S6 and the optical properties of Ba2P2S6 are mainly controlled by Ba–S interactions and P–S interactions.
The direct bandgap of KBiP2S6 was predicated to be 2.6 eV, which is slightly higher than the measured value of 2.3(1) eV (Fig. S18†). The HSE calculated bandgap is 2.37 eV, which agrees very well with the measured value (Fig. S7†). KBiP2S6 exhibits a comparable electronic structure to Ba2P2S6, which is shown in the middle of Fig. 2. The top of the valence band is mainly contributed by S 3p orbitals with almost equal contributions from K 4s orbitals, Bi 6p orbitals and P 3p orbitals. The bottom of the conduction band is dominantly contributed by Bi 6p orbitals and S 3p orbitals with significant contribution from P 3p orbitals. Interestingly, K 4s orbitals had a negligible contribution to the bottom of the conduction band. Compared with Ba2P2S6, [BiS7] polyhedra will be expected to significantly affect the optical properties of KBiP2S6, which was verified by DFT calculations (vide infra). The nonlinear optical process is mainly dominated by electrons transferring between these frontier orbitals. Within KBiP2S6, K–S interactions, Bi–S interactions and [P2S6] motifs all determine the optical properties of KBiP2S6. The stereochemically active lone pair electrons (SCALP) on Bi atoms are expected to significantly enhance the NLO properties of KBiP2S6, which is confirmed by experimental observations and DFT calculations (vide infra). Compared with the centrosymmetric Ba2P2S6, the mixed cations of K/Bi play a dual-role of shifting the arrangement of [P2S6] to form a NCS structure and shrinking the bandgap.
The dual-role of mixed cations is even more obvious in K2BaP2S6. The direct bandgap of K2BaP2S6 was predicated to be 2.6 eV and 2.5 eV by the tight bonding method and HSE calculation respectively (Fig. S9 and S10†). The experimentally measured bandgap of K2BaP2S6 is 4.1(1) eV, where the underestimation of the bandgap is normal for DFT calculations. The electronic structures of K2BaP2S6 exhibit certain high similarity to Ba2P2S6 and KBiP2S6. The top of the valence band is predominantly contributed by S 3p orbitals and small contents of contribution from K 4s orbitals and Ba 6s orbitals. The contribution from P 3p orbitals can be ignored at the top of the valence band. Inversely, the P 3p orbitals coupled with S 3p orbitals and Ba 6s orbitals dominantly contribute to the bottom of the conduction band, where K 4s orbitals have certain contributions. From the density of states, we can expect that [P2S6] motifs significantly shift the nonlinear optical properties of K2BaP2S6, while mixed cations of K/Ba forming ionic K–S interactions and Ba–S interactions are responsible for the large bandgap of K2BaP2S6. In addition to enlarging the bandgap of K2BaP2S6, mixed cations of K/Ba also account for the nearly parallel alignment of [P2S6] motifs, which is crucial for its superior optical performance (vide infra).
Bonding picture studies
The bonding pictures of Ba2P2S6 (Fig. 3a and b), KBiP2S6 (Fig. 3c and d), and K2BaP2S6 (Fig. 3e and f) were understood with the aid of the electron localization function (ELF) coupled with crystal orbital Hamilton population (COHP) calculations. For Ba2P2S6, there are no detectable attractors (ELF maxima isosurfaces) found between Ba–S interactions with attractors only surrounding S atoms, which suggests the dominant ionic character of Ba–S interactions. The Ba–S interactions exhibit certain strong bonding characteristics with –ICOHPs of 0.852 eV per bond and 0.409 eV per bond for 3.22 Å Ba–S interactions and 3.57 Å Ba–S interactions, respectively. An obvious attractor was located between P–P interactions with –ICOHPs of 2.947 eV per bond for 2.21 Å P–P interactions, which indicates the strong covalent bonding nature of P–P interactions. The strongest bonding character was found between P–S interactions. The –ICOHPs for 2.02 Å P–S interactions are 4.886 eV per bond. There were significant attractors located between P–S bonds with a small shift toward P atoms. All attractors surrounding S atoms show irregular spherical shapes, which indicates the polarized bonding nature of Ba–S interactions and P–S interactions. For KBiP2S6, K atoms play the role of cations with no attractors surrounding and existing between K–S interactions and low calculated –ICOHPs of 0.136 eV per bond for 3.19 Å K–S interactions, which demonstrates the weak ionic bonding nature of K–S interactions. The attractors capping the Bi atoms correspond to the 6s2 electron lone pairs on Bi atoms, which verifies that the oxidation states of Bi should be +3. The COHP calculations indicate the strong bonding interactions for 2.71 Å Bi–S bonding with –ICOHPs of 1.584 eV per bond. For Bi–S interactions, the attractors are solely surrounding S atoms with irregular shapes, which suggests the strong polarized dominantly ionic bonding characters of Bi–S interactions. The electronic studies predict that [BiS7] polyhedra will significantly contribute to NLO properties due to the presence of SCALP, which is confirmed by DFT calculations (vide infra). The P–P interactions and P–S interactions within KBiP2S6 exhibit comparable characters to Ba2P2S6. There are attractors located in the middle of P–P interactions with –ICOHPs of 2.698 eV per bond for 2.22 Å P–P interactions. There are attractors detected between P–S interactions with –ICOHPs of 5.071 eV per bond for 1.96 Å P–S interactions. Hence, both P–P interactions and P–S interactions exhibit strong covalent bonding characteristics.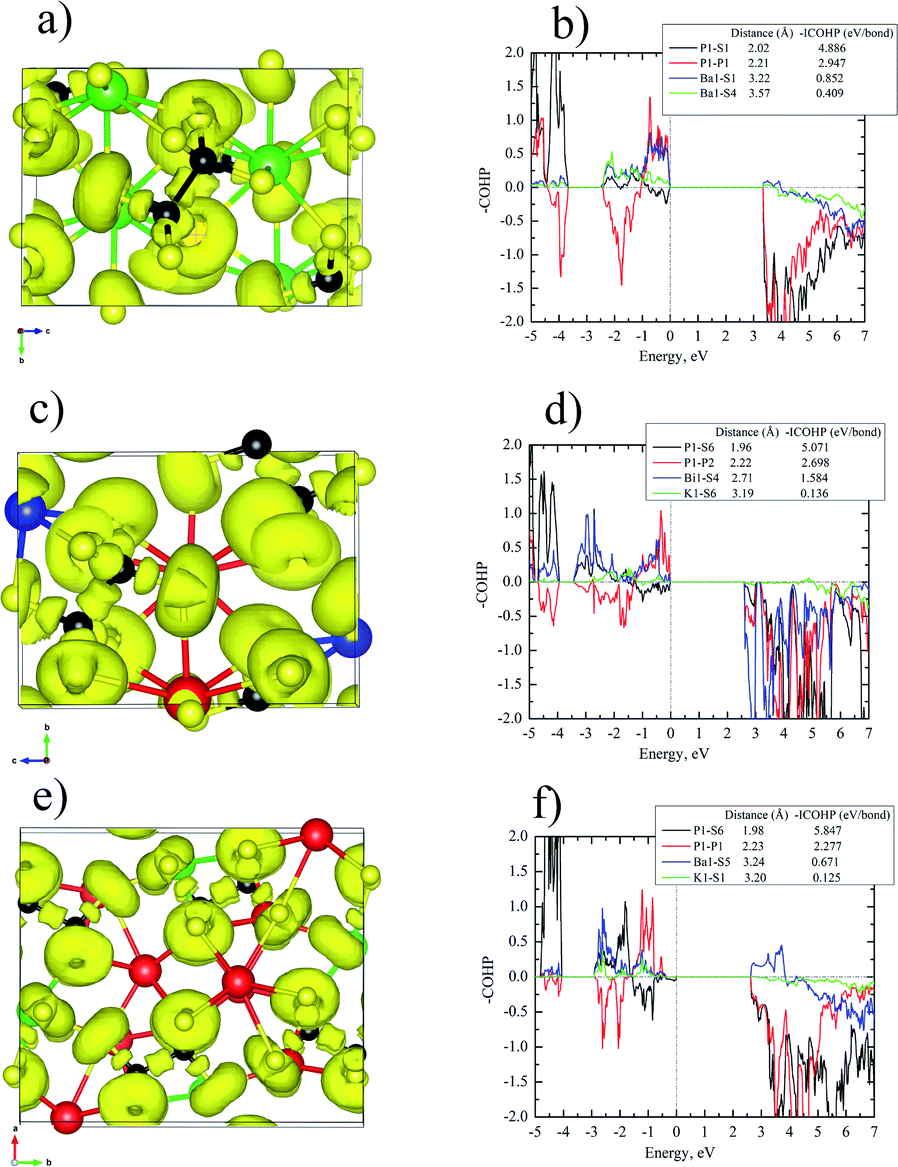 | ||
| Fig. 3 (a, c and e) 3D isosurfaces of the electron localization function (ELF) for Ba2P2S6 (a), KBiP2S6 (c), and K2BaP2S6 (e) with η = 0.75, Ba: green color, K: red color, Bi: blue color, P: Black color, S: yellow color, (b, d and f): Crystal Orbital Hamilton Population (COHP) analyses of the selected interactions within Ba2P2S6 (b), KBiP2S6 (d), and K2BaP2S6 (f). | ||
For K2BaP2S6, there are no ELF maxima isosurfaces residing between K–S interactions and Ba–S interactions. The calculated –ICOHPs for 3.24 Å Ba–S interactions are 0.671 eV per bond, which indicates that 3.24 Å Ba–S has certain strong bonding characteristics. The 3.20 Å K–S interactions have much weaker ionic bonding nature with –ICOHPs of 0.125 eV per bond as shown in Fig. 3f. The electronegativity difference between K (0.8) and Ba (0.9) may account for this slight bonding picture difference. The strong covalent bonding nature of P–P interactions and P–S interactions was verified by the presence of attractors between them and high –ICOHPs values of 5.847 eV per bond and 2.277 eV per bond for 1.98 Å P–S bonds and 2.23 Å P–P bonds, respectively. Similar to Ba2P2S6 and KBiP2S6, the K–S interactions and Ba–S interactions are also strongly polarized. When comparing Ba2P2S6, KBiP2S6, and K2BaP2S6 together, one very interesting observation is the decrease of bonding strengths in P–P interactions and the inverse trend of enhancement of bonding strengths in P–S interactions from Ba2P2S6 to KBiP2S6, and finally to K2BaP2S6. All our observations indicate the complex interactions happening between cations and anionic [P2S6] motifs.
Linear and nonlinear optical properties
The linear and nonlinear optical properties of KSbP2S6, KBiP2S6, and K2BaP2S6 were measured and are reported in Fig. 4. From Fig. 4a, Ba2P2S6, KBiP2S6, and K2BaP2S6 exhibited type-I phase-matching behavior, where the SHG response increases with particle size increasing. The calculated birefringence of KSbP2S6, KBiP2S6, and K2BaP2S6 is shown in Fig. S12–S14.† The moderate birefringence of KSbP2S6, KBiP2S6, and K2BaP2S6 accounts for their phase-matching capability, which is crucial for practical applications. For samples of particle size 225 μm, the SHG response of KSbP2S6, KBiP2S6, and K2BaP2S6 is 2.2× AGS, 1.8× AGS and 2.1× AGS, respectively. The measured bandgap of KSbP2S6, KBiP2S6, and K2BaP2S6 is 2.9(1) eV, 2.3(1) eV and 4.1(1) eV respectively. The high bandgap of K2BaP2S6 results in an extraordinarily high LDT of 8× AGS. KSbP2S6 and KBiP2S6 also possess excellent LDT of about 3× AGS for both samples. The type-I phase matching behavior, large bandgap, high SHG response, and high LDT make KSbP2S6, KBiP2S6, and especially K2BaP2S6 ideal candidates for middle IR NLO applications.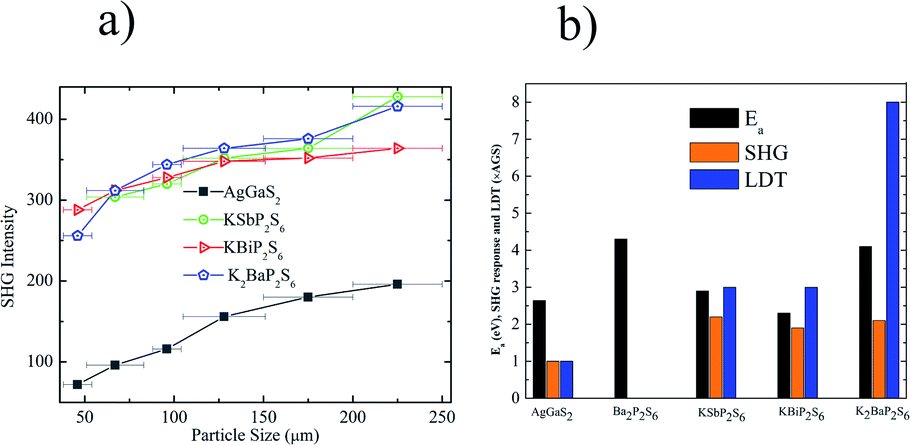 | ||
| Fig. 4 (a) SHG intensities of Ba2P2S6, KBiP2S6, K2BaP2S6, and AgGaS2 were measured on variable particle size samples utilizing a 2.09 μm laser. (b) The comparison of the bandgap (Ea), SHG response (×AgGaS2 based on 225 μm particle size samples) and LDT (×AgGaS2) between Ba2P2S6, KBiP2S6, K2BaP2S6, and AgGaS2. | ||
DFT calculations
To better understand the origin of the superior NLO properties of KSbP2S6, KBiP2S6 and K2BaP2S6, DFT calculations were carried out and are summarized in Table 3, Fig. 5 and S20–S22.† The calculations of KSbP2S6 are presented in Fig. S20.† As shown in Table 3, KSbP2S6 and KBiP2S6 were predicated to show a high SHG response, which agrees well with our experimental observations. The calculated χ222 for KSbP2S6 is 67.267 pm V−1 which is slightly higher than that of KBiP2S6 at χ222 = 63.404 pm V−1. The experimentally measured SHG response of KSbP2S6 is also slightly better than that of KBiP2S6. From Fig. 5a and b, the occupied virtual electrons (VE) states of KSbP2S6 and KBiP2S6 are mainly contributed by the [P2S6] motif and [XS7] (X = Sb or Bi) polyhedra due to the presence of SCALP, which is verified by the analysis of unoccupied virtual electrons states and virtual holes (VH) (Fig. S20 and S21†). These observations agree well with our previous electronic structure analysis.| KSbP2S6 | KBiP2S6 | K2BaP2S6 | |
|---|---|---|---|
| χ abc /pm V−1 | χ 112 = 1.647 | χ 112 = 11.234 | χ 113 = 0.171 |
| χ 123 = 16.114 | χ 123 = 1.890 | χ 333 = 0.678 | |
| χ 222 = 67.267 | χ 222 = 63.404 | — | |
| χ 233 = 16.691 | χ 233 = 0.4067 | — |
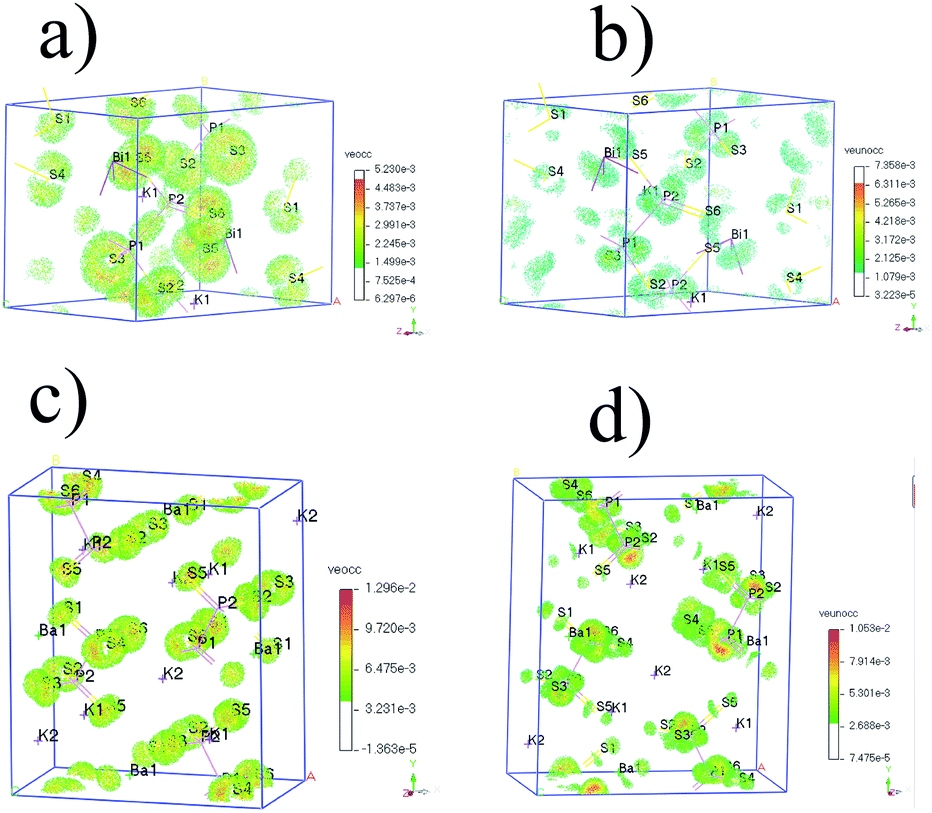 | ||
| Fig. 5 (a) SHG density of VE occupied (veocc) and (b) unoccupied (veunocc) states in KBiP2S6. (c) SHG density of VE occupied (veocc) and (d) unoccupied (veunocc) states in K2BaP2S6. | ||
For K2BaP2S6, the analysis of occupied and unoccupied states of VE and VH concluded that the [P2S6] motif plays a critical role in determining the SHG response, which agrees well with our electronic structure study. The calculated χ333 of K2BaP2S6 is only around 0.678 pm V−1, which is much lower than that of KSbP2S6 and KBiP2S6. The experimentally measured SHG response of K2BaP2S6 is comparable to that of KSbP2S6 and slightly better than that of KBiP2S6. This controversy can be explained by the alignment of [P2S6] motifs, as discussed in the crystal structure analysis. The nearly parallel alignment of [P2S6] motifs guarantees that the dipole moments originating from the [P2S6] motif do not cancel each other out during the nonlinear polarization process.67,68 For KSbP2S6 and KBiP2S6, the “imperfect” alignment of [P2S6] motifs is compensated by the presence of SCALP, which also results in a superior SHG response. In summary, K2BaP2S6 exhibits an excellent example of middle IR NLO materials with a good balance of a large bandgap (4.1(1) eV), high SHG coefficient (2.1× AGS), and large LDT (8× AGS) through a mixed cation routine. From centrosymmetric Ba2P2S6 to acentric KSbP2S6 and KBiP2S6, then finally to K2BaP2S6, our observations demonstrate the important dual-role of mixed cations: aligning structural motifs to form NCS structures and enlarging bandgaps. Our research provides a new direction for discovering novel middle IR NLO materials. To better evaluate the properties of (KX)P2S6 (X = Sb, Bi, Ba), obtaining high quality single crystals is essential. Our initial experiments of crystal growth of K2BaP2S6 utilizing salt flux have resulted in mm-sized crystals, which are shown in Fig. S23.† Type-I phase-matching capability, good air stability, and high SHG and LDT all indicate that (KX)P2S6 compounds (X = Sb, Bi, Ba) are excellent infrared nonlinear optical materials. The next phase for this system would be crystal growth of (KX)P2S6 compounds (X = Sb, Bi, Ba) and systemically studying their physical properties such as LDT and thermal conductivity, which are ongoing.
Conclusions
Three acentric thiophosphates KSbP2S6, KBiP2S6, and K2BaP2S6 were synthesized via a high-temperature solid-state method. KSbP2S6 is isostructural to KbiP2S6, which crystallizes in the β-KSbP2Se6 structure type. K2BaP2S6 was discovered for the first time and forms in a new structure type. KSbP2S6, KbiP2S6, and K2BaP2S6 were predicted to be direct bandgap semiconductors. The measured bandgaps for KSbP2S6, KBiP2S6, and K2BaP2S6 are 2.9(1) eV, 2.3(1) eV and 4.1(1) eV respectively. KSbP2S6, KBiP2S6, and K2BaP2S6 were discovered as superior middle IR NLO materials with excellent balance between the bandgap, second harmonic generation coefficient, and laser damage threshold. The superior NLO properties of KSbP2S6, KBiP2S6, and K2BaP2S6 originate from the comprehensive interactions between mixed cations and [P2S6] motifs. The mixed cations play a dual-role of aligning [P2S6] motifs and enlarging the bandgap in KSbP2S6, KBiP2S6, and K2BaP2S6, which is elucidated by a comprehensive study of crystal structure measurements, DFT calculations, and property tests. This study opens a new avenue for breaking the “3.5 eV wall” for middle IR NLO materials research.Data availability
Crystallographic data for K2BaP2S6 has been deposited at the CCDC under CCDC 2119928. The datasets supporting this article have been uploaded as part of the ESI,† DOI: 10.1039/d1sc06849k.Author contributions
J. W. conceived the project, designed the experiments and supervised the study. V. V. and J. B. performed the experiments and analysed the data. K. W. performed the SHG test. B. Zhang performed SHG theoretical calculations. V. V., J. B., K. W., B. Zhang., and J. W. discussed the data and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by start-up funds from Wichita State University. J. Wang thanks Arka Sarkar from Iowa State University for confirming the single crystal structure and Xiaocun Liu from Shandong University, China for help on the SEM test.References
- M. Yu, Y. Okawachi, A. G. Griffith, N. Picqué, M. Lipson and A. L. Gaeta, Nat. Commun., 2018, 9, 1869 CrossRef PubMed.
- A. V. Muraviev, V. O. Smolski, Z. E. Loparo and K. L. Vodopyanov, Nat. Photonics, 2018, 12, 209 CrossRef CAS.
- J. S. Dam, P. Tidemand-Lichtenberg and C. Pedersen, Nat. Photonics, 2012, 6, 788 CrossRef CAS.
- D. Pestov, X. Wang, G. O. Ariunbold, R. K. Murawski, V. A. Sautenkov, A. Dogariu, A. V. Sokolov and M. O. Scully, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 422 CrossRef CAS PubMed.
- A. K. Majumdar and J. C. Ricklin, Free-Space Laser Communications: Principles and Advances, Springer Science & Business Media, New York, NY, 2010, vol. 2 Search PubMed.
- S.-F. Li, X.-M. Jiang, Y.-H. Fan, B.-W. Liu, H.-Y. Zeng and G.-C. Guo, Chem. Sci., 2018, 9, 5700 RSC.
- A. Abudurusuli, J. Li and S. Pan, Dalton Trans., 2021, 50, 3155 RSC.
- Z. Li, J. Yao and Y. Wu, Cryst. Growth Des., 2020, 20, 7550 CrossRef CAS.
- X. Chen and K. M. Ok, Chem.–Asian J., 2020, 15, 3709 CrossRef CAS PubMed.
- D. Mei, W. Cao, N. Wang, X. Jiang, J. Zhao, W. Wang, J. Dang, S. Zhang, Y. Wu, P. Rao and Z. Lin, Mater. Horiz., 2021, 8, 2330 RSC.
- F. Liang, L. Kang, Z. Lin and Y. Wu, Cryst. Growth Des., 2017, 17, 2254 CrossRef CAS.
- I. Chung and M. G. Kanatzidis, Chem. Mater., 2014, 26, 849 CrossRef CAS.
- X. Cui, J. Xiao, Y. Wu, P. Du, R. Si, H. Yang, H. Tian, J. Li, W.-H. Zhang, D. Deng and X. Bao, Angew. Chem., Int. Ed. Engl., 2016, 55, 6708 CrossRef CAS PubMed.
- K. Wu, B. Zhang, Z. Yang and S. Pan, J. Am. Chem. Soc., 2017, 139, 14885 CrossRef CAS PubMed.
- L. Kang, M. Zhou, J. Yao, Z. Lin, Y. Wu and C. Chen, J. Am. Chem. Soc., 2015, 137, 13049 CrossRef CAS PubMed.
- C. Wu, X. Jiang, L. Lin, Y. Hu, T. Wu, Z. Lin, Z. Huang, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed. Engl., 2021, 60, 22447 CrossRef CAS PubMed.
- H. Lan, F. Liang, X. Jiang, C. Zhang, H. Yu, Z. Lin, H. Zhang, J. Wang and Y. Wu, J. Am. Chem. Soc., 2018, 140, 4684 CrossRef CAS PubMed.
- Y.-J. Jia, Y.-G. Chen, Y. Guo, X.-F. Guan, C. Li, B. Li, M.-M. Liu and X.-M. Zhang, Angew. Chem., Int. Ed. Engl., 2019, 58, 17194 CrossRef CAS PubMed.
- X. Du, X. Guo, Z. Gao, F. Liu, F. Guo, S. Wang, H. Wang, Y. Sun and X. Tao, Angew. Chem. Weinheim Bergstr. Ger., 2021, 133, 23508 CrossRef.
- G. Han, Y. Wang, X. Su, Z. Yang and S. Pan, Sci. Rep., 2017, 7, 1901 CrossRef PubMed.
- B.-W. Liu, H.-Y. Zeng, X.-M. Jiang and G.-C. Guo, CCS Chem., 2020, 2, 964 Search PubMed.
- W. Zhou, W.-D. Yao, Q. Zhang, H. Xue and S.-P. Guo, Inorg. Chem., 2021, 60, 5198 CrossRef CAS PubMed.
- K. Wu, Y. Chu, Z. Yang and S. Pan, Chem. Sci., 2019, 10, 3963 RSC.
- R.-A. Li, Z. Zhou, Y.-K. Lian, F. Jia, X. Jiang, M.-C. Tang, L.-M. Wu, J. Sun and L. Chen, Angew. Chem., Int. Ed. Engl., 2020, 59, 11861 CrossRef CAS PubMed.
- X. Luo, Z. Li, F. Liang, Y. Guo, Y. Wu, Z. Lin and J. Yao, Inorg. Chem., 2019, 58, 7118–7125 CrossRef CAS PubMed.
- Y.-N. Li, H. Xue and S.-P. Guo, Inorg. Chem., 2020, 59, 3546–3550 CrossRef CAS PubMed.
- H. Lin, W.-B. Wei, H. Chen, X.-T. Wu and Q.-L. Zhu, Coord. Chem. Rev., 2020, 406, 213150 CrossRef CAS.
- H. Huang, R. He, W. Yao, Z. Lin, C. Chen and Y. Zhang, J. Cryst. Growth, 2013, 380, 176–181 CrossRef CAS.
- Y. Wang, B. Zhang, Z. Yang and S. Pan, Angew. Chem., Int. Ed. Engl., 2018, 57, 2150–2154 CrossRef CAS PubMed.
- H. Huang, L. Liu, S. Jin, W. Yao, Y. Zhang and C. Chen, J. Am. Chem. Soc., 2013, 135, 18319–18322 CrossRef CAS PubMed.
- S. Jörgens, A. Mewis, R.-D. Hoffmann, R. Pöttgen and B. D. Mosel, Z. Anorg. Allg. Chem., 2003, 629, 429 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- Q. Wei, C. He, K. Wang, X.-F. Duan, X.-T. An, J.-H. Li and G.-M. Wang, Eur. J. Chem., 2021, 27, 5880–5884 CrossRef CAS PubMed.
- M. Luo, F. Liang, X. Hao, D. Lin, B. Li, Z. Lin and N. Ye, Chem. Mater., 2020, 32, 2615–2620 CrossRef CAS.
- R. Yin, C. Hu, B.-H. Lei, S. Pan and Z. Yang, Phys. Chem. Chem. Phys., 2019, 21, 5142–5147 RSC.
- H.-Y. Chang, S.-H. Kim, P. S. Halasyamani and K. M. Ok, J. Am. Chem. Soc., 2009, 131, 2426–2427 CrossRef CAS PubMed.
- W. Cai, Q. Jing and J. Zhang, New J. Chem., 2020, 44, 1228–1235 RSC.
- M. Yan, H.-G. Xue and S.-P. Guo, Cryst. Growth Des., 2021, 21, 698–720 CrossRef CAS.
- Bruker APEX2, Bruker AXS Inc., Madison, WI, 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- E. Parthé and L. M. Gelato, Acta Crystallogr., Sect. A: Found. Crystallogr., 1984, 40, 169 CrossRef.
- L. M. Gelato and E. Parthé, J. Appl. Crystallogr., 1987, 20, 139 CrossRef.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798 CrossRef CAS.
- O. Jepsen, A. Burkhardt and O. K. Andersen, The Program TB-LMTO-ASA, Version 4.7, Max-Planck-Institut für Festkörperforschung, Stuttgart, Germany, 1999. Search PubMed.
- U. Barth and L. Hedin, J. Phys. C: Solid State Phys., 1972, 5, 1629 CrossRef.
- B. G. Pfrommer, M. Côté, S. G. Louie and M. L. Cohen, J. Comput. Phys., 1997, 131, 233 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- A. M. Rappe, K. M. Rabe, E. Kaxiras and J. D. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 1227 CrossRef PubMed.
- J. D Breshears and M. G. Kanatzidis, J. Am. Chem. Soc., 2000, 122, 7839 CrossRef.
- V. Manrique, A. Galdámez, D. R. León, M. T. Garland and M. Jiménez, Z. Kristallogr. - New Cryst. Struct., 2003, 218, 163 Search PubMed.
- B. Ji, K. Pandey, C. P. Harmer, F. Wang, K. Wu, J. Hu and J. Wang, Inorg. Chem., 2021, 60, 10603 CrossRef CAS PubMed.
- F. Menzel, W. Brockner, W. Carrillo-Cabrera, H. G. Von Schnering and Z. Anorg, Allg. Chem., 1994, 620, 1081 CrossRef CAS.
- G. Cicirello, K. Wu and B. J. Wang, J. Solid State Chem., 2021, 296, 122017 CrossRef CAS.
- X. Wu, X. Gu, H. Pan, Y. Hu and K. Wu, Crystals, 2018, 8, 165 CrossRef.
- L. Nian, K. Wu, G. He, Z. Yang and S. Pan, Inorg. Chem., 2018, 57, 3434 CrossRef CAS PubMed.
- Y. Guo, F. Liang, M. Zhou, Z. Lin, J. Yao and Y. Wu, J. Alloys Compd., 2018, 762, 143 CrossRef CAS.
- G. Cicirello, A. Swindle and J. Wang, J. Solid State Chem., 2021, 296, 122017 CrossRef CAS.
- J. Wang, Y. He, N. E. Mordvinova, O. I. Lebedev and K. Kovnir, Chem, 2018, 4, 1465 CAS.
- J. Wang, J.-A. Dolyniuk, E. H. Krenkel, J. L. Niedziela, M. A. Tanatar, E. I. Timmons, T. Lanigan-Atkins, H. Zhou, Y. Cheng, A. J. Ramirez-Cuesta, D. L. Schlagel, U. S. Kaluarachchi, L.-L. Wang, S. L. Bud'ko, P. C. Canfield, R. Prozorov, O. Delaire and K. Kovnir, Chem. Mater., 2020, 32, 7932 CrossRef CAS.
- J. Dünner and A. Mewis, Z. Anorg. Allg. Chem., 1995, 621, 191 CrossRef.
- J. Wang, P. Yox, J. Voyles and K. Kovnir, Cryst. Growth Des., 2018, 18, 4076 CrossRef CAS.
- I. Pilchowski and A. Mewis, Z. Anorg. Allg. Chem., 1990, 581, 173 CrossRef CAS.
- J. Wang, K. Lee and K. Kovnir, Inorg. Chem., 2017, 56, 783 CrossRef CAS PubMed.
- B. Ji, E. Guderjahn, K. Wu, T. H. Syed, W. Wei, B. Zhang and J. Wang, Phys. Chem. Chem. Phys., 2021, 23, 23696 RSC.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192 CrossRef.
- N. A. Moroz, C. Bauer, L. Williams, A. Olvera, J. Casamento, A. A. Page, T. P. Bailey, A. Weiland, S. S. Stoyko, E. Kioupakis, C. Uher, J. A. Aitken and P. F. P. Poudeu, Inorg. Chem., 2018, 57, 7402 CrossRef CAS PubMed.
- Z. Li, Y. Liu, S. Zhang, W. Xing, W. Yin, Z. Lin, J. Yao and Y. Wu, Inorg. Chem., 2020, 59, 7614 CrossRef CAS PubMed.
- A. Abudurusuli, K. Wu, A. Tudi, Z. Yang and S. Pan, Chem. Commun., 2019, 55, 5143 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The crystallographic data, powder X-ray diffraction results, Tauc plots, DFT calculation, calculated birefringence, and the calculated SHG intensity map. CCDC 2119928. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06849k |
| This journal is © The Royal Society of Chemistry 2022 |