
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel type of donor–acceptor cyclopropane with fluorine as the donor: (3 + 2)-cycloadditions with carbonyls†
Haidong
Liu
a,
Lifang
Tian
 a,
Hui
Wang
a,
Zhi-Qiang
Li
a,
Chi
Zhang
a,
Fei
Xue
b and
Chao
Feng
*a
a,
Hui
Wang
a,
Zhi-Qiang
Li
a,
Chi
Zhang
a,
Fei
Xue
b and
Chao
Feng
*a
aTechnical Institute of Fluorochemistry (TIF), Institute of Advanced Synthesis (IAS), School of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, P. R. China. E-mail: iamcfeng@njtech.edu.cn
bInstitute of Material Physics & Chemistry, College of Science, Nanjing Forestry University, Nanjing 210037, China
First published on 15th February 2022
Abstract
gem-Difluorocyclopropane diester is disclosed as a new type of donor–acceptor cyclopropane, which smoothly participates in (3 + 2)-cycloadditions with various aldehydes and ketones. This work represents the first application of gem-difluorine substituents as an unconventional donor group for activating cyclopropane substrates in catalytic cycloaddition reactions. With this method, a wide variety of densely functionalized gem-difluorotetrahydrofuran skeletons, which are otherwise difficult to prepare, could be readily assembled in high yields under mild reaction conditions. Computational studies show that the cleavage of the C–C bond between the difluorine and diester moieties occurs upon a SN2-type attack of the carbonyl oxygen.
Introduction
Donor–acceptor cyclopropanes (DACs) constitute an important class of building blocks in modern organic synthesis, which participate in diverse chemical transformations such as ring-opening functionalization, cycloaddition and skeleton rearrangement reactions.1 The vicinal substituents with opposing electronic properties endow DACs with fascinating reactivities, which could be further activated via Lewis acid,2 transition-metal3 and small-molecule catalysis.4 The continuingly enriched reaction patterns as well as their predictable behaviour enable DAC-involved reactions to find broad application in asymmetric transformation,5 natural product synthesis and drug development as well.6While studying the correlation between the reactivity and structure, Werz and coworkers revealed that the properties of the donor group could exert more significant influence on the reaction efficiency, spanning more than four orders of magnitude.7 Generally, the donor group often involves carbon-,2,3b–g,4,8 nitrogen-2d,8f,g,9 and oxygen-based entities.8f–h,10 However, other heteroatom-based donor groups remain essentially unexplored. In this environment, the exploration of structurally novel DACs, especially those with unconventional heteroatom-based donor groups, is highly desirable for enriching DAC chemistry.
As the most electronegative element, fluorine has traditionally been treated as a type of potent σ-electron-withdrawing substituent. Because of its strong negative induction effect, the fluorine substituent always exhibits a strong destabilization effect on β-carbocations. Nevertheless, it is also revealed to display a decent resonance stabilization effect on α-carbocation, which is rationalized by p–p orbital interaction, thus resulting in positive charge delocalization by releasing an unshared electron pair of the fluorine substituent to the adjacent vacant p orbital of the cationic carbon center (termed the α-cation stabilizing effect of fluorine) (Scheme 1b).11 In this regard, the fluorine atom could thus be regarded as a special type of electron-donating group due to the fact that π-electron donation outcompetes its intrinsic negative inductive effect. Although this special electronic effect was uncovered as early as 1974, its application in synthetic organic chemistry remains underdeveloped since then. By leveraging this unique electronic effect, a set of elegant protocols were successfully developed by Ichikawa and coworkers,12 such as arylation of trifluoromethylated alkenes12a and Friedel–Crafts type cyclization of difluoroalkenes.12b Recently, our group has also developed a novel protocol for 1,3-fluoroallylation of aryl-substituted gem-difluorocyclopropane under photoredox catalysis.13 The fidelity of site-selective fluorine incorporation was rationalized by the cation stabilization capacity of the gem-difluorine substituent. Enlightened by these discoveries, we envisioned the possibility of extrapolating the α-cation stabilizing effect of fluorine by developing a new type of gem-difluorine-based DAC. If possible, a straightforward synthesis of CF2-embedded hetero/carbocycles could be readily realized through cycloaddition reactions with unsaturated π-systems. With our continuous interest in fluorine chemistry13,14 and enlightened by the well-developed cycloaddition of D–A cyclopropanes with carbonyls (Scheme 1c),15 we herein report our recent progress in the AlCl3-catalyzed (3 + 2)-cycloaddition of gem-difluorocyclopropane diester with aldehydes/ketones (Scheme 1d). By making use of this method, a large variety structurally diverse multi-substituted gem-difluorotetrahydrofurans, which could not be readily prepared by traditional methods, are efficiently constructed in high yields with good diastereoselectivities.
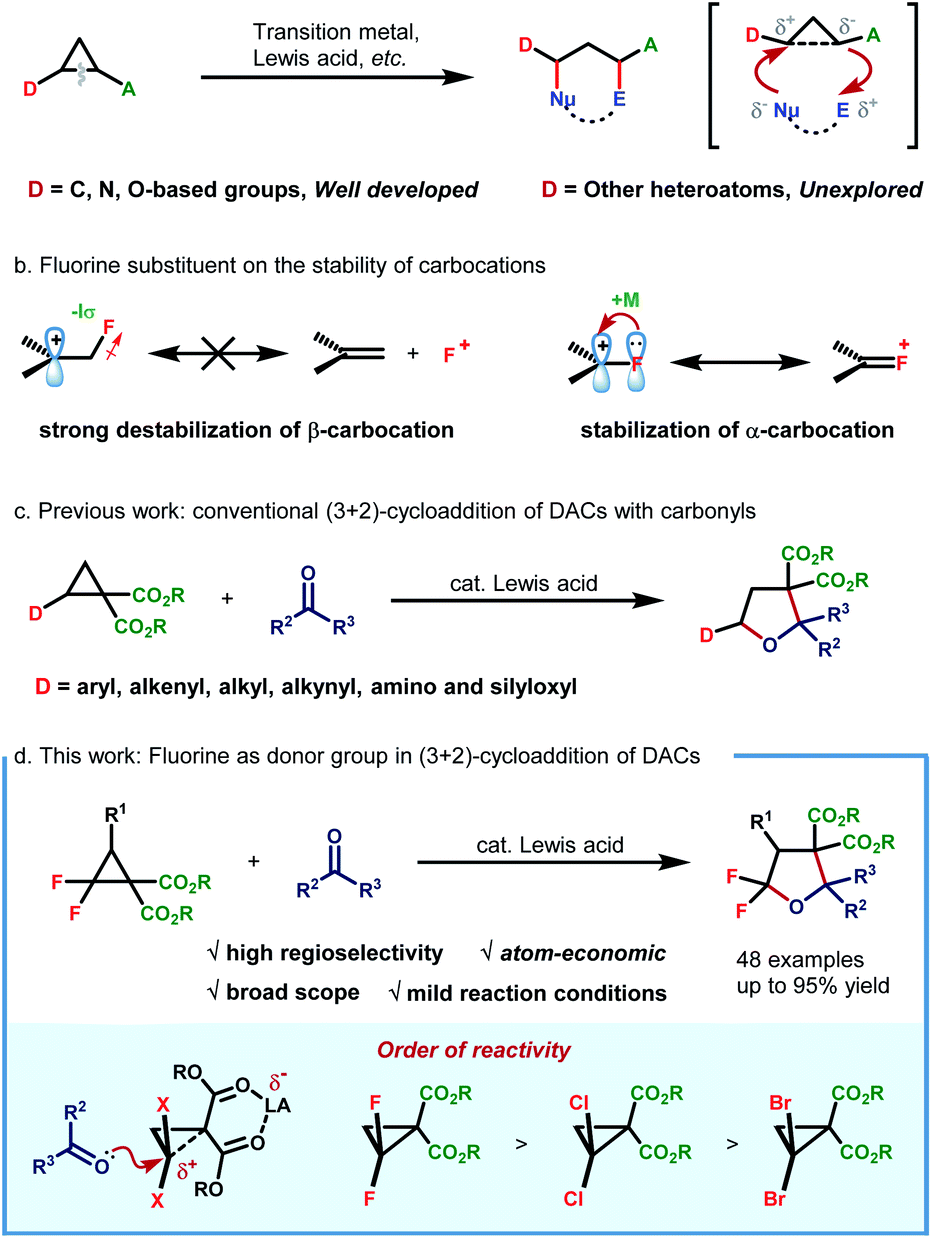 | ||
| Scheme 1 Ring-opening functionalization of donor–acceptor cyclopropanes. | ||
Results and discussion
At the outset, the starting DAC gem-difluorocyclopropane diester 1a was readily prepared following Chen's method using FSO2CF2CO2TMS as the difluorocarbene precursor with dibenzyl 2-methylenemalonate in 74% yield.16 Subsequently, the reaction of 1a with different unsaturated systems was examined.17 We found that the cycloadduct was only observed when an aldehyde or ketone was used as the dipolarophile for (3 + 2)-cycloadditions, while using 20 mol% AlCl3 as the Lewis acid and DCM as the solvent at room temperature. The desired product 3aa was detected in 65% yield (Table 1, entry 1). Other Lewis acids such as Zn(OTf)2 and Yb(OTf)3 all gave inferior results, whereas In(OTf)3 led to the formation of 3aa in comparable yield (entry 2–6). In view of the poor mass balance of substrate 1a, we surmised that product 3aa might degrade, to some extent, under the current reaction conditions. Consistent with our hypothesis, lowering the reaction temperature led to a significant enhancement in reaction yields (entry 7–8). Further promotion in reaction efficiency was observed when DCE was used as the solvent, resulting in the isolation of 3aa in 92% yield (entry 9). Finally, control experiments showed that the Lewis acid is of critical importance for product formation (entry 10).|
|
|||
|---|---|---|---|
| Entry | Lewis acid | Conversion of 1a (%) | Yield (%) |
| a Reaction preformed with 1a (0.1 mmol, 1.0 equiv.), 2a (0.15 mmol, 1.5 equiv.), Lewis acid (20 mol%), DCM (0.1 M), and 12 h. Yield was determined by 19F NMR analysis using 1-iodo-4-(trifluoromethyl)benzene as the internal standard. b Reaction preformed at 0 °C. c Reaction preformed at −20 °C. d Reaction preformed with DCE as solvent. e Isolated yield was given in parentheses. | |||
| 1 | AlCl3 | 97 | 65 |
| 2 | Zn(OTf)2 | 12 | 11 |
| 3 | Yb(OTf)3 | 47 | 34 |
| 4 | MgI2 | 65 | 0 |
| 5 | In(OTf)3 | 100 | 62 |
| 6 | Sn(OTf)2 | 100 | 0 |
| 7b | AlCl3 | 98 | 77 |
| 8c | AlCl3 | 99 | 82 |
| 9c,d,e | AlCl3 | 100 | 95(92) |
| 10c,d | — | 0 | 0 |
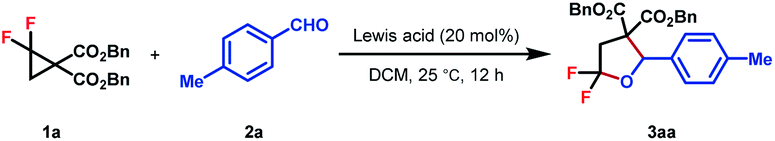
With the optimized conditions in hand, the generality of this (3 + 2)-cycloaddition was investigated with respect to different aldehydes by reacting with 1a (Table 2). Various substituted aromatic aldehydes were evaluated firstly. A range of functional groups with diverse electronic properties, either electron-rich (3aa–3ad) or deficient (3ae–3aj), were well tolerated under the standard reaction conditions to afford the corresponding products in moderate to good yields. The steric hindrance seemed to have no obvious effect on the reaction efficiency, and in the case of mesitylaldehyde 2b, product 3ab was obtained in 75% yield. The relatively low yield of cyano-substituted aldehydes might be caused by its Lewis basic nature, which may, to some extent, result in the attenuation of the reactivity of the catalyst (3ai). To our delight, heteroaromatic aldehydes performed equally well, which gave rise to biheterocyclic products in good yields (3ak–3al). In addition to aromatic aldehydes, aliphatic aldehydes, either as primary (3am–3an) or secondary (3ao–3ap) ones, were also viable substrates, which delivered the desired products in 53–65% yields. Pleasingly, the substrate scope could be further extended to acrolein and phenylpropargyl aldehydes to obtain gem-difluorotetrahydrofuran with extra alkene (3aq) or alkyne (3ar) structural motifs, which provided versatile handles for further synthetic elaboration. Finally, the reaction of p-phthalaldehyde with 2.5 equiv. of 1a also proceeded smoothly to give 3as in 83% yield as a mixture of two diasteroisomers.
| a Reaction conditions: 1 (0.2 mmol, 1.0 equiv.), 2 (0.3 mmol, 1.5 equiv.), AlCl3 (20 mol%), DCE (0.1 M), −20 °C, and 12 h. Diastereomeric ratios were determined by 1H NMR analysis of the crude reaction mixture. Isolated yield. b 1a (0.5 mmol, 2.5 equiv.) and terephthalaldehyde (0.2 mmol, 1.0 equiv.). |
|---|
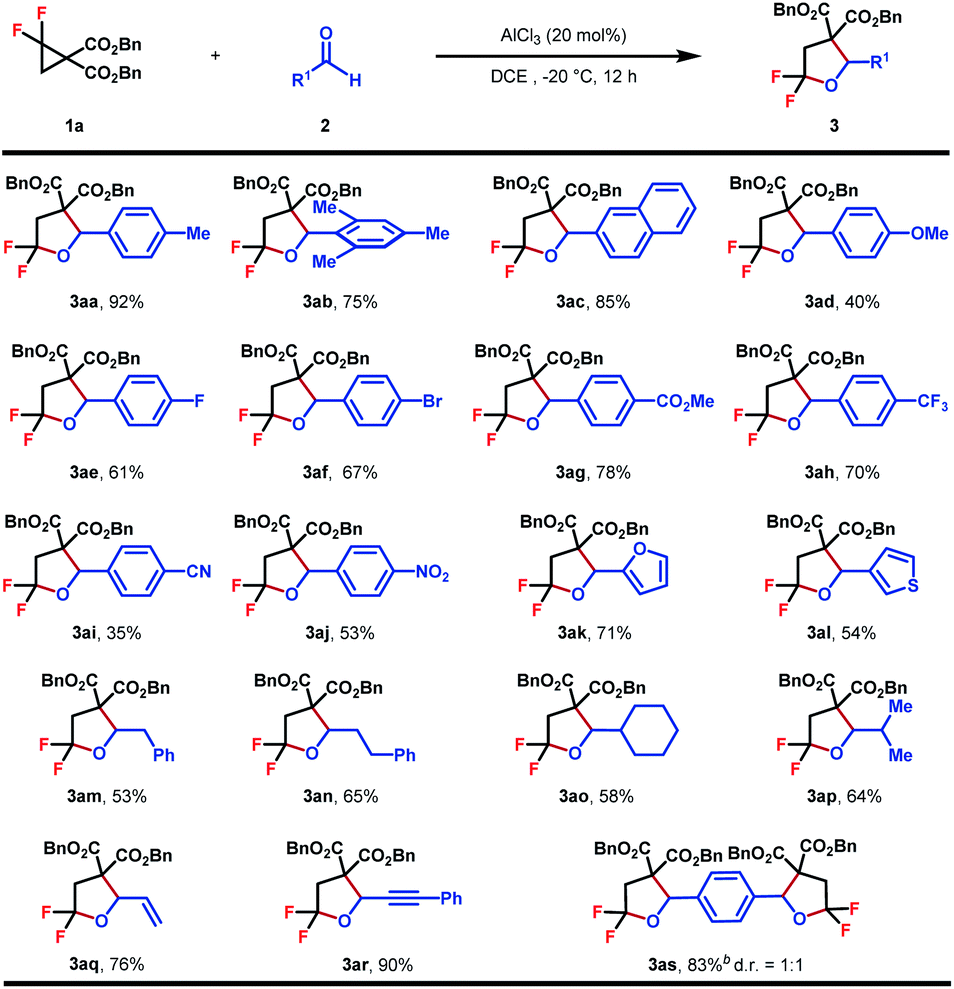
|
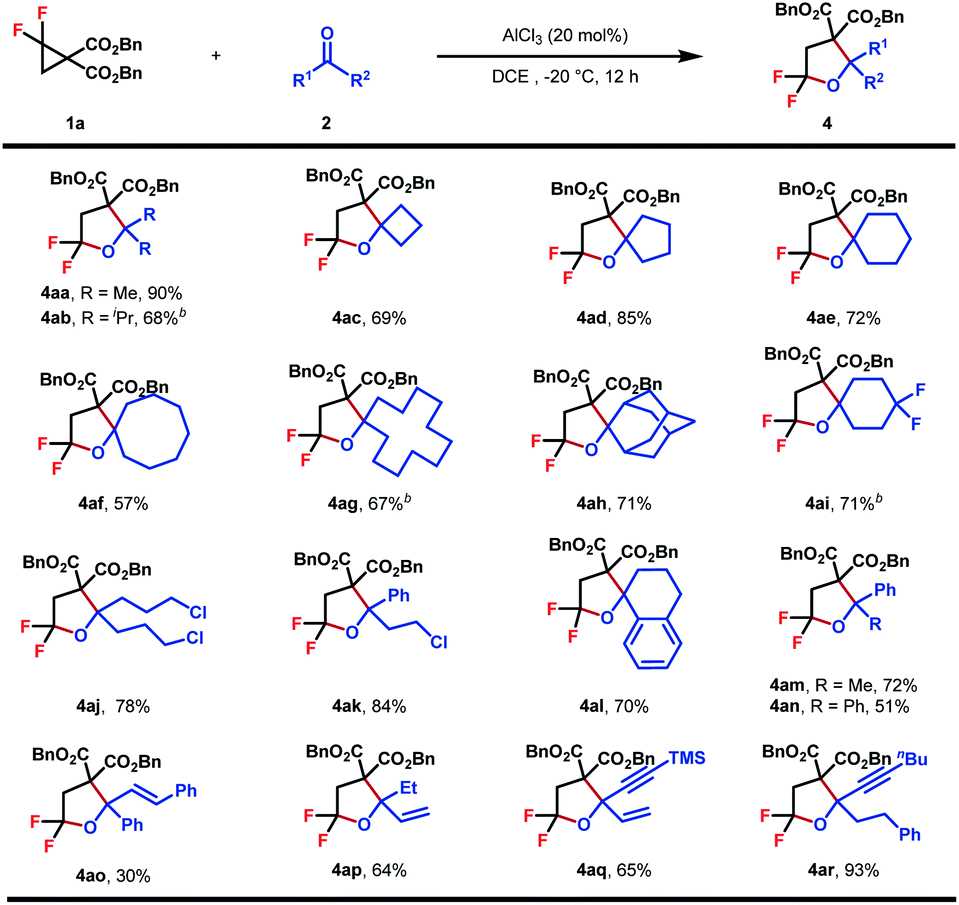
Encouraged by these outcomes, we turned to evaluate the scope of ketones as cycloaddition partners (Table 3). The reaction worked well with symmetrical aliphatic ketones (4aa–4aj), even those steric congested ones (4ab and 4ah). In some cases (4ab, 4ag, and 4ai), when AlCl3 was used as the Lewis acid catalyst, byproduct dibenzyl 2-(2-chloro-2,2-difluoroethyl)malonate could be detected, the formation of which was interpreted by a competitive nucleophilic attack of chloride ions originating from AlCl3. This issue, however, could be circumvented by replacing AlCl3 with In(OTf)3 to guarantee satisfactory yields. Simple unsymmetrical aliphatic and aromatic ketones also performed well to yield the corresponding products (4ak–4ar). A series of functional groups, including halogen (4ak), terminal and internal alkenes (4ap, 4aq, and 4ao) and internal alkynes (4aq and 4ar), proved to be compatible with the reaction conditions, showing the good compatibility of this reaction.
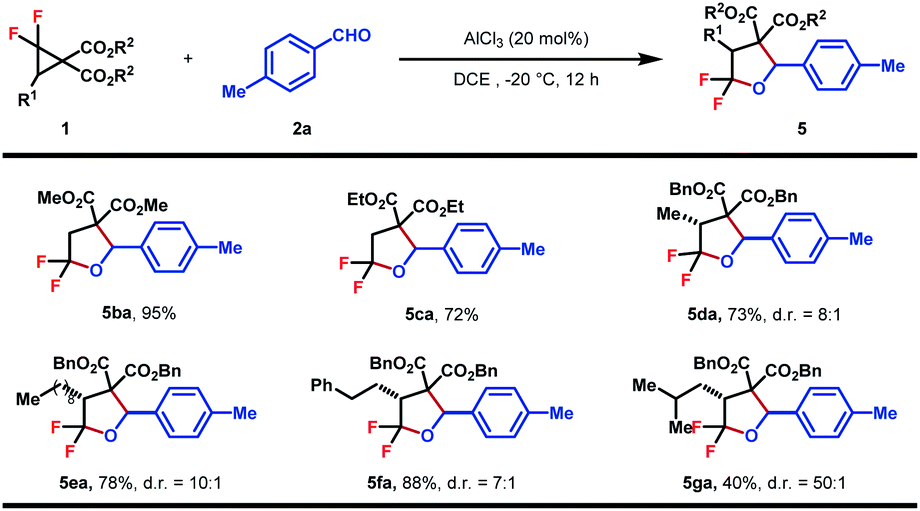
The substrate scope of DACs was then explored (Table 4). The switching of the benzylic ester to methyl ester did not affect the reaction efficiency and 5ba was obtained in a similar yield, higher than that of the ethyl ester analogue (5ca). A set of gem-difluorocyclopropane diesters with an additional alkyl substituent at C3 were synthesized and found to react well with 2a to afford the desired products in fair to good yields and high to excellent diastereoselectivities (5da–5ga). Increasing the steric hindrance of the alkyl substituent was beneficial to the stereoselectivity, albeit at the expense of yield (5ga). The trans-configuration of 5da was determined by NOESY of the major isomer and others were assigned in analogy. Note that in these cases, the bond between the gem-difluorine and diester group was cleaved selectively, indicating a more favored nucleophilic attack at the gem-difluorocarbon atom.
| a Reaction conditions: 1 (0.2 mmol, 1.0 equiv.), 2 (0.3 mmol, 1.5 equiv.), AlCl3 (20 mol%), DCE (0.1 M), −20 °C, and 12 h. Diastereomeric ratios were determined by 1H NMR analysis of the crude reaction mixture. Isolated yield. |
|---|
|
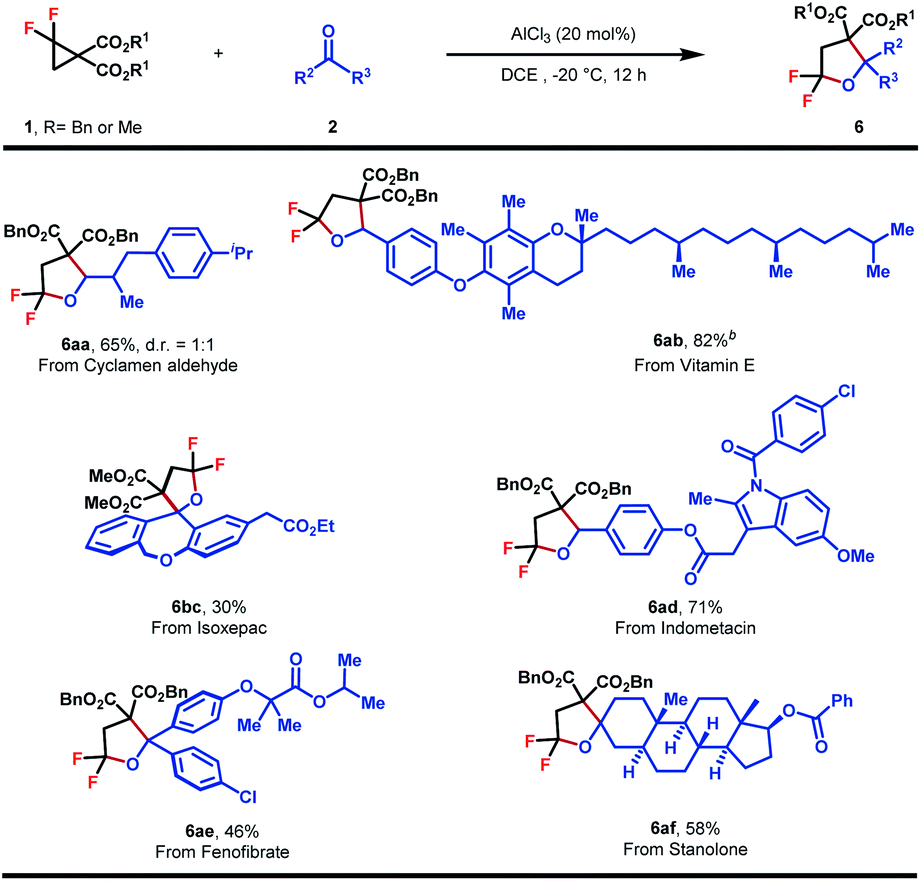
To further show the synthetic applicability, this method was applied to the late-stage functionalization of some natural products and pharmaceutically relevant molecules (Table 5). Cyclamen aldehydes and aldehydes derived from vitamin E and indometacin all proceeded well with 1a, providing 6aa, 6ab and 6ad in good to high yields. Furthermore, ketone reaction partners originating from fenofibrate, stanolone and isoxepac also underwent the (3 + 2)-cycloaddition uneventfully, affording hex-substituted tetrahydrofurans in moderate yields (6bc, 6ae and 6af). Taken together, the method developed herein could enable modular construction for the rapid installation of gem-difluoroheterocycle into molecular structures of interest.
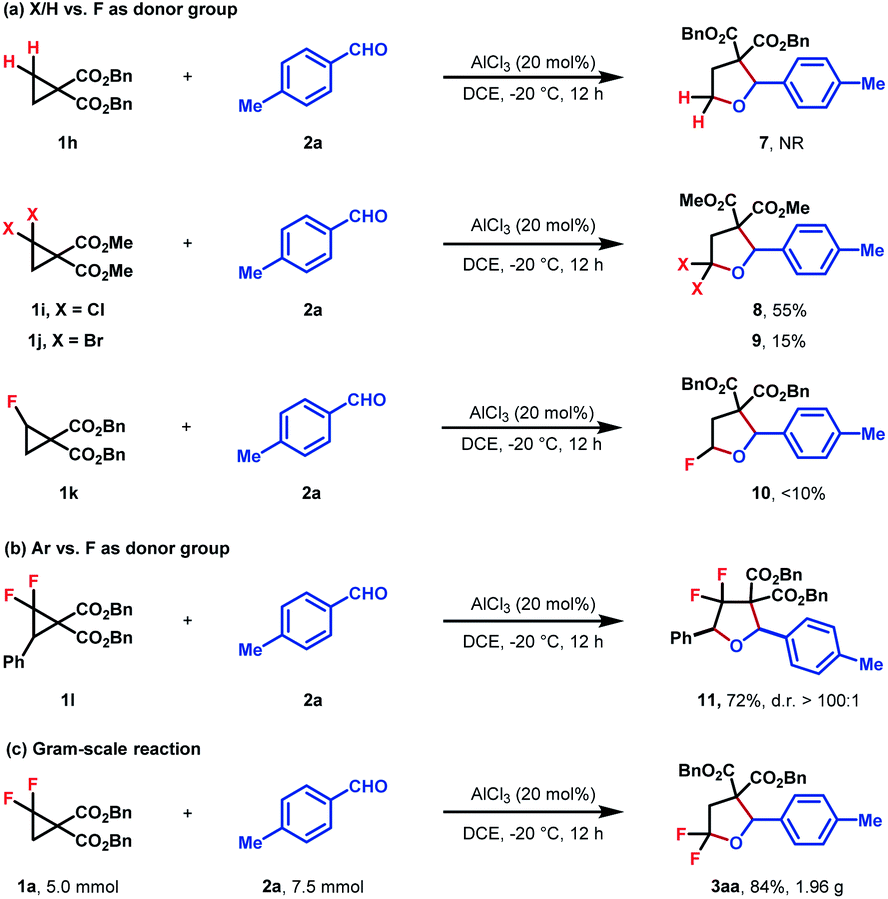
To better investigate the role of fluorine atoms in this reaction, a series of control experiments were then executed. The reaction of simple cyclopropane diester 1h with 2a under standard conditions resulted in the full recovery of 1h (Scheme 2a). Furthermore, gem-dichloro and gem-dibromocyclopropane diesters 1i and 1j were also prepared and subjected to the standard reaction conditions, which, however, delivered the desired products 8 and 9 in 55% and 15% yields, respectively (Scheme 2a). These two experiments revealed that the order of reactivity of cyclopropane entities in cycloadditions follows F > Cl > Br > H. The higher reactivity of F-based cyclopropane could be rationalized by the fact that the 2p-orbitals of fluorine are roughly the same size as that of the carbon atom, which leads to more effective resonance stabilization of positive charge developing at the α-carbon atom. When monofluorocyclopropane 1k was employed, the desired product 10 was detected in less than 10% yield, indicating the relatively weak π-electron donating ability of the mono-fluorine atom in activating the substrate for ring-opening functionalization. However, when gem-difluorocyclopropane diesters with an aryl group at C3 were subjected to the standard conditions, the oxygen of carbonyl exclusively attacked the C3 position and 3,3-difluoro substituted tetrahydrofuran 11 was obtained as the sole product, indicating that as compared with the gem-difluorine substituent, the aryl group is more apt to stabilize the adjacent positive charge (Scheme 2b). A gram-scale reaction using the model substrate was also performed, which produced the desired product 3aa without an obvious reduction in yield (Scheme 2c).
 | ||
| Scheme 2 Control experiments and gram-scale reaction. | ||
To gain more insight into the mechanism, density functional theory (DFT) calculation of this (3 + 2)-cycloaddition reaction was conducted with the Gaussian 09 program at the B3-LYP level of theory with the 6-31G(d, p) basis set in MeCN implicitly.18 The Gibbs free energy profile for the cycloaddition process is shown in Fig. 1. In the presence of AlCl3, the intermediate 1b-I was produced first upon coordination, with a Gibbs free energy downhill of 31.14 kcal mol−1. Then, a SN2 nucleophilic attack of aldehyde 2a to C1 of 1b-I occurred to generate the intermediate 1b-II. The activation barrier of this procedure was 15.1 kcal mol−1. Followed by a barrierless C1–C3 bond rotation, ensuing intramolecular cyclization afforded the final product, upon disassociation of AlCl3. According to the Gibbs free energy profiles, the SN2 ring-opening C–C bond cleavage was found to be the rate-determining step. Overall, the calculated reaction pathway is in good accordance with the model proposed by Johnson et al. of the Lewis acid catalyzed (3 + 2)-cycloaddition of D–A cyclopropanes with aldehydes.2b Furthermore, the electron distributions of 1b, 1b-I and 1b-TS1, in terms of Mulliken charge, were obtained respectively.17 Compared to 1b, the positive charges on the C1 of 1b-I increased from 0.610 to 0.649 after AlCl3 coordination. As expected, more positive charge on C1 was observed for the transition state TS1. In contrast, the C2 and C3 of 1b and 1b-I always showed negative charge. The length of the C–C bond between C1 and C2 was also found to be elongated from 1.5070 Å to 1.5639 Å after Lewis acid coordination. In addition, according to the theory of frontier molecular orbitals, the atom with a higher contribution to the LUMO is more susceptible to nucleophilic attack. The LUMOs of 1b and 1b-I were therefore calculated, which indicated the largest coefficients at C1 for both entities.17 Taken together, these results indicate that the attack of nucleophile 2a at the C1 of 1b is controlled by both the electrostatic effect and orbital interaction.
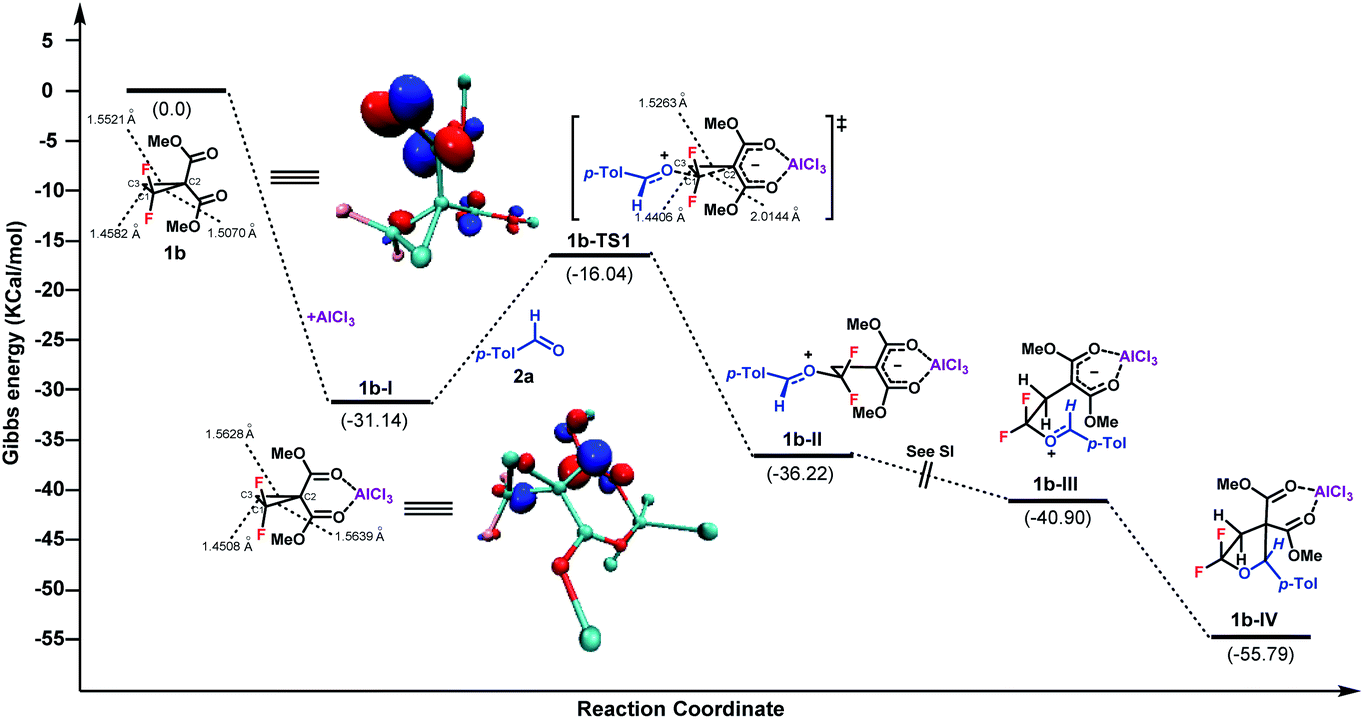 | ||
| Fig. 1 DFT calculation for the (3 + 2)-cycloaddition of gem-difluorocyclopropane and aldehyde. LUMO of 1b and 1b-I with an isovalue of 0.1 atomic units. | ||
Conclusions
In summary, by taking advantage of the π-electron donation properties of fluorine, a new class of DCAs is developed, which could participate in Lewis-acid catalyzed (3 + 2)-cycloadditions with aldehydes/ketones, thus enabling a straightforward construction of densely substituted gem-difluorotetrahydrofurans. This work represents the first example of using gem-difluorine as the donor group in DAC involved reactions, which also opens up a new reaction mode of gem-difluorocyclopropanes. The critical role of fluorine is probed by control experiments as well as DFT calculations. Further applications of the α-cation stabilizing effect of fluorine are underway in our laboratory.Data availability
Data for this work, including experimental procedures, characterization data for all new compounds, and DFT computational details are provided in the ESI.†Author contributions
C. F. conceived and directed the project. H. L., H. W. and Z.-Q. L. performed the experiment and collected the data. L. T. performed all DFT calculations. H. L., C. Z. and C. F. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the “Thousand Talents Plan” Youth Program, the “Jiangsu Specially-Appointed Professor Plan”, and the National Natural Science Foundation of China (21871138).Notes and references
- Reviews of D–A cyclopropanes: (a) P. Singh, R. K. Varshnaya, R. Dey and P. Banerjee, Adv. Synth. Catal., 2020, 362, 1447–1484 CrossRef CAS; (b) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS PubMed; (c) D. B. Werz and A. T. Biju, Angew. Chem., Int. Ed., 2020, 59, 3385–3398 CrossRef CAS PubMed; (d) A. U. Augustin and D. B. Werz, Acc. Chem. Res., 2021, 54, 1528–1541 CrossRef CAS PubMed; (e) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804–818 RSC; (f) F. de Nanteuil, F. De Simone, R. Frei, F. Benfatti, E. Serrano and J. Waser, Chem. Commun., 2014, 50, 10912–10928 RSC; (g) B. L. Pagenkopf and N. Vemula, Eur. J. Org. Chem., 2017, 2017, 2561–2567 CrossRef CAS.
- For selected examples, see: (a) I. S. Young and M. A. Kerr, Angew. Chem., Int. Ed., 2003, 42, 3023–3026 CrossRef CAS PubMed; (b) P. D. Pohlhaus, S. D. Sanders, A. T. Parsons, W. Li and J. S. Johnson, J. Am. Chem. Soc., 2008, 130, 8642–8650 CrossRef CAS PubMed; (c) S. Xing, W. Pan, C. Liu, J. Ren and Z. Wang, Angew. Chem., Int. Ed., 2010, 49, 3215–3218 CrossRef CAS PubMed; (d) A. U. Augustin, M. Sensse, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2017, 56, 14293–14296 CrossRef CAS PubMed; (e) D.-C. Wang, M.-S. Xie, H.-M. Guo, G.-R. Qu, M.-C. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2016, 55, 14111–14115 CrossRef CAS PubMed; (f) A. T. Parsons, A. G. Smith, A. J. Neel and J. S. Johnson, J. Am. Chem. Soc., 2010, 132, 9688–9692 CrossRef CAS PubMed.
- For the review, see: (a) J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110–139 CrossRef CAS PubMed; (b) B. M. Trost and P. J. Morris, Angew. Chem., Int. Ed., 2011, 50, 6167–6170 CrossRef CAS PubMed; (c) A. P. Dieskau, M. S. Holzwarth and B. Plietker, J. Am. Chem. Soc., 2012, 134, 5048–5051 CrossRef CAS PubMed; (d) Y. Miyake, S. Endo, T. Moriyama, K. Sakata and Y. Nishibayashi, Angew. Chem., Int. Ed., 2013, 52, 1758–1762 CrossRef CAS PubMed; (e) Q. Cheng, J.-H. Xie, Y.-C. Weng and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 5739–5743 CrossRef CAS PubMed; (f) J. Moran, A. G. Smith, R. M. Carris, J. S. Johnson and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 18618–18621 CrossRef CAS PubMed; (g) R. Tombe, T. Kurahashi and S. Matsubara, Org. Lett., 2013, 15, 1791–1793 CrossRef CAS PubMed; (h) F. de Nanteuil, E. Serrano, D. Perrotta and J. Waser, J. Am. Chem. Soc., 2014, 136, 6239–6242 CrossRef CAS PubMed.
- (a) J. Blom, A. Vidal-Albalat, J. Jørgensen, C. L. Barløse, K. S. Jessen, M. V. Iversen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2017, 56, 11831–11835 CrossRef CAS PubMed; (b) K. S. Halskov, F. Kniep, V. H. Lauridsen, E. H. Iversen, B. S. Donslund and K. A. Jørgensen, J. Am. Chem. Soc., 2015, 137, 1685–1691 CrossRef CAS PubMed; (c) E. Sanchez-Diez, D. L. Vesga, E. Reyes, U. Uria, L. Carrillo and J. L. Vicario, Org. Lett., 2016, 18, 1270–1273 CrossRef CAS PubMed; (d) J. Wallbaum, L. K. B. Garve, P. G. Jones and D. B. Werz, Chem.–Eur. J., 2016, 22, 18756–18759 CrossRef CAS PubMed.
- (a) V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS PubMed; (b) Y. Xia, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 9192–9204 CrossRef CAS PubMed.
- For reviews, see: (a) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051–3060 RSC; (b) B. M. Trost, W.-J. Bai, C. Hohn, Y. Bai and J. J. Cregg, J. Am. Chem. Soc., 2018, 140, 6710–6717 CrossRef CAS PubMed; (c) R. Frei, D. Staedler, A. Raja, R. Franke, F. Sasse, S. Gerber-Lemaire and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 13373–13376 CrossRef CAS PubMed; (d) F. De Simone, J. Gertsch and J. Waser, Angew. Chem., Int. Ed., 2010, 49, 5767–5770 CrossRef CAS PubMed; (e) P. Liu, Y. Cui, K. Chen, X. Zhou, W. Pan, J. Ren and Z. Wang, Org. Lett., 2018, 20, 2517–2521 CrossRef CAS PubMed; (f) B. Sun, J. Ren, S. Xing and Z. Wang, Adv. Synth. Catal., 2018, 360, 1529–1537 CrossRef CAS.
- (a) A. Kreft, A. Lücht, J. Grunenberg, P. G. Jones and D. B. Werz, Angew. Chem., Int. Ed., 2019, 58, 1955–1959 CrossRef CAS PubMed; (b) A. Kreft, P. G. Jones and D. B. Werz, Org. Lett., 2018, 20, 2059–2062 CrossRef CAS PubMed.
- For selected examples, see: (a) S. Haubenreisser, P. Hensenne, S. Schröder and M. Niggemann, Org. Lett., 2013, 15, 2262–2265 CrossRef CAS PubMed; (b) A. G. Smith, M. C. Slade and J. S. Johnson, Org. Lett., 2011, 13, 1996–1999 CrossRef CAS PubMed; (c) W. Zhu, J. Fang, Y. Liu, J. Ren and Z. Wang, Angew. Chem., Int. Ed., 2013, 52, 2032–2037 CrossRef CAS PubMed; (d) A. U. Augustin, M. Busse, P. G. Jones and D. B. Werz, Org. Lett., 2018, 20, 820–823 CrossRef CAS PubMed; (e) M. P. Sibi, Z. Ma and C. P. Jasperse, J. Am. Chem. Soc., 2005, 127, 5764–5765 CrossRef CAS PubMed; (f) J. Preindl, S. Chakrabarty and J. Waser, Chem. Sci., 2017, 8, 7112–7118 RSC; (g) L. K. B. Garve, P. Barkawitz, P. G. Jones and D. B. Werz, Org. Lett., 2014, 16, 5804–5807 CrossRef CAS PubMed; (h) S. Xing, Y. Li, Z. Li, C. Liu, J. Ren and Z. Wang, Angew. Chem., Int. Ed., 2011, 50, 12605–12609 CrossRef CAS PubMed.
- For selected examples, see: (a) F. Benfatti, F. d. Nanteuil and J. Waser, Org. Lett., 2012, 14, 386–389 CrossRef CAS PubMed; (b) F. Benfatti, F. de Nanteuil and J. Waser, Chem.–Eur. J., 2012, 18, 4844–4849 CrossRef CAS PubMed; (c) F. de Nanteuil and J. Waser, Angew. Chem., Int. Ed., 2011, 50, 12075–12079 CrossRef CAS PubMed; (d) S. Racine, F. de Nanteuil, E. Serrano and J. Waser, Angew. Chem., Int. Ed., 2014, 53, 8484–8487 CrossRef CAS PubMed; (e) D. Perrotta, M.-M. Wang and J. Waser, Angew. Chem., Int. Ed., 2018, 57, 5120–5123 CrossRef CAS PubMed; (f) B. Muriel, A. Gagnebin and J. Waser, Chem. Sci., 2019, 10, 10716–10722 RSC; (g) M.-C. Zhang, D.-C. Wang, M.-S. Xie, G.-R. Qu, H.-M. Guo and S.-L. You, Chem, 2019, 5, 156–167 CrossRef CAS.
- For selected examples, see: (a) H.-U. Reissig, H. Holzinger and G. Glomsda, Tetrahedron, 1989, 45, 3139–3150 CrossRef CAS; (b) H.-U. Reissig, Tetrahedron Lett., 1981, 22, 2981–2984 CrossRef CAS.
- (a) C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC; (b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (c) R. E. Banks and J. C. Tatlow, J. Fluorine Chem., 1986, 33, 227–346 CrossRef; (d) Organofluorine Chemistry, ed. B. E. Smart and J. C. Tatlow, Plenum Press, New York, 1994, pp. 57–88 Search PubMed; (e) K. Uneyama, Organofluorine Chemistry, Blackwell, Oxford, 2006 CrossRef; (f) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214–231 CrossRef CAS PubMed; (g) K. Fuchibe, Y. Mayumi, N. Zhao, S. Watanabe, M. Yokota and J. Ichikawa, Angew. Chem., Int. Ed., 2013, 52, 7825–7828 CrossRef CAS PubMed.
- (a) K. Fuchibe, H. Hatta, K. Oh, R. Oki and J. Ichikawa, Angew. Chem., Int. Ed., 2017, 56, 5890–5893 CrossRef CAS PubMed; (b) K. Fuchibe, H. Jyono, M. Fujiwara, T. Kudo, M. Yokota and J. Ichikawa, Chem.–Eur. J., 2011, 17, 12175–12185 CrossRef CAS PubMed.
- H. Liu, Y. Li, D.-X. Wang, M.-M. Sun and C. Feng, Org. Lett., 2020, 22, 8681–8686 CrossRef CAS PubMed.
- (a) H. Liu, L. Ge, D.-X. Wang, N. Chen and C. Feng, Angew. Chem., Int. Ed., 2019, 58, 3918–3922 CrossRef CAS PubMed; (b) H.-J. Tang, L.-Z. Lin, C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2017, 56, 9872–9876 CrossRef CAS PubMed; (c) H.-J. Tang, X. Zhang, Y.-F. Zhang and C. Feng, Angew. Chem., Int. Ed., 2020, 59, 5242–5247 CrossRef CAS PubMed; (d) C.-Q. Wang, Y. Zhang and C. Feng, Angew. Chem., Int. Ed., 2017, 56, 14918–14922 CrossRef CAS PubMed; (e) C. Zhu, M.-M. Sun, K. Chen, H. Liu and C. Feng, Angew. Chem., Int. Ed., 2021, 60, 20237–20242 CrossRef CAS PubMed; (f) L. Tang, Z.-Y. Liu, W. She and C. Feng, Chem. Sci., 2019, 10, 8701–8705 RSC; (g) P. Tian, C.-Q. Wang, S.-H. Cai, S. Song, L. Ye, C. Feng and T.-P. Loh, J. Am. Chem. Soc., 2016, 138, 15869–15872 CrossRef CAS PubMed; (h) C. Zhu, Z.-Y. Liu, L. Tang, H. Zhang, Y.-F. Zhang, P. J. Walsh and C. Feng, Nat. Commun., 2020, 11, 4860 CrossRef CAS PubMed; (i) C.-Q. Wang, Y. Li and C. Feng, Cell Rep. Phys. Sci., 2021, 2, 100461 CrossRef CAS.
- (a) P. D. Pohlhaus and J. S. Johnson, J. Am. Chem. Soc., 2005, 127, 16014–16015 CrossRef CAS PubMed; (b) A. T. Parsons and J. S. Johnson, J. Am. Chem. Soc., 2009, 131, 3122–3123 CrossRef CAS PubMed; (c) P. Yang, Y. Shen, M. Feng, G. Yang and Z. Chai, Eur. J. Org. Chem., 2018, 2018, 4103–4112 CrossRef CAS; (d) P. D. Pohlhaus and J. S. Johnson, J. Org. Chem., 2005, 70, 1057–1059 CrossRef CAS PubMed; (e) G. Yang, Y. Shen, K. Li, Y. Sun and Y. Hua, J. Org. Chem., 2011, 76, 229–233 CrossRef CAS PubMed; (f) G. Yang, Y. Sun, Y. Shen, Z. Chai, S. Zhou, J. Chu and J. Chai, J. Org. Chem., 2013, 78, 5393–5400 CrossRef CAS PubMed.
- F. Tian, V. Kruger, O. Bautista, J.-X. Duan, A.-R. Li, W. R. Dolbier and Q.-Y. Chen, Org. Lett., 2000, 2, 563–564 CrossRef CAS PubMed.
- See the ESI† for details.
- (a) M. J. Frischi, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucii, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01 Search PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (d) R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2011, 2, 2810–2817 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00302c |
| This journal is © The Royal Society of Chemistry 2022 |