
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaterial design and surface chemistry for advanced rechargeable zinc–air batteries
Soobeom
Lee†
abc,
Jinyeong
Choi†
abc,
Minsoo
Kim
abc,
Jihan
Park
abc,
Minjoon
Park
*abc and
Jaephil
Cho
 *d
*d
aDepartment of Nanoenergy Engineering, Pusan National University, 50, Busan daehak-ro 63 beon-gil 2, Geumjeong-gu, Busan 46241, Republic of Korea. E-mail: mjpark@pusan.ac.kr
bResearch Center of Energy Convergence Technology, Pusan National University, Busandaehak-ro 63beon-gil 2, Geumjeong-gu, Busan, 46241, Republic of Korea
cDepartment of Nano Fusion Technology, Pusan National University, Busandaehak-ro 63beon-gil 2, Geumjeong-gu, Busan, 46241, Republic of Korea
dDepartment of Energy Engineering, School of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan, Republic of Korea. E-mail: jpcho@unist.ac.kr
First published on 25th April 2022
Abstract
Zinc–air batteries (ZABs) have been considered as a next-generation battery system with high energy density and abundant resources. However, the sluggish multi-step reaction of the oxygen is the main obstacle for the practical application of ZABs. Therefore, bifunctional electrocatalysts with high stability and activity for the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER) are greatly required to promote the catalytic reaction. In this review, we first explain the reaction mechanism of the ZABs, mainly focusing on multiple oxygen intermediates. Then, the latest studies on bifunctional electrocatalysts for the air cathodes and their progress of the ZABs are discussed with following aspects: platinum group metal, metal-free, transition metal, and metal compound-derived electrocatalysts. Finally, we highlight the advanced ZAB systems with the design of the full-temperature range operation, the all-solid-state, and the newly reported non-alkaline electrolyte, summarizing the remaining challenges and requirements of the future research directions.
 Minjoon Park | Minjoon Park is Professor of Department of Nanoenergy Engineering at Pusan National University (Seoul, Republic of Korea). After receiving his Ph.D. from the School of Energy Engineering at Ulsan National Institute of Science and Technology (UNIST, Republic of Korea), he joined School of Engineering and Applied Science at Harvard University (MA, USA) and worked with Prof. Michael J. Aziz as a postdoctoral fellow. After then, he returned to Korea as Staff Researcher at Samsung SDI. His research is now focused on the development of electrocatalysts and electrode materials and investigating their electrochemical properties, particularly for redox flow, flexible Li-ion, and Al–air batteries for energy storage. |
 Jaephil Cho | Jaephil Cho (Ulsan National Institute of Science & Technology; UNIST) is a UNIST distinguished professor of Energy & Chemical Engineering. He studied at Iowa State University, Ames, and worked with Steve W. Martin at the latter institution for his Ph.D. (awarded in 1995). In 2009, he was made professor and Dean of Research Affairs of UNIST and Director of the Samsung SDI-UNIST Future Batteries Research Center. Cho's current research is focused on high energy density cathode and anode materials and their direct implantation in full-cell systems, as well as metal–air batteries and redox flow batteries for energy storage. |
1. Introduction
The global demand of sustainable and green energy storage technologies is significantly increasing to achieve carbon neutrality because of the increasing consumption of fossil fuels, which are rapidly deteriorating the global environment.1–3 Over the decade, many efforts have been conducted to address the security of energy supply, and the demand for sustainable green energy systems is increasing all around the world.4 Therefore, the environmentally friendly energy conversion and storage devices such as fuel cells, electrochemical water-splitting systems, and metal–air batteries have been widely studied due to their clean property.5,6 With respect to the metal–air batteries, this system can be operated without pre-storing oxygen in the cells. It is noteworthy to mention that oxygen can be taken directly from the ambient air. Because of this merit, this energy storage device shows the higher theoretical energy density compared to that of commercial lithium-ion batteries.7 Among them, the ZABs have attracted substantial attention in recent decades due to their high theoretical energy density (1084 W h kg−1, three to fourfold higher than commercial lithium-ion batteries), non-toxicity, low cost, and safety. The ZABs were originally developed as primary batteries; however, many different types of them have been demonstrated using a bifunctional electrocatalysts for recharging, flexible design, or solar-coupled rechargeable systems.8–17 Despite the high applicability of ZABs in various fields, the large overpotential at the air cathode during the charge and discharge cycles seriously hinders the practical application of the ZABs because of the sluggish kinetics of oxygen in the multi-step electron transfer process at the air cathode.4,5 This issue increases the full-cell overpotential of the ZAB system, causing significant energy loss.6 In order to address these limitations in the ZABs, it is urgent to develop low-cost, efficient electrocatalysts with long-term cycle stability.To date, Pt-based electrocatalysts have been most extensively used as ORR electrocatalysts. Similarly, IrO2 and RuO2 electrocatalysts have been employed for OER.18 However, platinum group materials (PGMs) do not satisfy the practical property of the ZABs because of their high cost, scarcity, and non-multifunctional electrocatalytic activities.19 With respect to the promising non-precious metal bifunctional electrocatalysts, the carbon-supported transition metal structure materials and metal-free carbon have been widely reported.18,19,23 The doping of heterogeneous non-metallic materials is considered as a good approach for modifying or optimizing the electronic structure of metallic materials.1,20 In particular, heteroatom doping including nitrogen, sulfur, phosphorous, and boron on the carbon matrix has received huge attention due to its high efficiency and tunability.10,21,22 Although previous reviews have well discussed the progress of the electrocatalysts according to their structures and materials, the recent intriguing topic for novel electrocatalysts and advanced ZAB systems have not been specified in detail. Thus, it is worth discussing the state-of-the art designs and strategies of ZABs in terms of atomic and electronic engineering and advanced cell designs.
In this review, the chemical mechanism of the ZABs and the multistep reaction of oxygen are firstly explained in detail. Then, we introduce the newly reported oxygen electrocatalysts with excellent oxygen reversibility in recent years in the following aspects: platinum group metals, metal-free, transition metals, and metal–organic framework-derived electrocatalysts. The bifunctional catalytic activity of electrocatalysts and their electrochemical performance when applied to ZABs are reviewed with surface electronic aspects. Notably, we highlight advanced ZABs with a unique design, including the full-temperature range operation, all-solid-state systems, and newly reported non-alkaline electrolytes. In the end, the future perspectives for existing challenges in electrocatalysts for ZABs will be discussed (Fig. 1).
 | ||
| Fig. 1 The schematic of bifunctional electrocatalyst design and advanced systems for rechargeable Zn–air batteries. | ||
2. Chemical mechanisms of ZABs
2.1 Working mechanisms of the ZABs
ZAB consists of four main components such as a zinc anode, a separator, an electrolyte, and an air cathode (Fig. 2a).24 In general, alkaline solutions such as KOH or NaOH solutions are used as aqueous electrolytes for ZABs. ZABs are operated by electrochemical reactions that occur at the zinc metal anode and the air cathode in an alkaline electrolyte during the charging and discharging process.25 In the electrochemical reaction of ZABs, the reaction of oxygen is dominant because of the sluggish ORR. During the discharge process, the zinc anode is firstly oxidized, then zinc is converted to zincate ions, Zn(OH)42− (eqn (1)). Subsequently, this Zn(OH)42− precipitates as ZnO powder (eqn (2)). At this time, the ORR occurs at the air cathode, and oxygen is reduced to OH− (eqn (3)). During the subsequent charging process, the OER reaction occurs at the air electrode. Contrary to the discharge process, zinc is deposited at the anode and O2 is created at the air cathode, respectively.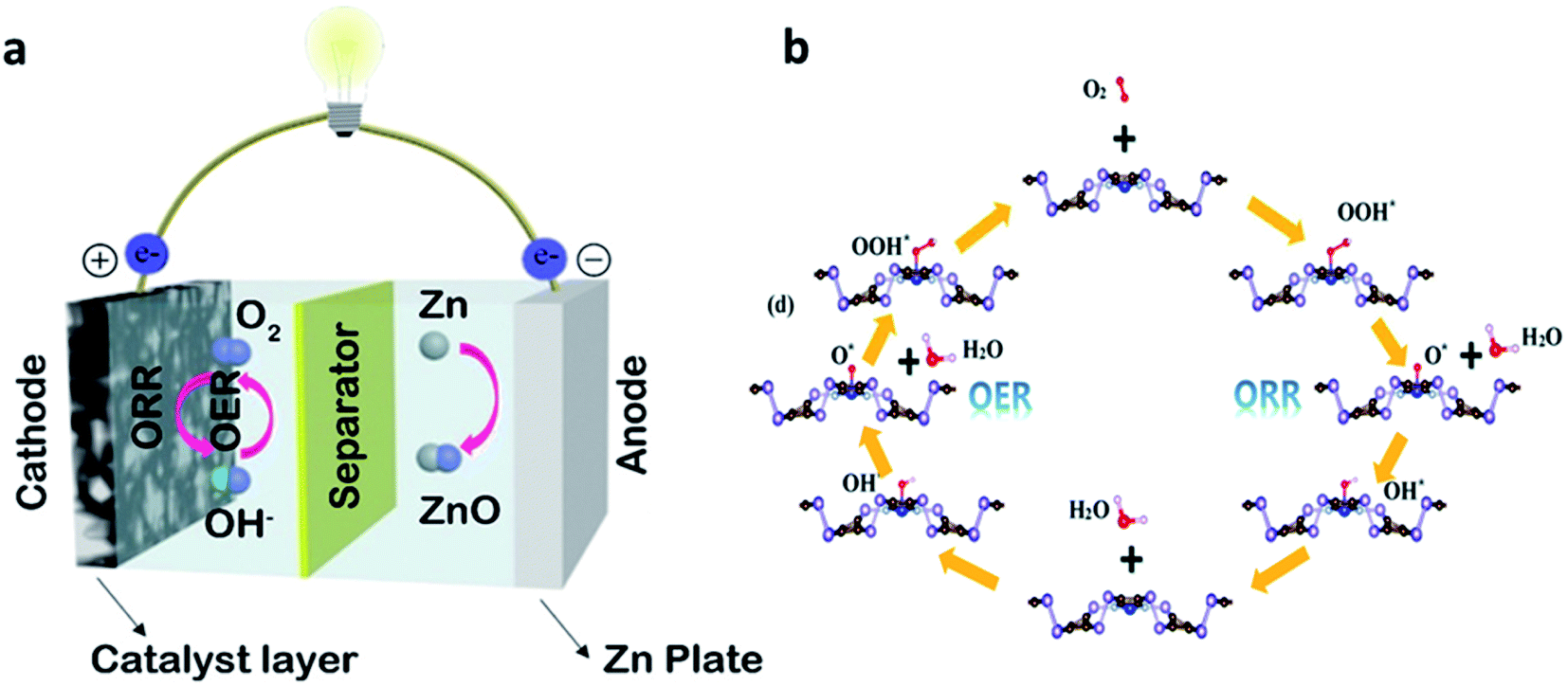 | ||
| Fig. 2 (a) Typical device of ZABs and air electrode of the ZAB structure. (a) Reprinted with permission from ref. 24. Copyright 2019, Elsevier. (b) The schematic of the OER and ORR path on the TM–N4 embedded b-PC monolayer. (b) Reprinted with permission from ref. 27. Copyright 2020, Royal Society of Chemistry. | ||
Zinc electrode reactions
| Zn + 4OH− ↔ Zn(OH)42− + 2e− (E = −1.25 V vs. SHE) | (1) |
| Zn(OH)42− ↔ ZnO + 2OH− + H2O | (2) |
Air electrode reaction
| O2 + H2O + 4e− ↔ 4OH− (E = 0.4 V vs. SHE) | (3) |
The overall reaction
| 2Zn + O2 ↔ 2ZnO (E = 1.65 V vs. SHE) | (4) |
Zinc can be electroplated in aqueous electrolytes; however, the reaction is easily passivated because of the local supersaturation of zincate ions in alkaline electrolyte, preventing the redistribution of zinc ions.26 Thus, the electrodeposition of zinc in the same position is hindered, which can cause the anode shape change or dendrite growth, degrading the battery performance and causing a short circuit in the cell. According to eqn (4), ZAB has a standard cell potential of 1.65 V; however, its practical discharge voltage can only reach about 1.2 V. More seriously, the charging voltage of the ZABs is usually higher than 2 V, which eventually reduces the energy efficiency of the ZABs. This large charge and discharge voltage gap is mainly caused by the unavoidable overpotentials of the ORR and OER by their slow kinetics. Therefore, many studies have been conducted on oxygen electrocatalysts to improve both the properties.27
2.2 Role of oxygen intermediates in the ZABs
The ORR in the ZABs is divided into four steps (Fig. 2b).28 Air diffuses through the pores of the air electrode, being adsorbed on the catalyst. After that, the O–O bond weakens and breaks. The adsorption and desorption of various oxygen species formed during the bond breakage affect the electrocatalyst performance.Oxygen reduction reactions
| O2 + 2H2O + 4e− + * → OOH* + OH− | (5) |
| OOH* + e− → O* + OH− | (6) |
| O* + H2O + e− → OH* + OH− | (7) |
| OH* + e− → 4OH− + * | (8) |
At each step, the reaction rate is greatly limited by the electron transfer rate by the multiple oxygen intermediates. The adsorption step of O2 is an effective standard for judging the ORR activity of the electrocatalyst. The multiple oxygen intermediates can be divided into *OO, *OOH, *O, and *OH (* is the active region). Among them, the formation energy of *OO is correlated with the length of the O–O bond. The longer the O–O bond length, the easier the oxygen dissociation reaction, following the adsorption of oxygen. Therefore, the overall reaction can be further activated by controlling and optimizing the adsorption energy of oxygen intermediates at the active sites.29 In addition, the catalytic activity trend of the oxygen can be confirmed from a thermodynamic point of view, which is correlated with the Gibbs free energy at each step.30 The reaction energy (ΔE) can be obtained through density functional theory (DFT) calculation, and the reaction free energy (ΔG) can be obtained through eqn (9).
ΔG = ΔH − TΔS − qU + kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln10 × (pH) ln10 × (pH) | (9) |
In general, the rate-determining step (RDS) in the ORR is the hydrogenation process of O2 to *OOH, and the ORR-activated electrocatalysis can lower the barrier to this reaction.31 The OER mechanism proceeds in the reverse direction of the ORR reaction (eqn (5)–(8)).
Oxygen evolution reactions
| 4OH− → O2 + 2H2O + 4e− | (10) |
According to the Sabatier principle, only an appropriate adsorption energy helps the overall OER/ORR reaction since a weak adsorption energy makes it difficult for the reactive species to bond with the catalysts.32
3. Design of electrocatalysts for ZABs
3.1 Conventional PGM electrocatalysts
The oxygen molecules in air can be diffused into the electrode, then reduced to OH−, which is the core reaction of the air cathode.33 The practical application of the ZAB has been severely hindered by the sluggish four-electron transfer kinetics of the ORR.34 To fabricate efficient ZABs, it is essential to develop bifunctional electrocatalysts with excellent ORR and OER performance. To date, noble metals such as Pt, Ir, and Ru have been considered as the most effective electrocatalysts for ORR.35 These PGMs have significantly high ORR activity. For example, platinum-supported carbon (Pt/C) has been regarded as the most powerful electrocatalyst for ORR, while RuO2 has been investigated as the electrocatalyst with the highest activity for OER.36,37 Nevertheless, the less active but more stable IrO2 is used as the benchmark electrocatalyst for application.38,39However, the conventional PGM electrocatalysts have the following intrinsic problems, such as scarcity of resources, low stability, detachment from support materials, and CO poisoning. Thus, the practical applications of ZABs have been greatly hindered, and the utilization for a large-scale energy storage system is also limited.33–36 To overcome these drawbacks, intensive efforts have been devoted to improving the catalytic activity using PGMs as the alloy or bimetallic nanoparticles with transition metals.33,34 The alloys and bimetallic nanostructures of transition metals and PGMs are good strategies to reduce the amount of precious metals, which can also improve the catalytic efficiency of the electrocatalysts.40–44 These methods can enhance the oxygen catalytic kinetics by increasing the active sites and the surface area of the electrocatalysts, providing stable characteristics against chemical oxidation due to the interaction of the d-orbital. In addition, the activation barriers of hydroxyl species in the limiting step can be changed.42,43,45 Moreover, they have resistance toward acidic environment because of the large formation enthalpy when forming the ordered phase after thermal treatment.45 Zhong et al. developed isolated bimetallic Pt and Fe single atoms anchored on nitrogen-doped carbon electrocatalysts using simple pyrolysis.33 The Pt–Fe bimetallic single atom showed a half-wave potential 0.895 V (E1/2), which was superior to the electrocatalyst using single Fe atoms (0.855 V). The DFT calculations revealed that the RDS including the breaking of the O–O bond and the formation of *OOH was improved in bimetallic single atom electrocatalysts than those of other electrocatalysts. In addition, the discharge characteristics of ZABs using PtFeNC were analyzed at various current densities and showed the most stable discharge voltage among the catalysts, including commercial Pt/C. This result was attributed to the high activity of bimetallic single atom sites, which consisted of four adjacent N atoms and one oxygen molecule. Gao et al. synthesized Pt–Fe pair single-site oxygen reduction electrocatalyst by loading atomic platinum on the α-Fe2O3(012) facets.46 Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) was employed to verify the distribution of single-site platinum dispersed in Fe2O3 (Fig. 3a). Fig. 3b shows the schematic diagram of the Pt–Fe electronic coupling effect. The iron d-band was matched with the platinum d-band to induce strong electrical coupling inside the Pt–Fe pair site. Electrical coupling gave the partially occupied dz2 orbital in platinum near the Fermi level, which was used as an O2 adsorption and activation site, showing excellent catalytic activity. Furthermore, the fully occupied eg orbital of the high-spin Fe3+ turned into a low-spin state, which was expected to be efficient for O2 adsorption and dissociation. The long term stability was evaluated using chronoamperometric response, and it retains more than 97% of the initial current density after 48 h (Fig. 3c). Fig. 3d shows the peak power density of 182 mW cm−2, which outperformed that of 20% Pt/C.
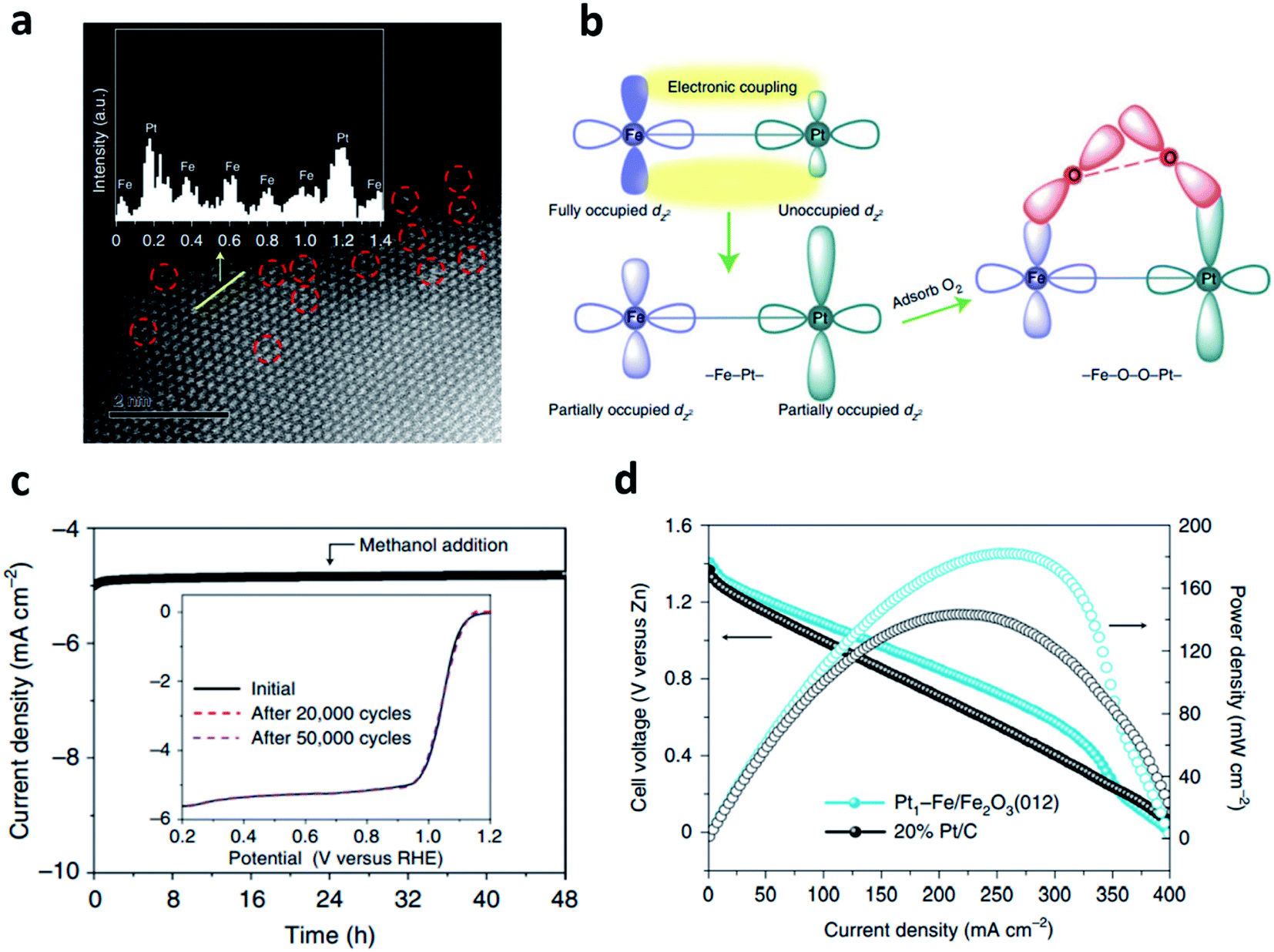 | ||
| Fig. 3 (a) An aberration-corrected HAADF-STEM image (6 × 6 nm2) of Pt1–Fe/Fe2O3 (012) with single-site platinum marked by red-dashed circles. (b) The schematic of the Pt–Fe electronic coupling effect for O2 activation. (c) Chronoamperometric responses obtained at 0.90 V versus RHE at 1600 rpm. with the addition of 10 vol% methanol. The inset shows ORR polarization curves of Pt1–Fe/Fe2O3 (012) before and after 20000 and 50000 voltammetry cycles. (d) Polarization and power density curves (cell voltage represents the potential versus zinc). (a–d) Reprinted with permission from ref. 45. Copyright 2021, Springer Nature. | ||
The optimization of the crystal structure and the surface morphology by heteroatom doping can also be an important strategy for activating OER electrocatalysts.39,47–49 Huang et al. reported the ruthenium-incorporated spinel Co3O4, which was rich in oxygen vacancies and had excellent oxygen mobility.47 They found lattice expansion where the diffraction peaks of certain planes were shifted to a low-angle orientation for ruthenium-doped contents. The author compared the ratio of Co2+/Co3+ for Ru doping through XPS quantitative analysis. It was confirmed that the Co3+ ions were converted into Co2+ ions by doping with Ru. The oxygen vacancies occur with the increase in Co2+ ions, which shows highly electrophilic properties and activates the adsorption of oxygen species.
3.2 Metal-free electrocatalysts
Carbon materials (such as graphene, carbon nanotube, and porous carbon) are of great interest due to their inherent advantages.50–56 Metal-free carbon-based electrocatalysts showed superior ORR performances in both acidic and alkaline electrolytes.57–59 In addition, it has emerged as a promising electrocatalyst owing to its advantages such as earth abundance, low price, environmental friendliness, and ease in tunability of their catalytic properties and structures.60,61Heteroatom doping strategies have been widely studied as an efficient way to improve the catalytic properties of metal-free carbon-based electrocatalysts.56,57,62–64 Many studies demonstrated that heteroatom doping can change the sp2 electronic structures of carbon materials and electrocatalytically active sites could be formed between elements with a different electronegativity.51,53,54,61,65 For example, Zhang et al. fabricated graphite-analogous nitrogen-doped carbon nanoflake using sandwich-like zeolitic imidazolate framework (ZIF)-8/MnO2 materials as the precursor.57 During the annealing process, ZIF-8 formed an edge-suffused graphite-analogous carbon nanoflake (GCF) intermediate on the surface of MnO2 as a carbon source. Subsequently, the edge-N-rich carbon nanoflakes (NCF) were synthesized using dicyandiamide (DCDA) (Fig. 4a). The irregular mesopores was observed in the basal plane of the NCF after nitrogen modification (Fig. 4b). These mesopores provided the in-plane pore edges to the basal plane, supplying a significant number of edge-N sites. The degree of graphitization can be confirmed by Raman spectroscopy as the peak intensity ratio (ID/IG) of the D-band (1350 cm−1) and the G-band (1580 cm−1) (Fig. 4c). It proved the high efficiency of MnO2 as a catalytic graphitization agent during the heat treatment. Fig. 4d shows the N 1s X-ray photoelectron spectra (XPS) of all the samples deconvoluted to 398.5, 400.0, 401.0, and 402.5 eV, corresponding to pyridinic-N, pyrrolic-N, graphitic-N, and oxidized-N, respectively. As a result, the total content of the main active sites, pyridinic-N and graphitic-N, increased from GCF (1.06 wt%) to NCF (1.64 wt%). It was found that the N content of NCF increased with the amount of pyridinic-N, which was preferentially located at the abundant edge-site. Also, the electronic synergism of pyridinic-N (Np) and graphitic-N (Ng) was demonstrated by the DFT calculations. The *OH adsorption overpotential was lower than that of Np and Ng single sites because of the dipole of Np and Ng. In addition, it was revealed that the catalytic activity of N-doped nanocarbon flakes was highly improved by the electronic synergism of Np and Ng.
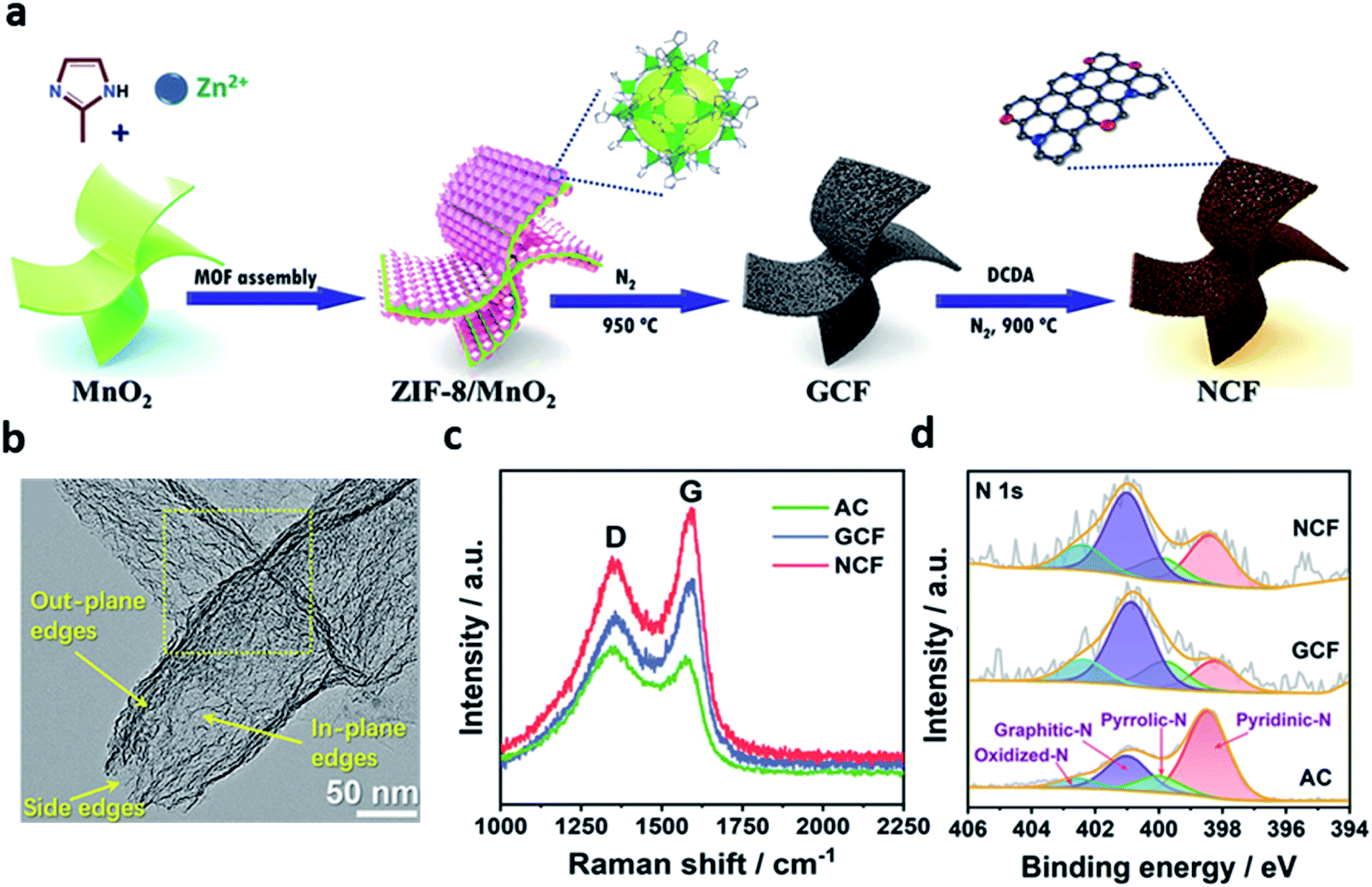 | ||
| Fig. 4 (a) Schematic for the synthesis of NCF. (b) TEM image of NCF. (c) Raman spectra; and (d) High-resolution N 1s XPS spectra for AC, GCF, and NCF. (a–d) Reprinted with permission from ref. 56. Copyright 2021, Wiley-VCH. | ||
Binary or more kinds of heteroatoms doping methods have been introduced for better catalytic activity compared to single heteroatom doping.51,58,65–67 For example, nitrogen single atom doping with additional sulfur doping enhanced the defective degree on the carbon plane, increasing the activation sites. The increase in the defective sites created vacant spots and formed pyridinic-N sites, which improved the ORR activity. It was explained that the redistribution of electron spin originated from a large radii difference between sulfur and carbon.55 More importantly, tri-doping can further enhance the catalytic activity by a synergistic effect between different dopants.59 Recently, Wang et al. synthesized N, S, and P tri-heteroatom-doped graphene (NSP-Gra) using thiourea and disodium phosphate as N, S, and P sources, respectively.54 Compared with N, S co-doped graphene, NSP-Gra exhibited excellent OER performance because of several defect sites and synergistic effect between N–S dopants. Similarly, Long et al. synthesized N, S, and P tri-doped holey carbon using thiocyanuric acid and phosphoric acid (PA) as the dopant sources.59 They found an increased number of pores with highly specific surface area in the electrocatalyst when the PA content increased. In addition, the defect density (R) of (ID′ + ID + IG′)/IG was derived, suggesting that multi-heteroatom doped sample with higher R value than other samples could induce more defective sites and increase the catalytic activities.
Besides the doping strategies, the compound electrocatalysts combining carbon-based materials with zero-dimensional quantum dots or covalent organic framework (COF) materials were reported.60,68,69 Carbon quantum dots are zero-dimensional materials with a size of less than 10 nanometers. Defective oxygen-containing moieties were randomly distributed in the basal plane and edge sites.60 Wang et al. effectively combined the graphene oxide quantum dots (GOQDs) into graphene/carbon nanotube (CNT) hybrid structures using GOQDs-dispersed solution.69 GOQDs with abundant oxygen containing functional groups and unsaturated bonds provided nitrogen and sulfur doping sites, which increased the doping level of electrocatalysts. Furthermore, the sample containing GOQDs exhibited the largest specific surface area and exceeded the commercial Pt/C electrocatalyst (4.74 mA cm−2) with a limiting current density of about 5.88 mA cm−2. Liu et al. developed van der Waals nanohybrid heterostructures (1D vdWHs) composed of a CNT core and a thienothiophene–pyrene COF shell with tunable thickness.68 The COFs are multidimensional and multifunctional organic network materials built by covalent bonds between light materials, which is similar to that of covalent organic polymers (COPs). This unique structure can act as a support material with many advantages such as high conductivity, porosity, defectivity, thermal and structural stability, and high specific surface area. They used CNT nanohybrid structures to solve the problem of spontaneous stacking of 2D-COFs, demonstrating the excellent catalytic effect through improved electron transport reaction. The DFT calculations revealed that delocalized electrons in the carbon substrate were provided in the COF shell, resulting in an N-doping effect. They manufactured the ZAB using a thickness-optimized COF, showing excellent potential difference (791 mV), high specific capacity (696 mA h gZn−1, J = 40 mA cm−2), and excellent cycle stability.
3.3 M–N–C electrocatalysts
Metal–nitrogen–carbon materials (hereafter referred to as M–N–C) are regarded as one of the most promising candidates due to their superior electrocatalytic activity, durability, abundant reserves, and low cost.71–75 This unique structure is realized by the coordination between nitrogen and transition metals in the carbon matrix.75 Nitrogen-doped carbon materials have been widely studied due to the favorable ORR catalytic activity and facile synthesis.76 Nitrogen can be easily doped into the carbon matrix owing to the similarity of the atomic radius of the nitrogen and carbon atoms when the nitrogen-based precursors such as dicyandiamide (CH4N4), urea (CH4N2O), and melamine (C3H6N6) in an inert atmosphere were pyrolyzed.77 Also, nitrogen has higher electronegativity (3.04) than that of carbon (2.55), which can facilitate the positively-charged neighboring carbon atoms by electron transfer from carbon to nitrogen.77Among the various candidates of transition metals (Fe, Co, Zn, Cu, Mn, Cr, and Mg), among single-atom site (SAS) catalysts, the iron-based SAS electrocatalysts have proved to be the most favorable materials because of their excellent ORR electrocatalytic activity and stability.70,71 Nitrogen doped with the coordinated iron atoms enables FeN4 active sites and graphitic N dopants to be formed with sufficient adsorption energy for oxygen-containing intermediates, thus enhancing the ORR activity.70,72 Wei et al. reported a Fe single atom site (SAS) catalyst enhanced by a graphene-encapsulated Fe3C nanocrystal (Fe3C@C).70 Fe3C@C-Fe-SAS was fabricated with glucosamine (carbon), melamine (nitrogen), and iron precursors through a one-step C3N4 confinement strategy. In addition, the spatial distance was extended by adding ZnCl2 to prevent the aggregation of iron precursors (Fig. 5a). The HAADF-STEM images confirmed a large amount of dispersed Fe SAS species (red rings) and Fe3C@C nanocrystals (yellow rings) (Fig. 5b). The high ratio of ID/IG indicated the abundance of defects and the degree of graphitization of the catalysts. The ID/IG ratio of Fe3C@C-Fe-SAS-glucose (0.94) and Fe3C@C-Fe-SAS-bulk (1.09) was smaller than those of Fe3C@C-Fe-SAS (1.31). This result implied that Fe3C@C nanoparticles had a significant level of graphitization (Fig. 5c). With respect to the electrochemical performance, the lowest Tafel slope (73.13 mV dec−1) indicated the excellent ORR catalytic activity. When compared with the RDS on the energy diagram by the DFT calculation, the lower the energy barrier, the more advantageous the accelerated *OH desorption. Notably, the introduction of Fe3C@C nanocrystals favored the adsorption and *OH desorption of O2 molecules in Fe SAS, accelerating the reaction kinetics and revealing the outstanding ORR activity. Fe3C@C–Fe SAS showed promising ORR activity and stability in neutral electrolytes than commercial Pt/C, a power density of 74.8 mW cm−2, and superior stability for 100 h. Shang et al. reported an atomically-dispersed iron (A-Fe-NC) catalyst supported on nitrogen-doped carbon using the ligand-stabilized pyrolysis synthesis.71 In the synthesis process, the sequestration of the chelating agent effectively acted as a mediator for converting iron ions into the atomic Fe–Nx species. The synthesized Fe–Nx species revealed that Fe–N bonds, Fe–N4, Fe–N5, Fe–N6, and Fe–N2+2 sites were formed through X-ray absorption near edge structure (XANES) (Fig. 5d). Also, it was revealed that Fe exhibited a coordination number of 5, indicating a change in the free energy of the lowest electron transport stage at the Fe–N5 site (Fig. 5e). ZAB with A-Fe-NC as an air cathode showed an open circuit voltage (OCV) of 1.45 V, which was higher than that of Pt/C (1.42 V).
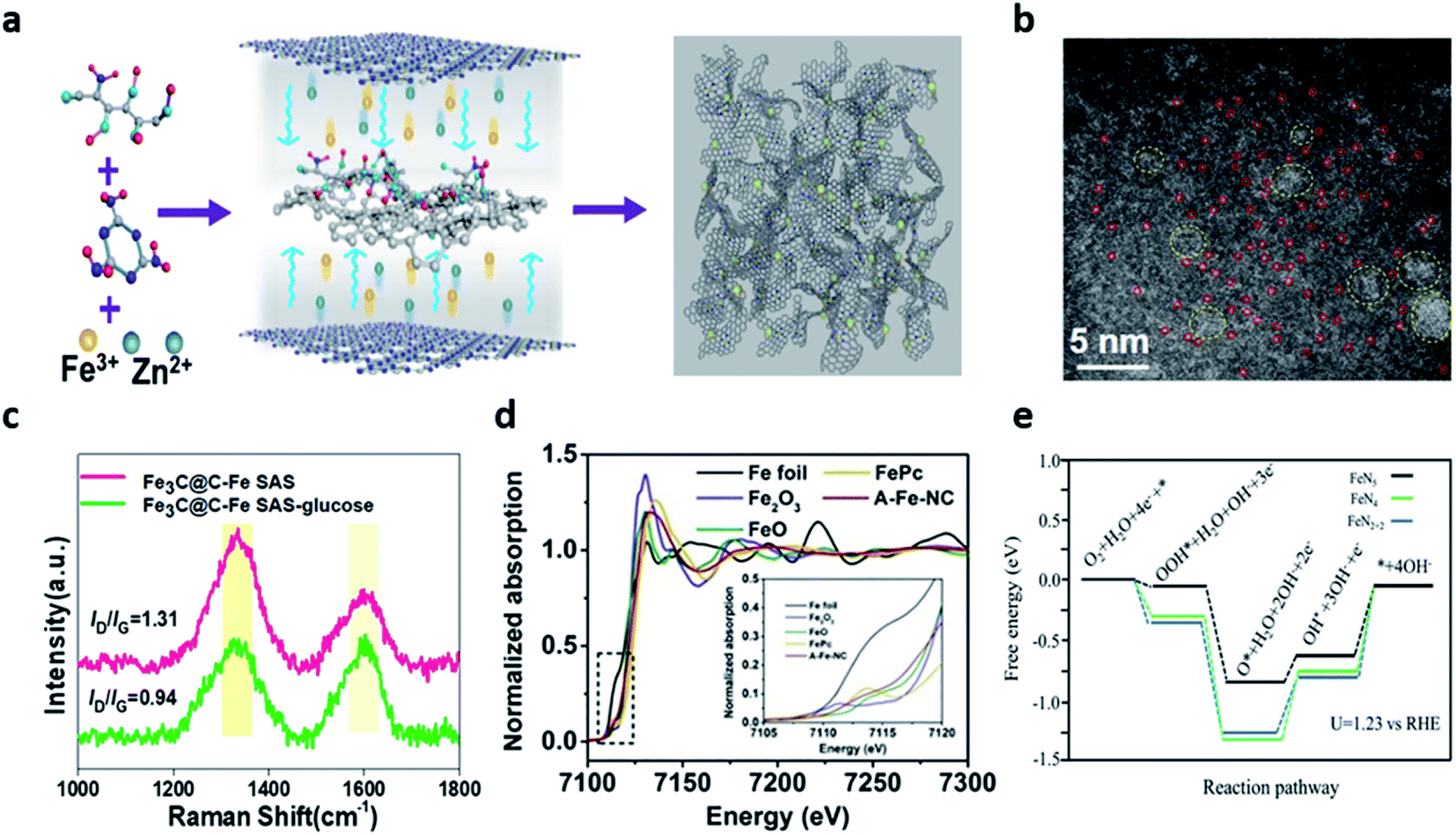 | ||
| Fig. 5 (a) Illustration of the synthesis process of Fe3C@C–Fe SAS. (b) AC-HAADF-STEM images of Fe3C@C–Fe SAS. (c) Raman spectra of Fe3C@C–Fe SAS and Fe3C@C–Fe SAS-glucose. (a–c) Reprinted with permission from ref. 69. Copyright 2021, Elsevier. (d) Fe-K-edge XANES spectra of A-Fe-NC. (e) Free energy paths of ORR on Fe–N4 sites, Fe–N2+2 sites, and Fe–N5 sites at U = 1.23 V and T = 298 K. (d and e) Reprinted with permission from ref. 70. Copyright 2020, Elsevier. | ||
In addition to the Fe–N4 site, Han et al. effectively implemented the embedded Fe–N–C sites by forming aligned mesoporous structures using highly ordered cubic silica Korea Advanced Institute of Science and Technology Number 6 (KIT-6) plates.72 The mass transport and electron transfer were enabled by an N-doped ordered mesoporous structure (N-OMC). TEM analysis showed that the high content (mass loading) of Fe and N in the porous structure of N-OMC enhanced the ORR active areas. Zhang et al. created the pores in the carbon matrix by the heat treatment of macrocyclic cucurbit[6]uril (CB[6]) and M–N–C precursors.76 They confirmed the N-doped carbon pore structure with Fe-bond through the HAADF-STEM images. Ma et al. reported that the intrinsic catalytic activity could be improved when the Fe–N4 site was doped with S.73 The S doping in the carbon matrix could decrease the d-band center of iron, thus enhancing the ORR kinetic current of per Fe–N4 sites compared with the pristine Fe–N–C catalysts. The single atom N–C catalyst studies based on the transition metals such as (Mn, Ni, and Co) have also been reported. Han et al. fabricated a Mn–N–C using a one-step heat-activated synthesis method.79 They demonstrated that the effectively formed Mn–N4 sites promote more electron transfer of the *OH electrons to *OH species in the atomically dispersed MnL+–N4 sites than in the Fe–N4 sites using the DFT calculations. Wang et al. synthesized the cobalt nanoparticles (Nps) coated with defective nitrogen-doped graphene (CoDNG800) by pyrolyzing the graphene–supermolecule complexes and encapsulating them in defective nitrogen-doped graphene.80 During the high-temperature heat treatment of defective nitrogen-doped graphene (DNG), the carbon shell structure was dislocated, and the broken fringes were formed by the evaporation of zinc species. Graphene embedded with Co Nps showed a lower work function, decreasing the electron transfer energy resulting from the formation of the *OOH species and catalyzing the ORR reaction. As a result, the CoDNG900-based ZABs exhibited a small charge–discharge voltage gap and excellent cycle life of 667 h, which is superior to that of commercial Pt + IrO2.
The alloy-based M–N–C catalyst materials have been reported by various approaches owing to the synergistic effect among heteronuclear metal atoms.77,78,81,82 The catalytic effect of alloy-based N–C can be determined by considering two factors including spin configuration and electron conduction. Among the transition metals, nickel is advantageous for improving the performance of electrocatalysts because the coupling of Ni 3d can control the spin magnetic moment to a modest level. When combined with iron, nickel reduces the localization of Fe 3d texture.77 Therefore, the FeNi-based alloy is regarded as a promising material for its excellent conductivity and tunable electronic structures.74,82 Yu et al. developed the FeNi SAS/NC catalyst by coating FeNi with polydopamine (PDA) and etching them to work in an alkaline environment.81 The XANES and DFT calculations proved that the Fe–Ni–N6 structure coordinated on a nitrogen-doped carbon support had a unique coordination structure and simultaneously shared the dual active sites, thereby improving the ORR activity. Zheng et al. directly grew an FeNi alloy on a nitrogen-doped CNTs using a self-jet vapor-phase growth method.82 When the cotton pad (CP) cluster was added during a carbonization process, a 3D porous framework was formed. The SEM images indicated that the hierarchical 3D porous network was composed of entangled rich carbon fibers, indicating that most of the metal nuclei were embedded in the tips of the CNTs. The TEM images showed a well-resolved lattice fringe of 0.209 nm FeNi alloy on bamboo-like N-doped CNTs (NCNTs) with a diameter of 20–30 nm (Fig. 6a). The Tafel slope for the FeNi@NCNT-CP cluster was smaller than that of other catalysts for the ORR and OER, including commercial Pt/C (82 mV dec−1) and IrO2 (136 mV dec−1). The ZAB with FeNi@NCNT-CP clusters exhibited a small voltage increase of 0.12 V after 250 cycles (250 h) at a current density of 10 mA cm−2, showing a stable performance compared to the previously reported ZAB performance based on FeNi@NCNT-CP cluster (Fig. 6b). Lai et al. fabricated the flexible 3D free-standing bifunctional electrode by the in situ coupling of NiFe nanoparticles and nitrogen-doped carbon nanofibers on carbon cloth (NiFe/NCNF/CC).74 The 3D spatial distribution of the NiFe Nps with N-doped carbon nanofibers provided sufficiently active areas and significantly increased the electrochemically active sites. In terms of the morphology, the NiFe Nps were grown on the N–C carbon fibers with the shape of a sesame candy (Fig. 6c). The light emitting diodes (LED) connected to a flexible electrode continued glowing even when the two batteries were bent to 90° (Fig. 6d).
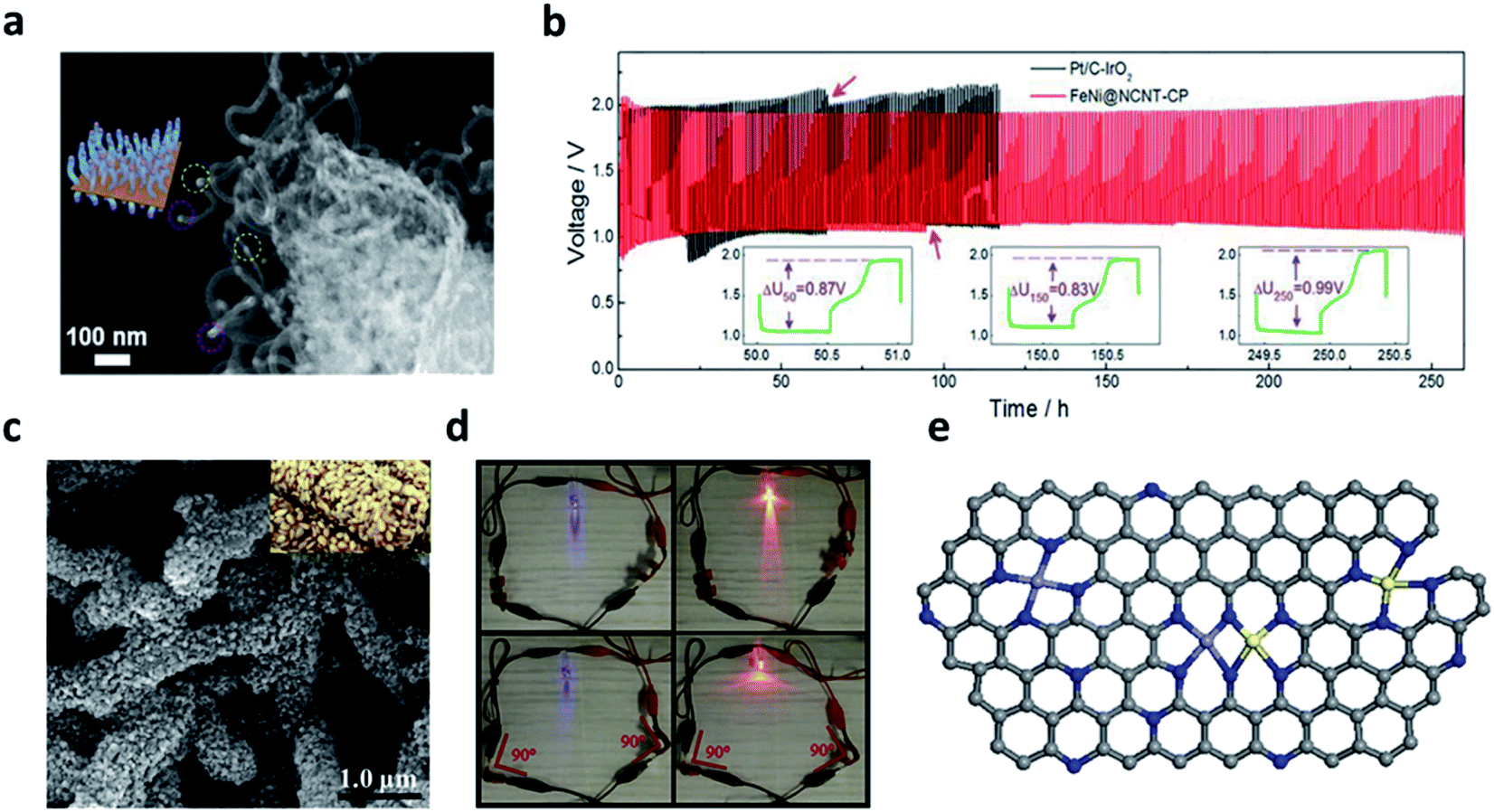 | ||
| Fig. 6 (a) SEM image of the FeNi@NCNT-CP clusters. (b) Cycling performance of ZAB based on the FeNi@NCNT-CP clusters and the commercial Pt/C–IrO2 (10 mA cm−2). (a and b) Reprinted with permission from ref. 81. Copyright 2020, Wiley-VCH. (c) The SEM images of NiFe/NCNF/CC; the inset in panel F is the digital picture of sesame seed candy. (d) The LEDs are lightened by rechargeable flexible zinc–air batteries in series. (c–d) Reprinted with permission from ref. 73. Copyright 2021, Elsevier. (e) The schematic structural model of Fe/Ni(1:3)-NG. Gray, blue, purple, and yellow represent C, N, Fe, and Ni, respectively. (e) Reprinted with permission from ref. 76. Copyright 2021, Elsevier. | ||
Ma et al. fabricated a bifunctional catalyst with atomically dispersed NiN4 and FeN4 in nitrogen-doped graphene (Fe/Ni(1:3)-NG) using a hydrothermal method.77 The properties of Fe 3d orbitals were related to the highly delocalized electrons in Fe-NG, leading to an undesirable electron conductivity. It was revealed that the introduction of Ni improved the charge transfer at the interface by reducing the delocalization of the Fe 3d texture. In addition, the structural model of the Fe/Ni (1:3)-NG catalyst consisted of active sites co-existing with FeN4, NiN4, and FeNi–N4 sites (Fig. 6e). Hao et al. developed the NPC/FeCo@NCNT catalyst with a carbon-based (NPC) coating that improved the electrical conductivity, stabilizing the FeCo alloy particles by a two-step calcination process.78 The DFT calculations showed that three layers of NPC, FeCo, and NCNTs improved the electronic state around the Fermi level compared to the two layers of FeCo and NCNTs. Also, it was revealed that electron transfer between the carbon materials and the FeCo alloy was accelerated by strong electrical coupling. Chen et al. developed the 0D–1D–2D hierarchical structure material of the FeCoNi alloy-N-rGO.75 This structure shared the N4 site between transition metals and effectively suppressed the overlap of 2D graphene sheets by the 0D–1D–2D hierarchical structure, exposing the more active sites. Furthermore, it was found that mass transfer between the reactant/catalyst/electrolyte multiphase interface was facilitated during the electrocatalytic reaction process. With respect to the electrochemical performance, the FeCoNi alloy-N-rGO showed a higher power density than Co–N–rGO, Pt/C–RuO2 (132.4 mW cm−2 at 0.64 V, 99.6 mW cm−2 at 0.67 V), and a specific capacity of 766 mA h gZn−1 (Co–N–rGO: 709 mA h gZn−1, Pt/C–RuO2: 695 mA h gZn−1).
3.4 Transition metal compound electrocatalysts
The sluggish and thermodynamically uphill reaction of the OER is considered as a key reaction for changing the metal–air batteries.83 Commercially available Ru or Ir oxide electrocatalysts are highly efficient for the OER process; however, the use of the PGMs electrocatalysts is seriously limited by the abovementioned disadvantages.84 Therefore, increasing efforts have been carried out to develop inexpensive and high-performance OER electrocatalysts. Transition metal-based electrocatalysts have been considered as promising alternatives due to their abundance of resources, various oxidation states, high stability, and catalytic activity.85,86 Recently, various transition metal compounds such as oxides, nitrides, sulfides, selenides, borides, and phosphides have been widely studied.83,85,87–90 Cobalt oxides have attracted much attention as efficient OER electrocatalysts due to their stability in cycle performances, excellent catalytic activities, and easy preparation properties.84,85,87,91–93For example, Doan et al. developed P, S dual-doped cobalt oxide nanosheets supported by Cu@CuS nanowires (P, S-CoxOy/Cu@CuS NWs).85 They fabricated highly conductive materials using the heterostructures of Cu@CuS nanowires and cobalt oxide. The morphology of the P, S-CoxOy/Cu@CuS is shown in Fig. 7a. The TEM images show the dense nanosheets grown on the surface of the Cu@CuS nanowires with a shell layer (Fig. 7b anb c). In particular, the selected area electron diffraction (SAED) pattern at the shell layer in Fig. 7b exhibited specific diffraction rings. These patterns corresponded to the d(200) plane of CoO and the d(220), d(222), and d(311) planes of Co3O4, indicating that the CoO and Co3O4 phases coexisted. The chemical state of the material surfaces was identified by the high-resolution XPS spectra, revealing the coexistence of metallic Cu and CuS by the high-resolution spectrum of Cu 2p (Fig. 7d and e). Specifically, the formation of the mixed-valence CoxOy was confirmed by analyzing Co3+ and Co2+ peaks in the high-resolution spectrum of Co 2p. After doping the heterogeneous elements, the number of Co2+ species was increased to downshift the density of the empty d-orbitals. In addition, the desorption of OH groups was promoted to activate the catalytic reaction. As a result of the rechargeable performance and stability test, the ZABs with Pt/C and RuO2 obviously exhibited an increase in its potential difference. However, the ZABs with CoxOy/Cu@CuS only showed a small potential difference of 0.09 V after 25 h (Fig. 7f).
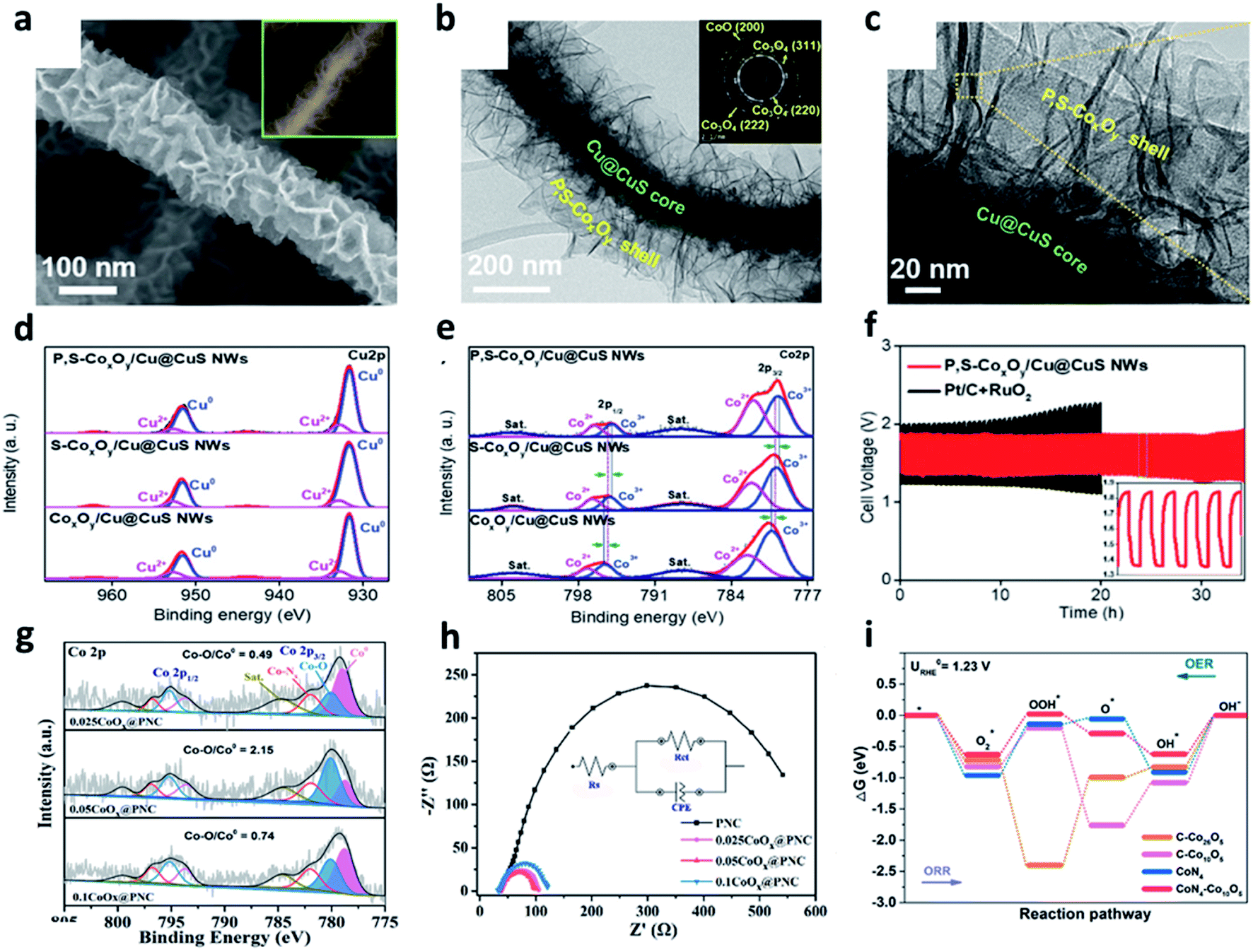 | ||
| Fig. 7 (a) SEM images of the P,S-CoxOy/Cu@CuS NWs. (b and c) TEM images of the P,S-CoxOy/Cu@CuS NWs; inset in (b) SAED pattern of the P,S-CoxOy shell layer. High-resolution XPS spectra of (d) Cu 2p and (e) Co 2p. (f) Galvanostatic discharge–charge cycling curves at 5 mA cm−2 of Zn–air batteries with P,S-CoxOy/Cu@CuS NWs or Pt/C + RuO2 as the air cathode (a–f). Reprinted with permission from ref. 84. Copyright 2020, Wiley-VCH. (g) Co 2p HR-XPS spectrum of 0.025CoOx@PNC, 0.05CoOx@PNC, and 0.1CoOx@PNC. (h) Electrochemical impedance spectroscopy. (i) The free energy diagrams of ORR and OER at the equilibrium potential on various catalysts model. (g–i) Reprinted with permission from ref. 86. Copyright 2021, Elsevier. | ||
Compared to the metal nanoparticles, metal nanoclusters with a smaller size can expose more active surfaces. In addition, the control of electrocatalysts at the sub-nanometer scale is much easier. Tan et al. fabricated a CoOx nanocluster-confined electrocatalyst (CoOx@PNC) on ZIF-8 derived N-doped carbon nanomaterials.87Fig. 7g shows the high-resolution Co 2p spectrum of 0.025CoOx@PNC, 0.05CoOx@PNC, and 0.1CoOx@PNC. Among these, 0.05CoOx@PNC showed the highest Co–O/Co0 ratio and high Co-based nanoclusters, indicating that Co nanoclusters with a more exposed surface area tended to be easily oxidized. 0.05CoOx@PNC showed a minimum semicircular diameter with the lowest interfacial charge transfer resistance (Rct) among all the prepared electrocatalysts during OER (Fig. 7h). The DFT calculations proved that cobalt oxide formed the coupling with Co–Nx nanoclusters, which promoted the orbital overlap of the Co element in cobalt oxide with N and O atoms, consequently reducing the reaction barrier of the ORR/OER oxygen catalytic reaction (Fig. 7i).
On the other hand, Rao et al. reported vanadium ions (V4+ and V5+) doped mesoporous Co3O4 nanorods.92 They used VCo-glycerate as a vanadium source to induce Co3+ ions in Oh sites toward an optimized eg occupancy. The catalytic effect of the spinel oxides depended on the eg occupancy of transition metal cations at the octahedral sites because there was a strong overlap between the eg orbitals and the O2p orbitals of the oxygen-related adsorbents. As an effective electrocatalyst for ORR and OER, the galvanostatic cycling performance was measured at a current density of 5 mA cm−2 for various catalysts, and the V-Co3O4 catalyst showed more than 1300 h of cycling performance, outperforming the benchmark combination of Pt–IrO2. This result was originated from the vacant d-orbitals formed by vanadium ions, exhibiting strong electrostatic attraction, which facilitated strong interaction between the electrocatalyst and the oxygen species.
To improve the electronic conductivity of the electrocatalysts, Balamurugan et al. reported CuMo-oxynitride@N-doped graphene nanohybrid materials.88 In the process of heat treatment in ammonia atmosphere, N atoms partially replaced O atoms. This process enabled the delocalization of the electron environment by changing the electron distribution of the metal. They also reported the transition metal-based selenide materials.89 The transition metal-based selenides have been studied as new electrocatalysts for energy storage systems because of their abundant nanoporous networks, excellent electrical conductivity, and superior catalytic activities. They firstly designed the 3D hierarchical molybdenum oxyselenide nanoarchitectures to solve the limited catalytic active sites and low stability. In this 3D structure, the OH-bonding energy was enhanced due to the holes generated in the valence band of Se 3d and O 1s, and the catalytic activity was increased by the formation of abundant oxygen vacancies. As a result, the rechargeable quasi solid-state ZAB exhibited a peak power density of 166.7 mW cm−2, providing a higher record than that of the benchmark Pt/C–IrO2-based air cathode (141.5 mW cm−2).
Borides are known to show excellent OER performance in alkaline solutions, and the synthesis of borides is facile compared to other catalysts. Jose et al. reported cobalt boride/N-doped carbon nanosheet hybrid structures.90 The interaction between boron and metal species could transfer electrons from B to the d band of Co, which was expected to be an advantage in electrochemical applications. They synthesized monometallic-boride/N-doped carbon nanostructure using a simple chemical reduction method using sodium borohydride as the boron source. This unique structure showed high chemical stability because the outer B-rich layer protected the inner metallic layer from oxidation. Also, the interaction of the electron transfer from B to the d band of Co occurred between boron and metallic cobalt, which was revealed by the shift of the peak of the boron–metal bond in the XPS spectrum.
Similarly, electrocatalysts based on transition metal sulfides and phosphides have been getting much attention.83 In general, transition metal phosphides have several shortcomings such as low specific surface area and insufficient conductivity. To address these problems, many studies have been conducted to induce an interaction between P and other transition metals or carbon materials.94,95 Guo et al. reported Cu3P/MoP nanospheres and confirmed that charge transfer occurred between the metal–phosphide bonds.95 The fabricated rechargeable ZABs exhibited a high power density of 156 mW cm−2 and high stability for 231 h, greatly outperforming the commercial Pt/C (131 mW cm−2, 157 h). These superior performances were correlated with the strong interaction between the Cu3P and MoP species. Wu et al. reported Co/Co2P nanocrystal heterojunction structures confined in N-doped carbon.94 They found that during the growth of NCNTs, the Co nanoparticles at the edges were easily converted to Co2P. Also, it was observed that the Co nanoparticles located in the central section of NCNTs were difficult to be phosphatized. This result revealed that the formed Co/Co2P heterojunctions directly catalyzed the growth of highly ordered NCNTs.
3.5 Metal–organic framework (MOF)-derived electrocatalysts
Recently, various nanostructured materials such as MOFs, COFs, and MXene have been used to achieve cost-effective and metal-free electrocatalysts and transition metal-based electrocatalysts.96,97 MOFs are composed of metal nodes and organic ligands in 3D porous networks that can be extended from micropores to mesopores. This porous structure can be synthesized without the addition of surfactants to create metal–organic compositions with high surface areas. In addition, its porosity can be controlled by different types of metal ions and organic ligands. The porous carbon body and metal oxide can be manufactured via heat treatment.98ZIFs are a subfamily of MOFs composed of metals and organic ligands with a zeolite structure. It has a high surface area and can produce nitrogen-doped porous carbon upon carbonization. Moreover, this unique structure can form metal oxides during the calcination process.99 It was reported that ZIF-67-derived Co@NC polyhedron exhibited bifunctional electrocatalytic activity for OER and ORR in alkaline electrolytes. ZIF-67 exhibited excellent compatibility with nanostructured materials. Gu et al. synthesized a noble metal-free trifunctional electrocatalyst by aligning Mo2C nanosheets vertical to the surface of the Co@NC structure (Fig. 8a).100 This heterostructure architecture greatly reduced the dependence on the porosity properties and the number of exposed active sites in Co@NC. The interfacial effect between the Mo2C nanosheets and the Co@NC polyhedron mediates the electronic structure to optimize intermediate adsorption, significantly enhancing the intrinsic activity. The discharge polarization curves showed that the peak power density of Mo2C/Co@NC (187.9 mW cm−2) was much higher than that of Pt/C (78.5 mW cm−2) (Fig. 8b). Therefore, they confirmed that the interfacial bonding effect between Co@NC and Mo2C could promote the ORR. Muthurasu et al. introduced cobalt vanadate integrated with cobalt oxide in an iron-doped metal–organic framework.101 Using the easy approach of a multistep solution process, the unique nanoflakes heterostructures were created, providing abundant pores, high specific surface area, and extremely large active sites (Fig. 8c). The unique hexagonal nanoflakes with cobalt hydroxy nanosheets and the coordination strategy with MOFs enabled the efficient doping of electrochemically activated iron. When Fe-doped MOF CoV@CoO nanoflakes were introduced into ZABs, which provided a maximum power density of 138 mW cm−2 much higher than that of Pt/C. They also demonstrated a power density of 138 mW cm−2 and good cycling stability (Fig. 8d). Vijayakumar E. et al. synthesized the CoP-NC@NFP electrocatalyst using a simple and low-cost precipitation method, followed by the hydrothermal process and the pyrophosphatization method (Fig. 8e).102 The bimetallic phosphide (NFP) nanoflakes were uniformly anchored to CoP-NC, forming a core-shell-like coral structure. The CoP-NC@NFPs multi-active sites effectively helped the formation of oxy/hydroxides by the proton and hydride active sites. Notably, the nanoflake-structured NiFeP on the surface provided improved electron transfer pathways by increasing the electrode and electrolyte contact. As a result, the power density of the ZAB with the CoP-NC@NFP cathode was superior to that of Pt/C benchmarked at 93 mW cm−2 at 145 mA cm−2 (Fig. 8f). Chen et al. synthesized a hierarchical Co–Ni–Fe spinel oxide–carbonitride hybrid (CoNiFeOx–NC) electrocatalyst using both Materials Institute Lavoisier (MIL) of Fe-MIL-101-NH2 and an ion exchange strategy of Co and Ni.103 Interestingly, the 2D ternary metal MOF shell layer was encapsulated in 3D octahedral MOF crystals. Also, the collapse of the MOF structure was prevented in the air calcination process, providing a highly porous structure and large surface area. Finally, the XPS and DFT calculations confirmed that the adsorption energy of oxygen species on the catalyst surface could be optimized by controlling the electronic structure through electron transfer between Co, Ni, and Fe. The optimized electronic structure lowered the OER energy barrier by controlling the oxygen-binding strength of the Ni–Co coordinated OH site, providing high OER efficiency under alkaline conditions.
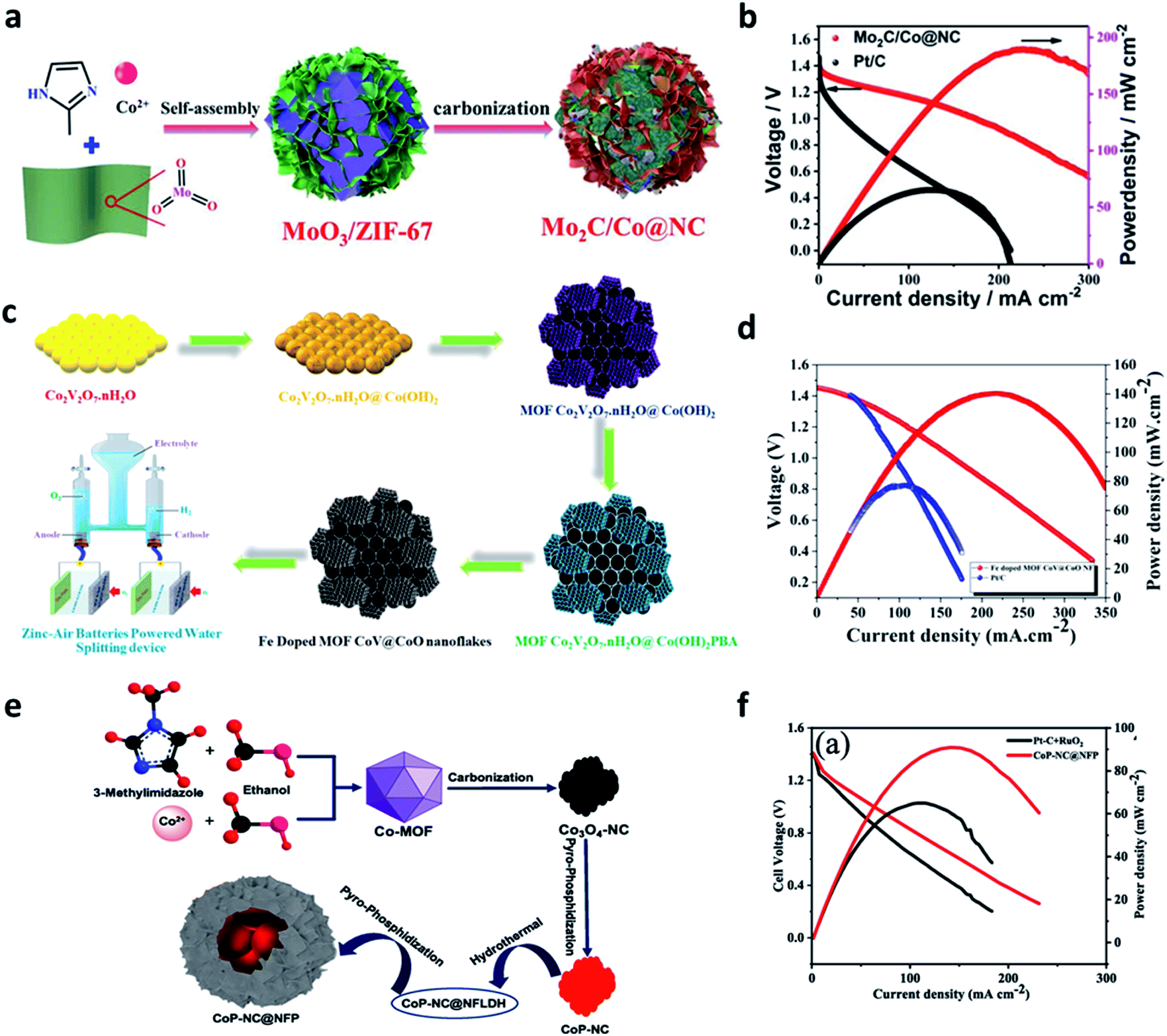 | ||
| Fig. 8 (a) Schematic illustration for the synthesis of Mo2C/Co@NC. (b) Discharge polarization curves and corresponding power density curves (a and b). Reprinted with permission from ref. 99. Copyright 2021, Elsevier. (c) Represents the schematic illustration of Fe-doped MOF CoV@CoO nanoflakes' synthetic strategy and self-powered zinc–air battery water splitting applications. (d) Discharge polarization curves and related power densities of Fe doped MOF CoV@CoO nanoflakes and Pt/C/IrO2 catalyst. (c and d) Reprinted with permission from ref. 100. Copyright 2021, Elsevier. (e) Preparation of the CoP-NC@NFP electrocatalyst. Performance of the zinc–air battery with the CoP-NC@NFP air-cathode: (f) power density curve and discharge curves compared with the state-of-the-art catalyst Pt–C + RuO2. (e and f) Reprinted with permission from ref. 101. Copyright 2021, Elsevier. | ||
In recent years, single-atom catalysts (SACs) have received much attention due to their high atom utilization efficiency and catalytic activity. Currently, many carbon-based SACs have been manufactured based on carbon derived from MOFs and ZIFs. Wang et al. developed large-scale synthesis method, in which Co–N4 sites (Co–SAs) and small Co-NPs (Co-SNPs) were dispersed atomically in an N-doped porous carbon nanocage (Fig. 9a).104 This approach was a facile strategy by simply controlling the Co species in the precursor. The DFT calculation confirmed that the strong interaction between the Co-SNP and Co–N4 sites properly increased the valence state of the active Co atoms of Co–SAs/SNPs@NC, modulating the free adsorption energy of the ORR intermediates. Furthermore, the downhill energy pathway of the free energy diagrams of the ORR indicated that the interaction between *OH and Co–N4 was quite strong, limiting the overall ORR reaction (Fig. 9b). The ZAB with the Co–SAs/SNPs@NC exhibited a high OCV of 1.493 V, and a higher power density than those of Co–SAs@NC and Pt/C catalysts (Fig. 9c). The long-term cell performance of Co–SAs/SNPs@NC + RuO2-based ZAB was also investigated by galvanostatic charge/discharge test at a current density of 5 mA cm−2 and was operated stably for 720 h in ambient air.
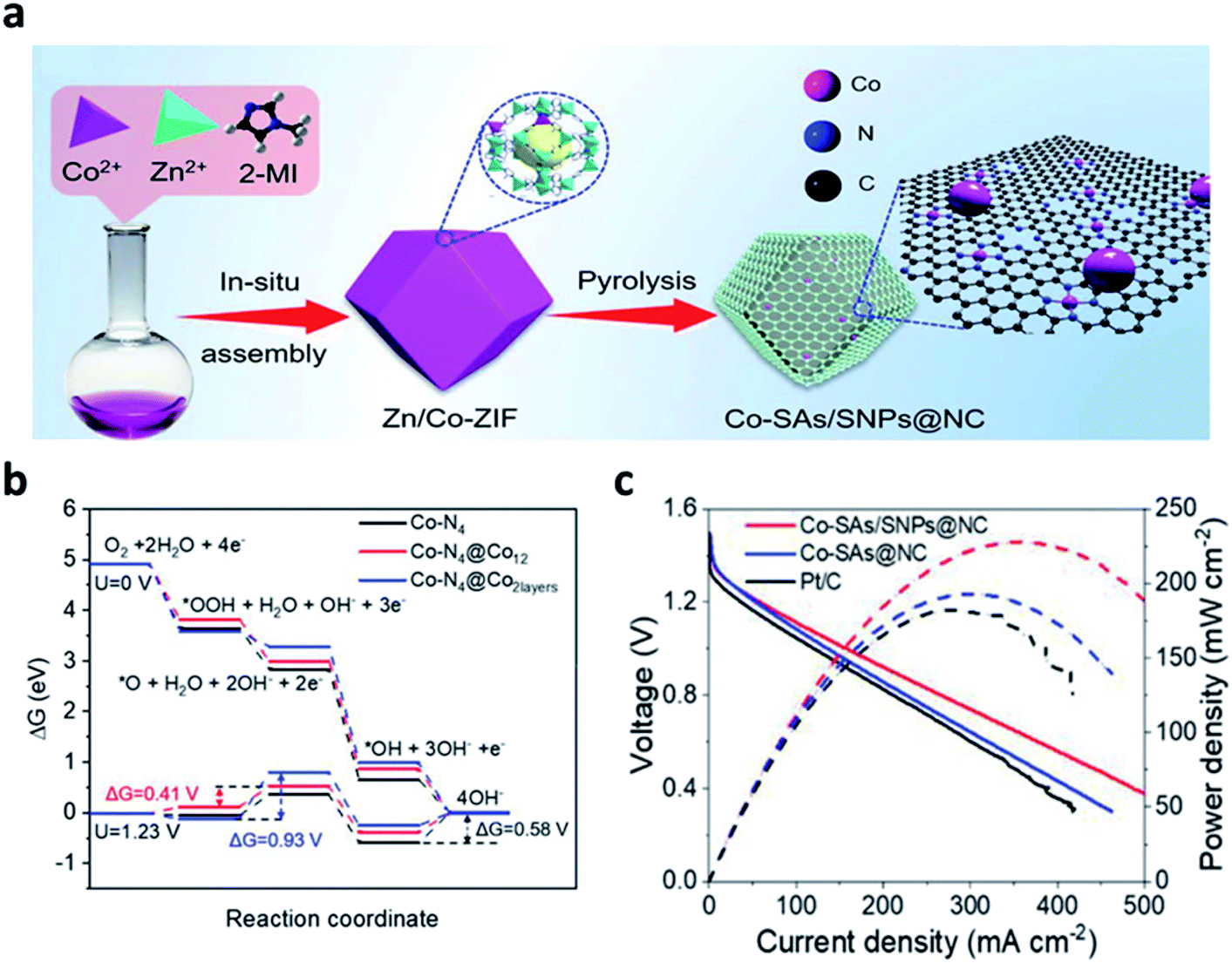 | ||
| Fig. 9 (a) Schematic illustration of the synthetic process for the Co–SAs/SNPs@NC catalyst, (b) ORR free energy diagrams for Co–N4 (black line), Co–N4@Co12 (red line), and Co–N4@Co2layers (blue line) at U = 0 V and 1.23 V along 4-electrons pathway; the Co atoms in Co–N4 moieties are regarded as adsorption sites for all the structures. (c) Discharge polarization curves and corresponding power density curves of the ZABs with Co–SAs/SNPs@NC, Co–SAs@NC, and Pt/C catalysts, respectively. (a–c) Reprinted with permission from ref. 103. Copyright 2021, Wiley-VCH. | ||
4. Progress and design for advanced ZABs
4.1 Wide operating temperature system
The performance of the ZABs with open air cathodes are easily affected by the temperature because the electrocatalytic reaction rate is very sensitive to the temperature changes, as described in the Arrhenius equation.105 The ORR and OER are particularly affected at lower temperatures because of the reaction mechanism by multiple oxygen intermediates and the aqueous electrolyte properties of the ZABs.106For wide temperature adaptability in flexible ZABs, Pei et al. fabricated an integrated fibrous carbon electrode with a large number of active sites.107 Using the stereoscopically modified planar electrolyte–electrocatalyst interface, they greatly improved the interfacial property among the solid catalyst, oxygen gas, and liquid electrolyte. They also used the interaction between water and terminal groups within the hydrogel backbones. With these two strategies, the full-temperature adaptive polyelectrolyte was demonstrated. The nanowire arrays were formed by the electrodeposition of polypyrrole (Ppy) on carbon cloth. Then, the nanowire arrays were impregnated, pyrolyzed, and phosphorized in Fe3+ and Co2+ salts to immobilize the active metal species on Ppy. The three-dimensional electrode efficiently enhanced the ORR by an almost direct 4e− reduction pathway with a half-wave potential of 0.85 V (E1/2) (Fig. 10a). Next, the carboxylate group was impregnated in alkalifine PAA (A-PAA) hydrogel in 30 wt% KOH solution for the flexible polyelectrolyte. The SEM image showed that the lyophilized A-PPA hydrogel had an abundance of interconnected pores that encapsulate the electrolyte, allowing the free drift of dissolved ions (Fig. 10b). Finally, ZAB with the stereoscopic FeCo–P/N–C–F electrode was assembled using an A-PAA-based polymer electrolyte to evaluate the performance according to the temperature change (Fig. 10c). At the current density of 2 mA cm−2, the cycling efficiencies were 65.9% at 25 °C, 69.9% at 80 °C, and 60.4% at −30 °C, implying that their ZAB was less sensitive to extreme temperature changes with a stable charge/discharge performance (Fig. 10d).
 | ||
| Fig. 10 (a) Comparison of ORR-OER bifunctional activity of the self-support electrodes. (b) SEM image showing the freeze-dried A-PAA hydrogel, scale bar: 10 μm. (c) Comparison of peak power density fluctuations of ZAB-Stereo and ZAB-Planar upon temperature changes. (d) Cycling tests of the two types of ZABs at 2 mA cm−2 and at different temperatures. (a–d) Reprinted with permission from ref. 106. Copyright 2021, Royal Society of Chemistry. | ||
In a similar study, Pei et al. proposed the highly active 1D bamboo-shaped fibrous catalysts (BFCs) for the low-temperature tolerant, flexible ZABs (Fig. 11a).108 For the synthesis of BFCs, polyacrylonitrile (PAN) solution and polyvinylpyrrolidone (PVP) solution were used as the external and internal fluids, respectively. The two solutions were separated into two layers by strong electrostatic tension because of the distinct difference in viscosity and surface tension. Also, the relatively low viscous PVP solution was shrunk into separate knots inside the fiber by Rayleigh–Taylor instability. The sample with only iron and cobalt salts were added to the PAN solution (BFC-FC), and the sample where the PVP solution was additionally added at a flow rate 0.2 mL h−1 was named as BFC-FC-0.2. In the N2 sorption isotherms of the BFC-FC and BFC-FC-0.2. It was confirmed that the specific surface area (711 m2 g−1) of the BFC-FC-0.2 sample was increased by approximately 2.24 times because of the introduction of PVP. In addition, the XRD pattern for the BFC-FC-0.2, the cubic phase FeCo alloy nanoparticles were observed, and the oxidized Co2+ (780.5 eV) was observed in the XPS spectrum of Co 2p, proving the oxidized surface of the FeCo alloy (Fig. 11b). The bifunctional property of the samples injected with PVP solution at various flow rates was tested in 0.1 M KOH solution. The BFC-FC-0.2 showed the excellent ORR onset potential of 1.03 V (Eonset) and a half-wave potential of 0.90 V (E1/2). In addition, BFC-FC-0.2 delivered the OER current density of 10 mA cm−2 at only 1.60 V (Ej=10), demonstrating the prominent ORR/OER catalytic activity with the lowest potential difference (ΔE = 0.70 V) among all the samples, including the Pt/C catalyst (ΔE = 0.98 V). Finally, the BFC-loaded cathode, A-PAA hydrogel electrolyte containing 30 wt% KOH, and zinc foil anode were stacked, and flexible ZAB was fabricated to measure the performance deterioration according to the temperature and mechanical deformation. ZAB-BFC-FC-0.2 achieved a capacity of 691 mA h g−1 and an energy density of 798 W h kg−1 at −20 °C, maintaining 92.7% and 87.2% of the initial performance, respectively, compared to the room temperature (Fig. 11c). Moreover, this system was stably operated in various deformations (folding and twisting) as well as mechanical bending more than 600 times at low temperatures, highlighting the firstly proposed flexible ZAB at low temperatures (Fig. 11d).
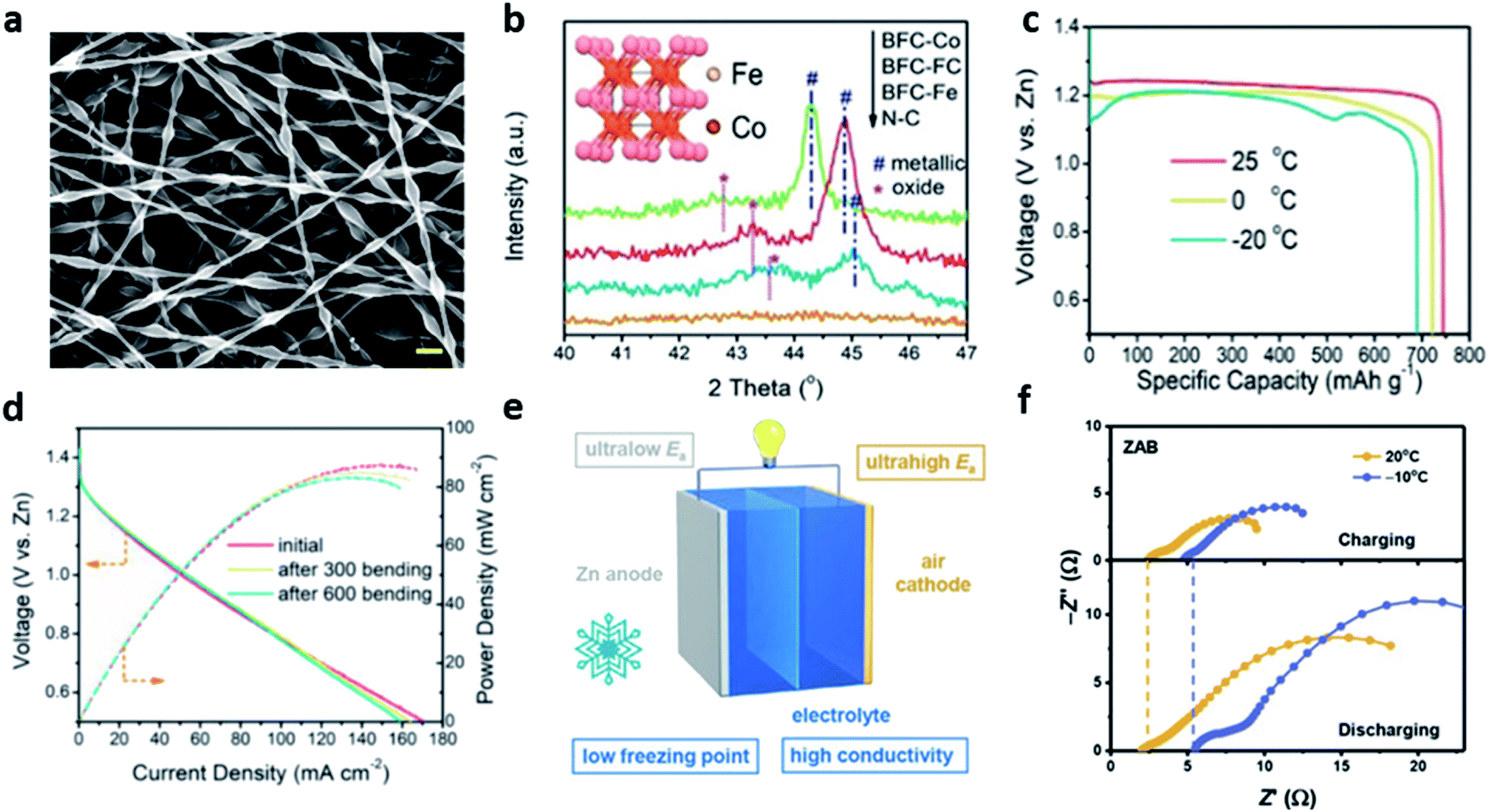 | ||
| Fig. 11 (a) SEM images of electrospun polymer fibers. (b) XRD patterns of different BFCs and the inset shows the molecular structure of FeCo alloy particles. (c) Temperature-dependent discharge capacity plots of ZAB-BFC-FC-0.2 recorded under the current density of 5 mA cm−2. (d) Polarization curves and corresponding power density plot of the battery at −20 °C after various times of bending (bent to 90°). (a–d) Reprinted with permission from ref. 108. Copyright 2020, Wiley-VCH. (e) Schematic diagram of the inherent advantages of ZABs for low-temperature energy storage. (f) EIS spectra of ZABs at 20 and −10 °C. (e and f) Reprinted with permission from ref. 107. Copyright 2021, Wiley-VCH. | ||
To identify the main bottleneck of low-temperature ZABs and to rationally improve the low-temperature performance, Zhao et al. systematically investigated the low-temperature responses of the anode, cathode, and electrolyte using the conventional cell configurations with Pt/C + Ir/C as the cathodic electrocatalysts and 6.0 M KOH + 0.2 M Zn(OAc)2 as aqueous electrolyte (Fig. 11e).109 The ionic conductivity decreased from 0.50 to 0.28 S cm−1 and the internal resistance increased by 3 Ω from the impedance of the blocking electrode at 20 and −10 °C, implying that the main limiting factor for low-temperature ZABs was the electrolyte's ionic conductivity (Fig. 11f). This limitation could be solved using CsOH-based electrolytes, regulating the solvation structure. In molecular dynamics (MD) simulation of CsOH-based electrolytes, it was observed that the ionic conductivity of the electrolyte at low temperature improved as the atomic number of the alkali metal increased. This result was explained by hydrogen bonding reinforced with the absence of cations in the anion solvation structure and the enhanced coulombic force between alkali metal cations and OH−. Finally, the performances of the ZAB using 6.0 M KOH-based and CsOH-based electrolyte were evaluated at −10 °C. Interestingly, the ZAB with CsOH-based electrolyte showed better discharge power density (99.3 mW cm−2), discharge voltage (25 mV higher than KOH-based), and lifetime characteristics (500 cycles) at a current density of 5 mA cm−2.
4.2 All-solid-state zinc–air pouch cells
In addition to the low-temperature concept, there has been an increasing demand on the safe and high-energy sustainable rechargeable ZABs to replace conventional lithium-based battery. All-solid-state zinc–air batteries have been proved to achieve both high energy density and power density with the high safety and multiple redox reactions of the air cathode. Despite the significant effort on the all-solid-state ZABs, the previous concept still has a limitation when scaling-up, only showing a laboratory scale stage.110–114Shinde et al. firstly reported a commercially available 1-Ah-scale all-solid-state zinc–air pouch cells (ZPCs), which was designed in a symmetrical structure with (101)-facet copper phosphosulfide (CPS(101)) cathode and anti-freezing chitosan-biocellulosics (CBC) electrolyte with a patterned zinc anode (Fig. 12a).115 This flexible ZPCs represented the high energy density (460 W h kgcell−1 and 1389 W h L−1), rate capability (5–200 mA cm−2), long cycle life (6000 cycles at 25 mA cm−2), and wide operating temperature range (−20 to 80 °C). They also found that their excellent electrochemical performance and mechanical robustness were attributed to the four design strategies (Fig. 12f–h). First, the 3D patterned zinc anode provided stability for zinc plating and stripping while facilitating the transport of ions and their reactions. Second, the dimensional stability and high ionic conductivity (86.7 mS cm−1 at 25 °C) of CBC conductors provided good robustness in contact with zinc metal. Third, a symmetrical configuration without the metallic current collectors and corrosive liquid electrolytes not only improved the specific and volumetric cell-level-energy densities but also improved the performance of ZPCs by constructing robust solid electrolyte interphase (SEI) between the patterned zinc anode and the CBC conductors (Fig. 12b). Lastly, the nanofiber framework of CPS(101) accommodated the volume change while preventing the mechanical fatigue of the cathode by constructing a continuous ion/electron three-dimensional transport path with excellent interfacial properties with the CBCs electrolyte. Copper phosphosulfide (CPxS1−x(101), x = 0.1, 0.3, 0.5, 0.7, 0.9) nanofibers, a multifunctional electrochemical catalyst, were fabricated using a two-step reaction of co-precipitation and autoclaving. The TEM images and XRD patterns were used to analyze the shape and nanostructure of 3D CPS sponges of various compositions. In the XRD pattern, the (101) orientation, which gave the best performance when the composition ratio was x = 0.5, was the most preferred. The lattice spacing of 0.466 nm was observed in the TEM image, showing an orthorhombic phase of the CPS with the well-defined (101) plane (Fig. 12c and d). With respect to the electrochemical performance, the ORR performance of CPS(101) showed an outstanding half-wave potential (E1/2 = 0.90 V) than that of Pt/C (20 wt%) and other CPS-based catalysts (Fig. 12e). In addition, in durability evaluation, there was almost no loss of half-wave potential (ΔE1/2 = 11 mV) even at 30000 cycles, which was superior to the durability protocols set by the US Department of Energy (loss of half-wave potential of <30 mV). The OER activity test of CPS(101) with RuO2 as the reference also showed the good durability of the lowest overpotential of 260 mV at 10 mA cm−2 and improved kinetics (the initial current of the OER decreases only 4% after 500 cycles).
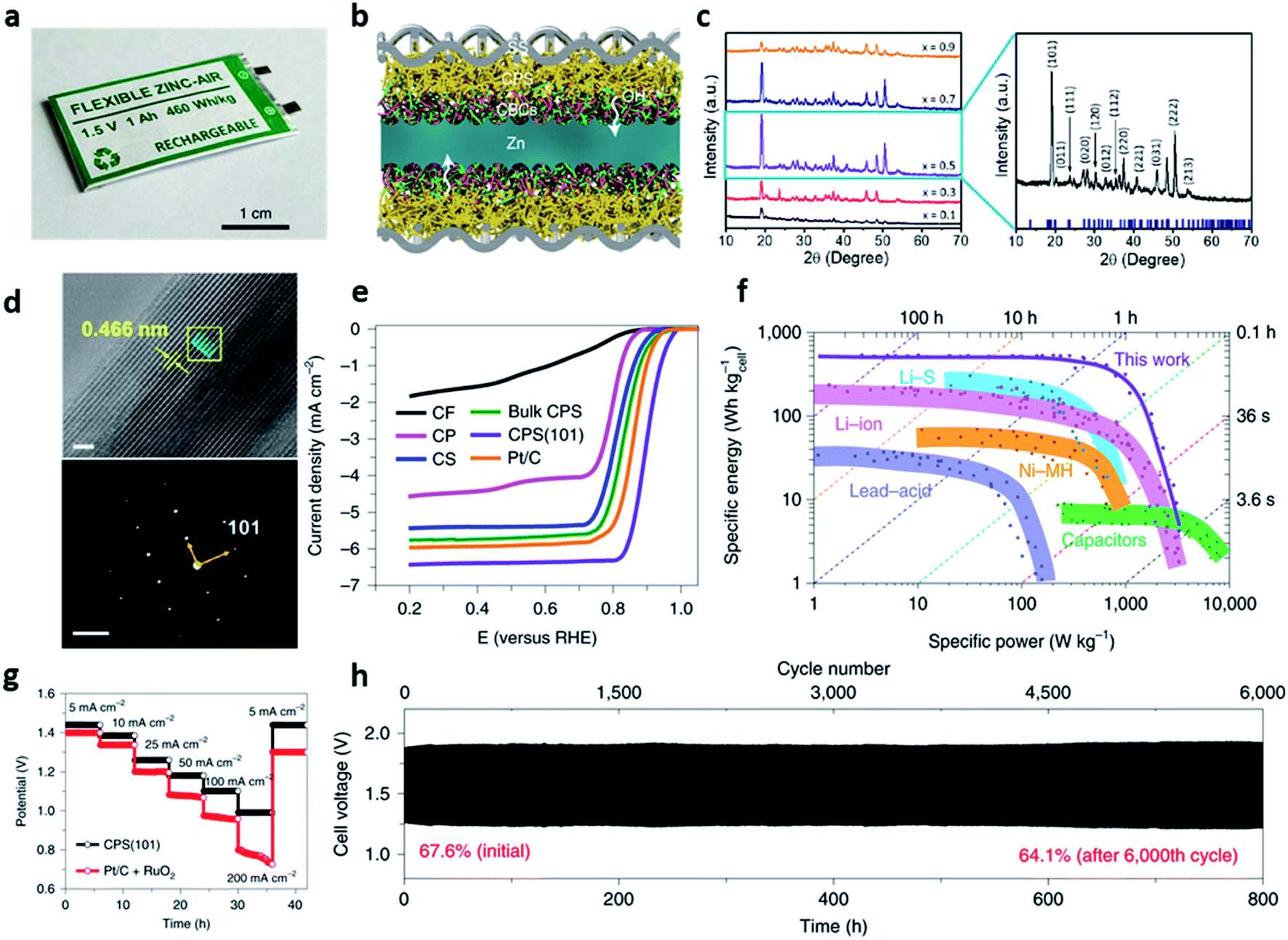 | ||
| Fig. 12 (a) A photograph of an all-solid-state zinc–air pouch cell. (b) A cross-sectional schematic of the symmetric bridging configuration [CPS(101)‖CBCs‖patterned zinc‖CBCs‖CPS(101)]. (c) XRD patterns of randomly oriented CPxS1−x series (x = 0.1 to 0.9) (left) and XRD pattern of the bulk CuP0.5S0.5 along the preferred (101) orientation (right). (d) High-resolution TEM images of CPS(101) (scale: 30 nm) and experimental SAED pattern with (101) orientation (below, scale: 2 nm−1). (e) Polarization ORR profiles for the fabricated copper-based cathodes in comparison with commercial Pt/C. CF, carbon fibre paper; CP, copper phosphide; CS, copper sulfide. (f) A comparison between the specific energies and powers of the fabricated ZPCs and the commercial capacitors and batteries previously reported. (g) Rate capabilities of CPS(101) and Pt/C + RuO2 cathodes with a discharge voltage range of 0.7–1.5 V for various current densities from 5 to 200 mA cm−2. (h) Galvanostatic cycling performance of CPS(101)‖CBCs‖patterned zinc‖CBCs‖CPS(101) for 25 mA cm−2 (realistic condition: E/C = ≤1, E/A = ≤2; time constraint = 8 min per cycle; DOD = 20%). The percentile values represent the voltaic efficiencies of the cells. (a–h) Reprinted with permission from ref. 114. Copyright 2021, Springer Nature. | ||
4.3 Non-alkaline electrolyte for the ZABs
Although the enhancement of the oxygen reversibility of cathode through bi-functional electrocatalyst is important, the electrolyte is also very important in terms of the cycling life characteristics of ZABs. In general, alkaline electrolytes have been widely used to fabricate ZABs with higher energy densities. In particular, KOH solvents have high ionic conductivity and low conductivity, providing excellent electrochemical properties.118 However, the discharge process of the ZABs in the strong alkaline electrolytes causes an insoluble product (Zn(OH)42−), which leads to ZnO precipitation.116,117| Zn + 4OH− → Zn(OH)42− + 2e− | (11) |
| Zn(OH)42− → ZnO + 2OH− + H2O | (12) |
On the other hand, there is another problem that the alkaline electrolyte at the anode is easily combined with CO2 in the air, forming the carbonates and reducing the porosity of the air electrode and the ion conductivity of electrolytes.118,119
| C + 6OH− → CO32− | (13) |
Thus, to achieve the high energy density and excellent cycle characteristics, the research on novel electrolytes has progressed. Recently, many studies on the electrolyte of the ZAB have focused on the neutral or near neutral electrolytes to remove the critical obstacles of the alkaline electrolytes. Although significant efforts have improved the stability of the Zn anode, the intrinsically irreversible and sluggish 4e−/oxygen (O2) chemistry could not be changed.120 Sun et al. reported a novel zinc-O2/zinc peroxide (ZnO2) chemical reaction to achieve a highly reversible redox reaction by replacing the alkaline electrolyte with non-alkaline water-based electrolyte in conventional ZAB configuration.121 The non-alkaline zinc trifluoro-methanesulfonate electrolyte (1 M mol kg−1 of Zn(OTf)2) showed a high zinc utilization ratio (ZUR) of 83.1% with high reversibility, while allowing stable operation in ambient air by eliminating the side reactions. On the other hand, the conventional alkaline electrolyte (6 mol kg−1 of KOH) exhibited a low ZUR of 8.1% because of the zinc anode corrosion (Fig. 13a). Interestingly, the high ZUR of Zn(OTf)2 was associated with the water-poor and zinc ion-rich inner Helmholtz layer (IHL) of the air cathode formed by the OTf− anions. Then, the ZnO2 formation mechanism was identified by analyzing the number of electrons participating in the reaction and the XRD pattern. Notably, the pressure of oxygen was twice as high as that of using Zn(OTf)2 electrolyte in the Zn–O2 battery using the ZnSO4 electrolyte. This result meant that 1.97e− electrons per O2 molecule moved (e−/O2) in the Zn(OTf)2 electrolyte and 3.79e− electrons per O2 molecule in ZnSO4 electrolyte (3.79e−/O2) (Fig. 13c). Meanwhile, the discharge products were analyzed using the XRD pattern and the SEM image of the air electrode obtained after discharge and recharge. The XRD patterns corresponding to ZnO2 in the Zn(OTf)2 electrolyte and zinc sulfate hydroxide [Zn4(OH)6SO4·0.5H2O (ZHS)] in the ZnSO4 electrolyte were detected, respectively. Upon recharging, the ZHS pattern was still present, but the ZnO2 pattern disappeared completely, confirming the high electrochemical reversibility of ZnO2 (Fig. 13b). The overall reaction of the ZAB chemistry of Zn(OTf)2 was expressed as the reversible formation and decomposition of ZnO2 (eqn (14)).
| Zn + O2 ↔ ZnO2 | (14) |
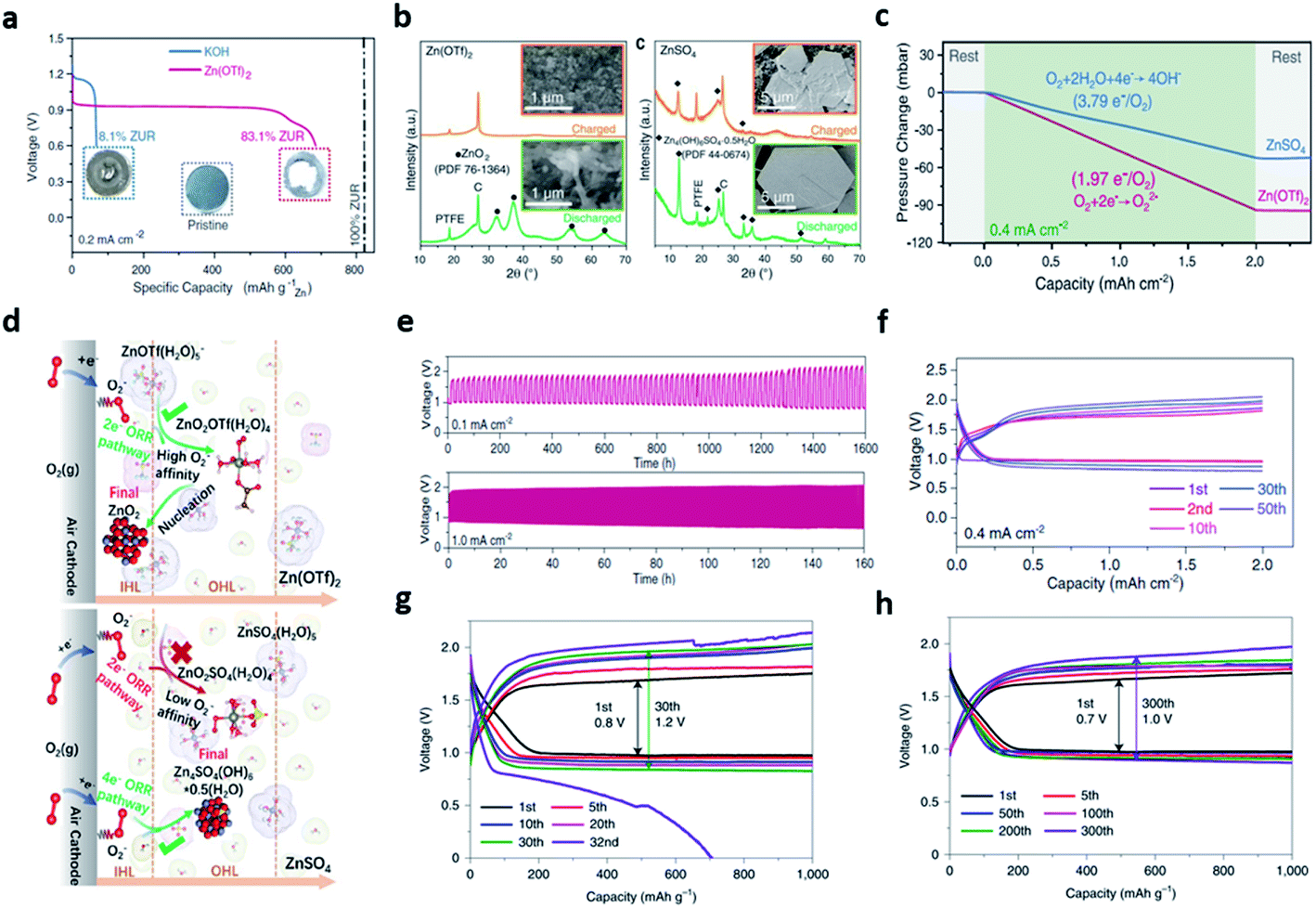 | ||
| Fig. 13 (a) Galvanostatic discharge profiles of Zn–air cells in KOH (blue) and Zn(OTf)2 (red). (b) XRD patterns and SEM images of air cathodes obtained after discharge and recharge in Zn(OTf)2 and ZnSO4 electrolytes. (c) Pressure change in the gas reservoir of Zn–O2 cells using Zn(OTf)2 and ZnSO4 electrolytes during a discharge process under neat O2 atmosphere. (d) Schematic illustration of the reaction processes in the IHL and outer Helmholtz layer (OHL) at the surface of the air cathode in Zn(OTf)2 and ZnSO4 electrolytes, respectively. (e) Galvanostatic voltage profiles of the Zn–air cell in a capacity fixed mode (fixed capacity: 2.0 mA h cm−2) at a current density of 0.4 mA cm−2. (f) Long-term cycling performance of Zn–air cells at current densities of 0.1 mA cm−2. (a–f) Reprinted with permission from ref. 116. Copyright 2021, American Association for the Advancement of Science. (g) Cycling performance of the Zn‖O2 batteries in 4 M Zn(OTf)2 (h) and 4 M Zn(OTf)2 + 0.5 M Me3EtOTf. (g and h) Reprinted with permission from ref. 117. Copyright 2021, Springer Nature. | ||
The cell reaction of the ZnSO4 electrolyte shows the formation and decomposition of reversible ZHS (eqn (15)).
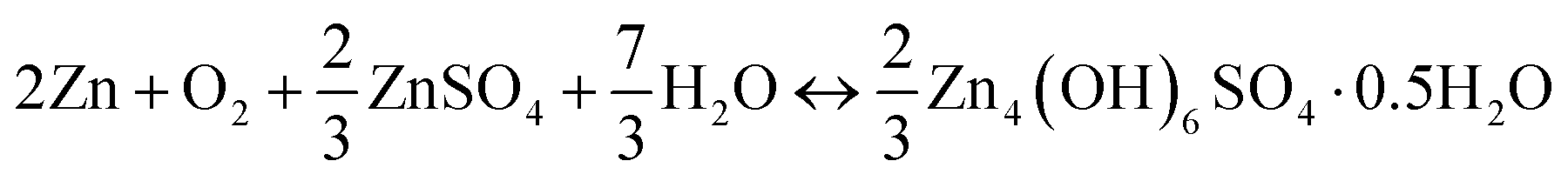 | (15) |
To understand the origin of the different mechanisms observed in the two electrolytes, multiscale simulations was employed, revealing that the difference originated from the hydrophobicity by the strongly hydrophobic CF3 group of OTf− (Fig. 13d). The dissimilarity of hydrophobicity led to a contrast electrochemical double-layer structure on the air cathode surface. It has been demonstrated with the interfacial accumulation density behavior, where the hydrophobic OTf− anion population formed an H2O-poor and Zn2+-rich IHL environment. Finally, a zinc–air cell fabricated with a zinc foil anode, Vucan-72R catalyst and Zn(OTf)2 electrolyte, maintained 10 h per cycle charge and discharge time and worked stably for 1600 h at a current density of 0.1 mA cm−2 (Fig. 13e). Furthermore, no significant polarization change was observed for 50 cycles even at a current density that is 4 times faster (Fig. 13f).
Inspired by the previous research results, Cao et al. added a trimethylethyl ammonium trifluoromethanesulfonate (Me3EtNOTf) salt to an aqueous Zn(OTf)2 electrolyte, which significantly inhibited the dendrite growth by the stable two-electron reaction.122 This unique reaction mainly caused by the in situ formed ZnF2-rich SEI enhanced the conductivity of Zn2+, while suppressing the hydrogen evolution reaction. The TEM image revealed that a ZnF2-rich interphase prevented the water reduction, while allowing Zn2+ transport. Furthermore, the inorganic ZnF2 was detected at ∼684.7 eV from the XPS spectra of the zinc electrode recovered from the Me3EtNOTf-containing electrolyte. To investigate the effects of composite interphase consisting of ZnF2 formed via alkylammonium salt on enhancing the zinc plating/stripping reversibility, the electrochemical stability window was measured using the non-active titanium (Ti) electrode. In 0.5 M Me3EtNOTf salt + 4 M Zn(OTf)2 electrolyte, both the onset potential for water reduction and the zinc plating potential were decreased by 201 and 97 mV, respectively, than in 4 M Zn(OTf)2 electrolyte. In addition, the hydrogen evolution current of 63 μA cm−2 generated at 0.4 V was removed. Finally, Zn‖O2 cell with 4 M Zn(OTf)2 electrolyte and Me3EtNOTf-containing electrolyte were fabricated, and the cycling test was conducted at 50 mA g−1 under a constant-capacity mode of 1000 mA h g−1 (the areal capacity of the cathode was 0.7 mA h cm−2). Interestingly, the overpotential of Zn‖O2 cell (Fig. 13g) increased 1.5 times after 30 cycles in the 4 M Zn(OTf)2 electrolyte (increasing from 0.8 to 1.2 V, and it increased significantly in the 32nd cycle). However, the overpotential of the Zn‖O2 cell increased only 1.43 times in the Me3EtNOTf-containing electrolyte (increasing from 0.7 to 1.0 V) even after 300 cycles (Fig. 13h).
5. Conclusion and outlook
In this review, we have shown the recent progress on bifunctional electrocatalysts for air cathodes, which is summarized in Table 1. This paper also reviewed the novel electrolyte system of ZABs with respect to the materials and the electrolyte aspects. The price competitiveness of the ZABs can be achieved by employing the PGM-free metal catalysts. However, the metal–air battery is a primary battery; thus, finding a suitable bifunctional electrocatalysts for the air cathode is important to effectively recharge the system. The requirements of the electrocatalysts and ZAB system are summarized in Fig. 14.| Catalyst | Current collector | E 1/2, ORR (V vs. RHE) | E j = 10, OER (V vs. RHE) | Potential difference ΔE (V) | OCV (V) | Power density (mW cm−2) | Cycle stablity @ j (mA cm−2) | Ref. |
|---|---|---|---|---|---|---|---|---|
| PtFeNC | Nickel foam | 0.895 | — | — | 1.492 | 148 | 24 h @ 10 (PtFeNC–IrO2) | 33 |
| SA-PtCoF | Nickel foam | 0.88 | 1.538 | 0.658 | 1.31 | 125 | 240 h @ 10 | 34 |
| Pt@CoS2-NrGO | Nickel foam | 0.85 | 1.465 | 0.615 | 1.41 | 114 | 55 h @ 10 | 36 |
| Pd–Cu/C | — | 0.84 | — | — | 1.43 | 219 | 40 h @ 10 | 41 |
| Ru@Co3O4 | Carbon cloth | 0.77 | 1.61 | 0.84 | — | 101.2 | 65 h @ 10 | 47 |
| NSP-Gra | Carbon paper | 0.82 | 1.76 | 0.94 | — | 225 | 15 h @ 5 | 54 |
| BP-CN-c | — | 0.84 | 1.58 | 0.74 | 1.47 | 168.3 | 300 h @ 10 | 61 |
| Co-NC-800 | Nickel foam | 0.93 | — | — | 1.44 | 109.5 | 33 h @ 5 | 62 |
| NPF-CNS | Carbon paper | 0.81 | 1.57 | 0.76 | 1.49 | 144 | 125 h @ 5 | 66 |
| Fe3C@C–Fe SAS | Carbon cloth | 0.91 | — | — | 1.38 | 74.8 | 110 h @ 10 | 70 |
| FeCoNi–N-rGO | Carbon cloth | 0.836 | 1.67 | 0.834 | 1.43 | 152.5 | 200 h @ 5 | 75 |
| Fe/Ni(1:3)-NG |
Carbon paper | 0.842 | 1.71 | 0.868 | 1.50 | 164.1 | 120 h @ 5 | 77 |
| CoDNG900 | Carbon paper | 0.864 | 1.614 | 0.75 | 1.45 | 205 | 667 h @ 10 | 80 |
| FeNi@NCNT-CP | Cotton pad | 0.85 | 1.58 | 0.73 | 1.551 | 200 | 250 h @ 10 | 82 |
| Cu@CuS NWs | Copper foam | 0.916 | 1.626 | 0.71 | 1.38 | 130 | 30 h @ 10 | 85 |
| CuMo2ON@NG | Nickel foam | 0.875 | 1.565 | 0.69 | 1.49 | 176.3 | 330 h @ 10 | 88 |
| Ni0.5Mo0.5OSe | CFC | 0.88 | 1.379 | 0.499 | 1.51 | 166.7 | 300 h @ 10 | 89 |
| V-Co3O4 | Carbon cloth | 0.821 | 1.581 | 0.76 | 1.45 | 120.3 | 1368 h @ 5 | 92 |
| Ru-Co3O4-1.0 | Carbon cloth | 0.77 | 1.61 | 0.84 | — | 101.2 | 65 h @ 10 | 93 |
| Ag/Ag2O@MCOF(Co) | — | 0.78 | 1.66 | 0.88 | 1.37 | 170.8 | 70 h @ 10 | 96 |
| Fe/Co-CNT@MXene-8 | Nickel foam | 0.86 | 1.59 | 0.73 | 1.52 | 138 | 375 h @ 10 | 97 |
| FeNiCO@NC-P | Carbon paper | 0.84 | 1.54 | 0.7 | 1.36 | 112 | 130 h @ 10 | 98 |
| CoP-NC@NFP | — | 0.831 | 1.55 | 0.719 | 1.44 | 93 | 200 h @ 2 | 102 |
| Co–SAs/SNPs@NC | Carbon paper | 0.898 | — | — | 1.482 | 223.5 | 720 h @ 5 | 104 |
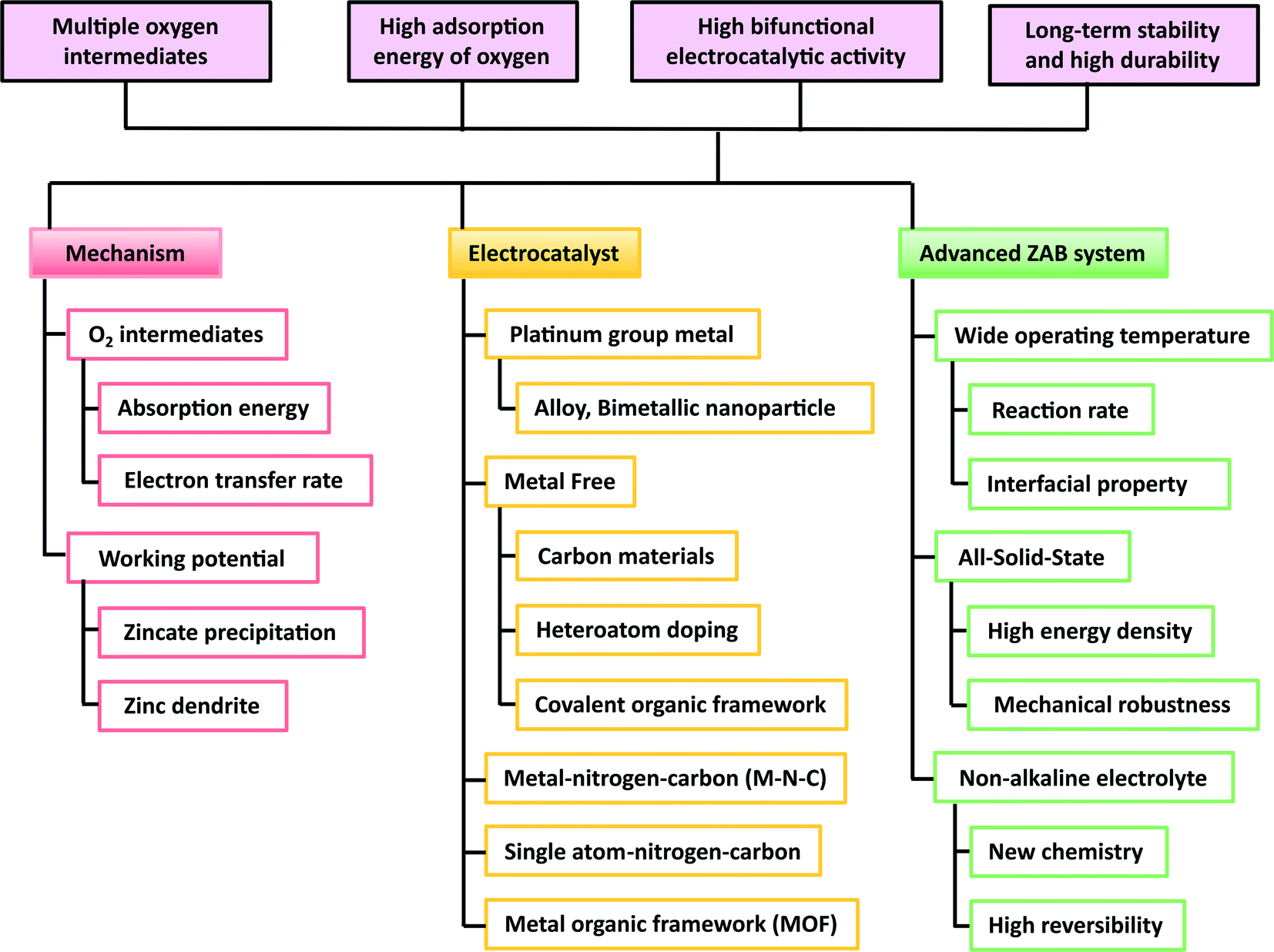 | ||
| Fig. 14 The requirement for the development of zinc–air batteries with respect to the mechanism, air cathode, and progress. | ||
In global urban environments, batteries with low cost as well as improved volumetric and gravimetric energy densities are required for applications in energy storage systems, mobile devices, and flexible devices. ZABs have excellent prospects because of their advantages, such as high energy density, low cost, and safety. However, for industrial applications, there are several challenges that should be overcome. It is necessary to develop air cathodes and electrocatalysts that can be used under various conditions such as aqueous/non-aqueous, voltage, and temperature. Thus, researchers should have an in-depth understanding of the reactions of complex oxygen intermediates and bi-functional mechanisms. In addition, there are many factors to consider, such as test environments, current density, and temperature, to apply ZABs from lab-scale results to practical applications.
Recently, the bifunctional performance of OER and ORR has become an essential element for the metal–air batteries. In general, the reversibility of oxygen is easily reduced because of the multi-step reactions between oxygen intermediates (*OO, *OOH, *O, and *OH). Therefore, it is necessary to improve the reversibility of oxygen with bifunctional electrocatalysts. Regarding this issue, we intensively discussed the reaction mechanism of the ZAB and its side reaction in alkaline electrolytes, as well as the effect of multiple oxygen intermediates. The PGM-based electrocatalysts are recognized as the best catalysts due to their excellent catalytic activity but they are limited by their low global storages and expensive cost. Metal-free electrocatalysts also have the problem of easy oxidation and structural decay. On the other hand, transition metals are inexpensive and have excellent catalytic activity for OER; thus, transition metals can be good alternatives for multi-functional electrocatalysts.
The performance of the rechargeable ZABs is not only dependent on the electrocatalysts but also strongly influenced by the interaction of the electrocatalysts with other components of the ZABs, for example separators, zinc metal anodes, and electrolytes. Commonly-used alkaline electrolytes can cause the formation of insoluble zinc oxide precipitates because of the formation of zincate ions and degradation of membranes. In addition, the alkaline solution can easily decompose the polymer binder of the air cathode, causing the loss of the active material and generating large interfacial impedance, which eventually increases the cell overpotential. Therefore, it is necessary to find alternative electrolytes to address the fundamental problem of using alkaline electrolytes. For instance, the use of a solidified liquid electrolyte is a good strategy to improve the electrolyte–electrode interface properties.
The cell structure optimization should be studied by considering the weight and volume of the ZABs to achieve high energy density. In addition, the oxygen circulation path in the air cathode of the ZABs should be considered because oxygen, the active material of the air cathode, is obtained from ambient air. Thus, a smart design providing a smooth oxygen transport path in a long cycle, high stability in various environments, and even applications in flexible devices is needed for the commercialization of ZABs.
On the other hand, to reach the commercialization level of rechargeable ZABs, there are still many challenges, such as the development of high-performance bifunctional catalysts and the improvement of the durability and oxidation resistance of the carbon support. Intrinsically, the carbon material in the air cathode possesses corrosion issues as the cycle continues, which should be addressed for its high cycle stability and catalytic property. The new electrolyte system has potential to address side reactants in the concentrated alkaline electrolyte, improving the battery efficiency and the cycle stability. Also, the stable bifunctional electrocatalysts and electrolytes for wide temperature operation in both aqueous and non-aqueous environment should be taken into account for the commercialization of ZABs. Finally, we believe that the ZAB technology with its low cost and high energy density has great potential as a next-generation energy storage device and an in-depth understanding toward the current challenges of ZABs can provide further insights into the development of better ZABs.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a 2-Year Research Grant of Pusan National University.References
- J. Han, H. Bao, J.-Q. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, Appl. Catal., B, 2021, 280, 119411 CrossRef CAS.
- J. Yu, Q. Zhou, X. Xue, H. Zhang, X. Li, F. Wang, Q. Chen and H. Zhu, New J. Chem., 2021, 45, 14608–14615 RSC.
- J. Wang, J. Xu, X. Guo, T. Shen, C. Xuan, B. Tian, Z. Wen, Y. Zhu and D. Wang, Appl. Catal., B, 2021, 298, 120539 CrossRef CAS.
- H. Wang, Y. Jiao, S. Wang, P. Ye, J. Ning, Y. Zhong and Y. Hu, Small, 2021, 2103517 CrossRef CAS PubMed.
- J. Yan, Y. Huang, Y. Zhang, W. Peng, S. Xia, J. Yu and B. Ding, Nano Lett., 2021, 21, 2618–2624 CrossRef CAS PubMed.
- G. Janani, S. Surendran, H. Choi, M.-K. Han and U. Sim, Small, 2021, 17, 2103613 CrossRef CAS PubMed.
- X. Han, X. Li, J. White, C. Zhong, D. Yida, W. Hu and T. Ma, Adv. Energy Mater., 2018, 8, 1801396 CrossRef.
- X. Li, Y. Guo, T. Gao, P. Li, Z. Jin and D. Xiao, ACS Appl. Mater. Interfaces, 2021, 13(48), 57411–57421 CrossRef CAS PubMed.
- L. Song, T. Zheng, L. Zheng, B. Lu, H. Chen, Q. He, W. Zheng, Y. Hou, J. Lian, Y. Wu, J. Chen, Z. Ye and J. Lu, Appl. Catal., B, 2022, 300, 120712 CrossRef CAS.
- H. Hu, J. Wang, B. Cui, X. Zheng, J. Lin, Y. Deng and X. Han, Angew. Chem., Int. Ed., 2022, 61, e202114441 CAS.
- N. Batool, W. Iqbal, X.-F. Han, W.-T. Wang, H.-T. Teng, X. Hao, R. Yang and J.-H. Tian, ACS Appl. Nano Mater., 2021, 4, 12365–12372 CrossRef CAS.
- X. Chen, B. Liu, C. Zhong, Z. Liu, J. Liu, L. Ma, Y. Deng, X. Han, T. Wu, W. Hu and J. Lu, Adv. Energy Mater., 2017, 7, 1700779 CrossRef.
- X. Liu, Y. Yuan, J. Liu, B. Liu, X. Chen, J. Ding, X. Han, Y. Deng, C. Zhong and W. Hu, Nat. Commun., 2019, 10, 4767 CrossRef PubMed.
- D. Zhu, Q. Zhao, G. Fan, S. Zhao, L. Wang, F. Li and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 12460–12464 CrossRef CAS PubMed.
- F. Gui, Q. Jin, D. Xiao, X. Xu, Q. Tan, D. Yang, B. Li, P. Ming, C. Zhang, Z. Chen, S. Siahrostami and Q. Xiao, Small, 2021, 2105928 Search PubMed.
- Z. Li, Q. Wang, X. Bai, M. Wang, Z. Yang, Y. Du, G. E. Sterbinsky, D. Wu, Z. Yang, H. Tian, F. Pan, M. Gu, Y. Liu, Z. Feng and Y. Yang, Energy Environ. Sci., 2021, 14, 5035–5043 RSC.
- C.-L. Huang, P.-Y. Wang and Y.-Y. Li, Chem. Eng. J., 2020, 395, 125031 CrossRef CAS.
- F. Liu, N. Yan, G. Zhu, Z. Liu, S. Ma, G. Xiang, S. Wang, X. Liu and W. Wang, New J. Chem., 2021, 45, 13004–13014 RSC.
- Y. Ying, K. Fan, X. Luo, J. Qiao and H. Huang, J. Mater. Chem. A, 2021, 9, 16860–16867 RSC.
- L. Hu, C. Dai, L. Chen, Y. Zhu, Y. Hao, Q. Zhang, L. Gu, X. Feng, S. Yuan, L. Wang and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 27324 CrossRef CAS PubMed.
- X. Zheng, X. Cao, Z. Sun, K. Zeng, J. Yan, P. Strasser, X. Chen, S. Sun and R. Yang, Appl. Catal., B, 2020, 272, 118967 CrossRef CAS.
- H. Liu, J. Guan, S. Yang, Y. Yu, R. Shao, Z. Zhang, M. Dou, F. Wang and Q. Xu, Adv. Mater., 2020, 32, 2003649 CrossRef CAS PubMed.
- G. Han, L. Li, X. Li, Y. Sun, C. Du, Y. Gao and G. Yin, Carbon, 2021, 174, 683–692 CrossRef CAS.
- W. Liu, J. Bao, L. Xu, M. Guan, Z. Wang, J. Qiu, Y. Huang, J. Xia, Y. Lei and H. Li, Appl. Surf. Sci., 2019, 478, 552–559 CrossRef CAS.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- M. B. Lim, T. N. Lambert and E. I. Ruiz, J. Electrochem. Soc., 2020, 167, 060508 CrossRef CAS.
- A. K. Worku, D. W. Ayele and N. G. Habtu, Mater. Today Adv., 2021, 9, 100116 CrossRef CAS.
- D. Chen, Z. Chen, Z. Lu, X. Zhang, J. Tang and C. V. Singh, Nanoscale, 2020, 12, 18721–18732 RSC.
- F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skúlason, T. Bligaard and J. K. Nørskov, Phys. Rev. Lett., 2007, 99, 016105 CrossRef CAS PubMed.
- Q. Liang, G. Brocks and A. Bieberle-Hütter, J. Phys.: Energy, 2021, 3, 026001 CAS.
- G. Yasin, S. Ibrahim, S. Ibraheem, S. Ali, R. Iqbal, A. Kumar, M. Tabish, Y. Slimani, T. A. Nguyen, H. Xu and W. Zhao, J. Mater. Chem. A, 2021, 9, 18222–18230 RSC.
- P. Sabatier, Ber. Dtsch. Chem. Ges., 1911, 44, 1984–2001 CrossRef CAS.
- X. Zhong, S. Ye, J. Tang, Y. Zhu, D. Wu, M. Gu, H. Pan and B. Xu, Appl. Catal., B, 2021, 286, 119891 CrossRef CAS.
- Z. Li, W. Niu, Z. Yang, N. Zaman, W. Samarakoon, M. Wang, A. Kara, M. Lucero, M. V. Vyas, H. Cao, H. Zhou, G. E. Sterbinsky, Z. Feng, Y. Du and Y. Yang, Energy Environ. Sci., 2020, 13, 884–895 RSC.
- L. Liang, H. jin, H. Zhou, B. Liu, C. Hu, D. Chen, J. Zhu, Z. Wang, H.-W. Li, S. Liu, D. He and S. Mu, J. Energy Chem., 2022, 65, 48–54 CrossRef.
- N. Logeshwaran, S. Ramakrishnan, S. S. Chandrasekaran, M. Vinothkannan, A. R. Kim, S. Sengodan, D. B. Velusamy, P. Varadhan, J.-H. He and D. J. Yoo, Appl. Catal., B, 2021, 297, 120405 CrossRef CAS.
- Y. Lee, J. H. Ahn, H.-Y. Park, J. Jung, Y. Jeon, D.-G. Lee, M.-H. Kim, E. Lee, C. Kim, Y. Kwon, H.-W. Lee, J. H. Jang, J. H. Lee and H.-K. Song, Nano Energy, 2021, 79, 105363 CrossRef CAS.
- R. K. Pittkowski, D. F. Abbott, R. Nebel, S. Divanis, E. Fabbri, I. E. Castelli, T. J. Schmidt, J. Rossmeisl and P. Krtil, Electrochim. Acta, 2021, 366, 137327 CrossRef CAS.
- S. Zhang, J. Zhang, P. Liang, C. Zhang, T. Kou and Z. Zhang, J. Power Sources, 2021, 497, 229895 CrossRef CAS.
- X. Deng, S. Yin, Z. Xie, F. Gao, S. Jiang and X. Zhou, Int. J. Hydrogen Energy, 2021, 46, 17731–17740 CrossRef CAS.
- M. Ma, W. Zhu, Q. Shao, H. Shi, F. Liao, C. Shao and M. Shao, ACS Appl. Nano Mater., 2021, 4, 1478–1484 CrossRef CAS.
- Y. Ma, A. N. Kuhn, W. Gao, T. Al-Zoubi, H. Du, X. Pan and H. Yang, Nano Energy, 2021, 79, 105465 CrossRef CAS.
- X. Zhu, L. Huang, M. Wei, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2021, 281, 119460 CrossRef CAS.
- Y. Chen, P. Xu, R. Wang and X. Pan, Mater. Lett., 2021, 286, 129266 CrossRef CAS.
- M. Gong, D. Xiao, Z. Deng, R. Zhang, W. Xia, T. Zhao, X. Liu, T. Shen, Y. Hu, Y. Lu, X. Zhao, H. Xin and D. Wang, Appl. Catal., B, 2021, 282, 119617 CrossRef CAS.
- R. Gao, J. Wang, Z.-F. Huang, R. Zhang, W. Wang, L. Pan, J. Zhang, W. Zhu, X. Zhang, C. Shi, J. Lim and J.-J. Zou, Nat. Energy, 2021, 6, 614–623 CrossRef CAS.
- C. Huang, Q. Ji, H. Zhang, Y. Wang, S. Wang, X. Liu, Y. Guo and C. Zhang, J. Colloid Interface Sci., 2022, 606, 654–665 CrossRef CAS PubMed.
- Y. Gu, Y. Min, L. Li, Y. Lian, H. Sun, D. Wang, M. H. Rummeli, J. Guo, J. Zhong, L. Xu, Y. Peng and Z. Deng, Chem. Mater., 2021, 33, 4135–4145 CrossRef CAS.
- Y. Zhu, J. Peng, X. Zhu, L. Bu, Q. Shao, C.-W. Pao, Z. Hu, Y. Li, J. Wu and X. Huang, Nano Lett., 2021, 21, 6625–6632 CrossRef CAS PubMed.
- J. Liu, C. Wang, Y. Song, S. Zhang, Z. Zhang, L. He and M. Du, J. Colloid Interface Sci., 2021, 591, 253–263 CrossRef CAS PubMed.
- S. Cao, W. Shang, G.-L. Li, Z.-F. Lu, X. Wang, Y. Yan, C. Hao, S. Wang and G. Sun, Carbon, 2021, 184, 127–135 CrossRef CAS.
- H. Yu, H. Zhang and Z. Zhang, ChemCatChem, 2021, 13, 397–406 CrossRef CAS.
- J. Deng, L. Wang, F. Jin and Y. H. Hu, J. Mater. Chem. A, 2021, 9, 10081–10087 RSC.
- Y. Wang, N. Xu, R. He, L. Peng, D. Cai and J. Qiao, Appl. Catal., B, 2021, 285, 119811 CrossRef CAS.
- H. Fan, T. Wang, H. Gong, C. Jiang, Z. Sun, M. Zhao, L. Song and J. He, Carbon, 2021, 180, 31–40 CrossRef CAS.
- L. Chen, Y. Chen, C. Xu, W. Wang, W. Fu, W. Hu, M. Zhou, B. He, Q. Chen, Z. Hou and W. Xu, Mater. Today Energy, 2021, 20, 100670 CrossRef CAS.
- L. Zhang, T. Gu, K. Lu, L. Zhou, D.-S. Li and R. Wang, Adv. Funct. Mater., 2021, 31, 2103187 CrossRef CAS.
- Y. Zan, Z. Zhang, B. Zhu, M. Dou and F. Wang, Carbon, 2021, 174, 404–412 CrossRef CAS.
- Y. Long, F. Ye, L. Shi, X. Lin, R. Paul, D. Liu and C. Hu, Carbon, 2021, 179, 365–376 CrossRef CAS.
- A. T. N. Nguyen and J. H. Shim, RSC Adv., 2021, 11, 12520–12530 RSC.
- X. Wang, R. K. M. Raghupathy, C. J. Querebillo, Z. Liao, D. Li, K. Lin, M. Hantusch, Z. Sofer, B. Li, E. Zschech, I. M. Weidinger, T. D. Kühne, H. Mirhosseini, M. Yu and X. Feng, Adv. Mater., 2021, 33, 2008752 CrossRef CAS PubMed.
- S. Zhao, L. Ban, J. Zhang, W. Yi, W. Sun and Z. Zhu, Chem. Eng. J., 2021, 409, 128171 CrossRef CAS.
- T. Lu, X. Hu, J. He, R. Li, J. Gao, Q. Lv, Z. Yang, S. Cui and C. Huang, Nano Energy, 2021, 85, 106024 CrossRef CAS.
- Z. Duan, G. Han, H. Huo, Z. Lin, L. Ge, C. Du, Y. Gao and G. Yin, ACS Sustainable Chem. Eng., 2021, 9, 1264–1271 CrossRef CAS.
- Q. Yu, J. Wang, H. Li, R. Li, S. Zeng, R. Li, Q. Yao, H. Chen, K. Qu and Y. Zheng, Chem. Eng. J., 2022, 429, 132102 CrossRef CAS.
- Y. Zheng, H. Song, S. Chen, X. Yu, J. Zhu, J. Xu, K. A. I. Zhang, C. Zhang and T. Liu, Small, 2020, 16, 2004342 CrossRef CAS PubMed.
- Y. Zheng, S. Chen, K. A. I. Zhang, J. Zhu, J. Xu, C. Zhang and T. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 13328–13337 CrossRef CAS PubMed.
- C. Liu, F. Liu, H. Li, J. Chen, J. Fei, Z. Yu, Z. Yuan, C. Wang, H. Zheng, Z. Liu, M. Xu, G. Henkelman, L. Wei and Y. Chen, ACS Nano, 2021, 15, 3309–3319 CrossRef CAS PubMed.
- B. Wang, P. Zhao, J. Feng, D. Chen, Y. Huang, L. Sui, H. Dong, S. Ma, L. Dong and L. Yu, J. Colloid Interface Sci., 2021, 588, 184–195 CrossRef CAS PubMed.
- X. Wei, S. Song, N. Wu, X. Luo, L. Zheng, L. Jiao, H. Wang, Q. Fang, L. Hu, W. Gu, W. Song and C. Zhu, Nano Energy, 2021, 84, 105840 CrossRef CAS.
- N. Shang, C. Wang, X. Zhang, S. Gao, S. Zhang, T. Meng, J. Wang, H. Wang, C. Du, T. Shen, J. Huang, Y. Qiao, Q. Wu and Y. Gao, Chem. Eng. J., 2021, 426, 127345 CrossRef CAS.
- J. Han, H. Bao, J.-Q. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, Appl. Catal., B, 2021, 280, 119411 CrossRef CAS.
- J. Ma, J. Li, R. Wang, Y. Yang, P. Yin, J. Mao, T. Ling and S. Qiao, Mater. Today Energy, 2021, 19, 100624 CrossRef CAS.
- C. Lai, J. Fang, X. Liu, M. Gong, T. Zhao, T. Shen, K. Wang, K. Jiang and D. Wang, Appl. Catal., B, 2021, 285, 119856 CrossRef CAS.
- X. Chen, D. Chen, G. Li, C. Gong, Y. Chen, Q. Zhang, J. Sui, H. Dong, J. Yu, L. Yu and L. Dong, J. Alloys Compd., 2021, 873, 159833 CrossRef CAS.
- S. Zhang, W. Yang, Y. Liang, X. Yang, M. Cao and R. Cao, Appl. Catal., B, 2021, 285, 119780 CrossRef CAS.
- Y. Ma, H. Fan, C. Wu, M. Zhang, J. Yu, L. Song, K. Li and J. He, Carbon, 2021, 185, 526–535 CrossRef CAS.
- X. Hao, Z. Jiang, B. Zhang, X. Tian, C. Song, L. Wang, T. Maiyalagan, X. Hao and Z.-J. Jiang, Adv. Sci., 2021, 8, 2004572 CrossRef CAS PubMed.
- X. Han, T. Zhang, W. Chen, B. Dong, G. Meng, L. Zheng, C. Yang, X. Sun, Z. Zhuang, D. Wang, A. Han and J. Liu, Adv. Energy Mater., 2021, 11, 2002753 CrossRef CAS.
- A. Wang, C. Zhao, M. Yu and W. Wang, Appl. Catal., B, 2021, 281, 119514 CrossRef CAS.
- D. Yu, Y. Ma, F. Hu, C.-C. Lin, L. Li, H.-Y. Chen, X. Han and S. Peng, Adv. Energy Mater., 2021, 11, 2101242 CrossRef CAS.
- X. Zheng, X. Cao, K. Zeng, J. Yan, Z. Sun, M. H. Rümmeli and R. Yang, Small, 2021, 17, 2006183 CrossRef CAS PubMed.
- G. Solomon, M. G. Kohan, M. Vagin, F. Rigoni, R. Mazzaro, M. M. Natile, S. You, V. Morandi, I. Concina and A. Vomiero, Nano Energy, 2021, 81, 105664 CrossRef CAS.
- A. Saad, D. Liu, Y. Wu, Z. Song, Y. Li, T. Najam, K. Zong, P. Tsiakaras and X. Cai, Appl. Catal., B, 2021, 298, 120529 CrossRef CAS.
- T. L. L. Doan, D. T. Tran, D. C. Nguyen, D. H. Kim, N. H. Kim and J. H. Lee, Adv. Funct. Mater., 2021, 31, 2007822 CrossRef CAS.
- Y. Lei, T. Xu, S. Ye, L. Zheng, P. Liao, W. Xiong, J. Hu, Y. Wang, J. Wang, X. Ren, C. He, Q. Zhang, J. Liu and X. Sun, Appl. Catal., B, 2021, 285, 119809 CrossRef CAS.
- Y. Tan, W. Zhu, Z. Zhang, W. Wu, R. Chen, S. Mu, H. Lv and N. Cheng, Nano Energy, 2021, 83, 105813 CrossRef CAS.
- J. Balamurugan, T. T. Nguyen, N. H. Kim, D. H. Kim and J. H. Lee, Nano Energy, 2021, 85, 105987 CrossRef CAS.
- J. Balamurugan, T. T. Nguyen, D. H. Kim, N. H. Kim and J. H. Lee, Appl. Catal., B, 2021, 286, 119909 CrossRef CAS.
- V. Jose, J. M. V. Nsanzimana, H. Hu, J. Choi, X. Wang and J.-M. Lee, Adv. Energy Mater., 2021, 11, 2100157 CrossRef CAS.
- W. Shang, W. Yu, Y. Ma, Y. He, Z. Zhao, M. Ni, H. Zhao and P. Tan, Adv. Mater. Interfaces, 2021, 8, 2101256 CrossRef CAS.
- Y. Rao, S. Chen, Q. Yue and Y. Kang, ACS Catal., 2021, 11, 8097–8103 CrossRef CAS.
- C. Huang, Q. Ji, H. Zhang, Y. Wang, S. Wang, X. Liu, Y. Guo and C. Zhang, J. Colloid Interface Sci., 2022, 606, 654–665 CrossRef CAS PubMed.
- M. Wu, G. Zhang, N. Chen, Y. Hu, T. Regier, D. Rawach and S. Sun, ACS Energy Lett., 2021, 6, 1153–1161 CrossRef CAS.
- M. Guo, M. Xu, Y. Qu, C. Hu, P. Yan, T. T. Isimjan and X. Yang, Appl. Catal., B, 2021, 297, 120415 CrossRef CAS.
- M. Wang, C. Wang, J. Liu, F. Rong, L. He, Y. Lou, Z. Zhang and M. Du, ACS Sustainable Chem. Eng., 2021, 9, 5872–5883 CrossRef CAS.
- C. Zhang, H. Dong, B. Chen, T. Jin, J. Nie and G. Ma, Carbon, 2021, 185, 17–26 CrossRef CAS.
- D. Ren, J. Ying, M. Xiao, Y.-P. Deng, J. Ou, J. Zhu, G. Liu, Y. Pei, S. Li, A. M. Jauhar, H. Jin, S. Wang, D. Su, A. Yu and Z. Chen, Adv. Funct. Mater., 2020, 30, 1908167 CrossRef CAS.
- X. Liu, Y. Ma, Y. Cai, S. Hu, J. Chen, Z. Liu and Z. Wang, RSC Adv., 2021, 11, 15722–15728 RSC.
- T. Gu, R. Sa, L. Zhang, D.-S. Li and R. Wang, Appl. Catal., B, 2021, 296, 120360 CrossRef CAS.
- A. Muthurasu, A. P. Tiwari, K. Chhetri, B. Dahal and H. Y. Kim, Nano Energy, 2021, 88, 106238 CrossRef CAS.
- E. Vijayakumar, S. Ramakrishnan, C. Sathiskumar, D. J. Yoo, J. Balamurugan, H. S. Noh, D. Kwon, Y. H. Kim and H. Lee, Chem. Eng. J., 2022, 428, 131115 CrossRef CAS.
- C. Chen, Y. Tuo, Q. Lu, H. Lu, S. Zhang, Y. Zhou, J. Zhang, Z. Liu, Z. Kang, X. Feng and D. Chen, Appl. Catal., B, 2021, 287, 119953 CrossRef CAS.
- Z. Wang, C. Zhu, H. Tan, J. Liu, L. Xu, Y. Zhang, Y. Liu, X. Zou, Z. Liu and X. Lu, Adv. Funct. Mater., 2021, 31, 2104735 CrossRef CAS.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. G. d. Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. D. Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- L. An, B. Huang, Y. Zhang, R. Wang, N. Zhang, T. Dai, P. Xi and C.-H. Yan, Angew. Chem., Int. Ed., 2019, 58, 9459–9463 CrossRef CAS PubMed.
- Z. Pei, L. Ding, C. Wang, Q. Meng, Z. Yuan, Z. Zhou, S. Zhao and Y. Chen, Energy Environ. Sci., 2021, 14, 4926–4935 RSC.
- Z. Pei, Z. Yuan, C. Wang, S. Zhao, J. Fei, L. Wei, J. Chen, C. Wang, R. Qi, Z. Liu and Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 4793–4799 CrossRef CAS PubMed.
- C.-X. Zhao, J.-N. Liu, N. Yao, J. Wang, D. Ren, X. Chen, B.-Q. Li and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 15281–15285 CrossRef CAS PubMed.
- H. Miao, B. Chen, S. Li, X. Wu, Q. Wang, C. Zhang, Z. Sun and H. Li, J. Power Sources, 2020, 450, 227653 CrossRef CAS.
- P. Jiang, S. Chen, C. Wang, D. Wang, J. Diao, Z. Cao, Z. Lin, Q. Luo, J. Lu, H. Huang, C. Zong, L. Hu and Q. Chen, Mater. Today Sustain., 2020, 9, 100039 CrossRef.
- D. Yang, D. Chen, Y. Jiang, E. H. Ang, Y. Feng, X. Rui and Y. Yu, Carbon Energy, 2021, 3, 50–65 CrossRef CAS.
- T. Jiang, P. Dai, W. Zhang and M. Wu, Electrochim. Acta, 2021, 373, 137903 CrossRef CAS.
- J. Deng, L. Wang, F. Jin and Y. H. Hu, J. Mater. Chem. A, 2021, 9, 10081–10087 RSC.
- S. S. Shinde, J. Y. Jung, N. K. Wagh, C. H. Lee, D.-H. Kim, S.-H. Kim, S. U. Lee and J.-H. Lee, Nat. Energy, 2021, 6, 592–604 CrossRef CAS.
- A. R. Mainar, O. Leonet, M. Bengoechea, I. Boyano, I. de Meatza, A. Kvasha, A. Guerfi and J. Alberto Blázquez, Int. J. Energy Res., 2016, 40, 1032–1049 CrossRef CAS.
- C.-Y. Jung, T.-H. Kim, W.-J. Kim and S.-C. Yi, Energy, 2016, 102, 694–704 CrossRef CAS.
- F. Allebrod, C. Chatzichristodoulou, P. L. Mollerup and M. B. Mogensen, Int. J. Hydrogen Energy, 2012, 37, 16505–16514 CrossRef CAS.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- C. Wang, J. Li, Z. Zhou, Y. Pan, Z. Yu, Z. Pei, S. Zhao, L. Wei and Y. Chen, EnergyChem, 2021, 3, 100055 CrossRef CAS.
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. Wang and M. Winter, Science, 2021, 371, 46–51 CrossRef CAS PubMed.
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Hou, K. Gaskell, J. T. Fourkas, X.-Q. Yang, K. Xu, O. Borodin and C. Wang, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |