
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Origin of the temperature dependence of 13C pNMR shifts for copper paddlewheel MOFs†
Zhipeng
Ke
 ,
Daniel M.
Dawson
,
Sharon E.
Ashbrook
* and
Michael
Bühl
*
,
Daniel M.
Dawson
,
Sharon E.
Ashbrook
* and
Michael
Bühl
*
School of Chemistry, EaStCHEM and Centre of Magnetic Resonance, University of St Andrews, St Andrews KY16 9ST, UK. E-mail: sema@st-andrews.ac.uk; mb105@st-andrews.ac.uk
First published on 3rd February 2022
Abstract
An efficient protocol for the calculation of 13C pNMR shifts in metal–organic frameworks based on Cu(II) paddlewheel dimers is proposed, which involves simplified structural models, optimised using GFN2-xTB for the high-spin state, and CAM-B3LYP-computed NMR and EPR parameters. Models for hydrated and activated HKUST-1 and hydrated STAM MOFs with one, two and three Cu dimers have been used. The electronic ground states are low-spin and diamagnetic, with pNMR shifts arising from thermal population of intermediate- and high-spin excited states. Treating individual spin configurations in a broken symmetry (BS) approach, and selecting two or more of these to describe individual excited states, the magnetic shieldings of these paramagnetic states are evaluated using the approach by Hrobárik and Kaupp. The total shielding is then evaluated from a Boltzmann distribution between the energy levels of the chosen configurations. The computed pNMR shifts are very sensitive to temperature and, therefore, to the relative energies of the BS spin states. In order to reproduce the temperature dependence of the pNMR shifts seen in experiment, some scaling of the calculated energy gaps is required. A single scaling factor was applied to all levels in any one system, by fitting to experimental results at several temperatures simultaneously. The resulting scaling factor decreases with an increasing number of dimer units in the model (e.g., from ∼1.7 for mono-dimer models to 1.2 for tri-dimer models). The approach of this scaling factor towards unity indicates that models with three dimers are approaching a size where they can be considered as reasonable models for the 13C shifts of infinite MOFs. The observed unusual temperature dependencies in the latter are indicated to arise both from the “normal” temperature dependence of the pNMR shifts of the paramagnetic states and the populations of these states in the thermal equilibrium.
Introduction
Solid-state Nuclear Magnetic Resonance (NMR) spectroscopy provides a very sensitive probe of the local, atomic-scale environment of the nuclear spins present in a sample without any requirement for long-range order.1–3 The sensitivity of the NMR parameters to small changes in geometry has resulted in this method being used to probe weak bonding interactions, host–guest interactions and both static and dynamic disorder. The NMR parameters can also depend on temperature; for diamagnetic materials, the chemical shifts are only weakly temperature dependent (in the absence of significant dynamics in the system), whereas for paramagnetic materials the changes can be much greater, and the control and monitoring of temperature during an NMR experiment is crucial in obtaining consistent spectra and in interpreting these correctly.4,5 Measuring the temperature dependence of paramagnetic NMR (or “pNMR”) shifts can provide additional information about the spin physics and electronic structure, ultimately aiding the assignment of resonances for materials where the shifts are often unusual or difficult to predict in advance.Metal–organic frameworks (MOFs) are well-known microporous materials, containing pores with diameters typically between 5 and 20 Å. These frameworks are charge neutral and composed of “nodes” of one or more metal ions coordinated by polytypic organic linkers, often based on benzene rings, such as benzene-1,3,5-tricarboxylate (BTC) and benzene-1,4-dicarboxylate (BDC). The versatile chemistry of MOFs, with many possible combinations of nodes and linkers, leads to a range of applications in fields as diverse as gas storage, catalysis and drug delivery.6–8 Solid-state NMR spectroscopy is frequently used to study MOFs, enabling the binding of guest molecules, the dynamics of guest species within the pores, and any structural changes that can result from these to be explored.9–12 MOFs that contain paramagnetic metal centres pose a particular challenge for NMR spectroscopy, but such materials often have interesting and useful physical and chemical properties that can be exploited. HKUST-1 is a MOF containing Cu(II) paddlewheel dimers linked by BTC (Fig. 1A), with guest molecules able to bind to the free coordination sites (Fig. 1C).13 Modification of the linker (Fig. 1D) leads to STAM-1 (linker L2), which contains both hydrophobic and hydrophilic pores (Fig. 1B)14 and the isoreticular STAM-17 series, (with L3, L4 or a range of linkers with larger alkyloxy groups).15 The STAM series of MOFs demonstrate an unusually high hydrolytic stability compared to the chemically similar but topologically different HKUST-1. This was shown to be related to a significant bulk structural rearrangement upon the interaction with water, likened to the dissipation of energy seen in the crumple zone of a car (and leading to the term “crumple-zone MOFs” for these materials).15,16 Dawson et al. demonstrated that fast (40–60 kHz) MAS and variable offset experiments were necessary to acquire undistorted, high-resolution 13C NMR spectra of these MOFs, which display signals over a shift range of −100 to +1000 ppm.17 However, isotopic labelling of specific C sites on the linker was required to unambiguously assign the resonances in HKUST-1 and STAM-1, which demonstrated that the most shifted signal did not result from the C closest to the Cu atoms, as might have been expected.17 For a mononuclear Cu(II) complex, the 13C isotropic shift (δiso) is expected to be linearly related to 1/T, with the extrapolation to T = 0 providing an estimate of the diamagnetic orbital shift (δiso(orb)), as shown in our previous work on Cu(II) oximes.18,19 However, for a Cu(II) benzoate dimer, the dependence of the experimental 13C δiso on 1/T was clearly non linear, leading to incorrect δiso(orb) shift predictions.16,20 In this work, we provide insight into the origin of the unusual temperature dependencies using a combination of NMR measurements and computational modelling.
 | ||
| Fig. 1 Crystal structures of (a) HKUST-1 13 and (b) STAM-1.14 (c) Structure of a copper paddlewheel dimer: green arrows indicate the axial guest binding sites (G). (d) The linkers and numbering schemes used in this work for HKUST-1 (L1), STAM-1 (L2), STAM-17-Me (L3) and STAM-17-OMe (L4). Additionally, numbering schemes are shown for the benzoate (L5) and dimethyl trimesate (L6) ligands, with only C species analogous to those in the MOFs numbered. The colouring scheme in parts (a)–(c) is Cu = blue, O = red, C = black, H = pale grey. | ||
For many materials, Density Functional Theory (DFT) provides a complementary approach to experimental NMR spectroscopy, helping to facilitate the interpretation, assignment and prediction of spectra.21,22 While now widely applied for diamagnetic materials (in an approach that is often referred to as NMR crystallography), such calculations are considerably more challenging for paramagnetic solids, although arguably of greater benefit owing to the significant and unusual shifts observed. In the solid state, periodic approaches for computing the NMR parameters (e.g., the Gauge-Including Projector Augmented Wave (GIPAW) method)23 have proven extremely useful. However, their extension to paramagnetic materials is still in its infancy.4,24 For isolated molecules with paramagnetic centres, a number of theoretical approaches for calculating NMR isotropic shifts have been developed,25–28 which contain temperature-dependent expressions related to the magnetic susceptibility of decoupled magnetic moments. However, in extended solids the magnetic moments of the individual paramagnetic sites may communicate and couple differently, complicating the modelling using a periodic approach.29 Moreover, the paramagnetic MOFs of interest in this work, HKUST-1 and the STAM family, have the additional complication of interacting spins on the paramagnetic centres in the paddlewheel dimer (and the possibility of a thermal equilibrium of differently (ferro- or antiferromagnetically) coupled states), in addition to potential couplings between dimers. In recent work we have shown that the experimental 13C δiso of a solid containing molecular Cu(II) paddlewheel dimers can be reproduced computationally assuming a thermal equilibrium between a diamagnetic ground state (with antiferromagnetic spin coupling on the Cu dimer) and an excited triplet state (with ferromagnetically coupled spins for a single molecule).20 In this work we will explore how pNMR parameters calculated for a series of increasingly complex isolated molecules can be used to provide insight into those measured experimentally for extended Cu(II) paddlewheel MOFs. We consider a set of models containing increasing numbers (and different arrangements) of paddlewheel dimer units and demonstrate that explicit consideration of the population of different spin configurations can explain both the experimentally observed shifts and the unusual temperature dependence of these in this interesting set of MOFs. The insights from this study will expand the applicability of NMR spectroscopy as a noninvasive tool to obtain information on the electronic and geometric structures of paramagnetic MOFs and related materials, for instance in the context of probing guest binding at the paramagnetic centres.
Methods
Synthesis
HKUST-1 and STAM-1 were prepared as described in ref. 17. STAM-17-Me and STAM-17-OMe were prepared as described in ref. 16.Solid-state NMR spectroscopy
13C magic angle spinning (MAS) NMR spectra were recorded using Bruker Avance III spectrometers with 14.1 or 20.0 T wide-bore superconducting magnets (13C Larmor frequencies of 150.9 and 213.8 MHz, respectively). Experimental details for the variable-temperature (VT) 13C NMR spectra of as-made HKUST-1 and STAM-1 (recorded at 20.0 T) can be found in ref. 30, and those for STAM-17-Me and STAM-17-OMe (recorded at 14.1 T) are given in ref. 16. The VT 13C NMR spectra of dehydrated (activated) HKUST-1 were recorded at 20.0 T. Activated HKUST-1 was packed into a 1.3 mm ZrO2 rotor and rotated at a MAS rate of 60 kHz. The data were obtained as two sub-spectra with different frequency offsets to provide higher sensitivity for the C1 and C2 signals (see Fig. 1D for numbering scheme). Spectra were acquired using a rotor-synchronised spin-echo pulse sequence, with τ = 16.7 μs and a recycle interval of 20 ms. Signal averaging was carried out for 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 960 transients (C2 and C3 subspectrum) or 131072 transients (C1 subspectrum). The temperature was monitored by an external thermocouple within the probe, and varied between 250 and 323 K. The temperature was allowed to equilibrate until a stability of ±0.1 K was achieved for a period of five minutes. The temperature was calibrated externally using KBr to account for the ∼20 K frictional heating arising from the rapid MAS. Additional calibration to account for the absolute sample temperature was not carried out and, therefore, there may be a small error in the temperature reported.‡ Shifts are reported in ppm relative to TMS using L-alanine (δ(CH3) = 20.5 ppm) as a secondary solid reference.
960 transients (C2 and C3 subspectrum) or 131072 transients (C1 subspectrum). The temperature was monitored by an external thermocouple within the probe, and varied between 250 and 323 K. The temperature was allowed to equilibrate until a stability of ±0.1 K was achieved for a period of five minutes. The temperature was calibrated externally using KBr to account for the ∼20 K frictional heating arising from the rapid MAS. Additional calibration to account for the absolute sample temperature was not carried out and, therefore, there may be a small error in the temperature reported.‡ Shifts are reported in ppm relative to TMS using L-alanine (δ(CH3) = 20.5 ppm) as a secondary solid reference.
Calculations
The structures of all models considered were optimised using GAUSSIAN 0931 at the PBE0-D3 level or using GFN2-xTB.32–37 In conjunction with PBE0-D3, an augmented Wachters basis set38,39 was used for Cu (8s7p4d) with the full contraction scheme 62111111/3311111/3111, while the 6-31G* basis set was used for the ligands (this combination of basis sets is labelled here as AE1). For model D1 (see Fig. 2 and Table 1), structural optimisation was carried out separately for each possible spin state (see below and ESI†) using unrestricted Kohn–Sham wavefunctions with a broken-symmetry (KS-BS) solution for the open-shell spin state (e.g., expectation values of the Ŝ2 operator of 1.985, 2.997 and 6.010 for the singlet, triplet and quintet states, respectively in model D1). As shown in the ESI (Table S11)† for di-dimer models, the optimised geometries for the different spin states are very similar, and so the structure of the high-spin state was used in pNMR calculations for all spin states. For GFN2-xTB calculations, the basis set was an augmented minimal basis. At this level, structures were optimised for the high-spin state only. The character of each stationary point was verified by the computation of the harmonic vibrational frequencies. The frequencies were also used to obtain thermodynamic corrections to relative enthalpies and free energies.
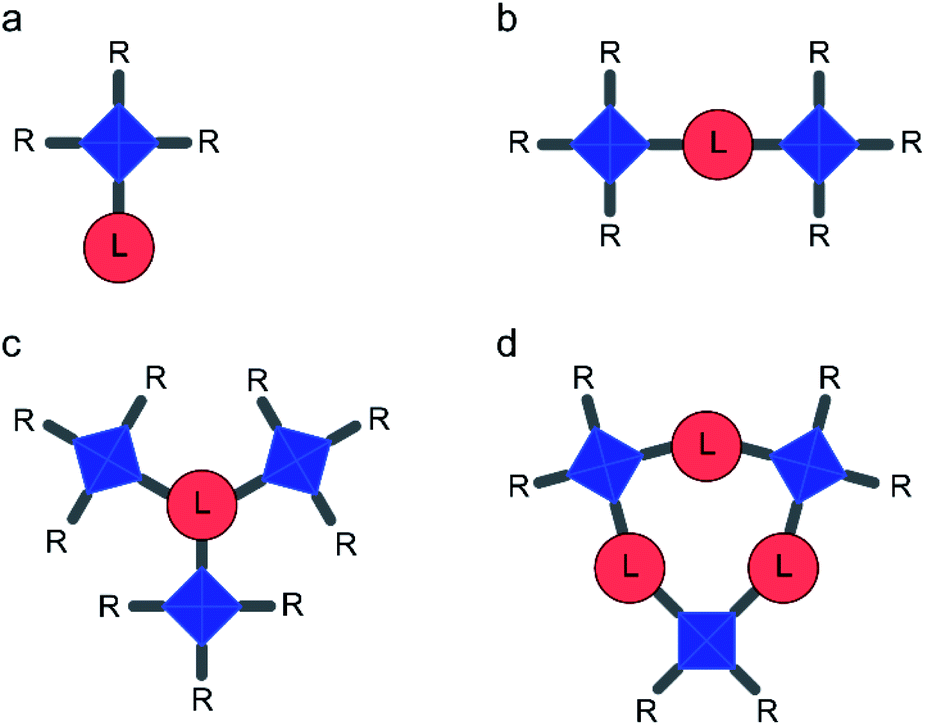 | ||
| Fig. 2 Schematic representation of models containing (a) one copper paddlewheel dimer (“mono-dimer” models, M) (b) two dimers (“di-dimer” models, D), (c) three dimers arranged radially around one linker (“radial tri-dimer” models, Ra) and (d) three dimers arranged in a ring (“ring tri-dimer” models, Ri). Copper dimers (Fig. 1C) are shown in blue and linkers (Fig. 1D) in red. Pendant R groups are either acetate or additional linkers, as detailed in Table 1. | ||
| Model | L | R groups | Axial guest | Formula |
|---|---|---|---|---|
| M1 | L5 | 3 × L5 | No | Cu2(L5)4 |
| M2 | L6 | 3 × L6 | No | Cu2(L6)4 |
| M3 | L5 | L5 + 2 × OAc | No | Cu2(L5)2(OAc)2 |
| M4 | L5 | 3 × OAc | No | Cu2(L5)(OAc)3 |
| D1 | L3 | 6 × OAc | No | Cu4(L3)(OAc)6 |
| D2 | L4 | 6 × OAc | No | Cu4(L4)(OAc)6 |
| D3 | L2 | 6 × OAc | No | Cu4(L2)(OAc)6 |
| D4 | L3 | 6 × OAc | 4 × H2O | Cu4(L3)(OAc)6·4(H2O) |
| D5 | L4 | 6 × OAc | 4 × H2O | Cu4(L4)(OAc)6·4(H2O) |
| D6 | L2 | 6 × OAc | 4 × H2O | Cu4(L2)(OAc)6·4(H2O) |
| Ra1 | L1 | 9 × OAc | No | Cu6(L1)(OAc)9 |
| Ra2 | L1 | 9 × OAc | 6 × H2O | Cu6(L1)(OAc)9·6(H2O) |
| Ri1 | L3 | 6 × OAc | No | Cu6(L3)(OAc)6 |
| Ri2 | L4 | 6 × OAc | No | Cu6(L4)(OAc)6 |
| Ri3 | L2 | 6 × OAc | No | Cu6(L2)(OAc)6 |
| Ri4 | L3 | 6 × OAc | 6 × H2O | Cu6(L3)(OAc)6·6(H2O) |
| Ri5 | L4 | 6 × OAc | 6 × H2O | Cu6(L4)(OAc)6·6(H2O) |
| Ri6 | L2 | 6 × OAc | 6 × H2O | Cu6(L2)(OAc)6·6(H2O) |
NMR and EPR parameters were computed for the optimised structures at the CAM-B3LYP40 level, employing a 9s7p4d (621111111/3311111/3111) basis set on Cu, which was constructed specifically for calculating accurate hyperfine coupling constants,41 and IGLO-basis II42 on the ligands (this combination of basis sets is labelled II). Orbital shieldings, σiso(orb), were computed using the GIAO (gauge-including atomic orbitals) implementation in Gaussian 09 for all spin states in a structure optimised for the high-spin state. The hyperfine coupling and g tensors were computed for all spin states (with the exception of the singlet) at the CAM-B3LYP/II level using ORCA.43 Energy differences, ΔE, with respect to the lowest energy configurations were evaluated at the CAM-B3LYP/II level using the broken-symmetry (BS) approach of Noodleman.44–46 The zero-field splitting (ZFS) parameters were calculated using the coupled-perturbed method of Neese47 at the BLYP/II level (for technical reasons only a non-hybrid functional could be used here). The structure and 13C σiso(orb) of the reference compound, TMS, was computed at the same level of theory.
Paramagnetic shieldings were calculated using the approach of Hrobárik and Kaupp48 with
| σiso = σiso(orb) − [S(S + 1)βe/3k(T − Θ)gNβN][geAFC + geAPC + ΔgisoAFC + (1/3)Tr(Δganiso·Adip)], | (1) |
| σiso = ∑fiσiso,i, | (2) |
| fi = giexp(−sΔEi/RT)/Σgiexp(−sΔEi/RT), | (3) |
Results and discussion
Models with a single copper paddlewheel dimer
The simplest models for HKUST-1 are based on an isolated paddlewheel dimer unit, as shown in Fig. 2 and Table 1. In the basic model (M1), four benzoate ligands are attached to the dimer, leaving two vacant coordination sites that could, in principle, be occupied by guest molecules. For HKUST-1, it is possible to consider an “activated MOF”, i.e., where these coordination sites remain vacant, which can be achieved experimentally by heating under vacuum to remove guest molecules.50 It should be noted that this is not possible for molecular benzoates (such as that studied in ref. 20), which would decompose or coordinate with other donors, or for the STAM-based crumple zone MOFs which undergo a structural phase transition15,16 (not considered in detail here). Model M1 can be adapted, as shown in Fig. 2 and Table 1, to more closely resemble HKUST-1 (as in M2 where ester functional groups are added in meta positions on each ring) or with the aim of increasing computational efficiency (as in M3 and M4 where two or three L5 are replaced by acetate, respectively). The structures of these four models, optimised using GFN2-xTB are shown in Fig. S2.†For a single paddlewheel dimer, three spin configurations are possible; two singlets and a triplet state (shown in Section S2 of the ESI† for model M4). The triplet state is well represented by a single configuration, but the singlet state is a linear combination of two configurations. The expression used to determine the pNMR shifts for the single dimer models is given in Section S2 of the ESI.†Fig. 3 shows calculated 13C shifts for models M1–M4, along with the experimental shifts of activated HKUST-1 (labelled using the scheme in Fig. 1D). In each case, the paramagnetic shifts of C2 and C1 are overestimated (too negative and too positive, respectively) when compared with experiment (open circles in Fig. 3). The addition of the ester groups (M2) more than doubles the CPU time (see Table S1†), with little, if any, improvement in the agreement between experimental and calculated shifts. Simplifying the dimer (M4) significantly speeds up the calculations (by ∼1.5 and ∼3 times for geometry optimisation and pNMR calculations respectively, Table S1†). The calculated shifts of these models appear to be slightly closer to experiment, but the disagreement is still significant. The changes seen for C1 and C2 mainly stem from the changes in triplet shielding values, which involve a large contribution from Fermi-contact shifts and become less pronounced (smaller in absolute value) as Ph groups are replaced with Me. The modifications of the initial dimer model result in a slight elongation of the distance between the two Cu atoms (of up to 0.02 Å), and a small change (between −3 and +11 cm−1) in ΔEST (see Table S2†).
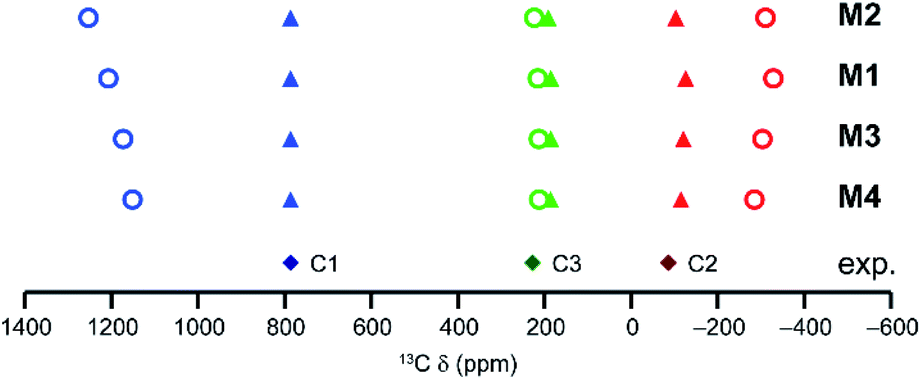 | ||
| Fig. 3 As-calculated (circles) and scaled (triangles) 13C isotropic shifts for models M1, M2, M3 and M4, at 298.2 K (CAM-B3LYP/II//GFN-xTB). The energy scaling factors are s = 1.86 (M1), 1.94 (M2), 1.78 (M3) and 1.73 (M4). The experimental data for activated HKUST-1 (298 K) are shown as diamonds. | ||
Although there is reasonable qualitative agreement between the calculated and experimental shifts, none of the models produce shifts in good quantitative agreement. For example, for C1 calculated isotropic shifts are between 1100 and 1250 ppm, compared to the experimental shift at ∼800 ppm. C1 signals above 1000 ppm were seen in previous work for a urea-loaded copper benzoate dimer, and in this case the computational and experimental shifts were in much better agreement.20 This perhaps suggests that models containing a single dimer work well for molecular solids, where the Cu paddlewheel units are so spatially remote that no interaction between them is expected. However, for MOFs, the dimer units are much closer and can interact via the aromatic organic linkers that joint them, suggesting more sophisticated models and a proper theoretical description of the magnetic coupling between dimers (see later) will be needed. This is supported by comparing the difference in the experimental 13C shifts for the aromatic CH species in hydrated HKUST-1 and hydrated STAM-1; signals are seen at δiso ≈ 227 ppm for the CH groups between two dimers in both MOFs, but the C4 signal in STAM-1 (an aromatic CH that is adjacent to one dimer only) has δiso = 178 ppm.
One possible additional reason for the poorer agreement between theory and experiment is the need to include macroscopic effects on the magnetic susceptibility (which enters the pNMR expression via a temperature-dependent pre-factor containing the Weiss constant Θ as shown in eqn (1)).24,51,52 A large negative Θ would decrease the pNMR contributions at a given temperature. While it would be possible to improve the agreement between calculated and observed shifts in Fig. 3 through empirical fitting of the Weiss constant (see Table S3† for resulting values for model M4 with Θ = −170 K), this was not employed routinely here for two reasons; firstly, the Weiss constants determined experimentally for MOFs of this type appear to be reasonably small (e.g., 4.7 K for HKUST-1 loaded with pyridine53) and secondly, the temperature dependence of the pNMR shifts is more complicated than the expression given in eqn (1), as it also depends on the thermal population of the paramagnetic states (see eqn (2)). A different Weiss constant would have to be fitted for each temperature separately, which is both unphysical and impractical. The dependence of the pNMR shifts on the population of the paramagnetic states (i.e., the triplet state for models with a single dimer), also leads to a possible source of error in the calculations, with an over- or underestimation resulting from an inaccurate determination of the energy gaps (i.e., ΔEST). While this energy was used as calculated in our previous work on the urea-loaded benzoate dimer (i.e., s = 1),20 this may not be suitable when attempting to model extended MOF systems, where communication between dimer units may raise the energy of the paramagnetic states, thereby reducing their contribution to the overall pNMR shifts. The ΔEST energy gap can be simply scaled, with the scaling factor s (eqn (3)) determined by minimising the mean absolute deviation (MAD) between experimental and theoretical shifts for the three 13C resonances in HKUST-1. This results in values of s between 1.7 and 2.0 for the four single dimer models and much improved agreement between experiment and theory, as shown in Fig. 3 (triangles). This indicates that the high spin states are populated to a lesser extent in the MOF than in the copper benzoates studied previously. Table S2† gives values of ΔEST for the models M1 to M4 with different scaling factors, and Table S4† gives values calculated for each model with their respective values of s (ranging between 1.73 and 1.94).
In principle, it would be possible to use experimental information such as exchange couplings from EPR measurements (see footnote a in Table S2†) instead of calculated and scaled ΔEST values, but this is undesirable for a number of reasons. These include loss of predictive power if information from experiment is vital for the calculations (assuming our empirical scaling factors are transferable between systems), and practicability, as separate such measurements would then be required for each and every system under scrutiny, e.g., upon loading with different guests.
Models with two copper paddlewheel dimers
The use of models with more than one copper dimer offers a potentially more accurate description of the long-range MOF structure, and the option to explore chemical differences between the Cu-based MOFs more easily. Models D1, D2 and D3, shown in Fig. 2B and Table 1 are based on L2, L3 or L4 (to model STAM-1, STAM-17-Me or STAM-17-OMe, respectively14–16), with two simplified paddlewheel dimers (R = acetate) attached. All three can also serve as potential models for HKUST-1, but more sophisticated models, where the linker is trimesate (L1) and the third substituent is another paddlewheel dimer (model Ra1, see later) can also be used. As noted in the methodology section above, optimised geometries of model D1 are very similar for all spin states at the PBE0-D3/AE1 level (see Table S11†). For the results described in this work, structures were optimised for the high spin state only using the GFN2-xTB semi-empirical method. From the mono-dimer models, it is clear that the pNMR shifts depend strongly on the energy gaps between the spin states, but that any inability of DFT to predict these accurately might be alleviated through the use of a scaling parameter. For the di-dimer models, where a greater number of spin states are possible, a single scaling factor, s, was applied to all energy differences (see Section S3 of the ESI† for more detail). When attempting to fit s using the pNMR parameters obtained at a single temperature, a spread of values was obtained depending on the temperature chosen. This approach is both unphysical and not particularly helpful for understanding or, perhaps more importantly, predicting experimental results. Therefore, s was determined for each model by obtaining the closest match between experimental and computed shifts for all temperatures simultaneously. In the following discussion, results are shown after this scaling has been applied. The spin states used (i.e., i in eqn (2) and (3)) can consist of more than one BS spin configuration. Information on how these were selected and the equations used to calculate the shieldings is given in Section S3 of the ESI† (note the combination in Fig. S5† and the resulting eqn S8† was adopted for the results shown below). | ||
| Fig. 4 Temperature dependence of experimental and calculated 13C shifts for (a) C1, (b) C3 and (c) C2 in activated HKUST-1, using models D1 and D3, with ΔEST of ∼219 cm−1, scaled by a factor of s = 1.305 and 1.330 (CAM-B3LYP/II//GFN2-xTB), respectively, employing eqn S8.† | ||
The non linear relationship between the 13C δiso and 1/T for the Cu(II)-based MOFs is in contrast to the behaviour seen for systems with a single spin centre (such as the complexes in ref. 18 and 19), and is a consequence of the temperature-dependent population of the paramagnetic states in thermal equilibrium. Scaling of the DFT-calculated ΔEST is required to ensure that this population is accurately reproduced. The effect of scaling on the computed shifts is illustrated in Fig. 5 for model D4 over the temperature range 20 to 698 K. The shifts initially show maxima or minima at 1000/T ≈ 5 K−1 (i.e., T = 200 K). Upon scaling ΔEST, the maximum and minimum shifts become less extreme and occur at T ≈ 250 K. It will depend on the system under study, therefore, whether the maxima/minima fall within a temperature range that is experimentally accessible under MAS (250–350 K). For activated HKUST-1, these are observed in experimental measurements (Fig. 4), but fall outside of the accessible temperature range for hydrated MOFs, such as hydrated HKUST-1 (as shown in Fig. 6).
Fig. 6 shows 13C isotropic shifts for model D6, computed using eqn S15† (see Tables S19 and S20†) with s = 1.295 (determined using the experimental shifts for STAM-1).30 The calculated shifts match experiment reasonably well for all carbon sites (see Fig. 1D for the numbering scheme). A version of this model without coordinated water molecules (D3) was used to compute shifts for activated HKUST-1 in the previous section, and so the computed shifts for this hydrated model can also be compared with the experimental values for hydrated HKUST-1 (also shown in Fig. 6). If the scaling factor for the computed shifts is determined not by fitting to experiments for STAM-1 but only to those for HKUST-1 (which has just three C sites), a small decrease (to 1.288) is observed, leading to the slightly different calculated shifts shown in Fig. 6.
 | ||
| Fig. 5 Plots of 13C δiso for model D4 as a function of inverse temperature, calculated using s = 1.374 (from fitting of the experimental data of STAM-17-Me),16 shown by solid lines and as-calculated (i.e., with s = 1) shown by dotted lines. The insets show the data in the range of 1000/T below 6 K−1 where 4–2.9 K−1 is the temperature range of 250–348 K (CAM-B3LYP/II//GFN2-xTB). | ||
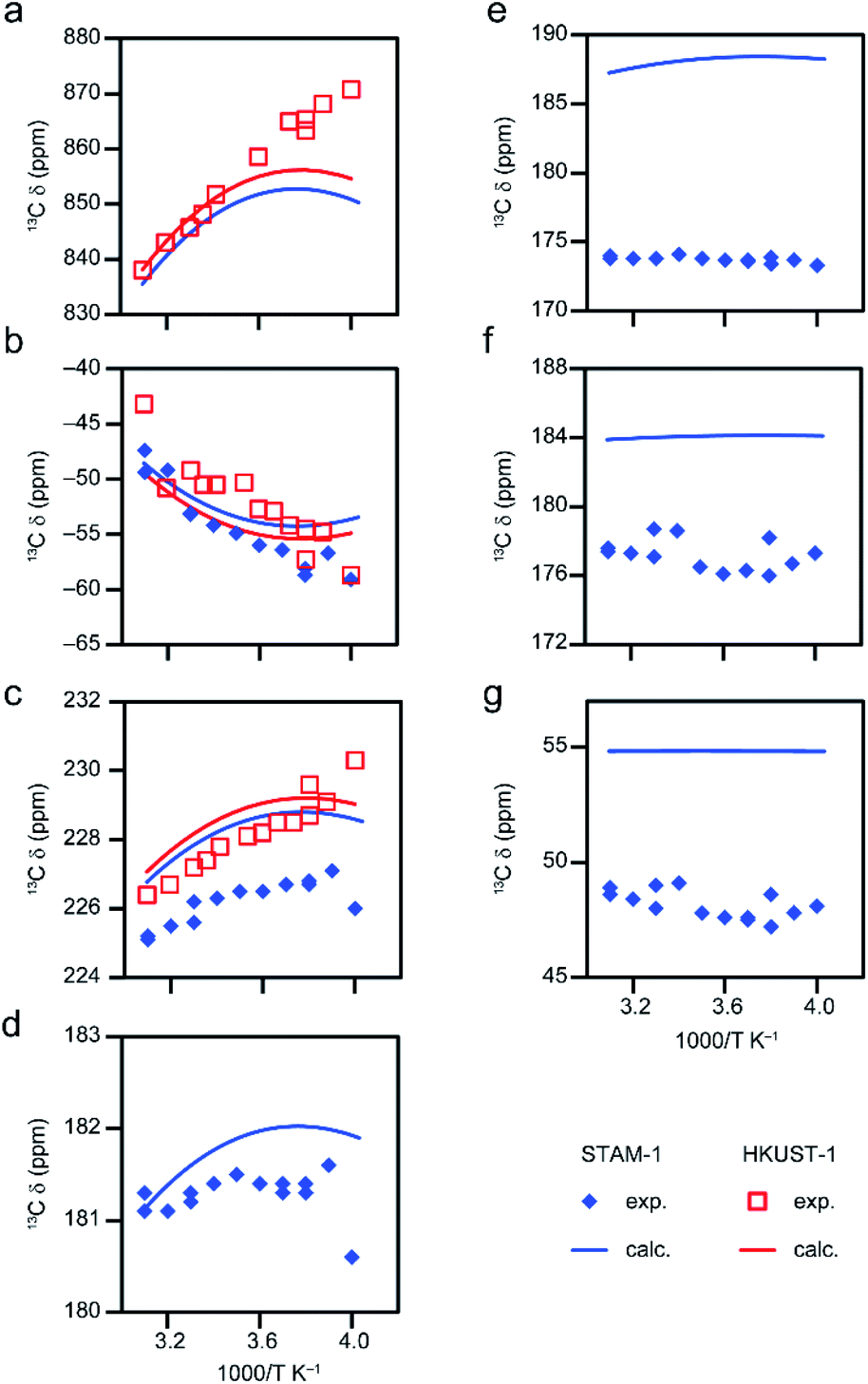 | ||
| Fig. 6 Temperature dependence of 13C shifts of hydrated STAM-1 and HKUST-1 from experiment14,30 and computation for model D6 with s = 1.295 and 1.288, respectively for STAM-1 and HKUST-1 for ΔEST ≈ 187 cm−1 (CAM-B3LYP/II//GFN2-xTB), using eqn S15.† Results are presented for (a) C1 in both MOFs (no experimental data for STAM-1 owing to the very broad resonance leading to unfeasibly long experimental times), (b) C2 in both MOFs, (c) C3 in both MOFs, (d) C4 in STAM-1, (e) C5 in STAM-1, (f) C6 in STAM-1 and (g) C7 in STAM-1 (See Fig. 1D for numbering schemes). | ||
Above 273 K (below 3.66 K−1) where chemically similar C environments exist for the two MOFs (i.e., C1, C2 and C3), the experimental shifts are very similar,17,30 validating the use of the same structural model for the two. However, below 273 K (above 3.66 K−1), these resonances seem to shift in different directions in Fig. 6B and C. For example, C2 (Fig. 6B) becomes more shielded for HKUST-1 while the shift of the same site in STAM-1 appears to vary little with temperature in this range. This flattening of the curve is captured in the computed shifts (see Table S20† for numerical data). It is unclear whether the observed deviations between the two samples at low temperature (up to ∼5 ppm in Fig. 6C) arise from (i) experimental uncertainties (suggested by the scatter of the experimental data), (ii) the structural differences between STAM-1 and HKUST-1 (which have two and three dimers, respectively, attached to each linker – see later discussion of models with three paddlewheel dimers) or (iii) the different behaviour of the extraframework water in the pores. Fully hydrated HKUST-1 is expected to contain free water molecules inside the pores, in addition to those bound to the metal centres, and these may behave differently below 273 K, i.e., on going from liquid-/gas-like to more restricted, crystal-like structures. For hydrated STAM-1, the network of water molecules is more confined, with a larger proportion expected to be bound to the metal sites, and the material has a more complex long-range structure with both hydrophobic and hydrophilic pores and a greater degree of framework flexibility.14 Models with a varying number of water molecules were explored computationally to study the effect of any extraframework hydration (see Section S4 of the ESI for more detail†). This shows that the experimental data for STAM-1 would be broadly compatible with models loaded with 4 and 5 water molecules. However, it would be difficult to draw any conclusions from these results regarding the hydration in the experimental sample, where significant dynamics of the water molecules is also likely to occur at room temperature.
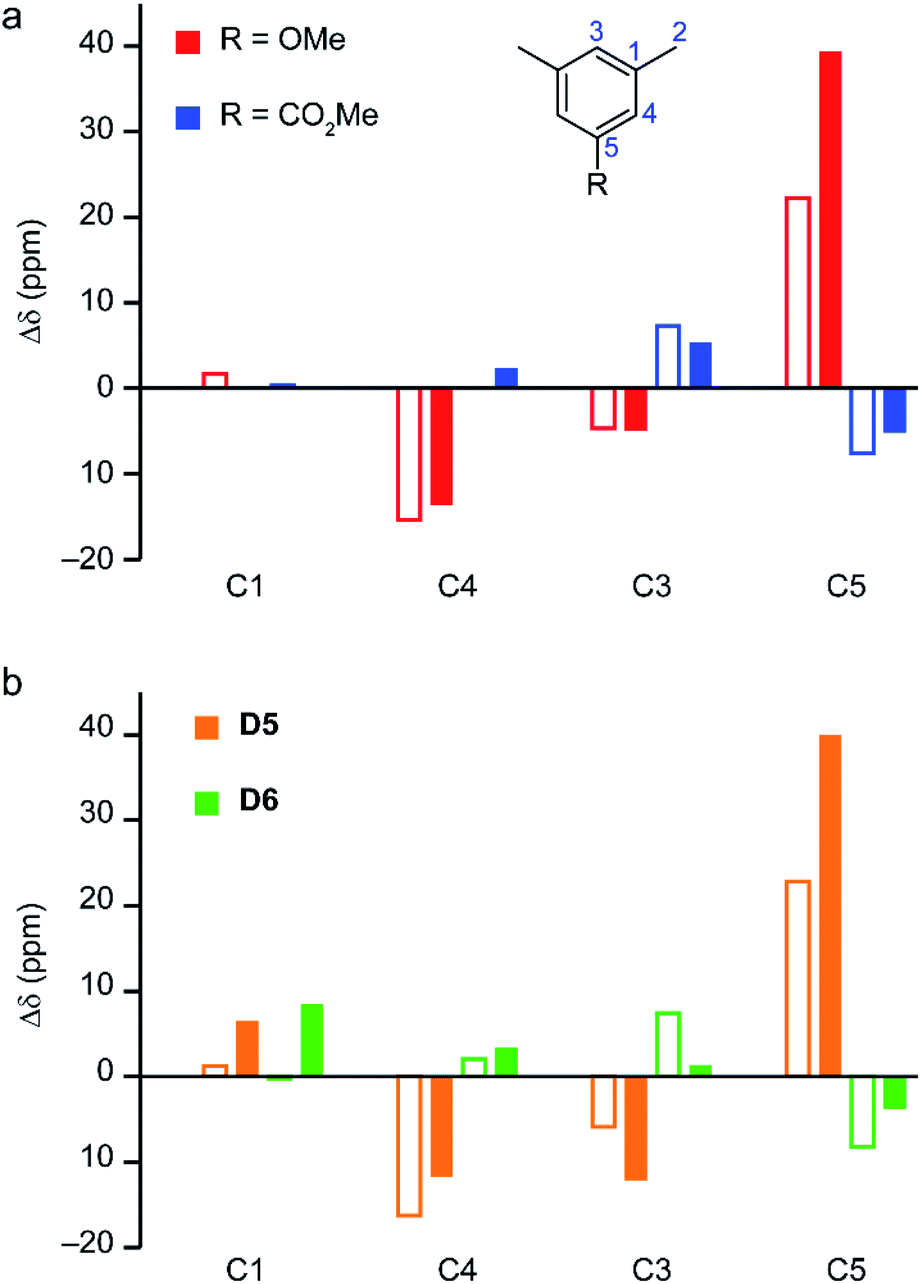 | ||
| Fig. 7 (a) Plot showing the substituent effect on the experimental 13C shifts of the MOFs14,16 at 298 K (filled bars) and their diamagnetic linker analogues (empty bars, see numbering scheme shown in the inset).55 For the diamagnetic molecules with R = OMe (red bars) and CO2Me (blue bars), the shifts are plotted relative to mesitylene (R = Me). For the paramagnetic MOFs, shifts are reported relative to STAM-17-Me. (b) The computed changes of the total (filled bars) and orbital (empty bars) shielding for 13C for models D5 (orange bars) and D6 (green bars) relative to D4, at 298 K (CAM-B3LYP/II//GFN2-xTB, s = 1.340, using eqn S15†). | ||
The calculated pNMR shifts are obtained as a sum of both paramagnetic and orbital shifts. The changes in calculated orbital shifts for hydrated di-dimer models with different substituents (models D5 and D6), shown in Fig. 7B (empty orange and green bars, respectively), are in good agreement with the variation in the experimental shifts for the molecular analogues in Fig. 7A. The effects are consistent with expectations based on the electron donating or withdrawing nature of the substituents (i.e., OMe and CO2Me, respectively). The paramagnetic contributions modulate these effects, substantially so for C5, giving variation in the total shifts that agrees better with those seen experimentally for the MOFs.
Models with three copper paddlewheel dimers

 | ||
| Fig. 9 (a) Temperature dependence of calculated 13C shifts for model Ri4 (Table S30†), using eqn S26,† with energy gaps scaled by 1.204 on an intradimer coupling of ∼184 cm−1 (CAM-B3LYP/II//GFN2-xTB). The results are compared with calculated 13C shifts for model D4 (Table S15†) and experimental values for hydrated STAM-17-Me.16 Note that, for C4 and C5, only a single resonance is observed experimentally and the blue points are the same in both plots. (b) Temperature dependence of calculated 13C shifts for model Ri5 (Table S32†), using eqn S26,† with energy gaps scaled by 1.183 on an intradimer coupling of ∼183 cm−1 (CAM-B3LYP/II//GFN2-xTB). The results are compared with calculated 13C shifts for model D5 (Table S18†) and experimental values for hydrated STAM-17-OMe.16 Note that the signal for C1 was not observed experimentally in ref. 16. | ||
The advantage of the tri-dimers over the di-dimer models is that they show better agreement with the experimental shifts observed at low temperatures. Adding the third dimer to the model increases the number of spin configurations and states. As the tri-dimer models have more intermediate and high spin states that can be populated, at lower temperature the tri-dimer models have relatively higher contributions from the paramagnetic shifts of these excited states than those of the di-dimer model, which apparently improves the agreement with experiment. These ring tri-dimer models would, thus, appear to be suitable for computing 13C shifts for the STAM-17 series.
Replacement of the Me/OMe groups with CO2Me gives a model for STAM-1 (model Ri6 in Fig. 2D and Table 1). As shown in Fig. S19,† the 13C shifts (calculated using eqn S26†) are in qualitative agreement with experiment (see Table S33† for calculated shieldings). For C2 there is a noticeable improvement when compared to the corresponding di-dimer model (D6) while, in contrast, for C3 and C4 the simpler model better reproduces the experimental temperature dependence of the shifts (see Fig. S19†), although these changes are smaller (2 to 5 ppm) within the temperature range considered.
In general, the 13C shifts of hydrated STAM MOFs are reproduced well using models containing two paddlewheel dimers or the Ri tri-dimer models. The assumption that shifts can be computed by Boltzmann averaging of selected spin states that are constructed from all possible spin configurations can lead to a remarkably good agreement with experiment. Although these tri-dimer models tend to perform better than di-dimer models, the latter can still lead to fairly good agreement with experimental VT NMR results. As these simpler models have far fewer spin configurations, their relatively lower cost makes them potentially useful and attractive models for further study. In contrast, however, a ring tri-dimer model appears to be less successful than the simpler di-dimer model for computing 13C shifts in HKUST-1 (see Fig. S18†). Although this framework does contain subunits of rings with three dimers, rings containing four dimers joined by four linkers are also present, and it may be that the ring model is not a sufficiently accurate representation of the complete structure. It could be possible to extend the investigation to consider ring models with four dimers (although at considerable cost given the larger number of spin states). However, reasonably good agreement with experiment is seen for the radial tri-dimer model Ra1, with much improved shifts for C1 (as shown in Fig. S16†). This model may provide a better picture of the local environment experienced by any one linker in HKUST-1, with any improvement in the description of the longer-range structure provided by a Ri tri-dimer model offset by only having two thirds of the required dimers bound to each linker. Thus, building larger molecular models for MOFs would not only require adding more dimers with an increasing number of spin configurations but the degree of success would also depend on the connectivity of the dimers in the material of interest.
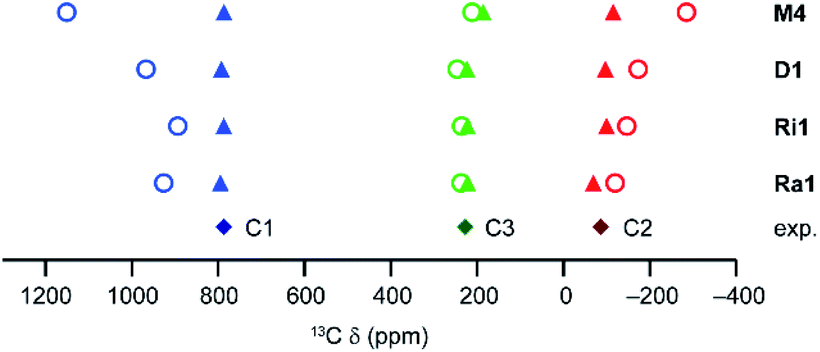 | ||
| Fig. 10 As-calculated (open circles) and scaled (filled triangles) computed 13C shifts for different molecular models of activated HKUST-1 at 298.2 K (CAM-B3LYP/II//GFN2-xTB). The energy scaling factors are 1.73, 1.30, 1.20 and 1.23 for the M4, D1, Ri1 and Ra1 models, respectively, determined as described above. Experimental points for activated HKUST-1 (298 K) are shown as filled diamonds. | ||
Conclusions
In this work, an efficient protocol for the calculation of pNMR shifts in materials containing multiple Cu(II) paddlewheel dimers is proposed. This neglects any effect of ZFS (which is shown to be very small), uses a cheap but reliable method for geometry optimisation (GFN2-xTB for the high-spin state), a simplified structural model and a promising DFT functional (CAM-B3LYP) for computing the NMR and EPR parameters.The use of molecular models containing two or three dimers has been validated by comparing calculated and experimental 13C pNMR shifts for hydrated and activated HKUST-1 and hydrated STAM MOFs. The success of this approach requires a consideration of the relevant spin configurations in a broken symmetry (BS) approach. High-spin configurations where all centres have the same spin can be well described using a single determinant; however there remains ambiguity over which BS determinants would contribute to the multi-determinantal wavefunctions of the low- or intermediate-spin states. The number of possible configurations grows rapidly with the number of dimer units in a model. In the absence of sophisticated configuration interaction calculations (which would provide information on the contribution of individual configurations to specific states, but at prohibitive computational cost), this work has explored a number of possible approaches by assuming plausible linear combinations of configurations for which magnetic shieldings can then be averaged. The total shielding is then evaluated from a Boltzmann distribution between the energy levels of the chosen configurations. The choice of the configurations that are combined affects the weightings in the expression for the final shielding and, thus, the overall calculated pNMR shift. Owing to thermal averaging the computed pNMR shifts are very sensitive to temperature and, therefore, to the relative energies of the BS spin states. However, in order to reproduce the temperature dependence of the pNMR shifts seen in experiment, some scaling of the as-calculated energy gaps is required. A single scaling factor was applied to all levels in any one system, by fitting to experimental results at several temperatures simultaneously. Even with such an approach, the predicted temperature dependence of the shifts do still depend to some extent on the choices of spin configurations that are combined, with best agreement found when only pairs of configurations are used. This leads to the use of eqn S8 and S19† to calculate 13C isotropic shifts for models with two and three dimers, respectively. Where possible, further simplifications can be introduced by exploiting the symmetry (exact or approximate) of a model to reduce the number of spin configurations considered (e.g., Cs and C3 symmetry for models with two and three dimers). This leads to the simplified expressions in eqn S15 and S26,† which provide results that are very similar to the more general expressions in eqn S8 and S19,† respectively.
This methodology provides good agreement between experimental shifts for hydrated HKUST-1 and MOFs in the STAM series, and for activated HKUST-1, with those predicted using models containing two Cu(II) dimers (note the phase change seen for crumple zone MOFs upon activation prevents any simple comparison for these materials). Although more computationally expensive, radial models with three dimer units on the same linker perform well for HKUST-1. For STAM MOFs, a ring model, with three Cu(II) dimers joined by three linkers (a structural motif found in the MOFs), showed improved agreement with experiment relative to a model with two dimer units (although at a higher computational cost). In contrast, however, this model performed less well for HKUST-1 (where the radial model was much better). Although this motif is present in HKUST-1, all linkers in the MOF have three dimer substituents, and these results suggest this contribution to the shifts is greater than that of a more accurate longer-range structure. It should be noted that the scaling factor required to improve agreement between calculation and experiment decreases with an increasing number of dimer units in the model (e.g., from ∼1.7 for mono-dimer models to 1.2 for tri-dimer models). The approach of this scaling factor towards unity indicates that models with three dimers are approaching a size where they can be considered as reasonable models for infinite MOFs, at least as far as the 13C shifts are concerned. The methodology we have introduced for accurate prediction of NMR parameters in systems with weakly coupled pairs of interacting spins has great potential for the study of more complex and challenging chemistry, including host–guest interactions, hydration, activation and phase transitions in MOFs. The insight we gain from the approaches introduced and validated here should also inform the future method development required for materials with more complex spin systems.
Data availability
The research data supporting this publication can be accessed at https://doi.org/10.17630/233aeedf-1dc4-49e4-b5b8-2c136b2e2365 [Ref. 57].Author contributions
S. E. A. and D. M. D. designed and carried out the experimental work, Z. K. and M. B. designed and carried out the computational work. All authors made significant contributions to the writing process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank EaStCHEM and the School of Chemistry for support. Calculations were carried out on a local Opteron PC cluster maintained by Dr H. Früchtl. This work was supported by the EPSRC through the Collaborative Computational Project on NMR Crystallography (CCP-NC), via EP/M022501/1. The UK 850 MHz solid-state NMR Facility used in this research was funded by EPSRC and BBSRC, as well as the University of Warwick including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). S. E. A. would also like to thank the Royal Society and Wolfson Foundation for a merit award. Z. K. would like to thank the Chinese Scholarship Council (CSC) for a scholarship (grant number 201708060464).Notes and references
- D. C. Apperley, R. K. Harris and P. Hodgkinson, Solid State NMR Basic Principles and Practice, Momentum Press, New York, 2012 Search PubMed.
- S. E. Ashbrook, D. M. Dawson and J. M. Griffin in Local Structure Characterisation, ed. D. W. Bruce, D. O'Hare and R. I. Walton, John Wiley & Sons, Oxford, 1st edn, 2014, vol. 1, ch. 1, pp. 1–88 Search PubMed.
- R. F. Moran, D. M. Dawson and S. E. Ashbrook, Int. Rev. Phys. Chem., 2017, 35, 39–115 Search PubMed.
- A. J. Pell, G. Pintacuda and C. P. Grey, Prog. NMR Spectrosc., 2019, 111, 1–271 CrossRef CAS PubMed.
- M. Bertmer, Solid State NMR, 2017, 81, 1–7 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. L. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. C. Zhou, Adv. Mater., 2018, 30, 1–35 Search PubMed.
- S. E. Ashbrook, D. M. Dawson and V. R. Seymour, Phys. Chem. Chem. Phys., 2014, 16, 8223–8242 RSC.
- B. E. G. Lucier, S. S. Chen and Y. N. Huang, Acc. Chem. Res., 2018, 51, 319–330 CrossRef CAS PubMed.
- M. Bertmer, Annu. Rep. NMR Spectrosc., 2020, 101, 1–64 CrossRef CAS.
- E. Brunner and M. Rauche, Chem. Sci., 2020, 11, 4297–4304 RSC.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- M. I. H. Mohideen, B. Xiao, P. S. Wheatley, A. C. McKinlay, Y. Li, A. M. Z. Slawin, D. W. Aldous, N. F. Cessford, T. Düren, X. Zhao, R. Gill, K. M. Thomas, J. M. Griffin, S. E. Ashbrook and R. E. Morris, Nat. Chem., 2011, 3, 304–310 CrossRef CAS PubMed.
- L. N. McHugh, M. J. McPherson, L. J. McCormick, S. A. Morris, P. S. Wheatley, S. J. Teat, D. McKay, D. M. Dawson, C. E. F. Sansome, S. E. Ashbrook, C. A. Stone, M. W. Smith and R. E. Morris, Nat. Chem., 2018, 10, 1096–1102 CrossRef CAS PubMed.
- D. M. Dawson, C. E. F. Sansome, L. N. McHugh, M. J. McPherson, L. J. McCormick McPherson, R. E. Morris and S. E. Ashbrook, Solid State Nucl. Magn. Reson., 2019, 101, 44–50 CrossRef CAS PubMed.
- D. M. Dawson, L. E. Jamieson, M. I. H. Mohideen, A. C. McKinlay, I. A. Smellie, R. Cadou, N. S. Keddie, R. E. Morris and S. E. Ashbrook, Phys. Chem. Chem. Phys., 2013, 15, 919–929 RSC.
- M. Bühl, S. E. Ashbrook, D. M. Dawson, R. A. Doyle, P. Hrobárik, M. Kaupp and I. A. Smellie, Chem. - Eur. J., 2016, 22, 15328–15339 CrossRef PubMed.
- D. M. Dawson, Z. Ke, F. M. Mack, R. A. Doyle, G. P. M. Bignami, I. A. Smellie, M. Bühl and S. E. Ashbrook, Chem. Commun., 2017, 53, 10512–10515 RSC.
- Z. Ke, L. E. Jamieson, D. M. Dawson, S. E. Ashbrook and M. Bühl, Solid State Nucl. Magn. Reson., 2019, 101, 31–37 CrossRef CAS PubMed.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azaïs, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733–5779 CrossRef CAS PubMed.
- S. E. Ashbrook and D. McKay, Chem. Commun., 2016, 52, 7186–7204 RSC.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- A. Mondal and M. Kaupp, J. Phys. Chem. Lett., 2018, 9, 1480–1484 CrossRef CAS PubMed.
- S. Moon and S. Patchkovskii, in Calculation of NMR and EPR Parameters: Theory and Applications, ed. M. Kaupp, M. Bühl and V. G. Malkin, Wiley, Weinheim, FRG, 2004, pp. 325–340 Search PubMed.
- P. Hrobárik, R. Reviakine, A. V. Arbuznikov, O. L. Malkina, V. G. Malkin, F. H. Köhler and M. Kaupp, J. Chem. Phys., 2007, 126, 024107 CrossRef PubMed.
- T. O. Pennanen and J. Vaara, Phys. Rev. Lett., 2008, 100, 133002 CrossRef PubMed.
- B. Martin and J. Autschbach, Phys. Chem. Chem. Phys., 2016, 18, 21051–21068 RSC.
- G. Mali and M. Mazaj, J. Phys. Chem. C, 2021, 125, 4655–4664 CrossRef CAS.
- D. M. Dawson, PhD Thesis, University of St Andrews, 2014 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, et al. Gaussian 09, Revision D.01, Gaussian Inc., 2013 Search PubMed.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. R. Johnson and A. D. Becke, J. Chem. Phys., 2006, 124, 174104 CrossRef PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- A. J. H. Wachters, J. Chem. Phys., 1970, 52, 1033–1036 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. Munzarova and M. Kaupp, J. Phys. Chem. A, 1999, 103, 9966–9983 CrossRef CAS.
- W. Kutzelnigg, U. Fleischer and M. Schindler, in NMR Basic Principles and Progress, Springer-Verlag, Berlin, 1990, vol. 23, pp. 165–262 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS.
- J. G. Norman, P. B. Ryan and L. Noodleman, J. Am. Chem. Soc., 1980, 102, 4279–4282 CrossRef CAS.
- F. Neese, Coord. Chem. Rev., 2009, 253, 526–563 CrossRef CAS.
- F. Neese, J. Chem. Phys., 2007, 127, 164112 CrossRef PubMed.
- P. Hrobárik, R. Reviakine, A. V. Arbuznikov, O. L. Malkina, V. G. Malkin, F. H. Köhler and M. Kaupp, J. Chem. Phys., 2007, 126, 024107 CrossRef PubMed.
- A. P. Ginsberg, J. Am. Chem. Soc., 1980, 102, 111–117 CrossRef CAS.
- R. Giovine, F. Pourpoint, S. Duval, O. Lafon, J. P. Amoureux, T. Loiseau and C. Volkringer, Cryst. Growth Des., 2018, 18, 6681–6693 CrossRef CAS.
- A. Mondal, M. W. Gaultois, A. J. Pell, M. Iannuzzi, C. P. Grey, J. Hutter and M. Kaupp, J. Chem. Theory Comput., 2018, 14, 377–394 CrossRef CAS PubMed.
- T. Wittmann, A. Mondal, C. B. L. Tschense, J. J. Wittmann, O. Klimm, R. Siegel, B. Corzilius, B. Weber, M. Kaupp and J. Senker, J. Am. Chem. Soc., 2018, 140, 2135–2144 CrossRef CAS PubMed.
- X. X. Zhang, S. S.-Y. Chui and I. D. Williams, J. Appl. Phys., 2000, 87, 6007–6009 CrossRef CAS.
- Y. Wu, A. Kobayashi, G. J. Halder, V. K. Peterson, K. W. Chapman, N. Lock, P. D. Southon and C. J. Kepert, Angew. Chem., Int. Ed., 2008, 47, 8929–8932 CrossRef CAS PubMed.
- https://nmrshiftdb.nmr.uni-koeln.de, accessed 28th October 2021.
- J. J. Led and S. B. Petersen, J. Magn. Reson., 1978, 32, 1–17 CAS.
- Z. Ke, D. M. Dawson, S. E. Ashbrook and M. Bühl, Origin of Temperature Dependence of 13C pNMR Shifts for Copper Paddlewheel MOFs, Dataset; University of St Andrews Research Portal, 2020, DOI:10.17630/233aeedf-1dc4-49e4-b5b8-2c136b2e2365.
Footnotes |
| † Electronic supplementary information (ESI) available: Complete ref. 31, additional detail on the methodology, and further computational results. See DOI: 10.1039/d1sc07138f |
| ‡ When comparing the spectra recorded at 20.0 T with in-house data, recorded using a more carefully calibrated temperature (using the phase transition temperature of DABCO at 351.1 K as a reference for the absolute temperature), we observed a maximum shift difference of ∼1 ppm for the C2 resonances in spectra of the same MOFs recorded at nominally the same temperature but at different fields. |
| This journal is © The Royal Society of Chemistry 2022 |