
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of trimetallic electrocatalysts by one-step co-electrodeposition and efficient CO2 reduction to ethylene†
Shuaiqiang
Jia
 ac,
Qinggong
Zhu
*b,
Haihong
Wu
*ac,
Shitao
Han
ac,
Mengen
Chu
ac,
Jianxin
Zhai
ac,
Xueqing
Xing
d,
Wei
Xia
ac,
Mingyuan
He
ac and
Buxing
Han
*abc
ac,
Qinggong
Zhu
*b,
Haihong
Wu
*ac,
Shitao
Han
ac,
Mengen
Chu
ac,
Jianxin
Zhai
ac,
Xueqing
Xing
d,
Wei
Xia
ac,
Mingyuan
He
ac and
Buxing
Han
*abc
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China. E-mail: hhwu@chem.ecnu.edu.cn; hanbx@iccas.ac.cn
bBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: qgzhu@iccas.ac.cn
cInstitute of Eco-Chongming, 20 Cuiniao Road, ChenjiaTown, Chongming District, Shanghai 202162, China
dBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, China
First published on 10th June 2022
Abstract
Use of multi-metallic catalysts to enhance reactions is an interesting research area, which has attracted much attention. In this work, we carried out the first work to prepare trimetallic electrocatalysts by a one-step co-electrodeposition process. A series of Cu–X–Y (X and Y denote different metals) catalysts were fabricated using this method. It was found that Cu10La1Cs1 (the content ratio of Cu2+, La3+, and Cs+ in the electrolyte is 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in the deposition process), which had an elemental composition of Cu10La0.16Cs0.14 in the catalyst, formed a composite structure on three dimensional (3D) carbon paper (CP), which showed outstanding performance for CO2 electroreduction reaction (CO2RR) to produce ethylene (C2H4). The faradaic efficiency (FE) of C2H4 could reach 56.9% with a current density of 37.4 mA cm−2 in an H-type cell, and the partial current density of C2H4 was among the highest ones up to date, including those over the catalysts consisting of Cu and noble metals. Moreover, the FE of C2+ products (C2H4, ethanol, and propanol) over the Cu10La1Cs1 catalyst in a flow cell reached 70.5% with a high current density of 486 mA cm−2. Experimental and theoretical studies suggested that the doping of La and Cs into Cu could efficiently enhance the reaction efficiency via a combination of different effects, such as defects, change of electronic structure, and enhanced charge transfer rate. This work provides a simple method to prepare multi-metallic catalysts and demonstrates a successful example for highly efficient CO2RR using non-noble metals.
:1:1 in the deposition process), which had an elemental composition of Cu10La0.16Cs0.14 in the catalyst, formed a composite structure on three dimensional (3D) carbon paper (CP), which showed outstanding performance for CO2 electroreduction reaction (CO2RR) to produce ethylene (C2H4). The faradaic efficiency (FE) of C2H4 could reach 56.9% with a current density of 37.4 mA cm−2 in an H-type cell, and the partial current density of C2H4 was among the highest ones up to date, including those over the catalysts consisting of Cu and noble metals. Moreover, the FE of C2+ products (C2H4, ethanol, and propanol) over the Cu10La1Cs1 catalyst in a flow cell reached 70.5% with a high current density of 486 mA cm−2. Experimental and theoretical studies suggested that the doping of La and Cs into Cu could efficiently enhance the reaction efficiency via a combination of different effects, such as defects, change of electronic structure, and enhanced charge transfer rate. This work provides a simple method to prepare multi-metallic catalysts and demonstrates a successful example for highly efficient CO2RR using non-noble metals.
Introduction
Electrocatalytic reduction of CO2 into value-added chemicals is emerging as a sustainable carbon-neutral approach to recycle CO2 and store intermittent renewable electricity.1,2 As an important C2 product of CO2RR, C2H4 is compatible with existing industrial infrastructure and can be used to produce a wide range of chemicals, particularly plastics and polymers.3–6 At present, C2H4 is mainly manufactured by thermal cracking of crude oil-derived naphtha and hydrogenation of CO via Fischer–Tropsch synthesis, and the selectivity is generally not high.7–9 Although electrocatalytic reduction of CO2 provides a straightforward way for C2H4 production, achieving high selectivity of C2H4 and high current density remains a challenge. To date, Cu-based catalyst is uniquely active to promote C–C coupling and yield C2+ products, but a single Cu catalyst still suffers from unsatisfied selectivity toward specified hydrocarbons.10–12 For that, several strategies have been proposed in constructing Cu-based catalysts, including surface reconstruction,13–15 hybridization,9,16 crystalline faceting,17,18 nano/meso-structuring,19,20 defect engineering,21,22 or creating multi-metallic structural motifs,23–25etc.Constructing multi-metallic structures has attracted considerable interest since it can create abundant defects to enhance the CO2-to-C2+ products activity through optimizing the binding energy among reactants, intermediates, and products with the multi-metallic surface at the nanoscale.26,27 So far, the most active doping metallic elements are still noble metals, because the presence of noble metals can easily modulate strains and lattice disorders of the Cu phase, and precisely steer the two pivotal steps towards C2H4 formation, including *CO formation and C–C coupling.28,29 Non-noble metal elements, however, still suffer from low to modest activity. Therefore, the attempts made to conduct efficient CO2-to-C2H4 electrocatalysts rely mostly on bimetallic materials involving noble metals, such as Cu–Au, Cu–Ag, Cu–Pd, etc.30–35 Unlike the bimetallic catalysts, integrating trimetallic nanostructures manifest great prospects in efficient CO2RR to C2+ products.36,37 To date, however, only Cu–Au/Ag nanoframes have shown promise for enhancing the efficiency of C2H4 production, in which the CO generation was promoted by the alloyed Ag/Au and the C–C coupling was facilitated by the highly strained and positively charged Cu domains (ESI Table S1†).36 However, conventional trimetallic catalysts still require the participation of noble metals and a complicated preparation process. Therefore, the rational design of non-noble trimetallic electrocatalysts with a facial synthesis strategy is highly desired for the practical deployment of electrochemical CO2RR.
Herein, we report a one-step strategy to synthesize trimetallic catalysts by co-electrodeposition. A series of trimetallic catalysts Cu–X–Y (X, Y = La, Cs, Zn, Co, Ag, Au) have been developed for CO2RR. It was discovered that co-electrodeposition can form a trimetallic composite structure that grown on CP. The presence of non-noble metals La and Cs could create abundant defects and modulate the electronic structure of the Cu phase, offering substantial active sites to stabilize the intermediates and promote C–C coupling to product C2H4. The as-synthesized trimetallic catalysts also provided large electrochemical surface area and facilitated charge transfer, which enhanced the reaction rate. The high CO2 electrocatalytic performance was demonstrated over Cu10La1Cs1 (the concentrations ratio of Cu2+, La3+, and Cs+ in the electrolyte is 10:1:1 in the deposition process) with up to 56.9% C2H4 selectivity with a current density of 37.4 mA cm−2 in an H-type cell, and a total C2+ selectivity of 70.5% with a current density of 485.5 mA cm−2 in a flow cell, respectively.
Results and discussion
To understand how trimetallic cooperation might tune the electrocatalytic activity, we prepared a group of trimetallic electrocatalysts (Cu–X–Y; X, Y = La, Cs, Zn, Co, Ag, Au). The non-noble trimetallic catalysts were grown on three-dimension CP (ESI Fig. S1†) through a one-step co-electroplating process, which was illustrated in Fig. 1a using trimetallic Cu–La–Cs as a representative example. Typically, a piece of CP with a geometric area of 1 cm2 and a Pt gauze were used for the cathodic and anodic electrodes with a gap of 1 cm, and the electrochemical experiments could be controlled by a DC Power supply. Before all the experiments, the CP was ultrasonically cleaned with acetone, ethanol, and deionized water. For the Cu10La1Cs1 electrode, the electrodeposition was carried out cathodically using a 50 mL solution of H2SO4 (10 mM), Cu(II) gluconate (100 mM), La(III) acetate (10 mM), Cs(I) acetate (10 mM), and 4-aminopyridine (10 mM). The deposition was carried out at a constant voltage of 4 V for 2 min. When the molar ratio of Cu(II), La(III), and Cs(I) in the electrolyte was 10:1:1, the as-synthesized catalyst was denoted as Cu10La1Cs1 with a total loading of 1.33 mg metals on 1.0 cm−2 CP (Table S2†), and the contents of Cu, La, and Cs in Cu10La1Cs1 were 93.92 wt%, 3.31 wt%, and 2.77 wt% (Table S3†), respectively, as determined by inductively coupled plasma optical emission spectroscopy (ICP-OES).
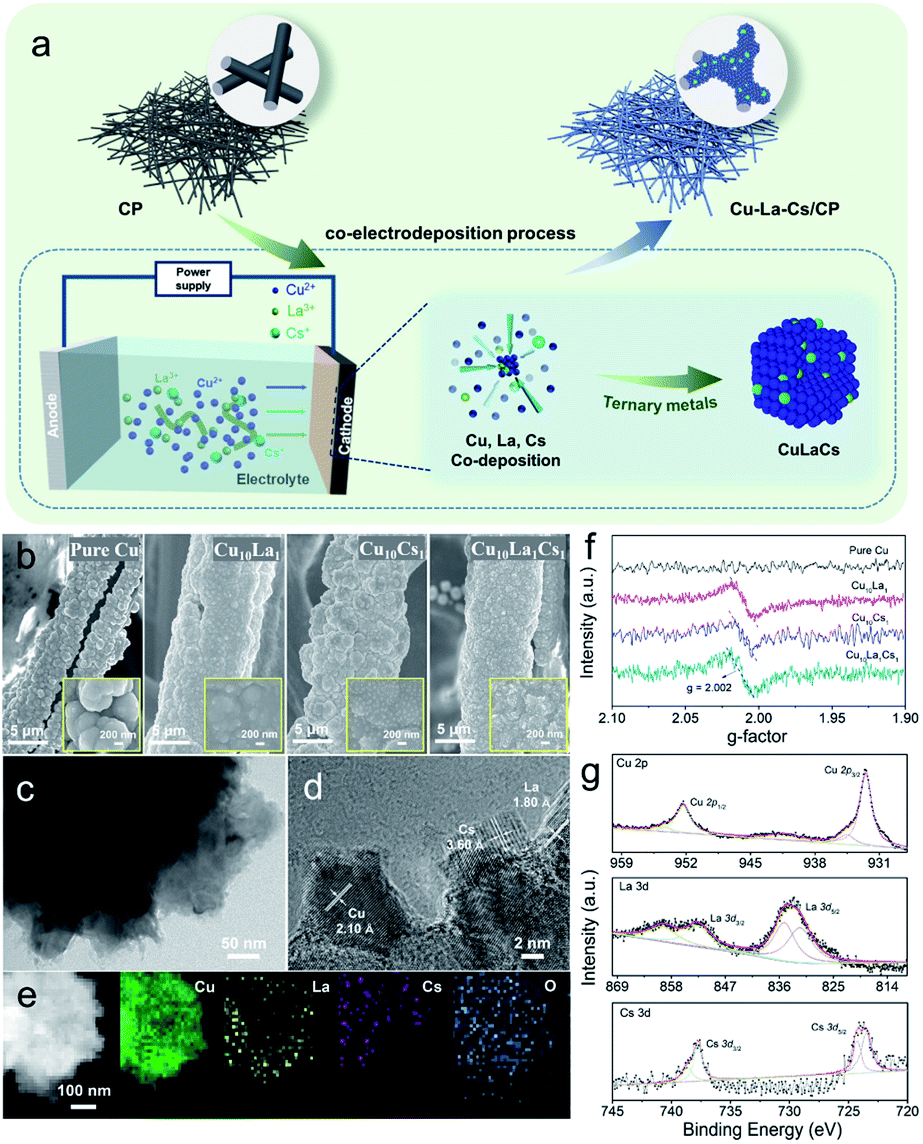 | ||
| Fig. 1 (a) Schematic illustration of the process to prepare pure Cu, bimetallic Cu–La or Cu–Cs, and trimetallic Cu–La–Cs catalysts; structural characterization of Cu–La–Cs catalysts: (b) SEM images of pure Cu, Cu10La1, Cu10Cs1, and Cu10La1Cs1 catalysts on a fiber of CP obtained after electrodeposition at a constant voltage of 4 V for 2 min (inset: high-magnification); (c, d) TEM images and HR-TEM image of Cu10La1Cs1 catalyst; and (e) elemental mappings images of Cu10La1Cs1 catalysts; (f) EPR spectra of pure Cu, Cu10La1, Cu10Cs1, and Cu10La1Cs1 catalysts at room temperature; (g) XPS spectra of Cu 2p spectra, La 3d spectra, and Cs 3d spectra in Cu10La1Cs1 catalyst. | ||
The scanning electron microscopy (SEM) images reveal that trimetallic Cu10La1Cs1 film grew uniformly on the fiber of the 3D CP substrate (Fig. 1b). Inset in Fig. 1b shows that the Cu, Cu10La1, Cu10Cs1, and Cu10La1Cs1 catalyst films had a rough surface. Besides, transmission electron microscopy (TEM) images confirm the formation of Cu10La1Cs1 trimetallic nanostructure, which was different from the pure Cu structure (Fig. 1c, d and S2†).
The high-resolution TEM (HR-TEM) image exhibit that the interplanar spacing was 2.10 Å, corresponding to the d spacing of (111) plane of Cu (Fig. 1d and S3a, b†), which is similar to pure Cu catalyst (Fig. S2†). In addition, the observed fringes with an interlayer spacing of 3.60 Å and 1.80 Å correspond to the (012) plane of Cs2O (JCPDS card no. 09-0104), and the (211) plane of La2O3 (JCPDS card no. 40-1284) (Fig. 1d and S3c–f†). The elemental distribution mapping (EDS) further confirmed the uniform dispersion of Cu, La, and Cs species in the trimetallic Cu10La1Cs1 catalysts (Fig. 1e). The abundant vacancies and lattice disorder could also be observed in Cu10La1Cs1 (Fig. S4 and S5†), which indicates that defect-rich nanostructure existed in the catalyst. Such defects result in high exposure of coordination-unsaturated Cu sites, which may change the electronic structure of Cu and influence its catalytic performance.38,39 The electron paramagnetic resonance (EPR) spectra of different samples were collected at room temperature (Fig. 1f). Compared with pure Cu, typical signals for oxygen vacancy appeared at g-value of 2.002 for Cu10La1, Cu10Cs1, and Cu10La1Cs1 catalysts, which indicates that introducing of La or Cs contributes to the formation of defects (Fig. 1f). The oxygen vacancy was also reported to optimize the adsorption energy of reactants on the catalyst surface, which reduces the reaction energy barrier and promotes molecular activation.40
The time-dependent X-ray diffraction (XRD) showed the gradual formation of trimetallic Cu10La1Cs1 catalysts (Fig. S6†). In detail, the representative peaks could be observed at 43.2°, 50.4°, 74.1°, 89.9°, and 37.0°, which can be indexed to the Cu (111), Cu (200), Cu (220), Cu (311), and Cu2O (111) planes. However, the diffraction peaks of La and Cs were not shown obviously for trimetallic Cu10La1Cs1, because their amounts were below the XRD detection threshold. The X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) analyses were performed to further investigate the surface chemical composition and elemental valence states of the catalyst. XPS results showed that the surface of Cu10La1Cs1 was composed of Cu, La, and Cs species (Fig. 1g and S7†). The peaks at 932.1 eV (Cu 2p3/2) and 952.0 eV (Cu 2p1/2) retained the characteristic feature of Cu species, which was further confirmed by the Cu 2p and Auger Cu LMM spectra that the Cu existed as Cu0 and Cu+, and Cu0 was predominant (Fig. S8†). In addition, the XPS spectra could also be fitted to include both La(III) and Cs(I) species, which correspond to peaks at 835.6 eV (La 3d5/2) and 852.5 eV (La 3d3/2), 723.9 eV (Cs 3d5/2) and 743.1 eV (Cs 3d3/2), respectively (Fig. 1g). These results showed that the Cu, La, and Cs coexisted in the Cu10La1Cs1 catalysts. To further study the electronic structures and chemical bonding of the Cu phase that was influenced by La and Cs atoms, we performed X-ray absorption spectroscopy (XAS) analysis. The Cu K-edge X-ray absorption near-edge spectra (XANES) spectra of Cu10La1Cs1 with the reference materials indicated that Cu10La1Cs1 were mainly composed of Cu0 and Cu+. From the XANES spectrum, it is obvious that the dominated Cu–Cu coordination at 2.23 Å existed in the catalyst, which is identical to that of Cu0 (Fig. S9†). This is in good agreement with the XRD and XPS analyses.
The as-synthesized catalysts were firstly tested for CO2RR in 0.1 M KCl aqueous electrolyte using a typical H-type cell. The low buffering capability of KCl aqueous electrolytes allowed the electrode surface pH to increase to a weakly basic range, which facilitated the formation of C2 products by combining with efficient electrocatalysts.41,42 In this study, the linear scanning voltammetry (LSV) curves over various catalysts were determined, including pure Cu, bimetallic Cu10La1, Cu10Cs1, and trimetallic Cu10La1Cs1 catalysts. As shown in Fig. 2a, over Cu10La1Cs1 catalyst, the current density (j) was much higher in CO2-saturated electrolyte than that in N2-saturated electrolyte from −0.5 V to −1.4 V vs. RHE, suggesting the reduction of CO2. Moreover, in CO2-saturated electrolyte, the current density over Cu10La1Cs1 was higher than that over other catalysts. The electrolysis performances at different potentials over various catalysts are displayed in Fig. 2b, c, and S10–S12.† Under the reaction conditions, the gaseous products were mainly composed of H2, CO, CH4, and C2H4, which were determined using gas chromatography (GC). The liquid products were evaluated by nuclear magnetic resonance (NMR), and only very small amounts of formic acid (<1%) were detected. The content was very low and was not considerable (Fig. S14†). Clearly, Cu10La1Cs1 had a much higher FE of C2H4 than other catalysts at all potentials (Fig. 2b). For pure Cu catalyst, the FE of C2H4 was only 33.0% at −1.2 V vs. RHE, which exhibited an inferior catalytic performance compared with other catalysts (Fig. 2b). For bimetallic Cu10La1 and Cu10Cs1, the FE of C2H4 increased consistently to 43.6% and 42.5% respectively. The FE of C2H4 over trimetallic Cu10La1Cs1 catalyst could reach 56.9% with a current density of 37.4 mA cm−2 at −1.2 V vs. RHE, suggesting a 2.7-fold increase in partial current density, compared to the pure Cu catalyst (Fig. 2b and S10–S12†). We also synthesized trimetallic Cu–La–Cs catalysts with different Cu–La–Cs ratios. Clearly, Cu10La1Cs1 had the highest catalytic CO2RR activity toward C2H4 production (Fig. S13†). Systematic comparisons to state-of-the-art catalysts revealed that the performance of the trimetallic Cu10La1Cs1 catalyst with rich-defects was one of the best ones in the H-type cell (Table S1†), including those over the catalysts consisting of Cu and noble metals. Long-term electrolysis was also performed to verify the stability of the catalyst. As shown in Fig. 2d, the current density and FE of C2H4 over Cu10La1Cs1 were not changed obviously at −1.2 V vs. RHE for 5 h, indicating the stability of the catalyst at the CO2RR condition. After the reaction, the XRD, EPR, and XPS analyses were performed and the results showed that the properties of the catalyst did not change noticeably (Fig. S15–S17†). The EXAFS at the Cu K-edge confirmed a well-retained Cu interaction in samples collected after the CO2RR test (Fig. S18†). These results also indicated the remarkable stability of the trimetallic Cu10La1Cs1 catalyst.
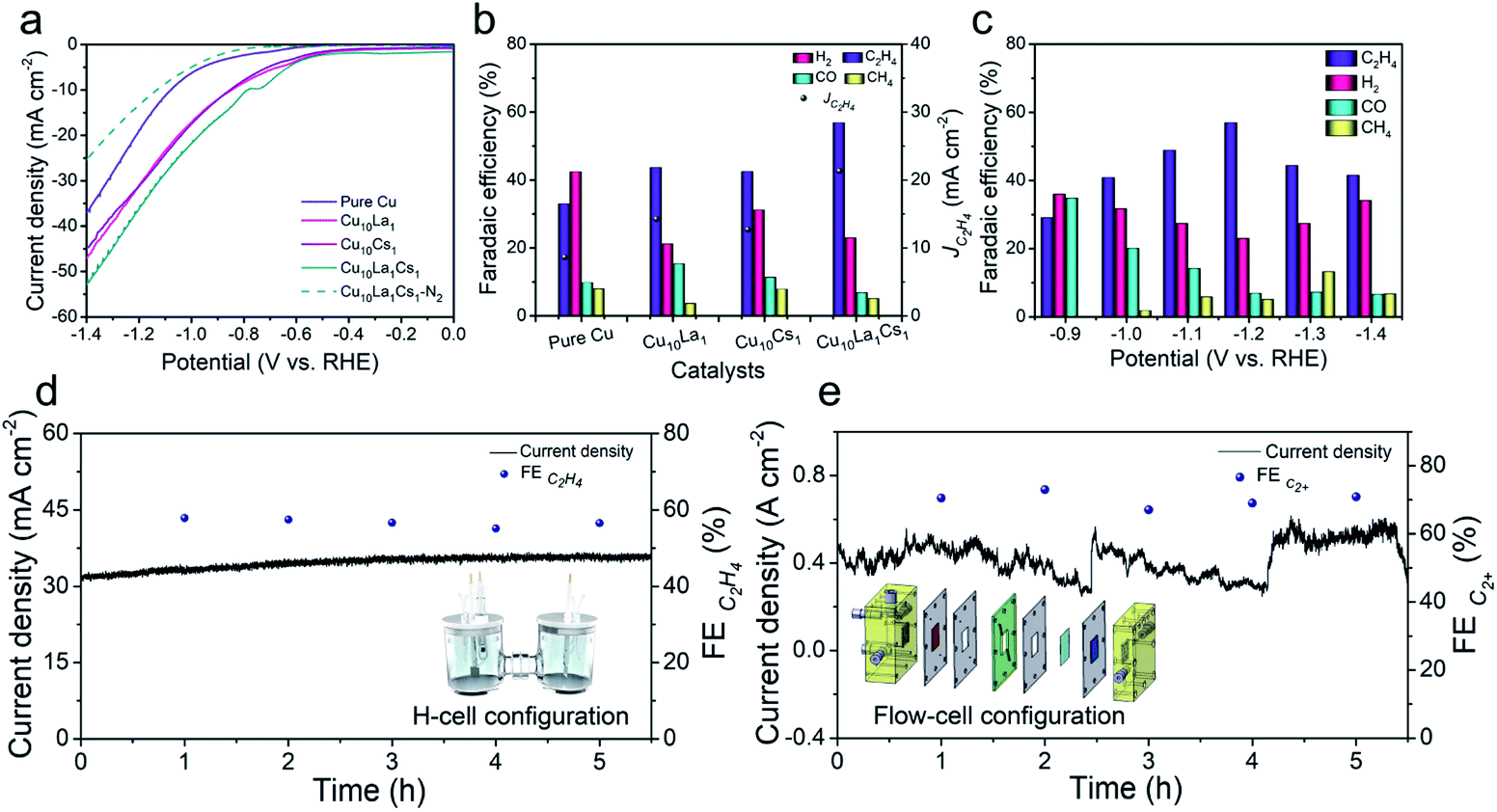 | ||
| Fig. 2 (a) LSV traces at a scan rate of 50 mV s−1 of pure Cu, Cu10La1, Cu10Cs1, and Cu10La1Cs1 catalysts in CO2-saturated and N2-saturated 0.1 M KCl aqueous electrolyte; (b) the distribution of reduction products and partial current densities for C2H4 at −1.2 V vs. RHE over pure Cu, Cu10La1, Cu10Cs1, and Cu10La1Cs1 catalysts; (c) the distribution of reduction products at different applied potentials over Cu10La1Cs1 catalysts; (d) electrochemical stability test of Cu10La1Cs1 film electrode at −1.2 V vs. RHE in an H-type cell; (e) electrochemical stability test of Cu10La1Cs1 film electrode at −0.97 V vs. RHE in a flow cell. | ||
In addition, to gauge the benefits of the trimetallic Cu–La–Cs catalyst for high-rate CO2RR, we translated the catalyst to a gas-diffusion environment (Fig. S19†). In this configuration, hydrophobic CP containing carbon black layer acted as a gas diffusion electrode (GDE). When using 1 M KOH as the electrolyte, we found that Cu10La1Cs1 could maintain C2+ selectivity up to 70.5% with a high current density of 485.5 mA cm−2 at a low reduction potential of −0.97 V vs. RHE. The FE of C2+ products were ethylene (42.1%), ethanol (20.8%), and n-propanol (7.6%), respectively (Fig. S20†). The configuration also showed a stable potential profile over 5 h without noticeable decay of the current density and C2+ product selectivity (Fig. 2e).
Considering the experimental observations above, we think that the excellent performance of the Cu10La1Cs1 catalyst resulted partially from the synergistic effect of the components in the trimetallic catalyst. We found that the surface roughness of the catalysts changed obviously with the introduction of La and Cs, which is beneficial for the increasing of active sites.43 The values of electrochemical double-layer capacitance (Cdl) were calculated from cyclic voltammograms (CV) curves (Fig. S21†) to evaluate the electrochemical active surface area (ECSA) of different catalysts. The Cdl value of Cu10La1Cs1 film was calculated to be 4.51 mF cm−2, which was obviously higher than that of others (Fig. S22†). It suggested that Cu10La1Cs1 film with rich-defects could provide more catalytic sites, leading to an increase in reaction rate during the electrocatalytic process. We also used electrochemical impedance spectroscopy (EIS) to study the interfacial properties of the catalysts at an open circuit potential in a CO2-saturated electrolyte (Fig. S23†). The Cu10La1Cs1 catalyst showed the lowest charge transfer resistance (Rct). Therefore, the charge transport is more facile on Cu10La1Cs1 catalyst, which is favorable to enhance the reaction rate.
To screen the synergistic effect of the components in the trimetallic catalysts, we also prepared a series of Cu10–X1–Y1 catalysts using the same method. When the molar ratio of metal ions for Cu, X, and Y in the electrodeposition electrolyte was 10:1:1, the as-synthesized catalyst was denoted as Cu10X1Y1. The compositions of these trimetallic catalysts, such as Cu10Ag1La1, Cu10Zn1La1, Cu10Ag1Cs1, Cu10Zn1Co1, and Cu10Ag1Au1, were then characterized by ICP-OES, and their element compositions are shown in Tables S3 and S4.† It indicates that the elemental compositions of the trimetallic catalysts varied with different doping components. The CO2RR performance tests of different trimetallic catalysts were conducted, and the results are shown in Fig. 3a. It indicates that the catalytic performances of the metals depended on the metals introduced into Cu. Some metal doping (e.g. Ag, Co, and Au) can promote the production of C1 products, while other metal doping (e.g. Zn, La, and Cs) can inhibit hydrogen evolution and promote C–C coupling. Among them, Cu10La1Cs1 yielded the highest FE of C2H4 and current density. The multi-metallic system could actively generate CO or inhibit cathodic hydrogen evolution reaction during the electrochemical reduction of CO2. The excess amount of CO molecules on the metal surface are expected to migrate to the adjacent Cu surface and then undergo C–C coupling for the formation of C2H4 products.6,30,44 The study indicated that the compositions not only significantly influenced the surface morphology of the trimetallic structures (Fig. S24†), but also changed the electronic properties of the catalyst. We then compare the effect of different defect/vacancies on the electronic properties, using Cu10La1Cs1 and Cu10Ag1Au1 as a comparing couple. The calculated density of states (DOS) of Cu10La1Cs1 and Cu10Ag1Au1 are shown in Fig. 3b. Comparing with the partial density of states (PDOS) for Cud orbitals, the electronic environment (the gap states) of the catalyst is constructed jointly by all the metals after doping with La/Cs or Ag/Au, which suggests that doping can significantly promote the electron transfer of the catalyst.45,46 It suggests that La/Cs doping has a similar effect as Ag/Au doping in regulating the electronic structure of Cu-based catalyst, which is beneficial to the charge transfer for CO dimerization.47–49 Therefore, we can conclude that the synergistic effect of the components in the trimetallic catalysts can be attributed to the change of different defects/vacancies on the electronic properties and surface structures.
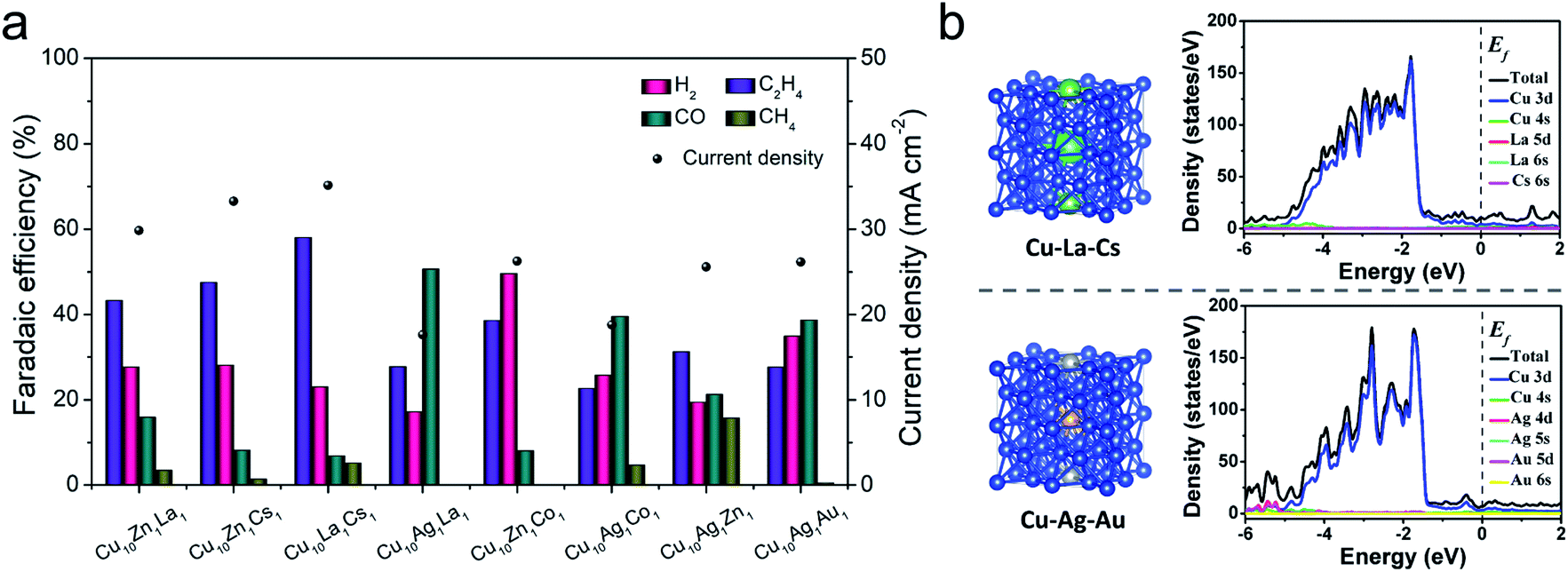 | ||
| Fig. 3 (a) The distribution of reduction products and current density at −1.2 V vs. RHE over different metal catalysts; (b) the structure models and the DOS of Cu10La1Cs1 and Cu10Ag1Au1 electrodes. (The atoms in blue, green, viridian, silvery, and brown represent Cu, La, Cs, Ag, and Au, respectively. The doping model shown is Cu 2 × 2 × 2 supercell-based substitution doping). | ||
It is worth mentioning that no obvious changes occurred in Cu phase during CO2RR. From the semi-in situ XAS characterization results presented in Fig. 4a–c, we can find that the surface of Cu10La1Cs1 was still mainly Cu0 sites with the increase of electrolysis time. These data further confirm that the La and Cs components could maintain the chemical state and the local coordination environment of the Cu phase was not changed under reaction conditions. The undercoordinated Cu sites are associated with outstanding C–C coupling.50–52 We then pursued theoretical insights into the study of the intrinsic property of the catalysts. As depicted in computational structure models in Fig. 4d and S25–S30,† the interactions and electronic structure among Cu, La, and Cs atoms of pure Cu, Cu10La1, Cu10Cs1, and Cu10La1Cs1 catalysts were different. For Cu10La1Cs1, the La and Cs atoms tend to delocalize charge by releasing electrons to the Cu atoms, manifesting the electron transfer effect. The optimized adsorption configurations of reaction intermediates on the simulated interface structures are displayed in Fig. S31.†Fig. 4e and f show the *CO and *OCCO adsorption configurations on the four simulated interface structures. We can find that the presence of La and Cs in Cu10La1Cs1 could effectively adjust the adsorption space position of *CO and *OCCO intermediates to an optimized state, which enables the lowest energy barrier for CO2 transformation to more reduced products with the multi-electron process. This result is obviously different from that over pure Cu catalyst, on which the energy barrier is mainly in the typical two steps of CO2 hydrogenation reduction to generate adsorbed carboxylic acid groups (CO2 + H+ + e− → *COOH) and CO molecular copolymerization (*2CO → *OCCO), requiring the high energy barrier.53 With the introduction of the Cs atom, the energy barrier of these two steps was reduced, and it becomes more noticeable with the further introduction of La atoms. Therefore, we consider that the synergistic effect between Cu, La, and Cs not only reduces the energy barrier for the CO2 hydrogenation reduction to form adsorbed carboxylic acid groups (CO2 + H+ + e− → *COOH) and CO molecules (*COOH + H+ + e− + H2O → *CO), but also promotes the C–C coupling process for C2H4 formation (*2CO → *OCCO).54–57 This suggests that the presence of La and Cs favors CO production and leads to higher CO* coverage around the active sites. The increased CO* coverage then improves the dimerization of neighboring *CO intermediates to generate *OCCO rather than desorbed. The above results, taken together, suggest that the synergistic interaction among Cu, La, and Cs can efficiently enhance the C2H4 selectivity via a combination of effects, including defects, change of electronic structure, fast charge transfer rate, and increase of active sites.
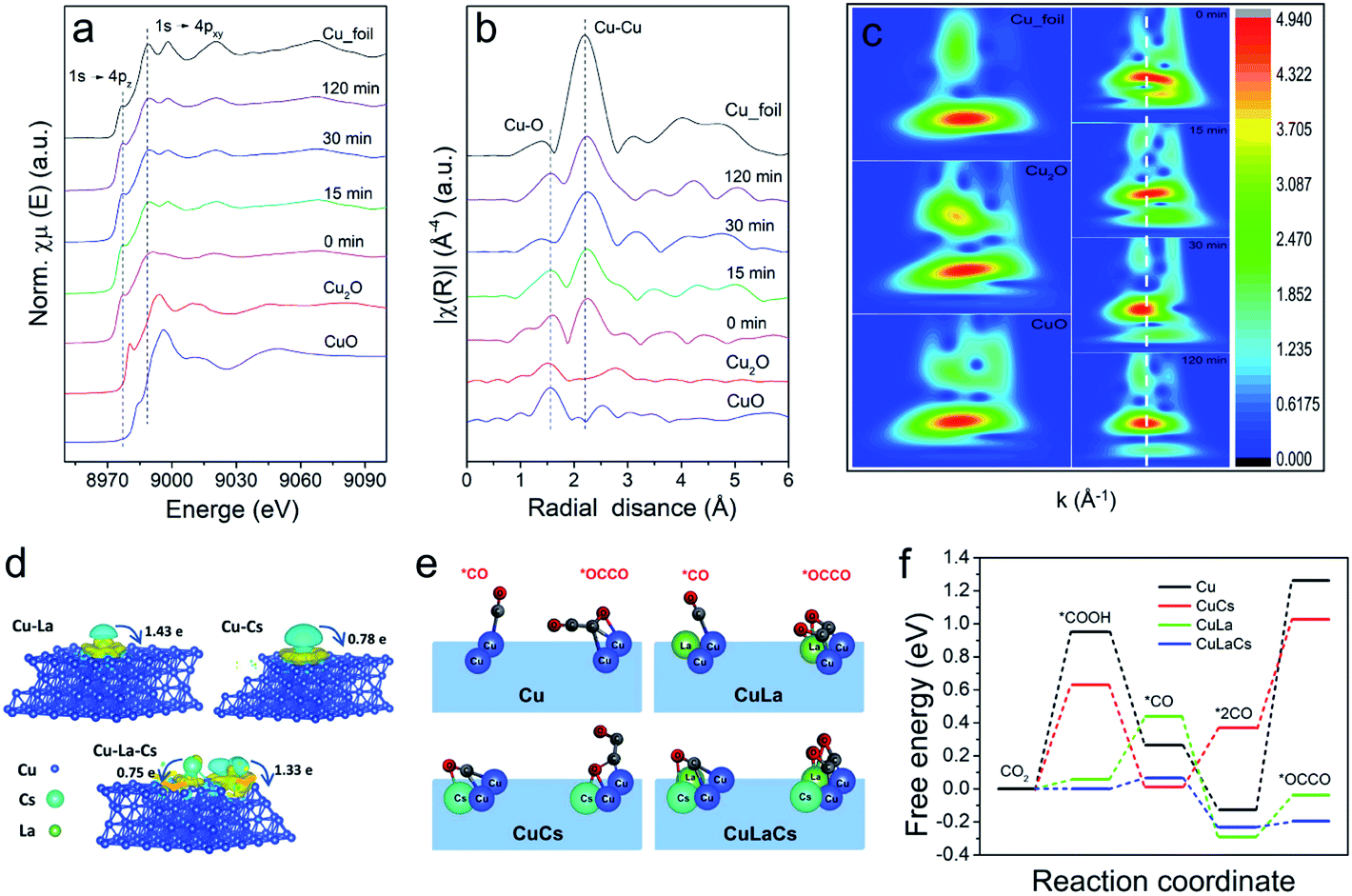 | ||
| Fig. 4 Semi-in situ XAS characterization and DFT calculations. (a) Normalized Cu K edge XANES spectra of Cu10La1Cs1 during CO2RR at −1.2 V vs. RHE; (b) corresponding k3-weighted FT-EXAFS spectra of Cu10La1Cs1 during CO2RR at −1.2 V vs. RHE; (c) Morlet WT of the k3-weighted EXAFS data for Cu10La1Cs1 during CO2RR at −1.2 V vs. RHE; (d) side views of the charge density difference of Cu10La1, Cu10Cs1, and Cu10La1Cs1 with an isosurface of 6 × 10−4, 2 × 10−3 and 2 × 10−3 e Å−3, respectively. (The charge accumulation is shown as the yellow region, and the charge depletion is shown as the cyan region); (e) the *CO and *OCCO adsorption configurations on pure Cu, Cu10La1, Cu10Cs1, and Cu10La1Cs1 interface structures; (f) the activation energy barrier of CO dimerization at different models. | ||
Conclusions
In summary, we find that a trimetallic catalyst prepared via a one-step co-electrodeposition strategy can act as a robust electrocatalyst for CO2RR to C2H4. In particular, over Cu10La1Cs1 catalyst, the C2H4 selectivity can reach 56.9% with a current density of 37.4 mA cm−2 in an H-type cell, and a total C2+ selectivity reaches 70.5% with a current density of 485.5 mA cm−2 in a flow cell, respectively. The outstanding electrocatalytic performance of the trimetallic Cu10La1Cs1 catalyst can be ascribed to the synergistic effect of Cu, La, and Cs. The abundant defects can modulate the electronic structure of the Cu phase, offering substantial potential active sites to stabilize the *CO intermediates and promote C–C coupling to produce C2H4. The as-synthesized trimetallic catalysts on 3D CP also result in a large electrochemical surface area and fast charge transfer, which enhance the reaction rate. We believe that the methodology to prepare multi-metallic catalysts by co-electrodeposition can also be used to design other efficient catalysts for CO2RR.Data availability
The authors declare that all data supporting the findings of this study are available within the paper [and its ESI†].Author contributions
S. Q. J., Q. G. Z., H. H. W. and B. X. H. proposed the project, designed the experiments, and wrote the manuscript. S. Q. J. performed the whole experiment. S. T. H., M. E. C., J. X. Z., X. Q. X. and W. X. performed the analysis of experimental data. Q. G. Z., H. H. W., M. Y. H. and B. X. H. co-supervised the whole project. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The work was supported by the National Key Research and Development Program of China (2020YFA0710201, 2017YFA0403102), the National Natural Science Foundation of China (22022307, 21890761, 22121002). The X-ray Absorption Fine Structure (XAFS) measurements were carried out at the 4B9A beamline of Beijing Synchrotron Radiation Facility (BSRF), China.Notes and references
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2020, 4, 20–27 CrossRef.
- A. Ozden, Y. Wang, F. Li, M. Luo, J. Sisler, A. Thevenon, A. Rosas-Hernández, T. Burdyny, Y. Lum, H. Yadegari, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, Joule, 2021, 5, 706–719 CrossRef CAS.
- X. F. Qiu, H. L. Zhu, J. R. Huang, P. Q. Liao and X. M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS PubMed.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen, D. Tan, X. Cheng, B. Han, L. Zheng and G. Mo, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- C. Choi, S. Kwon, T. Cheng, M. Xu, P. Tieu, C. Lee, J. Cai, H. M. Lee, X. Pan, X. Duan, W. A. Goddard and Y. Huang, Nat. Catal., 2020, 3, 804–812 CrossRef CAS.
- A. N. Kuhn, H. Zhao, U. O. Nwabara, X. Lu, M. Liu, Y. T. Pan, W. Zhu, P. J. A. Kenis and H. Yang, Adv. Funct. Mater., 2021, 31, 2101668 CrossRef CAS.
- R. Shi, Z. Wang, Y. Zhao, G. I. N. Waterhouse, Z. Li, B. Zhang, Z. Sun, C. Xia, H. Wang and T. Zhang, Nat. Catal., 2021, 4, 565–574 CrossRef CAS.
- J. Bu, Z. Liu, W. Ma, L. Zhang, T. Wang, H. Zhang, Q. Zhang, X. Feng and J. Zhang, Nat. Catal., 2021, 4, 557–564 CrossRef CAS.
- P. Shao, W. Zhou, Q. L. Hong, L. Yi, L. Zheng, W. Wang, H. X. Zhang, H. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16687–16692 CrossRef CAS PubMed.
- X. Yuan, S. Chen, D. Cheng, L. Li, W. Zhu, D. Zhong, Z. J. Zhao, J. Li, T. Wang and J. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS PubMed.
- H. Li, T. Liu, P. Wei, L. Lin, D. Gao, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 14329–14333 CrossRef CAS PubMed.
- H. S. Jeon, J. Timoshenko, C. Rettenmaier, A. Herzog, A. Yoon, S. W. Chee, S. Oener, U. Hejral, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2021, 143, 7578–7587 CrossRef CAS PubMed.
- M. G. Kibria, C. T. Dinh, A. Seifitokaldani, P. De Luna, T. Burdyny, R. Quintero-Bermudez, M. B. Ross, O. S. Bushuyev, F. P. Garcia de Arquer, P. Yang, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1804867 CrossRef PubMed.
- C. M. Gunathunge, X. Li, J. Li, R. P. Hicks, V. J. Ovalle and M. M. Waegele, J. Phys. Chem. C, 2017, 121, 12337–12344 CrossRef CAS.
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed.
- X.-Q. Li, G.-Y. Duan, J.-W. Chen, L.-J. Han, S.-J. Zhang and B.-H. Xu, Appl. Catal. B Environ., 2021, 297, 120471 CrossRef CAS.
- F. S. Roberts, K. P. Kuhl and A. Nilsson, Angew. Chem., Int. Ed., 2015, 54, 5179–5182 CrossRef CAS PubMed.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- X. Yan, C. Chen, Y. Wu, S. Liu, Y. Chen, R. Feng, J. Zhang and B. Han, Chem. Sci., 2021, 12, 6638–6645 RSC.
- C. Choi, T. Cheng, M. Flores Espinosa, H. Fei, X. Duan, W. A. Goddard 3rd and Y. Huang, Adv. Mater., 2019, 31, 1805405 CrossRef PubMed.
- T. Adit Maark and B. R. K. Nanda, J. Phys. Chem. C, 2017, 121, 4496–4504 CrossRef CAS.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS PubMed.
- D. Ren, B. S.-H. Ang and B. S. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- S. B. Varandili, D. Stoian, J. Vavra, K. Rossi, J. R. Pankhurst, Y. T. Guntern, N. López and R. Buonsanti, Chem. Sci., 2021, 12, 14484–14493 RSC.
- X. Wang, Z. Wang, T. Zhuang, C.-T. Dinh, J. Li, D.-H. Nam, F. Li, C.-W. Huang, C.-S. Tan, Z. Chen, M. Chi, C. M. Gabardo, A. Seifitokaldani, P. Todorović, A. Proppe, Y. Pang, A. R. Kirmani, Y. Wang, A. H. Ip, L. J. Richter, B. Scheffel, A. Xu, S.-C. Lo, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5186 CrossRef PubMed.
- Y. C. Li, Z. Wang, T. Yuan, D. H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. Pelayo García de Arquer, Y. Wang, C. T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- X. Wang, P. Ou, J. Wicks, Y. Xie, Y. Wang, J. Li, J. Tam, D. Ren, J. Y. Howe, Z. Wang, A. Ozden, Y. Z. Finfrock, Y. Xu, Y. Li, A. S. Rasouli, K. Bertens, A. H. Ip, M. Graetzel, D. Sinton and E. H. Sargent, Nat. Commun., 2021, 12, 3387 CrossRef CAS PubMed.
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS PubMed.
- T. T. H. Hoang, S. Verma, S. Ma, T. T. Fister, J. Timoshenko, A. I. Frenkel, P. J. A. Kenis and A. A. Gewirth, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- J. Gao, D. Ren, X. Guo, S. M. Zakeeruddin and M. Gratzel, Faraday Discuss., 2019, 215, 282–296 RSC.
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Gratzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed.
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS PubMed.
- R. Feng, Q. Zhu, M. Chu, S. Jia, J. Zhai, H. Wu, P. Wu and B. Han, Green Chem., 2020, 22, 7560–7565 RSC.
- L. Hou, J. Han, C. Wang, Y. Zhang, Y. Wang, Z. Bai, Y. Gu, Y. Gao and X. Yan, Inorg. Chem. Front., 2020, 7, 2097–2106 RSC.
- L. Xiong, X. Zhang, H. Yuan, J. Wang, X. Yuan, Y. Lian, H. Jin, H. Sun, Z. Deng, D. Wang, J. Hu, H. Hu, J. Choi, J. Li, Y. Chen, J. Zhong, J. Guo, M. H. Rummerli, L. Xu and Y. Peng, Angew. Chem., Int. Ed., 2021, 60, 2508–2518 CrossRef CAS PubMed.
- X. Wang, P. Ou, A. Ozden, S. Hung, J. Tam, C. M. Gabardo, J. Y. Howe, J. Sisler, K. Bertens, F. P. García de Arquer, R. K. Miao, C. P. O'Brien, Z. Wang, J. Abed, A. S. Rasouli, M. Sun, A. H. Ip, D. Sinton and E. H. Sargent, Nat. Energy, 2022, 7, 170–176 CrossRef CAS.
- L. R. L. Ting, R. García-Muelas, A. J. Martín, F. L. P. Veenstra, S. T.-J. Chen, Y. Peng, E. Y. X. Per, S. Pablo-García, N. López, J. Pérez-Ramírez and B. S. Yeo, Angew. Chem., Int. Ed., 2020, 59, 21072 CrossRef CAS PubMed.
- H. S. Jeon, S. Kunze, F. Scholten and B. Roldan Cuenya, ACS Catal., 2018, 8, 531 CrossRef CAS.
- A. Beniya and S. Higashi, Nat. Catal., 2019, 2, 590–602 CrossRef.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. D. Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- Y. Huang, C. W. Ong and B. S. Yeo, ChemSusChem, 2018, 11, 3299 CrossRef CAS PubMed.
- B. Roldan Cuenya, F. Scholten, K. C. Nguyen, J. P. Bruce and M. Heyde, Angew. Chem., Int. Ed., 2021, 60, 19169–19175 CrossRef PubMed.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- W. Guo, S. Liu, X. Tan, R. Wu, X. Yan, C. Chen, Q. Zhu, L. Zheng, J. Ma, J. Zhang, Y. Huang, X. Sun and B. Han, Angew. Chem., Int. Ed., 2021, 60, 21979–21987 CrossRef CAS PubMed.
- J. Wang, C. Cheng, B. Huang, J. Cao, L. Li, Q. Shao, L. Zhang and X. Huang, Nano Lett., 2021, 21, 980–987 CrossRef CAS PubMed.
- Z. Zeng, L. Y. Gan, H. Bin Yang, X. Su, J. Gao, W. Liu, H. Matsumoto, J. Gong, J. Zhang, W. Cai, Z. Zhang, Y. Yan, B. Liu and P. Chen, Nat. Commun., 2021, 12, 4088 CrossRef CAS PubMed.
- M. Li, B. Hua, L.-C. Wang, J. D. Sugar, W. Wu, Y. Ding, J. Li and D. Ding, Nat. Catal., 2021, 4, 274–283 CrossRef CAS.
- H.-Q. Liang, S. Zhao, X.-M. Hu, M. Ceccato, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2021, 11, 958–966 CrossRef CAS.
- X. Feng, K. Jiang, S. Fan and M. W. Kanan, ACS Cent. Sci., 2016, 2, 169–174 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed.
- X. Wang, K. Klingan, M. Klingenhof, T. Moller, J. Ferreira de Araujo, I. Martens, A. Bagger, S. Jiang, J. Rossmeisl, H. Dau and P. Strasser, Nat. Commun., 2021, 12, 794 CrossRef CAS PubMed.
- K. J. P. Schouten, Z. Qin, E. P. Gallent and M. T. M. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed.
- Z. Zhang, G. Wen, D. Luo, B. Ren, Y. Zhu, R. Gao, H. Dou, G. Sun, M. Feng, Z. Bai, A. Yu and Z. Chen, J. Am. Chem. Soc., 2021, 143, 6855–6864 CrossRef CAS PubMed.
- J. Y. Kim, D. Hong, J. C. Lee, H. G. Kim, S. Lee, S. Shin, B. Kim, H. Lee, M. Kim, J. Oh, G. D. Lee, D. H. Nam and Y. C. Joo, Nat. Commun., 2021, 12, 3765 CrossRef CAS PubMed.
- S. Ma, M. Sadakiyo, M. Heima, R. Luo, R. T. Haasch, J. I. Gold, M. Yamauchi and P. J. A. Kenis, J. Am. Chem. Soc., 2017, 139, 47–50 CrossRef CAS PubMed.
- W. Ren, X. Tan, J. Qu, S. Li, J. Li, X. Liu, S. P. Ringer, J. M. Cairney, K. Wang, S. C. Smith and C. Zhao, Nat. Commun., 2021, 12, 1449 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1sc06964k |
| This journal is © The Royal Society of Chemistry 2022 |