
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew insights into the folding–unfolding mechanism and conformations of cytochrome C†
Jiayu
Li
and
Hongbin
Li
 *
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: hongbin@chem.ubc.ca
First published on 30th May 2022
Abstract
Metalloproteins account for over one-third of all proteins in nature and play important roles in biological processes. The formation of the native structures of metalloproteins requires not only the correct folding of the polypeptide chains but also the proper incorporation of metal cofactors. Understanding the folding mechanism of metalloproteins has been challenging. Horse heart cytochrome C (cytc) is a classical model system for protein folding studies. Although a large number of ensemble studies have been carried out to characterize the folding mechanism of cytc, there is still a significant debate on the folding mechanism and the existence of the proposed “foldons”. Here, we used single-molecule optical tweezers to probe the mechanical folding–unfolding behaviors of cytc at the single-molecule level. By directly monitoring the folding and unfolding of holo-cytc, we revealed novel insights into the folding of cytc. Our results showed that the structural elements that are distant from the N- and C-termini can exist as a short-lived intermediate, a finding that contrasts with the general belief that the folding and packing of the N- and C-terminal helices are prerequisites for the folding of other structural elements in cytc. In addition, our results present strong evidence that apo-cytc, which has been long believed to be a random coil, is not a true random coil, and weak interactions within the unfolded polypeptide chain exist. Our results bring new insights into our understanding of the folding mechanisms of heme proteins as well as the role of heme in the folding process.
Introduction
Metalloproteins account for more than one-third of all proteins in nature and play a variety of important biological functions, ranging from electron transfer to catalyzing some of the most difficult chemical reactions that are challenging for the current synthetic chemistry. The folding of metalloproteins involves not only the folding of the polypeptide chain but also the constitution of the metal center through metal–ligand coordination.1 Elucidating the folding mechanisms of metalloproteins has been challenging, as the unfolding of many metalloproteins is often irreversible in vitro. This is particularly true for transition metal-containing metalloproteins of which the metal or metal cluster cofactors have high hydrolysis constants.2 Heme proteins are an important family of metalloproteins that contain a prosthetic heme group and play critical roles in many important cellular processes, such as oxygen transportation, drug detoxification and signal transduction.3 The cofactor heme, an Fe(II)-centered porphyrin, entails heme proteins with distinct spectroscopic characteristics. Among the heme proteins, horse heart cytochrome C (cytc) has long been used as a model system for protein folding studies.4 As an important electron transfer protein, the 104 amino acid (aa) residue protein is highly helical and contains a highly soluble c-type heme cofactor bounded to the polypeptide chain by two thioether bonds and two coordination bonds (Fig. 1).5 These unique features prevent the metal cofactor from hydrolysis and dissociation upon protein unfolding, making in vitro refolding studies possible. In addition, cytc is synthesized in the cytosol as an apoprotein and then translocated into the mitochondria, where it associates with the heme prosthetic group to form holo-cytc.6 During the translocation process, cytc needs to be mechanically unfolded and translocated by the translocation machinery. | ||
| Fig. 1 The structure of horse heart cytc. (A) Three-dimensional structure of horse heart cytc (PDB code: 1hrc). Cytc is a 104 aa helical protein containing a c-type heme cofactor. The heme is covalently bound to the polypeptide chain through two thioether bonds, and the heme iron forms axial Fe–N and Fe–S coordination bonds with a histidine and a methionine residue. (B) Schematics of the structure of cytc. The black circle indicates the porphyrin ring, the spirals indicate the helices, and the blue dotted lines represent the coordination bonds. (C) Schematic of the optical tweezer experiment to investigate the mechanical unfolding–folding of cytc. | ||
The folding–unfolding mechanisms of apo- and holo-forms of cytc have been studied extensively using various experimental techniques at the ensemble level. Early equilibrium chemical denaturation studies suggested that cytc exhibits two-state folding behavior,7 while more recent spectroscopic and calorimetric studies provide strong evidence showing the existence of both on-pathway and off-pathway folding intermediate states.8–12 In addition to equilibrium studies, kinetic studies on cytc have also confirmed the complexity of the folding process and identified possible misfolded conformations either involving the non-native coordination to the heme iron of wrong histidines, lysines, and even the N-terminal amino group, or arising from proline isomerization.13–16 The hydrogen exchange method has also been used to study the folding–unfolding of cytc, and a foldon dependent hierarchical multistep folding–unfolding mechanism was proposed.17–19 In contrast to holo-cytc, the heme-free apo-form cytc (apo-cytc) has long been considered as a random coil with no folded structures based on results from multiple spectroscopic experiments.20,21 It is thus believed that the heme plays a decisive role in transforming the random coil structure into the folded globular conformation of cytc, and the flexible and disordered conformation of apo-cytc is believed to facilitate the accepting and enveloping of the heme cofactor during the folding of holo-cytc.20,21 Despite this progress, the detailed folding mechanism of holo-cytc is still under debate, and the random coil conformation of apo-cytc remains to be substantiated.
Over the last two decades, single-molecule force spectroscopy (SMFS) techniques, including atomic force microscopy (AFM), optical tweezers (OT) and magnetic tweezers, have evolved into powerful tools to probe protein folding–unfolding mechanisms and nucleic acid conformational dynamics at the single-molecule level.22–29 By mechanically stretching/relaxing a protein from its two chosen residues, one can use SMFS techniques to probe the folding–unfolding reaction of a protein in real-time along a well-defined reaction coordinate at the single-molecule level, revealing unique insights into the protein folding–unfolding mechanism. These features have enabled SMFS techniques to become an important new tool to probe the folding–unfolding mechanism of metalloproteins that are otherwise difficult to study using traditional biophysical methods in vitro.30–39 Here, we used the single-molecule OT technique to investigate the mechanical folding–unfolding behaviors of the holo- and apo-forms of horse heart cytc. On the one hand, OTs enable one to investigate the unfolding/folding of apo-cytc in a mechanical setting that partially mimics that of the translocation process; on the other hand, OTs allow investigation of the unfolding/folding of holo-cytc along a well-defined reaction coordinate set by the applied stretching force. Our results showed that holo-cytc is mechanically stable and unfolds following two distinct pathways: a two-state unfolding pathway and a three-state unfolding pathway involving an intermediate state. In contrast, the folding of holo-cytc followed an apparent two-state pathway without the accumulation of any observed intermediate state. Moreover, our results showed that apo-cytc demonstrates some intrachain interactions and may form structures that exhibit low mechanical resistance. Our results revealed some new insights into the conformation of apo-cytc and the folding–unfolding mechanism of holo-cytc and help elucidate the role played by the heme in the folding of cytc.
Materials and methods
Protein engineering
The original plasmid of pBTR(hCc), which was a gift from Gary Pielak (Addgene plasmid # 61026), encodes both horse heart cytc and yeast heme lyase, thus enabling expression of holo-form horse heart cytc in Escherichia coli.40 The pre-existing BglII (A′GATCT) restriction site in the plasmid was removed via standard site-directed mutagenesis methods, and the plasmid was modified by polymerase chain reaction (PCR) to carry a cysteine upstream to cytc, as well as a new 3′ BglII, a 3′ KpnI (G′GTACC) restriction site, and a cysteine downstream to cytc. The gene of the protein NuG2 was then inserted between the BglII and KpnI restriction sites by digestion and ligation.The gene of cytc (C14, 17A) was obtained by standard site-directed mutagenesis methods. The genes of cytc and cytc (C14, 17A) were amplified using standard PCR to carry 5′ BamHI (G′GATCC) and 3′ KpnI restriction sites. They were then subcloned into two modified pQE80L (Qiagen, Valencia, CA) expression vectors, respectively, which allow for adding a cysteine and a cysteine together with an NuG2, to both termini of the protein. All the sequences were confirmed by direct DNA sequencing.
All the recombinant proteins were overexpressed in the Escherichia coli strain BL21 (DE3) at 37 °C. To express holo-cytc, 5 mL of preculture was inoculated in 2 L of rich medium (12 g L−1 tryptone, 24 g L−1 yeast extract, 4 mL L−1 glycerol, 2.3 g L−1 KH2PO4, and 12.5 g L−1 K2HPO4) containing 100 μg mL−1 ampicillin, and the protein expression continued for 30 h.40 To express apo-cytc, 3 mL of preculture was inoculated in 200 mL of 2.5% Luria–Bertani media containing 100 mg mL L−1 ampicillin, and when the OD600 of the culture reached ∼0.7, protein overexpression was induced with 0.5 mM isopropyl-b-D-1-thiogalactopyranoside (Thermo Fisher Scientific, Waltham, MA) and continued for 4 h. Both the cells were pelleted by centrifugation at 4000g for 10 min at 4 °C and resuspended in 10 mL of phosphate-buffered saline (PBS) buffer (10 mM, pH 7.4). After adding 10 μL of protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO), 100 μL of 50 mg mL−1 lysozyme from egg white (Sigma-Aldrich, St. Louis, MO), 1 mL of 10% (w/v) Triton X-100 (VWR, Tualatin, OR), and 50 μL of 1 mg mL−1 DNase I (Sigma-Aldrich, St. Louis, MO) and RNase A (Bio Basic Canada Inc, Markham, ON), cells were lysed for 40 min on ice. Cell debris was then removed by centrifugation at 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g at 4 °C, and the supernatant was loaded into a Co2+ affinity chromatography column (Takara Bio USA Inc, Mountain View, CA). After washing the column with 50 mL of washing buffer (10 mM PBS, 300 mM NaCl, 7 mM imidazole, and pH 7.4), the protein was eluted with 2 mL of elution buffer (10 mM PBS, 300 mM NaCl, 250 mM imidazole, and pH 7.4).
000g at 4 °C, and the supernatant was loaded into a Co2+ affinity chromatography column (Takara Bio USA Inc, Mountain View, CA). After washing the column with 50 mL of washing buffer (10 mM PBS, 300 mM NaCl, 7 mM imidazole, and pH 7.4), the protein was eluted with 2 mL of elution buffer (10 mM PBS, 300 mM NaCl, 250 mM imidazole, and pH 7.4).
Preparation of DNA-protein chimera
Double-strand DNA (dsDNA) handles with the 5′-end modified with biotin or digoxigenin and the 3′-end modified with NH2 were prepared via the methods described previously.41 The dsDNA handles were then allowed to react with 4-(N-maleimidomethyl) cyclohexanecarboxylic acid N-hydroxysuccinimide ester (SMCC, Sigma-Aldrich, St. Louis, MO) overnight, which resulted in the functionalization of dsDNA handles with the maleimide group. 50 μM of the freshly expressed proteins were reduced with 1 mM tris(2-carboxyethyl) phosphine (TCEP) (Sigma-Aldrich, St. Louis, MO) for 1 hour, and the remaining TCEP was removed by using Zeba desalting columns (7k molecular weight cutoff, Thermo Fisher Scientific, Waltham, MA). Then the reduced proteins were diluted to ∼3 μM with Tris buffer (20 mM Tris, 150 mM NaCl, and pH 7.4). 1 μL of the diluted protein was added into 1 μL of mixed dsDNA handles (both are at 3 μM). The thiol-maleimide reaction was carried out at room temperature overnight. The formed dsDNA-protein chimera was diluted with Tris buffer (20 mM Tris, 150 mM NaCl, and pH 7.4) to ∼10 nM and ready for the optical tweezer experiment.OT-based SMFS experiments
The OT based-SMFS experiments were carried out with a Minitweezers setup (http://tweezerslab.unipr.it/cgi-bin/mt/home.pl), which was described previously.41 The liquid chamber of optical tweezers was filled with Tris buffer (20 mM Tris, 150 mM NaCl, and pH 7.4) to provide the working environment, unless otherwise noted. In a typical experiment, 1 μL of 0.5% streptavidin modified polystyrene beads (1% w/v 1 μm, Spherotech Inc, Lake Forest, IL) was diluted to 3 mL and injected into the fluid chamber. A single streptavidin modified polystyrene bead was captured by a laser beam and then held with a micropipette tip within the chamber. 1 μL of 5 nM DNA-protein chimera was allowed to react with 5 μL of 0.1% antidigoxigenin modified polystyrene beads (0.5% w/v, 2 μm, Spherotech Inc, Lake Forest, IL) for 30 min at room temperature. The mixture was then diluted to 3 mL and injected into the chamber. A single anti-digoxigenin (anti-Dig) modified polystyrene bead was captured by the laser trap. The laser trap controlled the movement of the anti-dig bead against the streptavidin modified polystyrene bead fixed on the pipette tip to carry out the force–extension experiments. The stretching–relaxing experiments in this work were carried out at a constant speed of 50 nm s−1.Calculation of the kinetics of unfolding/folding of proteins
We used the method proposed by Oesterhelt et al. to extract the folding and unfolding rate constants at different forces from the force–distance curves.42 Briefly, the curves were divided into time windows (Δt) that are small enough so that the force can be considered constant within the time window. The rate constant of protein folding/unfolding at the force F can be calculated using P(F) = N(F)/(M(F) × Δt), where the N(F) is the total number of all the folding or unfolding events at the force of F, and M(F) is the total number of time windows at the force of F. The mechanical chevron plot, that is the force-dependence of the unfolding and folding rate constants, was fitted to the Bell–Evans model to extract the unfolding and folding rate constants at zero force (α0 and β0) and the unfolding and folding distances to the transition state (Δxu and Δxf). The Bell–Evans model assumes that the transition state does not move with the applied stretching force.43,44 Since the force-dependences of lnα0 and lnβ0 do not show any obvious curvature, we used the Bell–Evans model for fitting the data. In doing so, we did not need to assume a certain shape of the free energy profile for the mechanical unfolding/folding of cytc, which is required for other models, such as the Dudko–Hummer–Szabo model.45
Results
Mechanical unfolding of holo-cytc using optical tweezers
To investigate the unfolding and folding of holo-cytc using OTs, we engineered Cys-holo-cytc-NuG2-Cys, in which the well-characterized protein NuG2 serves as a fingerprint to facilitate the identification of single-molecule stretching events. NuG2 is a computationally designed fast folding protein, and its mechanical folding–unfolding has been well characterized in our previous studies.46,47 The unfolding of NuG2 occurs at ∼20–40 pN and folding occurs at ∼8 pN with a contour length change ΔLc of ∼18 nm (Fig. S1, ESI†). These fingerprints, together with the overstretching transition of dsDNA handles, allow us to readily identify single molecule stretching in OT measurements.33,36,46 The two cysteine residues at both termini allow for the attachment of dsDNA handles for OT experiments. We cloned the gene encoding Cys-cytc-NuG2-Cys into the pBTR vector, which encodes the gene of yeast heme lyase. The co-expression of Cys-cytc-NuG2-Cys and yeast heme lyase allowed for the covalent attachment of the heme prosthetic group to the expressed Cys-cytc-NuG2-Cys.40Fig. 2A shows the UV-Vis spectra of the purified reduced form of Cys-holo-cytc-NuG2-Cys and the control holo-cytc. The distinctive absorption peaks at ∼414, ∼521, and ∼550 nm correspond to the γ, β and α absorption bands of the reduced holo-cytc. Based on the ratio of the absorption at 414 nm and 280 nm,48 we estimated that more than 70% of our Cys-cytc-NuG2-Cys sample contained holo-cytc. | ||
| Fig. 2 UV-Vis spectra of purified holo-cytc and Cys-holo-cytc-NuG2-Cys proteins expressed using the modified pBTR vector. The absorption at ∼410, ∼520, ∼550 nm is characteristic of the reduced holo-cytc. | ||
For OT experiments, we coupled two dsDNA handles with Cys-holo-cytc-NuG2-Cys via thiol-maleimide coupling chemistry to create the DNA-protein-DNA chimera. Stretching Cys-holo-cytc-NuG2-Cys allowed us to stretch the reduced holo-cytc between its N- and C-termini. Fig. 3A shows the representative force–distance curves of the protein-DNA chimera at a pulling speed of 50 nm s−1. Each curve displayed two distinct unfolding/folding events. As expected, the fingerprint domain NuG2 (colored in cyan) unfolded at ∼20–40 pN and folded at ∼8 pN with a ΔLc of 18 nm. Hence, the unfolding/folding events of holo-cytc (colored in red and blue) can be readily identified from the force–distance curves. Evidently, the unfolding of cytc mostly occurred between ∼25 and 30 pN. The great majority of the native holo-cytc unfolded via an apparent two-state pathway (∼92%, 258 out of 279 events), and a small percentage occurred following a three-state pathway involving one short-lived intermediate state (∼8%, 21 out of 279 events) (Fig. 3A). Fitting the force–extension relationships of holo-cytc using the worm-like chain model (WLC) of polymer elasticity yielded a ΔLc of 34.6 ± 1.2 nm (average ± standard deviation) for the complete unfolding of holo-cytc (Fig. 3B).49 For the three-state unfolding pathway, cytc displayed a ΔLc1 of ∼15 nm (from the native to the intermediate state) and a ΔLc2 of ∼20 nm (from the intermediate state to the unfolded state). Based on the structure of holo-cytc, it is expected that the complete mechanical unfolding of holo-cytc should lead to a ΔLc of 35.7 nm (104 aa × 0.36 nm/aa − 1.7 nm = 35.7 nm, where 0.36 nm/aa is the length of an aa residue and 1.7 nm is the distance between the N- and C-termini). This value is in close agreement with the experimentally determined ΔLc of ∼34.6 nm, confirming that the two-state and three-state unfolding events we observed indeed correspond to the complete unfolding of holo-cytc.
 | ||
| Fig. 3 Mechanical folding–unfolding signatures of holo-cytc. (A) Representative force–distance curves of Cys-holo-cytc-NuG2-Cys at a pulling speed of 50 nm s−1. The unfolding of holo-cytc is colored in red and green, the refolding of holo-cytc is colored in blue, and the unfolding and folding of NuG2 are colored in cyan. An unfolding intermediate can be observed when holo-cytc unfolds at relatively low forces, while the folding is always two-state. (B) Force–extension relationships of unfolding and refolding of holo-cytc (red and blue). The WLC fit to the experimental data reveals a persistence length of 0.8 nm and a ΔLc of 34.6 ± 1.2 nm (red curve) between the folded and unfolded states of holo-form cytc. (C) Unfolding (red and green) and refolding (blue) force histograms of holo-cytc at a pulling speed of 50 nm s−1. The I–U (intermediate state to unfolded state) event (green) can only be observed when holo-cytc unfolds at relatively low forces (<∼22 pN). The bin size is 1 pN for both unfolding histograms, and for clarity, the I–U unfolding data are shown as a line chart. It is noteworthy that the lower end of the refolding force distribution is close to the limit of detecting refolding events reliably in the force–distance curves. (D) Force-dependent refolding–unfolding rates for holo-cytc. The solid lines are the fits of the Bell–Evans model to the experimental data with the kinetic parameters: the spontaneous unfolding rate constant α0 = (1.83 ± 1.13) × 10−8 s−1, unfolding distance Δxu = 2.74 ± 0.10 nm, spontaneous folding rate constant β0 = (4.94 ± 2.00) × 102 s−1, and folding distance Δxf = 6.91 ± 0.53 nm. | ||
It is important to note that in our OT experiments, cytc is stretched from its N- and C-termini. Both termini pack against each other in the folded structure of cytc, effectively shielding the rest of the cytc structure from the stretching force (Fig. 1A). Due to this feature, it is apparent that in order to extend cytc, its N- and C-termini must be separated (and/or unfolded) in the first step. In the three-state of unfolding, the mechanical unfolding of cytc into its unfolding intermediate shows a ΔLc of ∼15 nm, suggesting that the N- and C-termini are separated by ∼15 nm. Hence, in the mechanical unfolding intermediate state, the structure formed by N- and C-terminal helices must have been separately and likely unravelled. However, it remains unknown if other parts of cytc are unravelled or not.
Unfolded holo-cytc can refold efficiently
The mechanically unfolded holo-cytc consists of an extended polypeptide chain and a covalently attached heme cofactor. This makes the in vitro folding of holo-cytc possible, as the metal cofactor does not hydrolyze or dissociate upon protein unfolding. After unfolding, we relaxed the mechanically unfolded holo-cytc back to 0 pN under a constant speed to allow holo-cytc to refold. In the total of 316 relaxation traces that we recorded, the vast majority (∼93%, 294 out of the 316 relaxation traces recorded from 15 different molecules) showed a 2-state refolding event of holo-cytc at ∼3 pN with a ΔLc of ∼35 nm (Fig. 3A and C).Most of these refolded holo-cytc molecules (265 out of the 294 refolding events) subsequently unfolded at ∼20–30 pN with a ΔLc of ∼35 nm, in the same way as the pristine holo-cytc unfolded, suggesting that these refolded holo-cytc molecules were correctly folded into their native states in these cases. To obtain the spontaneous unfolding/folding rate constants and unfolding/folding distances, we measured the unfolding and folding rate constants as a function of force using the Oesterhelt method.42 Fitting the experimental data to the Bell–Evans model (Fig. 3D) estimated a spontaneous unfolding rate constant α0 of (1.83 ± 1.13) × 10−8 s−1 and a folding rate constant β0 of (4.94 ± 2.00) × 102 s−1 at zero force.43 It is worth noting that the spontaneous unfolding rate constant α0 of cytc is extremely small (1.83 × 10−8 s−1), which is consistent with that measured from chemical denaturation studies (∼3 × 10−10 s−1),10 yet cytc can be readily unfolded at ∼28 pN, an acceleration of ∼109 times. This significant acceleration is achieved by the extremely malleable native state, which is characterized by a large unfolding distance ΔLc of cytc (∼2.7 nm).
It is important to note that, in these refolding events, holo-cytc did not always refold successfully into its native state. We observed that in ∼10% of the refolding events (29 out of 294), their subsequent unfolding occurred at forces lower than 15 pN, significantly lower than that of the native holo-cytc (∼28 pN) (Fig. 4A, cycle 1), suggesting that these molecules folded into a structure that is mechanically much more labile than the native state. In addition, about 5% of the relaxation traces (17 out of 316) showed a refolding event of holo-cytc with shorter folding ΔLcs (Fig. 4A, cycle 2); and in rare cases (∼2%, 5 out of the 316 relaxation traces), cytc did not fold at all (i.e. no folding event in the relaxation curve, and no unfolding event in the subsequent stretching curve) (Fig. 4A, cycle 3). These phenomena indicated that, besides folding back into its native state, unfolded holo-cytc may also misfold into non-native structures or not fold at all during the relaxation. The refolded holo-cytc with a lower mechanical unfolding force (<15 pN) or a shorter ΔLc is classified as misfolded holo-cytc. It is worth noting that refolding and misfolding can occur in the same molecule (Fig. 4B left panel), and the misfolding occurs at a low frequency (Fig. 4B right panel). In addition, after the unfolding of a misfolded holo-cytc, holo-cytc can refold back to its native state and regain its mechanical stability (with an unfolding force of ∼28 pN). These results suggest that the misfolding of holo-cytc is not irreversible, and the misfolded state can be mechanically unfolded to allow its subsequent correct refolding. Similarly, not being able to fold in a few cycles does not render a permanent loss of the ability of the unfolded holo-cytc to refold, as the unfolded holo-cytc could refold into its native state in the subsequent relaxation cycles (Fig. 4C). We also found that, after the holo-cytc started to misfold or did not fold, extending the folding time at 0 pN to up to 60 s did not change the folding probability of cytc to its native state significantly (Fig. 4C), implying that such states were not productive folding intermediate states.
 | ||
| Fig. 4 Unfolded holo-cyt may misfold or fail to fold during consecutive stretching–relaxing cycles. (A) Representative force–distance curves of Cys-holo-cytc-NuG2-Cys that misfolds (Cycle 2–3) and does not fold (Cycle 4) at a pulling speed of 50 nm s−1 (cyan events: unfolding and folding of NuG2; red and blue events: unfolding and folding of holo-cytc). Cycle 1 shows an unfolding event of native holo-cytc for comparison. (B) Unfolding force of one holo-cytc molecule in consecutive stretching–relaxing cycles (left panel), and the unfolding force histogram of holo-cytc in such consecutive stretching–relaxation experiments (right panel). The waiting time between consecutive curves was zero. The black line is drawn to separate the unfolding events of natively folded (above the line) and misfolded (below the line) holo-cytc. (C) Three consecutive force–distance curves of a Cys-holo-cytc-NuG2-Cys molecule at a pulling speed of 50 nm s−1 (cyan events: unfolding and folding of NuG2; red and blue events: unfolding and folding of holo-cytc). The curves are offset horizontally relative to each other. In (curve 1, cytc remained unfolded. After being relaxed to zero force (curve 2), cytc refolded, giving rise to the unfolding event at ∼25 pN in (curve 3). (D) Representative force–distance curves of two Cys-holo-cytc-NuG2-Cys molecules which misfolded (curve 1–3) and did not fold (curve 4–6) at a pulling speed of 50 nm s−1 (red and blue events: unfolding and folding of holo-cytc). The molecules were held at 0 pN for 60 s after relaxation. The curves are offset horizontally relative to each other. | ||
Previous OT studies50 showed the molten globule of apo-myoglobin, in which most of the secondary structures have formed in a native-like geometry but the tertiary contact is not fully formed,51,52 displayed similar mechanical unfolding behaviors as misfolded holo-cytc, i.e. significantly lower unfolding forces but the same ΔLc as that of the native state. Due to this similarity, we cannot completely rule out the possibility that the “misfolded” conformation of holo-cytc with a lower stability but the same ΔLc is the molten globule state of holo-cytc. However, the molten globule state usually leads to the correct folding of the native state. Our observation that this misfolded conformation of cytc can persist for some extended time without folding into its native state suggests that this misfolded conformation is likely to be a misfolded state or a kinetically trapped off-pathway folding intermediate, rather than the molten globule state. Nonetheless, the molecular mechanism underlying this misfolding behavior in the mechanical unfolding–folding experiments at the single-molecule level is unknown. It is possible that proline isomerization and the mis-ligation of heme play some roles, as proposed for the misfolding of cytc in the chemical folding–unfolding process.17
Apo-cytc is not a random coil
To elucidate the role the heme plays in the folding process of holo-cytc, we then set to investigate the folding and conformation of apo-cytc without the heme cofactor. We expressed cytc alone without the yeast lyase, so that the heme moiety cannot be covalently attached to cytc, and the resultant cytc should be apo-cytc. Indeed, the UV-Vis spectrum of cytc lacked the characteristic absorption peaks at 414, 521 and 550 nm, confirming that the as-prepared protein was indeed apo-cytc. We then used far-UV CD spectroscopy to characterize its secondary structure. As shown in Fig. 5B, the CD spectrum of apo-cytc was characterized by the band at ∼200 nm, indicating that apo-cytc lacked secondary structures, consistent with what was reported in previous studies.21 | ||
| Fig. 5 (A) UV-Vis spectrum of apo-cytc. The protein lacks the characteristic absorption peak from the incorporation of heme and is in its apo-form. (B) CD spectra of holo-cytc and apo-cytc. Apo-cytc shows characteristic features of a random coil, which is in sharp contrast to those of the folded holo-cytc. | ||
We then used OT to examine the mechanical response of apo-cytc to further examine this seemingly random coil. Apo-cytc has two free endogenous cysteine residues (Cys 14 and Cys 17), which may react with two extra dsDNA handles in the coupling process and result in an altered mechanical response of the protein or only a part of the protein being stretched in the OT experiments. To mitigate these potential complications, we mutated both Cys residues to Ala and engineered Cys-NuG2-cytc (C14, 17A)-NuG2-Cys for OT experiments, so that apo-cytc would only be stretched from its N- and C-termini. Since cytc (C14, 17A) is flanked by two NuG2 fingerprint domains, force–distance curves that contain the unfolding/folding signatures of two NuG2 domains must contain the mechanical response of apo-cytc (C14, 17A), thus allowing for the unambiguous identification of the mechanical features of apo-cytc (C14, 17A).
Previous SMFS studies showed that stretching random coils resulted in monotonically increased force versus extension, and no “unfolding” or “folding”-like events were present in force–extension curves.53–55 These include real random coiled proteins, such as the apo-form RTX domain of CyaA,26 and unfolded globular proteins that are not able to refold, such as protein MJ0336 with a bounded BiP domain.56 In our OT experiments, ∼77% (325 out of 423 events measured from 11 different molecules) of the stretching–relaxing cycles of single Cys-NuG2-apo-cytc-NuG2-Cys molecules showed only the unfolding and folding events of two NuG2 domains (Fig. 6A, cycle 1), suggesting that apo-cytc did behave like a random coil in these stretching–relaxing cycles with no detectable intrachain interactions along the polypeptide chain. However, intriguingly, in 23% (98 out of 423) of the trajectories, some “unfolding”-like rupturing events were observed at forces below ∼10 pN (Fig. 6A, cycle 2–5), in addition to the unfolding and folding events of two NuG2 domains. This observation suggests that some apo-cytc molecules displayed some intrachain interactions that give rise to the observed “rupturing” events. Moreover, these low-force rupturing events were observed in all the 11 apo-cytc molecules that we measured, indicating that such deviations from the behaviors of random coils is a general feature among all apo-cytc molecules. Looking closely at these rupturing events, we found that these events occurred with different ΔLcs and different rupturing behaviors, including one-step and multiple-step rupturing behaviors. Plotting the force against the ΔLc of each rupturing event (Fig. 6B) showed that there are no clear clusters of data points present, which may represent the rupturing of a specific interaction. This result indicates that, although there are some weak intrachain interactions, the amino acid residues in apo-cytc do not have a well-defined interaction mode, consistent with the previous observations from ensemble experiments that apo-cytc lacks a folded structure.21 In addition, the fact that the observed ΔLc is always smaller than the contour length of cytc indicates that these interactions are intrinsic to the apo-cytc polypeptide chain itself, but not due to any interaction between apo-cytc and dsDNA handles (Fig. S3, ESI†). Nevertheless, the formation of such interactions did not result in any “folding”-like events in the relaxation curves, implying that the weak intrachain interactions can form only when the polypeptide chain is relaxed close to zero force.
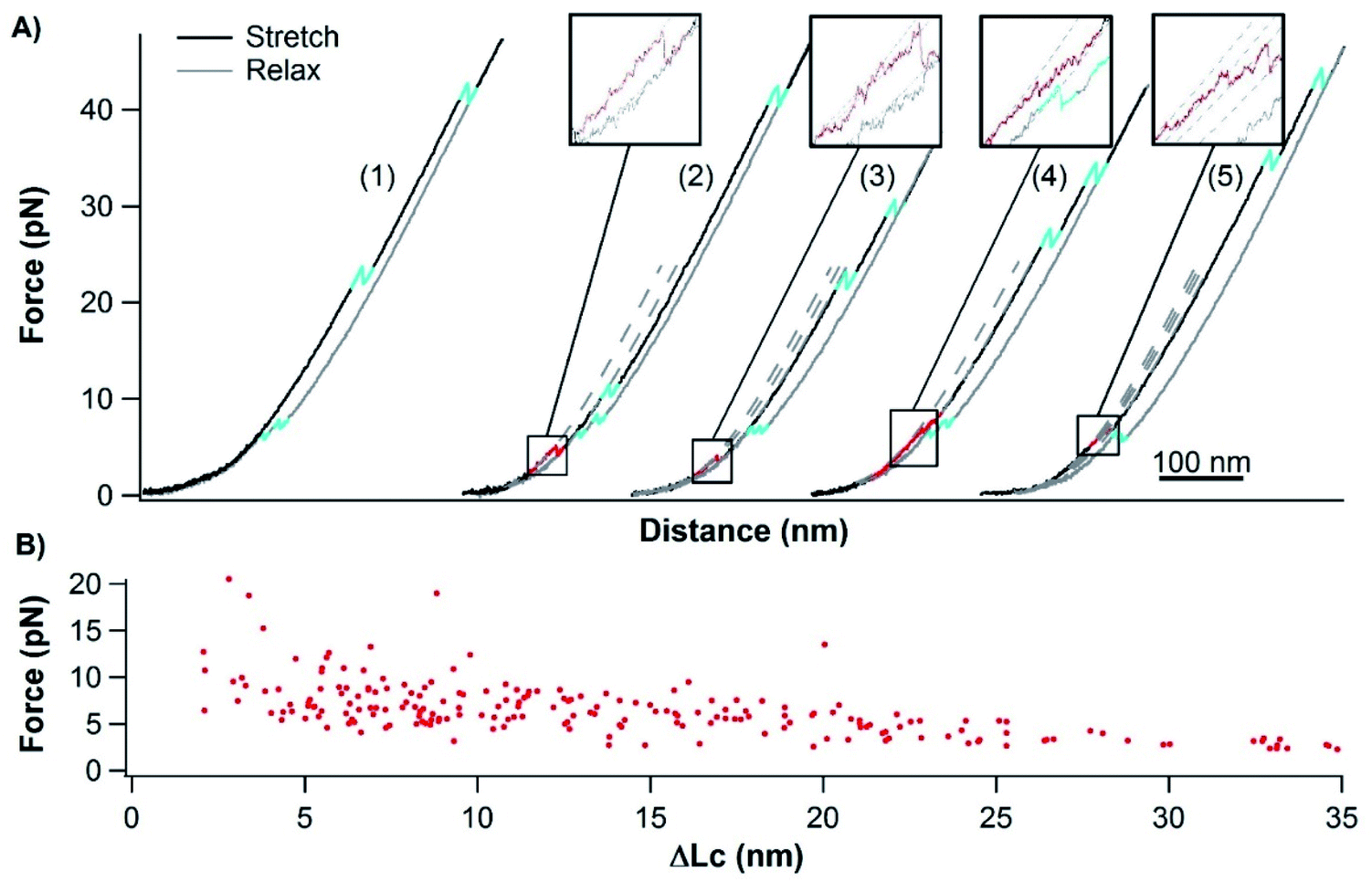 | ||
| Fig. 6 Mechanical behaviors of apo-cytc. (A) Representative force–distance curves of Cys-NuG2-apo-cytc-NuG2-Cys at a pulling speed of 50 nm s−1 (cyan events: unfolding and folding of NuG2; red events: rupturing of apo-cytc). ∼77% of the cycles contain only unfolding and folding events of 2 NuG2 domains (cycle 1), and ∼23% of the cycles show rupturing events of apo-cytc (cycle 2–5). (B) Force–ΔLc relationship of individual rupture events of apo-cytc. | ||
Although the use of fingperprint domains is a widely used method in SMFS studies on proteins, including intrinsically disordered proteins (IDPs),57–60 fusing NuG2 to apo-cytc raised the question if the weak intrachain interactions observed in apo-cytc could be due to the interactions between NuG2 and apo-cytc. To address this issue, we engineered dsDNA-apo-cytc-dsDNA for OT measurements. In the force–distance curves of dsDNA-apo-cytc-dsDNA (Fig. S2 in the ESI†), we observed similar unfolding-like events occurring at low forces, suggesting that there exist weak, nonlocal intrachain interactions in apo-cytc. This finding is similar to those of dsDNA-NuG2-apo-cytc-NuG2-dsDNA, corroborating our conclusion that the weak intrachain interactions indeed originate from the polypeptide chain of apo-cytc itself.
Discussion
The folding–unfolding mechanism of holo-cytc at the single-molecule level
By combining single-molecule OT and protein engineering techniques, here we investigated the mechanical folding–unfolding behavior of a heme-containing metalloprotein, horse heart cytochrome C. When stretched from its N- and C-termini, holo-cytc unfolds following two parallel pathways, either in an all-or-none fashion from its native state directly to the unfolded state, or following a three-state pathway involving an unfolding intermediate state. The unfolding force of native holo-cytc has a narrow distribution centered at ∼28 pN. In most cases, the mechanically unfolded holo-cytc can fold back to its native state in a two-state fashion at ∼3 pN, while a small fraction of cytc molecules misfold or are not able to fold (Fig. 7). Our results provide some new insights into the folding–unfolding mechanism of holo-cytc at the single-molecule level, which are not available from ensemble studies.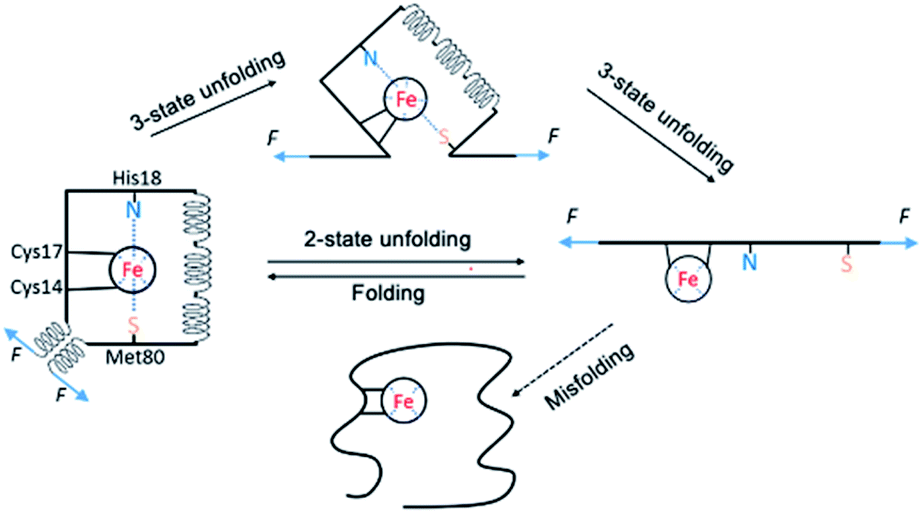 | ||
| Fig. 7 Schematic of the folding–unfolding mechanism of holo-cytc. Holo-cytc unfolds in either a two-state fashion, or a three-state fashion. Mechanically unfolded holo-cytc mostly folds back to its native state in a two-state fashion, but misfolds or does not fold occasionally. In the unfolding intermediate state, the N- and C-terminal helices must have been separated and extended, and likely unravelled. However, its structure remains unknown. The structure of the intermediate shown in the figure is highly schematic and meant for illustration purpose only. | ||
The mechanism of the chemical folding of holo-cytc has been studied extensively by using hydrogen exchange techniques.17–19 It was found that holo-cytc is composed of five cooperative folding units (termed foldons), and the folding and unfolding processes go through the same foldon-dependent native-like intermediates in a reversible fashion. In the unfolding process, the substructures in the middle of the protein's sequence unfold first, followed by the unfolding of the foldon containing the N- and the C-terminal helices; and in the folding process, the N- and the C-terminal helices bind first on the millisecond time scale, and the other structures form subsequently in ∼2 s.17 It was concluded that the folded substructure formed by the N- and the C-terminal helices is more thermodynamically stable and has faster folding kinetics than the other part of holo-cytc, and the folding of the substructures in the middle of the protein's sequence may rely on the correct folding of the N- and the C-terminal helices.
However, in the mechanical folding–unfolding experiments, the unfolding–folding mechanism of cytc may well differ from that of chemical ones due to the directional nature of the mechanical unfolding–folding experiments. In the OT experiments, the folded substructure formed by the N- and the C-terminal helices of cytc is directly subject to the stretching force and acts as the force-bearing structural unit. Upon stretching, detachment of the N- and the C-helices and/or unraveling these two helices are the first steps of the mechanical unfolding of cytc. In the mechanical two-state pathway, it seems that this first step directly led to the complete unfolding of the whole protein without populating any “foldon” structure. In the mechanical folding pathway, we did not observe any formation of foldon structures, other than the ultimate folding of cytc, which is indicated by the formation of N- and C-terminal helices. These results suggested that if these foldons existed in the mechanical unfolding/folding pathway, their existence depended on the formation of the foldon formed by the N- and C-terminal helices. From a mechanical perspective, our results again highlighted the critical importance of the N- and C-terminal helices in the folding of cytc. Moreover, this insight also raises an intriguing question if the observed “misfolded” cytc (with a much weaker mechanical stability) corresponds to a conformation in which the N-, and C-terminal helices are fully formed while the internal helices (or foldons) are yet to form. Future protein engineering work will be needed to test this possibility.
Another novel insight from our OT results is the observation of the short-lived unfolding intermediate state, in which the N- and C-terminal helices are unraveled. In previous chemical (un)folding studies of cytc, the formation and packing of the N- and C-terminal helices is the first step in the folding process while their unfolding is the last step in the unfolding step.12,17,18 In other words, the folding of the N- and C-helices is the prerequisite for the folding of other structural elements. In contrast to this view, our results clearly showed that it is possible that other structural elements can exist in the absence of the N- and C-terminal helices, revealing new structural information of cytc in the absence of chemical denaturants.
There are intrachain interactions in apo-cytc
Apo-cytc has been long considered as a random coil.20,21 It is intriguing that our OT experiments revealed that there exist weak, nonlocal intrachain interactions in apo-cytc, which allow apo-cytc to fold into an ensemble of different conformations with low mechanical stabilities. Clearly, apo-cytc exhibits behaviors that deviate from those of random coils, which have not been detected in previous spectroscopic measurements.Intrinsically disordered proteins behave as random coils, and chemically denatured proteins are often assumed to be random coils too.61–63 Although a variety of techniques have been used to characterize random coils,62,64 it remains difficult to detect and characterize residual structures in unfolded polypeptide chains, due to the insensitivity of these techniques to the residual and/or transient structures in polypeptide chains. For example, the hydrodynamic radius, which can be measured by different techniques, such as fluorescence correlation spectroscopy and dynamic light scattering, is often used to characterize the random coil behaviors of unfolded proteins. However, the hydrodynamic radius is insensitive to the native contacts/transient structures in unfolded polypeptide chains.62,65 In fact, a random coil-like radius can be generated in a protein by randomizing only 8% of its native contacts.66 These prior studies clearly showed that observation of a random coil-like radius in an unfolded polypeptide chain does not necessarily rule out the possibility of transient structural formation. As a result, residual folded structures may not be detected in some spectroscopic measurements, leading to the mischaracterization of random coils.67,68
Nuclear magnetic resonance (NMR) spectroscopy is a sensitive technique that allows for the detection of residual structures in unfolded proteins.64,69 For example, NMR studies revealed the formation of hydrophobic clusters in the urea-denatured 434 repressor which causes intense medium-range interactions suggested by the NMR nuclear Overhauser effect, hydrophobic interactions between aromatic sidechains that keep a β-hairpin secondary structure in a 16-residue peptide from protein G, and interactions between charged sidechains stabilize helices in the S-peptide from ribonuclease A.70–72 Although these proteins exhibit random coil-like properties in several spectroscopic measurements, they are not true random coils as they contain residual structures and/or intrachain interactions as revealed by NMR.
In the case of apo-cytc, its conformation has been considered as a random coil since the early 1970s based on results from multiple spectroscopic methods, including CD spectroscopy, intrinsic viscosity, sedimentation coefficients, and the UV absorption, reactivity and ionization of certain amino acid residues.20,21 However, these methods do not characterize the fine details of the interactions between residues. Different from these prior studies, our single-molecule OT measurements have yielded the evidence that apo-cytc is not a true random coil. Instead, there exist intrachain interactions in apo-cytc, leading to the formation of mechanically labile but detectable structures. It is likely that due to hydrophobic interactions, apo-cytc folds into an ensemble of collapsed conformations with weak non-local interactions in the polypeptide chain. This finding is similar to the results of some recent SMFS studies on several IDPs, including α-synuclein and the neuronal RNA binding protein Orb2,73–75 which showed that these IDPs are not true random coils and weak, nonlocal interactions exist in these polypeptide chains. This common feature among these supposedly random coiled proteins revealed new features of IDPs and highlighted the unique suitability of the SMFS technique as an effective tool to evaluate the conformations of these proteins from a mechanical perspective at the single-molecule level, making SMFS and NMR the few available techniques that can detect residual/transient structures in unfolded polypeptide chains.
The role of heme in the folding of holo-cytc
As apo-cytc was believed to be a random coil, the interactions between the heme and the polypeptide chain were considered to introduce the intrachain interactions and induce the folding of holo-cytc. By revealing the deviation from random coiled behavior of apo-cytc, our results revealed some new insights into the understanding of the folding mechanism of holo-cytc. Instead of being a completely random coil, apo-cytc has a nonnegligible tendency to fold by itself to form an ensemble of mechanically labile structures. The incorporation of the heme greatly enhances the intrachain interactions and the folding tendency of the polypeptide chain and helps guide cytc to fold into its native structure. The role of the heme in the folding process of holo-cytc is distinguished from those of the metal cofactors in some other metalloproteins of which the folding mechanisms have been revealed previously, such as rubredoxin, in which the binding of the ferric ion only enhances the conformational stability but does not alter the apo-form conformation;16 or the C-terminal RTX domain of CyaA, whose apo-form is intrinsically disordered and the folding is entirely induced by and dependent on the binding of calcium ions;36 or calmodulin, whose apo-form folds, but the binding of calcium ions leads to a new folded holo-form structure.35 In other words, in holo-cytc, the metal binding transforms a molten globule structure into a folded structure, instead of bringing no effect to the conformation (as in rubredoxin), or transforming a random coil into a folded structure (as in RTX), or transforming a folded structure into another folded structure (as in calmodulin). The heme plays a critical role in the folding of holo-cytc by turning the irregular intrachain interactions in the polypeptide chain into specific interaction modes that drive the protein to fold into a well-defined folded structure. This is also consistent with the previous finding that, in vivo, the polypeptide chain of cytc folds after the ligation of the heme, unlike the case for hemoglobins and some other cytochromes, and most apo-form cytc are directly degraded if the heme is not attached.76 Our results reveal a new folding mechanism of metalloproteins and again highlight the complexity in the folding process of metalloproteins arising from the intertwining effects of polypeptide folding and metal coordination.Conclusions
Using single-molecule OTs, here we revealed the complete folding–unfolding mechanism of holo-cytc and demonstrated the mechanical response of apo-cytc. Holo-cytc mechanically unfolds in a two-state fashion or via an intermediate and folds in a two-state fashion. Apo-cytc can fold into an ensemble of collapsed conformations, and the incorporation of the heme cofactor greatly enhances the folding tendency and increases the folding fidelity to the native structure. Our results not only deepen and revise our understanding of the folding mechanism of an important type of metalloprotein, holo-cytc, but also identify the deviation of random coil behavior of apo-cytc which was missed or overlooked in previous spectroscopic measurements.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
H. L. conceived the project, J. L. and H. L. designed the experiments and wrote the manuscript, J. L. did the measurements and analyzed the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC). J. L. acknowledged the fellowship support from NSERC NanoMat CREATE Program. We thank Mr G. Chen for technical assistance.Notes and references
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823–830 CrossRef CAS PubMed.
- D. W. Barnum, Inorg. Chem., 1983, 22, 2297–2305 CrossRef CAS.
- T. Shimizu, A. Lengalova, V. Martínek and M. Martínková, Chem. Soc. Rev., 2019, 48, 5624–5657 RSC.
- F. R. Salemme, Annu. Rev. Biochem., 1977, 46, 299–329 CrossRef CAS PubMed.
- I. Bertini, G. Cavallaro and A. Rosato, Chem. Rev., 2006, 106, 90–115 CrossRef CAS PubMed.
- C. Garrido, L. Galluzzi, M. Brunet, P. E. Puig, C. Didelot and G. Kroemer, Cell Death Differ., 2006, 13, 1423–1433 CrossRef CAS PubMed.
- J. A. Knapp and C. N. Pace, Biochemistry, 1974, 13, 1289–1294 CrossRef CAS PubMed.
- B. S. Russell and K. L. Bren, J. Biol. Inorg Chem., 2002, 7, 909–916 CrossRef CAS PubMed.
- R. F. Latypov, K. Maki, H. Cheng, S. D. Luck and H. Roder, J. Mol. Biol., 2008, 383, 437–453 CrossRef CAS PubMed.
- A. K. Bhuyan and J. B. Udgaonkar, J. Mol. Biol., 2001, 312, 1135–1160 CrossRef CAS PubMed.
- A. K. Bhuyan and R. Kumar, Biochemistry, 2002, 41, 12821–12834 CrossRef CAS PubMed.
- S.-R. Yeh and D. L. Rousseau, Nat. Struct. Biol., 1998, 5, 222–228 CrossRef CAS PubMed.
- G. A. Elöve, A. K. Bhuyan and H. Roder, Biochemistry, 1994, 33, 6925–6935 CrossRef PubMed.
- W. Colón, L. P. Wakem, F. Sherman and H. Roder, Biochemistry, 1997, 36, 12535–12541 CrossRef PubMed.
- M. M. Pierce and B. T. Nall, J. Mol. Biol., 2000, 298, 955–969 CrossRef CAS PubMed.
- J. R. Winkler, Curr. Opin. Chem. Biol., 2004, 8, 169–174 CrossRef CAS PubMed.
- W. Hu, Z. Y. Kan, L. Mayne and S. W. Englander, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3809–3814 CrossRef CAS PubMed.
- H. Maity, M. Maity and S. W. Englander, J. Mol. Biol., 2004, 343, 223–233 CrossRef CAS PubMed.
- Y. Bai, T. R. Sosnick, L. Mayne and S. W. Englander, Science, 1995, 269, 192–197 CrossRef CAS PubMed.
- W. R. Fisher, H. Taniuchi and C. B. Anfinsen, J. Biol. Chem., 1973, 248, 3188–3195 CrossRef CAS PubMed.
- E. Stellwagen, R. Rysavy and G. Babul, J. Biol. Chem., 1972, 247, 8074–8077 CrossRef CAS PubMed.
- K. C. Neuman and A. Nagy, Nat. Methods, 2008, 5, 491–505 CrossRef CAS PubMed.
- Y. Javadi, J. M. Fernandez and R. Perez-Jimenez, Physiology, 2013, 28, 9–17 CrossRef CAS PubMed.
- T. Hoffmann and L. Dougan, Chem. Soc. Rev., 2012, 41, 4781–4796 RSC.
- C. Bustamante, L. Alexander, K. Maciuba and C. M. Kaiser, Annu. Rev. Biochem., 2020, 89, 443–470 CrossRef CAS PubMed.
- M. L. Hughes and L. Dougan, Rep. Prog. Phys., 2016, 79, 076601 CrossRef PubMed.
- H. Li and P. Zheng, Curr. Opin. Chem. Biol., 2018, 43, 58–67 CrossRef CAS PubMed.
- R. Petrosyan, A. Narayan and M. T. Woodside, J. Mol. Biol., 2021, 433, 167207 CrossRef CAS PubMed.
- Z. Yu, V. Gaerig, Y. Cui, H. Kang, V. Gokhale, Y. Zhao, L. H. Hurley and H. Mao, J. Am. Chem. Soc., 2012, 134, 5157–5164 CrossRef CAS PubMed.
- P. Zheng and H. Li, J. Am. Chem. Soc., 2011, 133, 6791–6798 CrossRef CAS PubMed.
- H. Lei, Y. Guo, X. Hu, C. Hu, X. Hu and H. Li, J. Am. Chem. Soc., 2017, 139, 1538–1544 CrossRef CAS PubMed.
- J. Li and H. Li, J. Phys. Chem. B, 2018, 122, 9340–9349 CrossRef CAS PubMed.
- J. Li and H. Li, Nanoscale, 2020, 12, 22564–22573 RSC.
- A. E. M. Beedle, A. Lezamiz, G. Stirnemann and S. Garcia-Manyes, Nat. Commun., 2015, 6, 7894 CrossRef CAS PubMed.
- J. Stigler, F. Ziegler, A. Gieseke, J. C. Gebhardt and M. Rief, Science, 2011, 334, 512–516 CrossRef CAS PubMed.
- H. Wang, X. Gao and H. Li, J. Am. Chem. Soc., 2019, 141, 20498–20506 CrossRef CAS PubMed.
- J. Perales-Calvo, A. Lezamiz and S. Garcia-Manyes, J. Phys. Chem. Lett., 2015, 6, 3335–3340 CrossRef CAS PubMed.
- L. F. Milles, E. M. Unterauer, T. Nicolaus and H. E. Gaub, Nat. Commun., 2018, 9, 4764 CrossRef PubMed.
- G. Yuan, H. Liu, Q. Ma, X. Li, J. Nie, J. Zuo and P. Zheng, J. Phys. Chem. Lett., 2019, 10, 5428–5433 CrossRef CAS PubMed.
- C. N. Patel, M. C. Lind and G. J. Pielak, Protein Expression Purif., 2001, 22, 220–224 CrossRef CAS PubMed.
- X. Zhang, K. Halvorsen, C. Z. Zhang, W. P. Wong and T. A. Springer, Science, 2009, 324, 1330–1334 CrossRef CAS PubMed.
- L. Oberbarnscheidt, R. Janissen and F. Oesterhelt, Biophys. J., 2009, 97, L19–L21 CrossRef CAS PubMed.
- E. Evans, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 105–128 CrossRef CAS PubMed.
- G. I. Bell, Science, 1978, 200, 618–627 CrossRef CAS PubMed.
- O. K. Dudko, G. Hummer and A. Szabo, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15755–15760 CrossRef CAS PubMed.
- H. Lei, C. He, C. Hu, J. Li, X. Hu, X. Hu and H. Li, Angew. Chem., Int. Ed. Engl., 2017, 56, 6117–6121 CrossRef CAS PubMed.
- C. He, C. Hu, X. Hu, X. Hu, A. Xiao, T. T. Perkins and H. Li, Angew. Chem., Int. Ed. Engl., 2015, 54, 9921–9925 CrossRef CAS PubMed.
- L. A. Abriata, A. Cassina, V. Tórtora, M. Marín, J. M. Souza, L. Castro, A. J. Vila and R. Radi, J. Biol. Chem., 2009, 284, 17–26 CrossRef CAS PubMed.
- J. F. Marko and E. D. Siggia, Macromolecules, 1995, 28, 8759–8770 CrossRef CAS.
- P. J. Elms, J. D. Chodera, C. Bustamante and S. Marqusee, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3796–3801 CrossRef CAS PubMed.
- M. Arai and K. Kuwajima, Adv. Protein Chem., 2000, 53, 209–282 CrossRef CAS PubMed.
- E. Judy and N. Kishore, Biophys. Rev., 2019, 11, 365–375 CrossRef CAS PubMed.
- L. Zhang, C. Wang, S. Cui, Z. Wang and X. Zhang, Nano Lett., 2003, 3, 1119–1124 CrossRef CAS.
- H. Li, W. Zhang, X. Zhang, J. Shen, B. Liu, C. Gao and G. Zou, Macromol. Rapid Commun., 1998, 19, 609–611 CrossRef CAS.
- H. Li, W. Zhang, W. Xu and X. Zhang, Macromolecules, 2000, 33, 465–469 CrossRef CAS.
- M. P. Ramírez, M. Rivera, D. Quiroga-Roger, A. Bustamante, M. Vega, M. Baez, E. M. Puchner and C. A. M. Wilson, Protein Sci., 2017, 26, 1404–1412 CrossRef PubMed.
- M. Sandal, F. Valle, I. Tessari, S. Mammi, E. Bergantino, F. Musiani, M. Brucale, L. Bubacco and B. Samori, PLoS Biol., 2008, 6, e6 CrossRef PubMed.
- R. Hervas, L. Li, A. Majumdar, C. Fernandez-Ramirez Mdel, J. R. Unruh, B. D. Slaughter, A. Galera-Prat, E. Santana, M. Suzuki, Y. Nagai, M. Bruix, S. Casas-Tinto, M. Menendez, D. V. Laurents, K. Si and M. Carrion-Vazquez, PLoS Biol., 2016, 14, e1002361 CrossRef PubMed.
- R. Hervas, J. Oroz, A. Galera-Prat, O. Goni, A. Valbuena, A. M. Vera, A. Gomez-Sicilia, F. Losada-Urzaiz, V. N. Uversky, M. Menendez, D. V. Laurents, M. Bruix and M. Carrion-Vazquez, PLoS Biol., 2012, 10, e1001335 CrossRef CAS PubMed.
- H. Li, A. F. Oberhauser, S. D. Redick, M. Carrion-Vazquez, H. P. Erickson and J. M. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10682–10686 CrossRef CAS PubMed.
- C. J. Oldfield and A. K. Dunker, Annu. Rev. Biochem., 2014, 83, 553–584 CrossRef CAS PubMed.
- J. E. Kohn, I. S. Millett, J. Jacob, B. Zagrovic, T. M. Dillon, N. Cingel, R. S. Dothager, S. Seifert, P. Thiyagarajan, T. R. Sosnick, M. Z. Hasan, V. S. Pande, I. Ruczinski, S. Doniach and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12491–12496 CrossRef CAS PubMed.
- L. J. Smith, K. M. Fiebig, H. Schwalbe and C. M. Dobson, Folding Des., 1996, 1, R95–R106 CrossRef CAS PubMed.
- M. Brucale, B. Schuler and B. Samori, Chem. Rev., 2014, 114, 3281–3317 CrossRef CAS PubMed.
- K. Chattopadhyay, S. Saffarian, E. L. Elson and C. Frieden, Biophys. J., 2005, 88, 1413–1422 CrossRef CAS PubMed.
- N. C. Fitzkee and G. D. Rose, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12497–12502 CrossRef CAS PubMed.
- U. B. Choi, J. J. McCann, K. R. Weninger and M. E. Bowen, Structure, 2011, 19, 566–576 CrossRef CAS PubMed.
- K. W. Plaxco and M. Gross, Nat. Struct. Biol., 2001, 8, 659–660 CrossRef CAS PubMed.
- R. Mohana-Borges, N. K. Goto, G. J. Kroon, H. J. Dyson and P. E. Wright, J. Mol. Biol., 2004, 340, 1131–1142 CrossRef CAS PubMed.
- D. Neri, M. Billeter, G. Wider and K. Wüthrich, Science, 1992, 257, 1559–1563 CrossRef CAS PubMed.
- F. J. Blanco, G. Rivas and L. Serrano, Nat. Struct. Biol., 1994, 1, 584–590 CrossRef CAS PubMed.
- R. L. Baldwin, Biophys. Chem., 1995, 55, 127–135 CrossRef CAS PubMed.
- M. Sandal, F. Valle, I. Tessari, S. Mammi, E. Bergantino, F. Musiani, M. Brucale, L. Bubacco and B. Samorì, PLoS Biol., 2008, 6, e6 CrossRef PubMed.
- R. Hervas, J. Oroz, A. Galera-Prat, O. Goni, A. Valbuena, A. M. Vera, A. Gomez-Sicilia, F. Losada-Urzáiz, V. N. Uversky and M. Menéndez, PLoS Biol., 2012, 10, e1001335 CrossRef CAS PubMed.
- K. Neupane, A. Solanki, I. Sosova, M. Belov and M. T. Woodside, PLoS One, 2014, 9, e86495 CrossRef PubMed.
- R. G. Kranz, C. Richard-Fogal, J. S. Taylor and E. R. Frawley, Microbiol. Mol. Biol. Rev., 2009, 73, 510–528 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01126c |
| This journal is © The Royal Society of Chemistry 2022 |