
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceValence-controlled protein conjugation on nanoparticles via re-arrangeable multivalent interactions of tandem repeat protein chains†
Hyeongjoo
Choi
and
Yongwon
Jung
 *
*
Department of Chemistry, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon 34143, Republic of Korea. E-mail: ywjung@kaist.ac.kr; Fax: +82-42-350-2810; Tel: +82-42-350-2817
First published on 8th June 2022
Abstract
Precise control of the number of conjugated proteins on a nanoparticle surface has long been a highly challenging task. Here, we developed a one-pot, purification-free strategy for valency-controlled conjugation of tandem repeat protein chains on gold nanoparticles. Protein chains were designed to contain multiple, regularly spaced binding modules, which can multivalently interact with coating molecules on nanoparticle surfaces. We discovered that a slow increase of this interaction strength facilitates full participation of repeated binding modules on a protein chain for surface binding (as well as dynamic rearrangement) on a single nanoparticle, which resulted in stable protein chain wrapping around nanoparticles. By varying the protein chain length, a defined number of protein chains were conjugated on gold nanoparticles with difference sizes. Various high-order nanoparticle structures were accurately assembled with these valence-controlled protein–particle conjugates. The present strategy offers a highly dynamic but controlled protein coating approach on solid surfaces of diverse nanostructures. In addition, this work also provides a valuable clue to understand dynamic binding processes of multivalent repeat proteins.
Introduction
Researchers have conjugated biomolecules to nanoparticles with the goals of utilizing unique nanoparticle properties in biology and providing additional functions to these nanostructures.1 While diverse conjugation strategies have been developed against various nanoparticles, one of the major remaining challenges is to precisely control the number of conjugated biomolecules (valence) on a particle surface. The necessity and the importance of precise valence control in various nanoparticles (including monovalent conjugation) have been raised in many fields such as unperturbed imaging,2–4 high-order nanostructure assembly,5–7 and accurate targeting/sensing.8–10 For example, monovalent nanoparticles are recognized as powerful and necessary probes to track cell surface receptors without uncontrolled receptor cross-linking that might result from multivalent probes.3,11 In addition, high-order assemblies of discrete monovalent and multivalent nanoparticles offer various hierarchical nanoparticle architectures, including those with atom-like geometries.12Synthetic DNA has been most widely and successfully used to prepare valence-controlled nanoparticles.13 The early attempts entailed simply mixing particles and thiolated DNA strands in a stoichiometric manner, yielding mixed DNA valences, and isolating particles with desired valences by a gel electrophoretic or chromatographic technique.14–16 Valence-controlled nanoparticles with more diverse and even asymmetric DNA patterns could also be fabricated by using pre-assembled DNA scaffolds such as DNA tile or origami structures.17–20 Alternatively, DNA valence has been controlled by synthesizing nanoparticles directly on various DNA templates, which contain functional moieties to guide the in situ growth of DNA-templated nanoparticles.7,21–23 The ability of a single poly-adenine DNA strand to specifically bind and wrap around a single gold nanoparticle was successfully used to produce monovalent DNA–gold nanoparticles.12,24,25 Particle wrapping by long poly-adenine prevented further binding of additional DNA strands, and a single-step fabrication strategy offered a high-production yield without the need for purification from valence mixtures. This wrapping method was also similarly used to fabricate monovalent DNA–quantum dots, where a phosphorothioates modified DNA strand wrapped a quantum dot.3
Unlike DNA, which shows rather simple and stable structures as well as predictable properties such as uniform charge distributions, proteins have highly diverse structures and numerous properties that are largely unpredictable, and they also could become unstable on some particle surfaces. Therefore, conjugation methods are highly varied for different proteins and particles,1,26,27 and further controlling protein–particle valence has been extremely difficult and the processes to achieve this are not well developed. The valence control of protein–nanoparticle conjugation has mostly relied on simple stoichiometric conjugation and valence-dependent purification.2,11,28,29 Otherwise, proteins are coupled with pre-made monovalent particles with modified DNA or reactive chemical moieties.30,31 Considering the ample diversities of structures and functions of proteins, facile and direct control of protein valences on nanoparticles will allow greatly widened and more precise use of nanoparticles in bio- and nano-sciences.
Here, we designed modular tandem repeat protein chains that contain multiple hexa-histidine tags (6His) and thereby multivalently bind to Ni-NTA(nitrilotriacetic acid)-coated gold nanoparticles. We found that the slow increase of binding affinities between 6His and Ni-NTA by slowly lowering competing imidazole concentrations resulted in stable wrapping of only a single protein chain on a single AuNP (Scheme 1), producing monovalent protein–AuNP conjugates without any purification steps. In addition, nanoparticles could be conjugated with a defined number of protein chains (one to four) depending on the repeat protein chain length (and thus the number of 6His on the chain) and particle sizes. Protein valences were validated by fusing binding proteins such as a nanobody to protein chains and conjugating to AuNPs with defined valences (Scheme 1). Expected high-order AuNP assemblies were valence-dependently formed.
 | ||
| Scheme 1 Schematic representation of valence-controlled protein chain wrapping around gold nanoparticles. Under slowly decreasing imidazole concentration, a protein chain wraps around a gold nanoparticle via dynamic and re-arrangeable binding of repeated 6His tags on a particle surface. A functional protein (e.g. nanobody), repeated 6His tags (red line) on protein chains and Ni-NTA coating (translucent gray cover) on gold nanoparticles are indicated. | ||
Results and discussion
Repeat protein chain binding to gold nanoparticles via multivalent 6His/Ni-NTA interactions
Interaction-driven wrapping of long protein chains on nanoparticles could be a powerful strategy for potentially one-step, purification-free particle–protein conjugation with defined valences (Scheme 1), as demonstrated with synthetic DNAs.3,12 A protein chain must have repeated and regularly-spaced interaction moieties, which can cooperatively bind to a particle surface without overlaps. Ideally, this interaction would be reversible and affinity controllable to flexibly dictate mono- and multi-valent interactions on surfaces. We used the specific interaction between Ni-NTA and the 6His peptide tag. With their small sizes and easy ways to be introduced to various surfaces and proteins, proteins fused with 6His have been stably immobilized on Ni-NTA-covered surfaces.32,33 Importantly, the 6His–Ni-NTA binding affinity can be easily and precisely controlled by simply varying the concentration of competing imidazole. We prepared Ni-NTA covered AuNPs (Ni-NTA–AuNPs) through self-assembled monolayer (SAM) formation on gold surfaces and subsequent Ni ion charge. Only proteins with 6His can specifically bind to Ni-NTA–AuNPs, showing a clear AuNP band shift in an agarose gel (Fig. 1a and S1†). | ||
| Fig. 1 Construction of repeat modular protein chains with multiple 6His for multivalent binding to Ni-NTA–AuNPs. (a) 6His-fused protein (6His–GFP) binding to 5 nm Ni-NTA–AuNPs. 5 nm Ni-NTA–AuNPs were mixed with 6His–GFP or 6His-cleaved GFP, and the resulting particles were analyzed on an agarose gel. Ctrl: 5 nm Ni-NTA–AuNP. (b) Construction of 6His–SC inserted ST-repeat protein chains. ST-repeat backbone scaffolds (STn) were reacted with excess 6His–SC, and unbound 6His–SC was removed by 180 mM imidazole washing. A SDS-PAGE data for 6His–SC–ST10–ALFA construction is shown in the right. (c) Agarose gel shift analysis of 5 nm Ni-NTA–AuNP binding to 6His–SC monomer (left) or to 6His 10mer (right) under different imidazole concentration from 0 to 300 mM. Ctrl: 5 nm Ni-NTA–AuNP. | ||
To prepare regularly spaced 6His tags on a protein, we designed flexible protein chains with tandemly repeated modular (cassette like) units, where 6His-tagged proteins can be inserted, by using the specific isopeptide bond formation between the SpyTag (ST) peptide and the small SpyCatcher (SC) protein34 (Fig. 1b). ST peptides were tandemly repeated on protein backbone scaffolds, and 6His-fused SC proteins (6His–SC) were covalently inserted. Protein chain lengths and resulting 6His numbers were regulated by varying the number of ST repeats. Compared to direct 6His repeating on backbone protein chains, our modular method will allow facile variation of (inserted) repeating protein units, offering different properties such as sizes and charge distributions. Moreover, 6His tags on modular ST/6His–SC chains will be better exposed for Ni-NTA interactions than internal 6His tags on backbones and thus allow full participation of 6His tags for efficient chain wrapping around particles.
ST peptides (13 residues) were tandemly repeated with a glycine (G) and aspartate (D) rich peptide linker (DGDGDGDGDGLVPRGS, 16 residues). We used glycines to provide flexibility for efficient particle wrapping35 and negative aspartates to provide repulsion for minimal overlaps during wrapping, with the aim of mimicking a DNA strand. In addition, this linker with negative DG repeats will preferentially be in extended forms, which are beneficial for 6His–SC insertion and also for 6His spacing. Monovalent functional protein units can be added to the N- or C-terminus of ST repeat scaffolds (STn; n = the number of ST) (Fig. 1b). For saturated 6His–SC insertion on STn scaffolds, excess 6His–SC proteins were reacted with scaffolds, and the mixture was subjected to Ni-NTA resin purification. Unreacted 6His–SC proteins with only one 6His were removed by washing with fairly high concentration imidazole (180 mM), and 6His–SC–conjugated STn chains with many 6His were eluted by 500 mM imidazole with a high purity (Fig. 1b right). 6His–SC–STn chains with various lengths (n = 7, 10, and 15) and with various (N- or C-terminal) functional proteins were successfully prepared (Fig. S2†). For example, a strong binding tag pair, ALFA-tag and interacting nanobody (NbALFA),36 were fused to the C- and N-termini of STn chains, respectively, to provide binding abilities to protein chain-conjugated AuNPs.
Binding of 6His–SC–STn chains with multiple 6His to Ni-NTA–AuNPs was examined in the presence of varying amount of imidazole. For monomeric 6His–SC, only 50 mM imidazole significantly inhibited binding of this 1mer 6His protein to Ni-NTA–AuNPs (5 nm) (Fig. 1c). On the other hand, the band shift by 6His–SC–ST10–ALFA (6His 10mer) (C-terminal ALFA-tag-fused chain) binding to AuNPs was maintained with up to 100 mM imidazole. Even with 300 mM imidazole, most Ni-NTA–AuNPs were still conjugated with 6His 10mer, although multiple binding conjugates were formed. We also examined how bound 6His proteins on Ni-NTA–AuNPs were detached by imidazole. Again, 50 mM imidazole can easily dissociate monomeric 6His–SC from AuNPs, while 6His–SC–ST10 stably remained on AuNPs at the same condition (Fig. S3†). Stable particle binding of 6His 10mer (but not 6His 1mer) in the presence of imidazole was also similarly observed with larger 15 nm AuNPs (Fig. S3†). These binding patterns clearly suggest that binding strengths of monomeric or multimeric 6His proteins to Ni-NTA–AuNP surfaces can be widely varied by controlling imidazole concentrations.
Valence-controlled wrapping of protein chains on gold nanoparticles
To examine protein chain wrapping on gold nanoparticles, 5 nm Ni-NTA–AuNPs were mixed with an equal amount of long 6His 15mer (6His–SC–ST15–NbALFA) first in the absence of imidazole, and analyzed by transmission electron microscopy (TEM). Without competing imidazole, in which even a single 6His tag can stably bind to Ni-NTA–AuNPs (Fig. 1), a significant portion of nanoparticles were clustered, likely due to binding of more than one particle to a single protein chain (Fig. 2a). When proteins and particles were mixed with 50 mM imidazole, however, particle clustering was clearly reduced. Nevertheless, even with 50 mM imidazole a noticeable degree of particle clusters were observed in TEM images. It is possible that 15 6His tags on 6His–SC–ST15–NbALFA can still bind to more than one particle, despite 50 mM competing imidazole, via multivalent (thus strong) interactions of 15 6His tags with multiple Ni-NTA-covered particles. | ||
Fig. 2 Valence-controlled wrapping of protein chains on gold nanoparticles. (a) TEM images of 5 nm Ni-NTA–AuNP binding to 6His 15mer without (left) or with (right) 50 mM imidazole. Potential protein chain wrapping routes are schematically described at the top. Scale bars: 100 nm. (b) TEM images of 5 nm Ni-NTA–AuNP binding to 6His 15mer with slowly decreasing imidazole concentration (from 1 M to 25 mM) at protein![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :particle ratios 1:1 (left) or at 0.5:1 (right). Scale bars: 100 nm. (c) Agarose gel shift analysis of 5 nm Ni-NTA–AuNP binding to 6His 15mer (6His–SC–ST15–NbALFA) at varying protein:particle ratios. (d) Representative TEM images of 5 nm Ni-NTA–AuNPs without protein binding (left), with 6His 15mer (6His–SC–ST15–ALFA) binding (middle), and large 6His 15mer (6His–SC–mCherry–ST15–ALFA) binding (right). Scale bars: 10 nm. (e) Agarose gel shift analysis of 5 nm Ni-NTA–AuNP binding to 6His 15mer (6His–SC–ST15–ALFA; left), 6His 10mer (middle), or 6His 7mer (right) at varying protein:particle ratios. (f) Representative TEM images of 5 nm AuNP assemblies between monovalent NbALFA–AuNP and monovalent ALFA–AuNP (left), divalent ALFA–AuNP (middle), or trivalent-ALFA–AuNP (right). Scale bars: 20 nm. AuNP (yellow circle), Ni-NTA coating (light gray), protein chains (dark gray), and fused NbALFA (red) or ALFA (blue) are schematically indicated. :particle ratios 1:1 (left) or at 0.5:1 (right). Scale bars: 100 nm. (c) Agarose gel shift analysis of 5 nm Ni-NTA–AuNP binding to 6His 15mer (6His–SC–ST15–NbALFA) at varying protein:particle ratios. (d) Representative TEM images of 5 nm Ni-NTA–AuNPs without protein binding (left), with 6His 15mer (6His–SC–ST15–ALFA) binding (middle), and large 6His 15mer (6His–SC–mCherry–ST15–ALFA) binding (right). Scale bars: 10 nm. (e) Agarose gel shift analysis of 5 nm Ni-NTA–AuNP binding to 6His 15mer (6His–SC–ST15–ALFA; left), 6His 10mer (middle), or 6His 7mer (right) at varying protein:particle ratios. (f) Representative TEM images of 5 nm AuNP assemblies between monovalent NbALFA–AuNP and monovalent ALFA–AuNP (left), divalent ALFA–AuNP (middle), or trivalent-ALFA–AuNP (right). Scale bars: 20 nm. AuNP (yellow circle), Ni-NTA coating (light gray), protein chains (dark gray), and fused NbALFA (red) or ALFA (blue) are schematically indicated. | ||
To ensure strict single particle-to-a single protein chain conjugation, we mixed proteins with particles at high imidazole (1 M) and slowly reduced imidazole concentration to 25 mM by dialysis. Under this condition, protein chain–particle conjugates will first form only through the highest-valent 6His–Ni-NTA interactions, which likely require wrapping of a full protein chain on a single particle (Scheme 1). In addition, we also expect constant association and dissociation of individual 6His units during this wrapping under a high but slowly decreasing imidazole concentration. This constant re-arrangement of 6His units might also allow a homogeneous and well-dispersed distribution of 6His–SC repeats on a particle surface for effective wrapping and thus full covering of a gold nanoparticle with a protein chain. Indeed, 6His–SC–ST15–NbALFA conjugation to 5 nm Ni-NTA–AuNPs with dialyzed imidazole resulted in well dispersed protein–particle conjugates in a TEM image without noticeable particle clusters (Fig. 2b and S4†). When protein chains were mixed with particles in a 0.5:1 protein:particle ratio, however, ∼35% of particles were observed in dimeric or higher multimeric forms (Fig. 2b and S4†), likely because excess particles can participate in ongoing protein–particle wrapping. The distance between observed AuNP dimers was 3–4 nm, which roughly matches twice the thickness of a NTA layer on AuNPs. The AuNP gel band was homogeneously and similarly shifted by protein chain binding at both protein:particle ratios (1:1 and 0.5:1) (Fig. 2c). Protein–AuNP dimers might migrate in a manner similar to protein–AuNP monomers. Interestingly, even when excess protein chains were mixed (protein:particle ratios 2:1 and 3:1), protein chain–AuNP bands were nearly identical to that of the 1:1 protein–particle mixture, suggesting mostly a single 6His–SC–ST15–NbALFA chain binding (wrapping) to a single 5 nm Ni-NTA–AuNP.
Based on the estimated surface area of 5 nm Ni-NTA–AuNP and the covering area of a 6His SC/ST-linker repeating unit, we can approximate that up to 18 6His units can bind to a single AuNP surface, assuming no overlapping between these units (Fig. S5†). This calculation might explain the observed binding saturation by a single 6His–SC–ST15–NbALFA chain (15 units) on a 5 nm Ni-NTA–AuNP (Fig. 2c). Through dynamic re-arrangement of 6His units on a Ni-NTA surface, 15 6His–SC/ST repeat units can be uniformly scattered on a particle surface, which might prevent further binding of additional protein chains to the particle. Protein chain wrapping on AuNPs induced a strong blue shift of particle absorbance spectra (∼9 nm maximum absorbance wavelength shift) (Fig. S6a†), indicating heavy protein coating on particles. More importantly, fairly homogeneous and well-dispersed protein coating on AuNPs (likely by protein chain wrapping) was clearly visualized when comparing negative-stained TEM images of bare 5 nm Ni-NTA–AuNPs and protein chain wrapped particles (Fig. 2d and S6b†). The protein coating thickness by 6His–SC–ST15–NbALFA wrapping was ∼5 nm, which is well-matched with the SC–ST complex size (Fig. S5†). We further examined well-dispersed protein chain wrapping by using a larger protein repeat unit. 6His–SC was additionally fused with fluorescent protein mCherry (SC ∼10 kDa, mCherry ∼30 kDa), and the resulting 6His–SC–mCherry proteins were inserted onto the ST15 backbone scaffold to generate 6His–SC–mCherry–ST15–NbALFA. Again, TEM images clearly show well-dispersed protein chain wrapping on Ni-NTA–AuNPs with over 10 nm coating thickness by this 6His 15mer with large repeating units (Fig. 2d and S6b†). Fluorescent proteins on repeating units were also fluorescently active on AuNP surfaces (Fig. S6c†), suggesting that various functional proteins can be inserted on repeating units for particle coating. Size change by protein chain wrapping was also examined by dynamic light scattering (DLS) analysis. The hydrodynamic particle sizes of free 5 nm AuNP, Ni-NTA–AuNP, and protein chain wrapped AuNP are 6.5 nm, 10.4 nm, and 13.2 nm, respectively with narrow size distributions (Fig. S7†). In addition, when AuNPs were conjugated to protein chains without imidazole, the measured particle size increased to 42 nm, likely by particle clustering (Fig. 2a).
We also envisioned that more than one protein chain could be discretely conjugated to single AuNP if protein chain lengths (ST repeat numbers) were properly shortened. 5 nm Ni-NTA–AuNPs were mixed with three length varied, ALFA-tag-fused protein chains (6His–SC–STn–ALFA; n = 15, 10, 7) at increased protein/particle ratios (Fig. 2e). The binding gel patterns of ALFA-tag-fused 6His 15mer were similar to those of NbALFA-fused 6His 15mer, saturated after a 1:1 ratio (Fig. 2c and e). On the other hand, particle bands were clearly further shifted by increased amounts of short protein chains (6His–SC–ST10–ALFA and 6His–SC–ST7–ALFA). Band shift (saturation) patterns suggest that a 5 nm Ni-NTA covered particle can be stably and homogeneously wrapped by two 6His–SC–ST10–ALFA strands or by three 6His–SC–ST7–ALFA strands under the slowly lowered imidazole condition. Band shift patterns by this chain length-dependent (thus valence controlled) particle conjugation were consistently observed with other protein chains with various fused functional proteins (Fig. S8†).
To validate the protein chain valence on AuNPs, we used a specific and strong interaction between ALFA-tag and its binding nanobody NbALFA.36 Potentially monovalent, divalent, and trivalent ALFA-tag-conjugated AuNPs were prepared simply by mixing 5 nm Ni-NTA–AuNPs with 6His–SC–ST15–ALFA at a 1:1 protein:particle ratio, 6His–SC–ST10–ALFA at a 2:1 protein:particle ratio, and 6His–SC–ST7–ALFA at a 3:1 protein:particle ratio, respectively (Fig. 2d). The resulting ALFA-tag-conjugated AuNPs were then reacted with potentially monovalent NbALFA-conjugated AuNPs, which were prepared by mixing 5 nm Ni-NTA–AuNPs with 6His–SC–ST15–NbALFA at a 1:1 protein:particle ratio (Fig. S4†). When monovalent ALFA-tag–AuNPs were mixed with monovalent NbALFA–AuNPs at an equal ratio, over 70% of ALFA-tag–AuNPs formed AuNP dimers with NbALFA–AuNPs (Fig. 2f and S9†), supporting mono-valences of these protein chain–AuNP conjugates. Incomplete dimer formation likely results from a low particle concentration (∼50 nM), uneven particle concentrations due to difficulties in accurately determining particle concentration, and a strong but reversible ALFA-tag–NbALFA interaction,36 which could also be weakened by conjugated heavy AuNPs.37 When divalent ALFA-tag–AuNPs were mixed with monovalent NbALFA–AuNPs at a 1:2 ratio, over 53% divalent ALFA-tag–AuNPs formed clear AuNP trimers (Fig. 2f and S9†). This trimer formation yield (requiring two ALFA-tag–NbALFA bindings) is well explained with 70% of the dimer formation yield (requiring one ALFA-tag–NbALFA binding). AuNP tetramers were also distinctly formed by interactions between trivalent ALFA-tag–AuNPs and monovalent NbALFA–AuNPs (mixing ratio = 1:3) (Fig. 2f and S9†).
These gel band shift and TEM analysis data strongly support the valence-controlled formation of protein chain–particle conjugates by stimulating effective protein chain wrapping and varying protein chain lengths. The conjugation is a simple and purification-free process, which is conducted simply by protein–particle mixing, imidazole dialysis, and centrifugation to remove unbound proteins. Moreover, the present method does not require precise stoichiometric conjugation ratios between proteins and particles, which are often difficult to obtain. In fact, we used approximately 1.2–1.5 fold excess protein chains against AuNPs for a single protein chain-to-a single particle wrapping. This flexibility in binding ratios greatly enhances reproducibility of our particle–protein conjugation strategy. In addition, protein chain wrapping on AuNP was highly stable even against metal chelator EDTA (1 mM) (Fig. S10†).
We also examined whether protein chains can wrap directly on other nanoparticles without surface Ni-NTA since 6His can bind with metal ions presenting on the nanoparticle surface (e.g. quantum dots38). Commercially available, mercaptoundecanoic acid (MUA) covered CdZe–ZnS quantum dots (Qdots) (∼5 nm) were further stabilized with additional MUA to prevent particle aggregation during chain conjugation in high imidazole concentrations (Fig. S11a†), and conjugated to NbALFA-fused 6His 15mer. Although purchased Qdots were not individually dispersed (clustered from monomer to trimer), Qdots were not further clustered by protein chain conjugation (Fig. S11b and c†). Moreover, when NbALFA–Qdots were mixed with monovalent 5 nm ALFA-tag–AuNPs, Qdots mostly bound with one monomeric AuNP (Fig. S12†). These data suggest the feasibility of protein chain wrapping directly on a Qdot surface, although these Qdots must be properly protected and individually dispersed.
Protein chain conjugation to particles with different sizes
We next examined larger particles for observed particle wrapping by a discrete number of protein chains. Ni-NTA-covered 10 nm AuNPs and 15 nm AuNPs were prepared and conjugated with 6His 15 mer (6His–SC–ST15–NbALFA). Again, protein chain–particle conjugations without dialyzing imidazole resulted in significant clustering of 10 nm Ni-NTA–AuNPs, while no AuNP clusters were observed with slowly lowering imidazole (Fig. S13†). 10 nm AuNP gel band shifts by 6His 15 mer conjugations with varying protein:particle ratios indicated binding saturation by about two 15 mer protein chains to a 10 nm particle (Fig. 3a). On the other hand, as many as 4 6His–SC–ST15–NbALFA could bind to single 15 nm Ni-NTA–AuNP. Observed maximal conjugation valences are also well-explained with estimated surface areas of 10 nm and 15 nm Ni-NTA–AuNPs, which are approximately two- and four-times larger than that of 5 nm Ni-NTA–AuNPs (Fig. S5†).
 | ||
| Fig. 3 Valance-controlled protein chain conjugation to particles with different sizes. (a) Agarose gel shift analysis of 10 nm Ni-NTA–AuNP (left) or 15 nm Ni-NTA–AuNP (right) binding to 6His 15mer (6His–SC–ST15–ALFA) at varying protein:particle ratios. Band shifts by additional protein chain binding are indicated with arrows. (b) Representative TEM images of AuNP assemblies between monovalent 5 nm NbALFA–AuNP and divalent 10 nm ALFA–AuNP (top) or tetravalent 15 nm ALFA–AuNP (middle). Assemblies between divalent 10 nm NbALFA–AuNP and divalent 10 nm ALFA–AuNP are also shown in the bottom. Scale bars: 20 nm. | ||
Again, the protein valences of these NbALFA–chain-conjugated particles were validated by reacting with ALFA-tag-conjugated particles. Divalent 10 nm NbALFA–AuNPs were prepared by mixing 10 nm Ni-NTA–AuNPs with 6His–SC–ST15–NbALFA at a 1:2 particle:protein ratio and reacted with monovalent 5 nm ALFA-tag–AuNPs at a 1:2 10 nm:5 nm particle ratio. TEM images showed that almost 65% of 10 nm AuNPs were bound with two 5 nm AuNPs (Fig. 3b and S14†). In addition, when tetravalent 15 nm NbALFA–AuNPs were mixed with monovalent 5 nm ALFA-tag–AuNPs (mixing ratio = 1:4), nearly 20% of 15 nm AuNPs were clearly assembled with four 5 nm AuNPs (Fig. 3b and S14†). As statistically expected, a larger portion of 15 nm AuNPs were assembled with two (28%) or three (40%) 5 nm AuNPs. These assembly yields were again well-supported by the observed yield (∼70%) of a single ALFA-tag/NbALFA interaction on particle surfaces. Additionally, divalency of protein chains on a 10 nm particle (divalent 10 nm NbALFA–AuNP) was further examined by reacting with divalent 10 nm ALFA-tag–AuNPs, which could lead to successive oligomeric particle assemblies. Many long and mostly linear high-order particle strings were observed in TEM images (Fig. 3b and S15†). This successive linear assembly indicates that both mixed particles are divalent (by ALFA-tag or NbALFA) and two binding moieties (ALFA-tag or NbALFA) are well separated on a particle surface. In addition, almost no branched particle assembly further supports divalency of these protein chain-wrapped particles.
Protein targeting with valence-controlled nanoparticles
We next examined protein targeting abilities of valence-controlled (particularly monovalent) nanoparticles, which were fused with various binding moieties. Specific protein labeling with nanoparticles with well-defined valences and geometries is highly challenging but desirable for accurate protein functionalization with unique properties of nanoparticles. For example, unbiased single protein tracking has required protein labeling with monovalent particle probes.4,11,39 We first prepared monovalent biotin-fused AuNPs by mixing 5 nm Ni-NTA–AuNPs and 6His–SC–ST15–AP, where the AP peptide is site-specifically biotinylated (Fig. S6 and S16†). Protein labeling with monovalent AuNPs was investigated with multimeric target proteins, which will induce a multimeric AuNP assembly. Monovalent 5 nm biotin–AuNPs were treated to natural dimeric avidin protein, rhizavidin,40 which contains two biotin binding domains. Dimeric AuNP assemblies were clearly observed (Fig. 4a and S16†), supporting the mono-valency as well as a protein labeling ability of biotin–AuNPs. Nearly 64% of monovalent biotin–AuNPs were assembled into dimers, despite the small size of RA, which can cause steric hindrance between large particles during the dimeric assembly. Rhizavidin was also mixed with divalent biotin–AuNPs, which were prepared by conjugation between 10 nm Ni-NTA–AuNP and two 6His–SC–ST15–AP chains. Dimeric rhizavidin and divalent biotin–AuNPs were linearly assembled (Fig. 4a and S16†), as demonstrated with assemblies between divalent ALFA-tag–AuNPs and divalent NbALFA–AuNPs (Fig. 3b).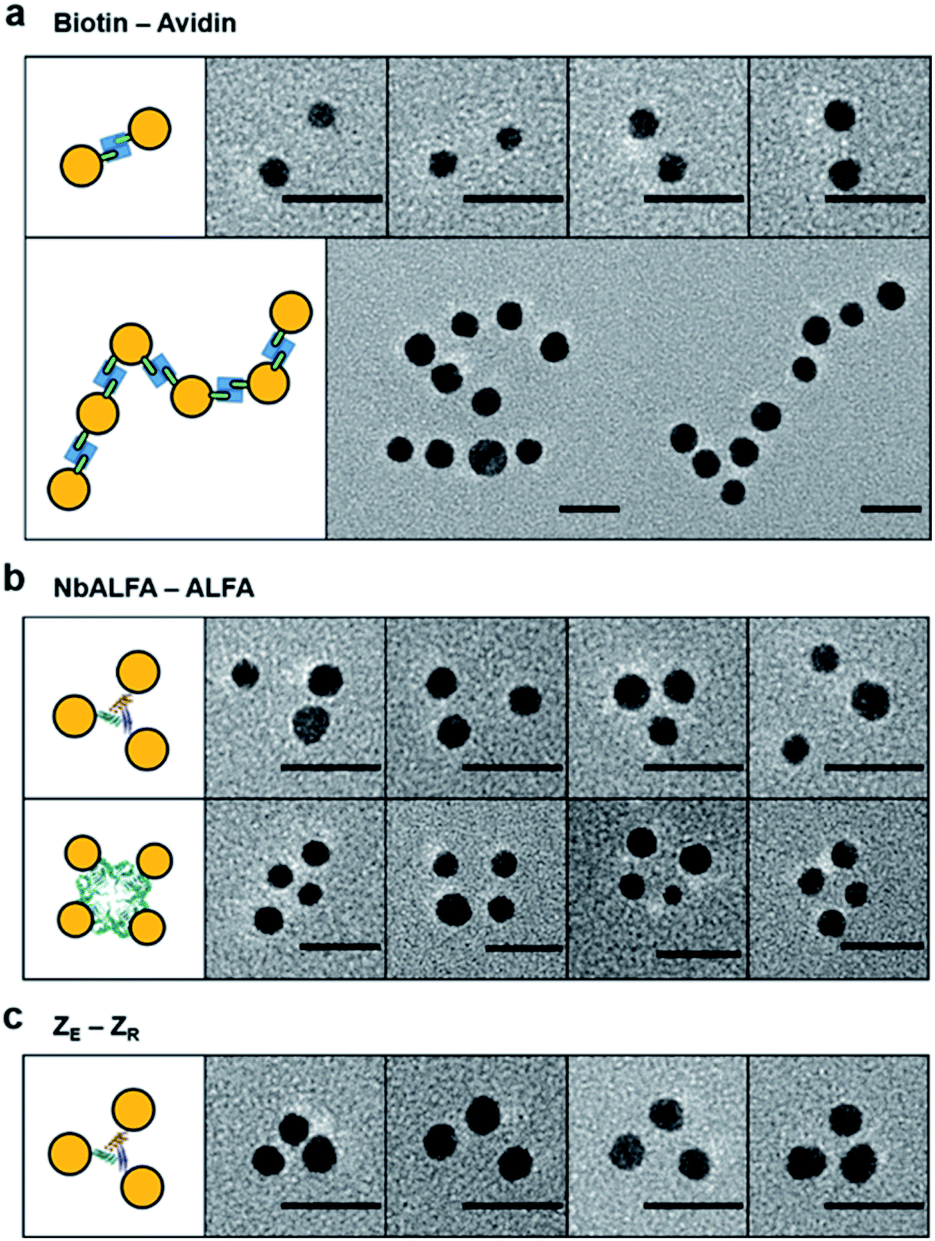 | ||
| Fig. 4 Multimeric protein targeting with valence-controlled AuNPs. (a) Representative TEM images of monovalent 5 nm biotin–AuNP (top) or divalent 10 nm biotin–AuNP (bottom) binding to dimeric rhizavidin. (b) Representative TEM images of monovalent 5 nm NbALFA–AuNP binding to trimeric ALFA–5K7V (top) or tetrameric ALFA–Gab (bottom). (c) Representative TEM images of monovalent 5 nm ZR–AuNP binding to trimeric ZE–5K7V. Scale bars: 20 nm. | ||
We also used monovalent 5 nm NbALFA–AuNP to target ALFA-tag-fused multimeric proteins. When ALFA-tag-fused trimeric (5K7V)41 and tetrameric (GAB)42 protein multimers were mixed with monovalent NbALFA–AuNP, AuNP trimers and tetramers were clearly observed (Fig. 4b and S17†). Protein labelling with monovalent particles was also conducted by using a well-established and strong peptide binding pair, leucine zipper (ZE and ZR).43 Monovalent ZR–AuNPs were prepared by mixing 5 nm Ni-NTA–AuNPs with 6His–SC–ST15–ZR and treated to ZE-fused trimeric 5K7V. A large portion of AuNPs were assembled onto 5K7V trimers (Fig. 4c and S18†). Overall, however, full AuNP assemblies on protein multimers with triangular or rectangular geometries were less efficient compared to monovalent 5 nm AuNP binding to other AuNPs with various valences. This is likely due to relatively small separations between binding tags on protein multimers and resulting steric hindrances. More importantly, however, random and large AuNP clusters were not observed, supporting the monovalency of a protein moiety on AuNPs. Protein multimers also contain their own multiple 6His tags, which can potentially bind to Ni-NTA surfaces of AuNPs. A lack of high degree AuNP clustering during these particle–protein assemblies implies that unwanted AuNP bindings by 6His tags on protein multimers are likely blocked by protein chain wrapping around AuNPs under our binding condition (50 mM imidazole).
Conclusions
We developed a facile and reliable method for valence-controlled protein conjugation to gold nanoparticles by introducing a unique strategy to wrap protein chains on nanoparticles. Tandem repeat protein chains with regularly spaced 6His units allowed dynamic multivalent interactions on Ni-NTA surfaces on AuNPs. In particular, protein chain wrapping on AuNPs via constant surface protein rearrangement was induced by slowly decreasing competing imidazole concentrations. TEM images showed homogeneously dispersed protein wrapping chains around particles. Here, slow removal of excess imidazole favored surface binding of all repeating units in protein chains (wrapping with maximal cooperativity), which will be unaffected by the numbers of repeating units or particle sizes. Therefore, various lengths of protein chains could be stably wrapped on AuNPs with different sizes, allowing us to find optimal protein chain lengths for valence controlled conjugation to particles. Diverse functional proteins, from a nanobody domain, leucine zippers, to an even small AP tag (15 residues), were conjugated to AuNPs with defined valences. These proteins also retained their binding abilities after conjugation, indicating that binding moieties at the end of protein chains are well exposed from AuNP surfaces after chain wrapping.Cooperative chain wrapping during our one-pot conjugation also alleviated the strictness in mixing ratios between conjugated proteins and particles, increasing experimental reliability by avoiding the need for (often difficult) accurate particle concentration measurement. Conjugation through multiple 6His and Ni-NTA interactions were highly stable (no dissociation after several months at 4 °C). However, size increases by protein chain coating (∼5 nm thickness) must be considered when precise manipulations are required. The present method will be applicable to other nano-structures with proper surface NTA modifications. Moreover, other protein interaction pairs different from 6His–NTA can also be used for protein chain wrapping. Developed protein coated nanoparticles with defined functional protein valences will allow the wider use of protein–particle conjugates in various fields such as precise high-order nanostructure assembly and biomolecule sensing/imaging. We also believe that multivalent surface interactions of repeated protein units on modular protein chain scaffolds offer a highly stable but dynamic new protein coating strategy on solid surfaces.
Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
H. Choi conducted all experiments. H. Choi and Y. Jung designed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by BioNano Health Guard Research Center funded by the Ministry of Science and ICT (MSIT) as Global Frontier Project (H-GUARD_2014M3A6B2060507 (1711073453)) and the National Research Foundation of Korea (NRF) grant funded by MSIT (NRF-2019R1A2C2008558). Dr Kibeom Hong provided the genes for the ST tandem repeat scaffold proteins. Professor Joonseok Lee advised the methods for quantum dot surface modifications.Notes and references
- K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- M. Howarth, W. Liu, S. Puthenveetil, Y. Zheng, L. F. Marshall, M. M. Schmidt, K. D. Wittrup, M. G. Bawendi and A. Y. Ting, Nat. Methods, 2008, 5, 397–399 CrossRef CAS PubMed.
- J. Farlow, D. Seo, K. E. Broaders, M. J. Taylor, Z. J. Gartner and Y. W. Jun, Nat. Methods, 2013, 10, 1203–1205 CrossRef CAS PubMed.
- D. Bhatia, S. Arumugam, M. Nasilowski, H. Joshi, C. Wunder, V. Chambon, V. Prakash, C. Grazon, B. Nadal, P. K. Maiti, L. Johannes, B. Dubertret and Y. Krishnan, Nat. Nanotechnol., 2016, 11, 1112–1119 CrossRef CAS PubMed.
- J. W. Kim, J. H. Kim and R. Deaton, Angew. Chem., Int. Ed., 2011, 50, 9185–9190 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E. M. Roller, A. Hogele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed.
- G. Tikhomirov, S. Hoogland, P. E. Lee, A. Fischer, E. H. Sargent and S. O. Kelley, Nat. Nanotechnol., 2011, 6, 485–490 CrossRef CAS PubMed.
- D. Seo, K. M. Southard, J. W. Kim, H. J. Lee, J. Farlow, J. U. Lee, D. B. Litt, T. Haas, A. P. Alivisatos, J. Cheon, Z. J. Gartner and Y. W. Jun, Cell, 2016, 165, 1507–1518 CrossRef CAS PubMed.
- M. Kwak, W. Gu, H. Jeong, H. Lee, J. U. Lee, M. An, Y. H. Kim, J. H. Lee, J. Cheon and Y. W. Jun, Nano Lett., 2019, 19, 3761–3769 CrossRef CAS PubMed.
- T. Lee, M. Mohammadniaei, H. Zhang, J. Yoon, H. K. Choi, S. Guo, P. Guo and J. W. Choi, Adv. Sci., 2020, 7, 1902477 CrossRef CAS PubMed.
- Y. H. Liao, C. H. Lin, C. Y. Cheng, W. C. Wong, J. Y. Juo and C. L. Hsieh, ACS Nano, 2019, 13, 10918–10928 CrossRef CAS PubMed.
- G. Yao, J. Li, Q. Li, X. Chen, X. Liu, F. Wang, Z. Qu, Z. Ge, R. P. Narayanan, D. Williams, H. Pei, X. Zuo, L. Wang, H. Yan, B. L. Feringa and C. Fan, Nat. Mater., 2020, 19, 781–788 CrossRef CAS PubMed.
- L. Song and Z. Deng, ChemNanoMat, 2017, 3, 698–712 CrossRef CAS.
- D. Zanchet, C. M. Micheel, W. J. Parak, D. Gerion and A. P. Alivisatos, Nano Lett., 2001, 1, 32–35 CrossRef CAS.
- T. Pellegrino, R. A. Sperling, A. P. Alivisatos and W. J. Parak, J. Biomed. Biotechnol., 2008, 2007, 26796 Search PubMed.
- S. A. Claridge, H. W. Liang, S. R. Basu, J. M. Frechet and A. P. Alivisatos, Nano Lett., 2008, 8, 1202–1206 CrossRef CAS PubMed.
- K. Suzuki, K. Hosokawa and M. Maeda, J. Am. Chem. Soc., 2009, 131, 7518–7519 CrossRef CAS PubMed.
- T. G. Edwardson, K. L. Lau, D. Bousmail, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2016, 8, 162–170 CrossRef CAS PubMed.
- N. Xie, S. Liu, H. Fang, Y. Yang, K. Quan, J. Li, X. Yang, K. Wang and J. Huang, ACS Nano, 2019, 13, 4174–4182 CrossRef CAS PubMed.
- J. Shen, Q. Tang, L. Li, J. Li, X. Zuo, X. Qu, H. Pei, L. Wang and C. Fan, Angew. Chem., Int. Ed., 2017, 56, 16077–16081 CrossRef CAS PubMed.
- H. Xing, Y. Bai, Y. Bai, L. H. Tan, J. Tao, B. Pedretti, G. A. Vincil, Y. Lu and S. C. Zimmerman, J. Am. Chem. Soc., 2017, 139, 3623–3626 CrossRef CAS PubMed.
- T. Trinh, C. Liao, V. Toader, M. Barlog, H. S. Bazzi, J. Li and H. F. Sleiman, Nat. Chem., 2018, 10, 184–192 CrossRef CAS PubMed.
- Z. Dai, Y. Tan, K. He, H. Chen and J. Liu, J. Am. Chem. Soc., 2020, 142, 14023–14027 CrossRef CAS PubMed.
- H. Pei, F. Li, Y. Wan, M. Wei, H. Liu, Y. Su, N. Chen, Q. Huang and C. Fan, J. Am. Chem. Soc., 2012, 134, 11876–11879 CrossRef CAS PubMed.
- G. Yao, H. Pei, J. Li, Y. Zhao, D. Zhu, Y. Zhang, Y. Lin, Q. Huang and C. Fan, NPG Asia Mater., 2015, 7, e159 CrossRef.
- W. Ma, A. Saccardo, D. Roccatano, D. Aboagye-Mensah, M. Alkaseem, M. Jewkes, F. Di Nezza, M. Baron, M. Soloviev and E. Ferrari, Nat. Commun., 2018, 9, 1489 CrossRef PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- F. Aldeek, M. Safi, N. Zhan, G. Palui and H. Mattoussi, ACS Nano, 2013, 7, 10197–10210 CrossRef CAS PubMed.
- L. Z. Ma, F. Li, T. Fang, J. T. Zhang and Q. B. Wang, ACS Appl. Mater. Inter., 2015, 7, 11024–11031 CrossRef CAS PubMed.
- D. J. Nieves, N. S. Azmi, R. Xu, R. Levy, E. A. Yates and D. G. Fernig, Chem. Commun., 2014, 50, 13157–13160 RSC.
- J. W. Kim, D. Seo, J. U. Lee, K. M. Southard, Y. Lim, D. Kim, Z. J. Gartner, Y. W. Jun and J. Cheon, Nat. Protoc., 2017, 12, 1871–1889 CrossRef CAS PubMed.
- R. Wieneke and R. Tampe, Angew. Chem., Int. Ed., 2019, 58, 8278–8290 CrossRef CAS PubMed.
- D. Wasserberg, J. Cabanas-Danes, J. Prangsma, S. O'Mahony, P. A. Cazade, E. Tromp, C. Blum, D. Thompson, J. Huskens, V. Subramaniam and P. Jonkheijm, ACS Nano, 2017, 11, 9068–9083 CrossRef CAS PubMed.
- A. H. Keeble and M. Howarth, Chem. Sci., 2020, 11, 7281–7291 RSC.
- X. Chen, J. L. Zaro and W. C. Shen, Adv. Drug Deliv. Rev., 2013, 65, 1357–1369 CrossRef CAS PubMed.
- H. Götzke, M. Kilisch, M. Martínez-Carranza, S. Sograte-Idrissi, A. Rajavel, T. Schlichthaerle, N. Engels, R. Jungmann, P. Stenmark and F. Opazo, Nat. Commun., 2019, 10, 4403 CrossRef PubMed.
- J. L. Swift, R. Heuff and D. T. Cramb, Biophys. J., 2006, 90, 1396–1410 CrossRef CAS PubMed.
- A. R. Clapp, E. R. Goldman and H. Mattoussi, Nat. Protoc., 2006, 1, 1258–1266 CrossRef CAS PubMed.
- H. Shen, L. J. Tauzin, R. Baiyasi, W. Wang, N. Moringo, B. Shuang and C. F. Landes, Chem. Rev., 2017, 117, 7331–7376 CrossRef CAS PubMed.
- S. H. Helppolainen, K. P. Nurminen, J. A. Määttä, K. K. Halling, J. P. Slotte, T. Huhtala, T. Liimatainen, S. Ylä-Herttuala, K. J. Airenne, A. Närvänen, J. Jänis, P. Vainiotalo, J. Valjakka, M. S. Kulomaa and H. R. Nordlund, Biochem. J., 2007, 405, 397–405 CrossRef CAS PubMed.
- J. A. Fallas, G. Ueda, W. Sheffler, V. Nguyen, D. E. McNamara, B. Sankaran, J. H. Pereira, F. Parmeggiani, T. J. Brunette, D. Cascio, T. R. Yeates, P. Zwart and D. Baker, Nat. Chem., 2017, 9, 353–360 CrossRef CAS PubMed.
- M. R. Chance, A. R. Bresnick, S. K. Burley, J. S. Jiang, C. D. Lima, A. Sali, S. C. Almo, J. B. Bonanno, J. A. Buglino, S. Boulton, H. Chen, N. Eswar, G. He, R. Huang, V. Ilyin, L. McMahan, U. Pieper, S. Ray, M. Vidal and L. K. Wang, Protein Sci., 2002, 11, 723–738 CrossRef CAS PubMed.
- J. R. Moll, S. B. Ruvinov, I. Pastan and C. Vinson, Protein Sci., 2001, 10, 649–655 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1sc06993d |
| This journal is © The Royal Society of Chemistry 2022 |