
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvent-modulated proton-coupled electron transfer in an iridium complex with an ESIPT ligand†
Siin
Kim
ab,
Jungkweon
Choi
 ab,
Dae Won
Cho
c,
Mina
Ahn
d,
Seunghwan
Eom
ab,
Jungmin
Kim
ab,
Kyung-Ryang
Wee
*d and
Hyotcherl
Ihee
*ab
ab,
Dae Won
Cho
c,
Mina
Ahn
d,
Seunghwan
Eom
ab,
Jungmin
Kim
ab,
Kyung-Ryang
Wee
*d and
Hyotcherl
Ihee
*ab
aDepartment of Chemistry and KI for the BioCentury, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
bCenter for Advanced Reaction Dynamics, Institute for Basic Science, Daejeon 34141, Republic of Korea. E-mail: hyotcherl.ihee@kaist.ac.kr
cDepartment of Advanced Materials Chemistry, Korea University, Sejong Campus, Sejong 30019, Korea
dDepartment of Chemistry and Institute of Natural Science, Daegu University, Gyeongsan 38453, Republic of Korea. E-mail: krwee@daegu.ac.kr
First published on 5th March 2022
Abstract
Proton-coupled electron transfer (PCET), an essential process in nature with a well-known example of photosynthesis, has recently been employed in metal complexes to improve the energy conversion efficiency; however, a profound understanding of the mechanism of PCET in metal complexes is still lacking. In this study, we synthesized cyclometalated Ir complexes strategically designed to exploit the excited-state intramolecular proton transfer (ESIPT) of the ancillary ligand and studied their photoinduced PCET in both aprotic and protic solvent environments using femtosecond transient absorption spectroscopy and density functional theory (DFT) and time-dependent DFT calculations. The data reveal solvent-modulated PCET, where charge transfer follows proton transfer in an aprotic solvent and the temporal order of charge transfer and proton transfer is reversed in a protic solvent. In the former case, ESIPT from the enol form to the keto form, which precedes the charge transfer from Ir to the ESIPT ligand, improves the efficiency of metal-to-ligand charge transfer. This finding demonstrates the potential to control the PCET reaction in the desired direction and the efficiency of charge transfer by simply perturbing the external hydrogen-bonding network with the solvent.
Introduction
Proton-coupled electron transfer (PCET), which involves both proton transfer (PT) and electron transfer (ET), plays important roles in chemical and biological processes relevant to energy conversion, such as oxygen evolution in photosystem II.1–11 Naturally, researchers have long endeavored to understand the mechanism of PCET and harness it in molecular systems synthesized to achieve higher energy conversion efficiencies by mimicking natural biological systems.12–19 Previous studies on PCET discovered that PCET occurs through three representative mechanisms; (i) stepwise ET-PT, (ii) stepwise PT-ET, and (iii) concerted PCET.5,7,11,20,21 Moreover, to gain further insight into strategies for designing artificial photosynthetic devices, the photoinduced PCET has been intensively studied.2,22Recently, several researchers have employed PCET in various metal complexes to provide optimal photophysical properties for photochemical devices.23–25 Metal complexes with strong spin–orbit coupling (SOC) and tunable photophysical properties modulated by ligands have garnered significant attention as materials applicable to photocatalysts, solar cells, organic light-emitting diodes, and photosensitizers. Accordingly, such metal complexes have been extensively used in various photoelectric devices and photocatalytic reactions.26–29 In this regard, the strategy of using PCET in metal complexes has great potential to further improve the photophysical properties of metal complexes.2,9,12,30 Nevertheless, the photophysical properties of PCET-based metal complexes are yet to be explored because of their complicated reaction dynamics compared to those of general metal complexes.13,31 Moreover, to apply PCET-based metal complexes to biological systems and photochemical devices, it is critical to understand the solvent dependence of their photophysical properties, including PCET dynamics. In particular, the effect of external hydrogen bonding, which significantly affects the proton transfer process, must be studied.
Among various metal complexes, cyclometalated Ir complexes have attracted considerable interest owing to their usage in various fields, which is, in turn, owing to their synthetic versatility, tunable photophysical properties, and excellent thermal and photochemical stability.27–29,32–36 Despite the advantages of Ir complexes, the excited-state dynamics of PCET-based Ir complexes have been less studied than ruthenium and rhenium complexes.37 The study of photoinduced PCET in Ir complexes is scarce.
To address this issue, we designed and synthesized an Ir complex, Ir-PIPN, as a model system using 2-(2,4-difluorophenyl)pyridine (dfppy) as the main cyclometalating ligand and 1-(1-phenyl-1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl)naphthalen-2-ol (PIPN) as the ancillary ligand (Fig. 1a and S1†). PIPN, which undergoes the excited-state intramolecular proton transfer (ESIPT) process from the enol form to the keto form according to a recent report38 on its excited-state relaxation dynamics, was used as the ancillary ligand to avoid the complexity of the intermolecular PCET. The NMR spectrum of Ir-PIPN demonstrates that Ir-PIPN exists as the enol form in the ground state (Fig. S1†). To understand how PCET is modulated by external hydrogen bonding perturbation, we investigated the PCET process in Ir-PIPN in aprotic and protic solvents using femtosecond transient absorption (fs-TA) spectroscopy, and density functional theory (DFT) and time-dependent DFT (TDDFT) calculations. The data reveal solvent-modulated PCET in which the temporal order of PT and charge transfer (CT) is reversed by the solvent environment. Here, CT refers to one full ET or partial ET,39,40 and the CT in Ir complexes means metal-to-ligand CT (MLCT), which occurs from Ir to the ESIPT ligand. In an aprotic solvent such as dichloromethane (DCM), PT precedes CT. In contrast, in a protic solvent, such as ethanol, CT is followed by PT. Furthermore, the efficiency of CT decreases in a protic solvent where PT occurs later than CT. In contrast, CT can occur efficiently with PT preceding in an aprotic solvent, indicating that PT can facilitate CT. These results raise the possibility of controlling the reaction pathway and efficiency of CT in metal complexes by modulating the solvent environment. We also investigated the effect of different ESIPT ligands on the dynamics of the Ir complexes. It was reported that 2-(1-phenyl-1H-imidazo[4,5-f][1,10]phenanthrolin-2-yl)phenol (PIPP-H) has remarkably different dynamics compared with PIPN owing to the structural difference between two molecules.38 We synthesized Ir-PIPP-H and investigated the dynamics of Ir-PIPP-H for comparison with Ir-PIPN. The NMR spectrum demonstrated that Ir-PIPP-H exists as an enol form in the ground state, as in Ir-PIPN (Fig. S2 and S19a†). It turned out that the dynamics of Ir-PIPP-H are not significantly different from those of Ir-PIPN, whereas the characteristics of the steady-state emission spectrum differ from those of Ir-PIPN.
Results and discussion
Absorption/emission spectra and emission lifetimes
Because Ir-PIPN exists as the enol form in the ground state (1GSE), as supported by its NMR spectrum (Fig. S1†), the absorption spectrum originates from the enol form. The absorption spectrum of Ir-PIPN in DCM generally follows that of the free PIPN ligand, except that some of the fine features of PIPN are broadened in Ir-PIPN, as shown in Fig. 1d. Although the free dfppy ligand exhibits no absorption at wavelengths longer than 310 nm,41 Ir-PIPN has absorption up to 550 nm. In comparison with the free PIPN ligand, Ir-PIPN shows a weakened vibronic feature, particularly at approximately 375 nm. This spectral difference between the free ligands and Ir-PIPN provides evidence of the interaction between Ir and the ligand. The absorption from 350 to 390 nm of the free PIPN ligand corresponds to the π–π* transition of the ESIPT ligand (PIPN), and thus the absorption in the same wavelength range of Ir-PIPN should correspond to the transition of the PIPN ligand of Ir-PIPN. Considering that only the enol form predominantly exists in the ground state, this absorption band corresponds to the transition from the ground state of the enol form (1GSE) to the singlet ligand-centered state of the enol form (1LCE).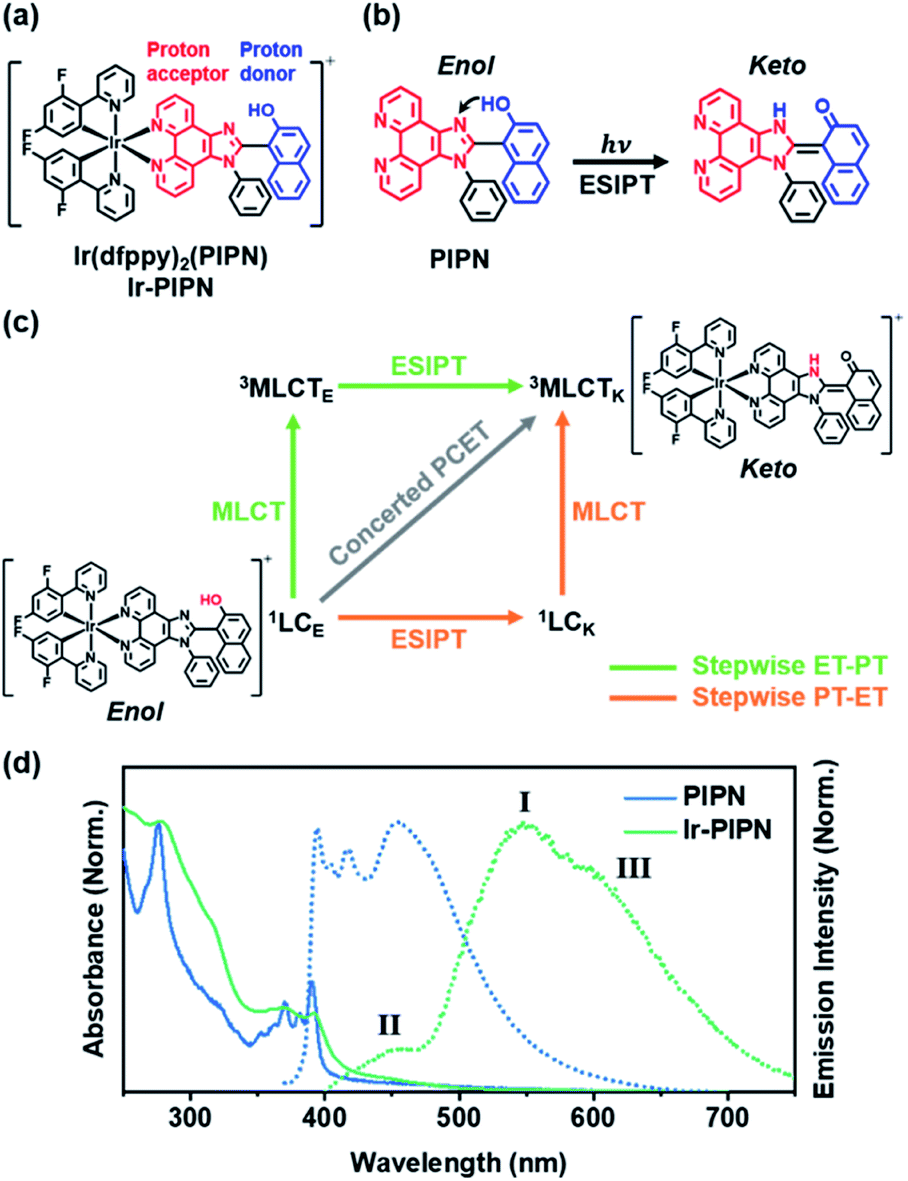 | ||
| Fig. 1 (a) Molecular structure of Ir-PIPN. The proton-accepting part and donating parts are marked in red and blue, respectively. (b) ESIPT reported to occur in the free PIPN. The proton-accepting part and donating parts are marked in red and blue, respectively. (c) Schematic representation of three possible pathways of PCET. The stepwise ET-PT pathway and stepwise PT-ET pathway are marked in green and orange, respectively. The concerted PCET pathway is marked in grey. LC and MLCT represent ligand-centered and metal-to-ligand charge transfer states, respectively. The subscripts E and K refer to the enol and keto forms, respectively. The superscripts indicate the spin multiplicities. (d) Normalized steady-state absorption (solid line) and emission spectra (dotted line) of the free PIPN ligand and Ir-PIPN in DCM with the 350 and 365 nm excitation, respectively. I, II, and III represent the major emission band, minor band, and shoulder peak, respectively. | ||
To explore the ESIPT dynamics of Ir-PIPN, the steady-state emission spectrum was obtained with excitation at 365 nm, which induced the transition from 1GSE to 1LCE. Ir-PIPN exhibits a broad and intense emission band at approximately 550 nm (I) with a shoulder peak (III) at 625 nm, and a minor band (II) with a low intensity at approximately 450 nm (Fig. 1d). These spectral features imply that Ir-PIPN has at least two emissive states. To characterize the emission bands, we measured the emission lifetimes in the absence and presence of oxygen (Fig. S5†). The determined emission lifetimes are summarized in Table S1.† The lifetimes of the emission bands (I) at approximately 510 nm are significantly affected by oxygen, revealing that the emission is from a triplet state. It is known that in Ir complexes, intersystem crossing (ISC) from singlet manifolds (singlet ligand-centered (1LC) or 1MLCT states) to the 3MLCT state occurs efficiently because of the large SOC of the metal, and the lowest emissive state is the 3MLCT state.29,41–43 Thus, we assign the main emission band (I) observed at approximately 550 nm to the phosphorescence from the 3MLCT state.
To further elucidate the origin of the emission bands, we also measured the time-resolved emission spectra. The minor band (II), which is visible in the steady-state emission spectrum, is not observed in the time-resolved emission spectra, even at the earliest time delay (10 ns) (Fig. S6†). This result indicates that the minor band (II) must have decayed within 10 ns. To determine the emission lifetime of the minor emission band (II), we used the time-correlated single-photon counting (TCSPC) technique (Fig. S7†). The determined emission lifetimes are in the range of 1–2 ns (for instance, is 2.1 ± 0.02 ns at 475 nm), and such a short emission lifetime suggests that this band corresponds to fluorescence (Fig. S7†). Among the candidate singlet states responsible for the fluorescence, 1MLCT and 1LC, one can rule out the 1MLCT state because the excited molecules in the 1MLCT state quickly transit to the 3MLCT state through ISC within 100 fs because of the strong SOC of the Ir atom. Thus, the emission observed at approximately 450 nm (II) is assigned to fluorescence from the 1LC state.
A comparison with the emission spectrum of the free PIPN ligand provides additional clues on the origin of the minor emission band (400–475 nm, II). In the same wavelength region as the minor emission (II) of Ir-PIPN, the free PIPN ligand exhibits emission bands assigned to the excited singlet state. Therefore, we infer that the minor emission band (II) in the 400–475 nm region of Ir-PIPN originates from the excited singlet state of the PIPN ligand of Ir-PIPN, which is the 1LC state (Fig. 1d). In addition, it has been reported that the longer-wavelength (∼450 nm) and shorter-wavelength (∼420 nm) emissions of the free PIPN ligand originate from the keto and enol forms, respectively.38 Accordingly, we can infer that the band at ∼450 nm and the tail at ∼420 nm correspond to the emissions from the singlet excited state of the keto form (1LCK) and the enol form (1LCE), respectively. As shown in Fig. 1d, the emission intensity of Ir-PIPN at ∼450 nm is significantly greater than that at ∼420 nm, indicating that the minor emission band (II) of Ir-PIPN is mainly a characteristic of the keto form of PIPN than that of the enol form. This result indicates that efficient ESIPT from the enol form to the keto form occurs in Ir-PIPN and that the emitting state of Ir-PIPN is dominantly in the keto form.
We now consider how the keto and enol forms contribute to the other bands (I and III). For instance, it is plausible that the major emission band (I) and shoulder peak (III) are 3MLCT bands corresponding to the enol and keto forms (or the keto and enol forms), respectively. In such cases, changing the ratio of the enol and keto forms should modulate the ratio of the peak intensities of the major emission band (I) and shoulder peak (III). Moreover, because the ratio of the peak intensities of the enol and keto forms in minor band (II) would change, the shape of minor band (II) would also change. To verify this hypothesis, we measured the spectral change in the Ir-PIPN emission by adding a protic solvent, which is known to disturb the PT process from the enol form to the keto form because of hydrogen bonding between the solute and the solvent.44,45 In particular, the steady-state emission spectra were measured while increasing the ratio of ethanol (EtOH), a protic solvent, by adding EtOH to DCM (Fig. 2). As the ratio of EtOH increases, the relative intensity of the shoulder peak (III) at approximately 625 nm decreases compared with the intensity of the major emission (I) at 550 nm (Fig. 2). These results indicate that the reduced emission peak (III) at 625 nm and the major emission band (I) at 550 nm correspond to the emissions from the keto (3MLCTK) and enol (3MLCTE) forms, respectively. The increase in the relative ratio of the emission intensity of 3MLCTE with respect to 3MLCTK indicates that the relative concentration of the enol form increases in the excited state as the ratio of EtOH increases, and the enol form becomes dominant over the keto form in DCM/EtOH (20/80). In addition to the change in relative peak intensities, the overall intensities of the emission bands (I and III) decrease as the proportion of EtOH increases (Fig. 2). The decreased emission intensities of the emission bands (I and III) reflect a decrease in the MLCT efficiency. Emission band (II) also exhibits a change in emission intensity according to the ratio of EtOH. As the ratio of EtOH increases, the relative intensity of the emission at approximately 450 nm decreases in minor band (II) spanning 400–475 nm. This result supports that minor emission (II) of Ir-PIPN observed in the presence of a large amount of EtOH is mainly a characteristic of the enol form instead of the keto form. Overall, the data with varying EtOH ratios reveal that ESIPT efficiently occurs even in an Ir complex, and MLCT as well as PT can be modulated by the external hydrogen-bonding network with the solvent.
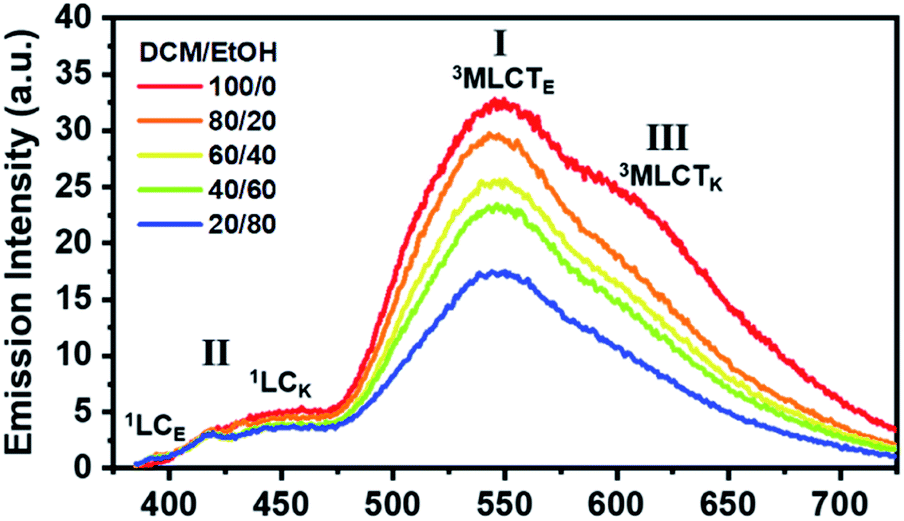 | ||
| Fig. 2 Emission spectra of Ir-PIPN in DCM/EtOH mixtures with various ratios (100/0, 80/20, 60/40, 40/60, and 20/80). The excitation wavelength is 375 nm. When the relative portion of EtOH increases, the band corresponding to the emission of the keto form of PIPN (1LCK) and the shoulder corresponding to the emission of 3MLCTK decays. The subscripts E and K refer to the enol and keto forms, respectively. I, II, and III represent the major emission band, minor band, and shoulder peak, respectively. | ||
We also measured the absorption spectra for various DCM and EtOH compositions (Fig. S8†). Unlike the steady-state emission spectra, the absorption spectra did not exhibit significant dependence on the EtOH ratio. This result indicates that the ground state is not significantly affected by hydrogen bonding with the solvent, and that the enol form is the dominant species in the ground state, regardless of the addition of a protic solvent.
Excited-state dynamics
The experimental results presented in the previous section strongly support that both PT and MLCT occur in Ir-PIPN. In other words, PCET is operational. To study the mechanism of PCET and the excited-state dynamics of Ir-PIPN in DCM, we performed fs-TA measurements by pumping the absorption band at approximately 350 nm. The excitation at 350 nm resulted in the transition from 1GSE to 1LCE, as discussed in the previous section. The TA spectra of Ir-PIPN in DCM exhibit a broad positive signal corresponding to excited-state absorption (ESA) from 400 to 700 nm (Fig. 3a). The time profiles are well fitted with a time constant of 6.4 ± 1.1 ps (Fig. 3b and Table S2†). To extract information on the excited-state dynamics of Ir-PIPN in DCM, we performed a global kinetic analysis of time-resolved TA spectra based on singular value decomposition (SVD). The SVD analysis identified two significant singular components, suggesting that the two components mainly contribute to the excited-state dynamics in DCM (Fig. S9†). The two significant right singular vectors (rSVs) from SVD can be satisfactorily fitted by single-exponential functions with a shared relaxation time of 10.8 ± 2.1 ps, which is similar to the time constant (6.4 ps) obtained from the fitting of time profiles at specific wavelengths (Fig. 3b and S9b†). Based on the results of the SVD analysis, we constructed a sequential kinetic model of I1 → I2 involving two components (I1 and I2) with a time constant of 10.8 ps for the global kinetic analysis. The fitted TA spectra reconstructed by linear combinations of the two species-associated difference spectra (SADSs) exhibit good agreement with the experimental TA spectra, confirming that the employed sequential kinetic model is suitable for describing the experimental data (Fig. 3c). The TA signal intensities after the dynamics of 10.8 ps do not converge to zero and exhibit a constant value, suggesting the existence of a long-lived component. The SADS of the long-lived component, I2, resembles the previously reported TA feature of the ESA band corresponding to T1 → Tn transition of the keto form of a free PIPN ligand (Fig. 3d).38 Therefore, we attribute the TA spectra of the long-lived component of Ir-PIPN to the T1 → Tn transition of the keto form of the ESIPT ligand moiety, indicating that the long-lived component is a triplet ligand-centered state (3LC). Accordingly, I2 can be assigned to 3LC. Generally, the lowest triplet excited states of Ir complexes are 3MLCT states, which can emit phosphorescence with a high quantum yield at room temperature.41 In contrast, the lowest triplet states of Ir complexes with strong π-conjugated ligands are often not 3MLCT state, but 3LC, resulting in the decrease of the emission intensity from 3MLCT state due to the nonradiative transition from the 3MLCT state to the 3LC state.46–48 Considering that PIPN has a large π-conjugation compared with the dfppy ligand, we expected that the lowest triplet state of Ir-PIPN is 3LC. To explore this possibility, we performed DFT and TDDFT calculations of the enol and keto forms of Ir-PIPN for the relative energy levels between the excited states. As shown in Fig. S11 and S12,† the T1 state corresponding to the lowest triplet excited state exhibits electron densities localized on the ESIPT ligand, indicating that the calculation results support that the lowest triplet excited state is not 3MLCT, but 3LC. Therefore, we can infer that the excited molecules in the 3MLCT state undergo a nonradiative process to 3LC, resulting in a decrease in their emission quantum yield. Indeed, Ir-PIPN exhibits a low emission quantum yield of 0.021, supporting the occurrence of a nonradiative process from 3MLCT to 3LC.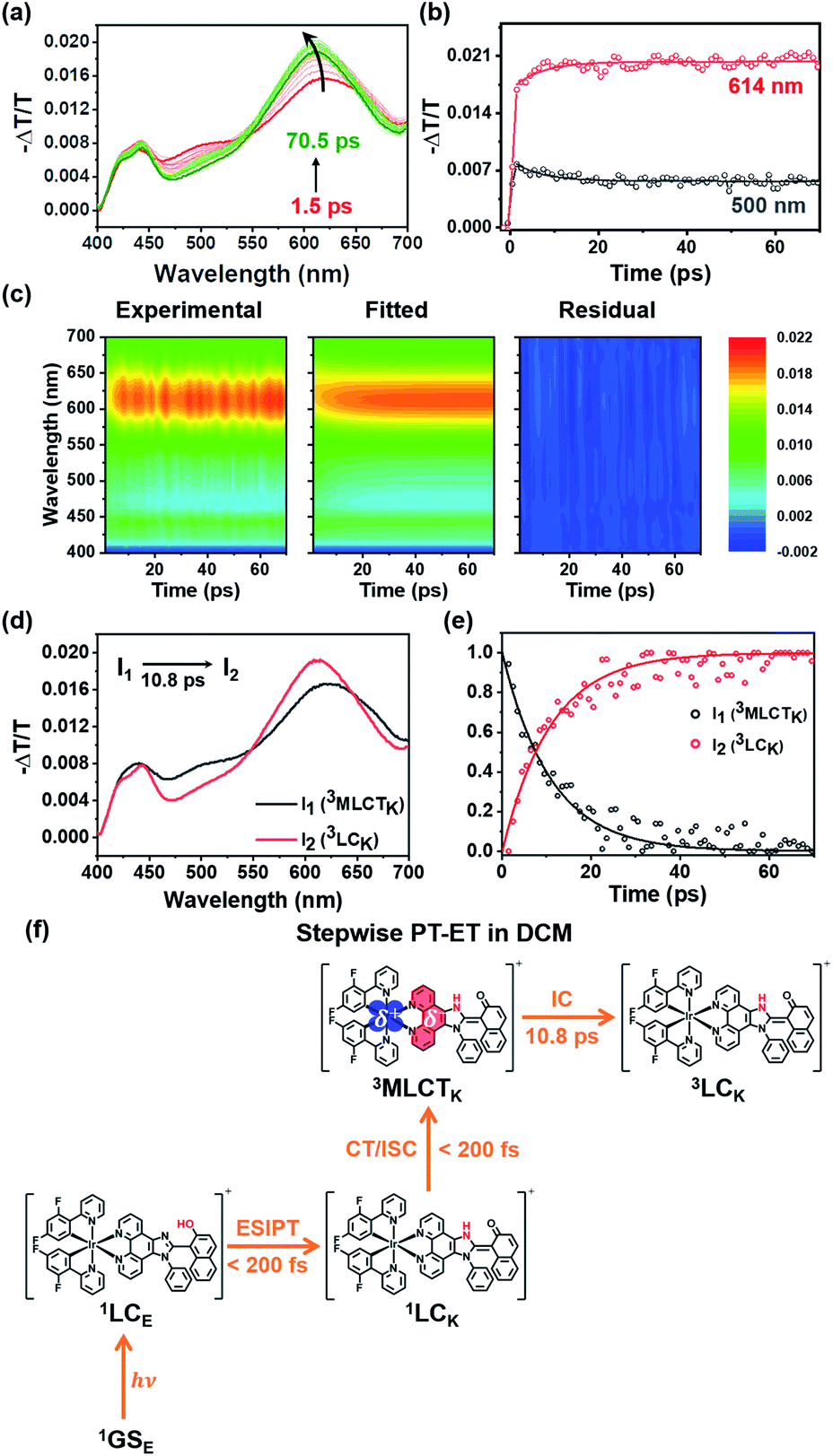 | ||
| Fig. 3 Time-resolved TA spectra and kinetic analysis of Ir-PIPN in DCM with a sequential kinetic model of I1 → I2. (a) TA spectra of Ir-PIPN in DCM. The excitation wavelength is 350 nm. (b) Time profiles monitored at two different wavelengths of Ir-PIPN in DCM. The solid lines are the fits with a single exponential function. (c) Experimental TA spectra (left), fitted spectra (middle) by a linear combination of the three species-associated difference spectra (SADS), and the residuals (right), which are obtained by subtracting the fitted data from the experimental data. All plots share a color scale representing −ΔT/T. (d) SADSs determined from the kinetic analysis of TA spectra based on the sequential kinetic model. (e) Time-dependent population changes. The solid lines are the concentrations obtained from the kinetic analysis. The open circles correspond to the optimized populations obtained by fitting the experimental curve at each time point with a linear combination of SADSs for I1 and I2. (f) Stepwise PT-ET mechanism of Ir-PIPN in DCM. 1GSE indicates the ground state, which is singlet and the enol form. | ||
It is known that in Ir complexes, the transition from 1LC to the 1MLCT state is ultrafast (<100 fs); subsequently, the 1MLCT → 3MLCT transition occurs through a fast ISC within 100 fs because of the large SOC of the Ir atom.29,46,49–52 Thus, we can infer that the broad ESA band observed within the temporal resolution of our TA system is due to the absorption from the resulting state (3MLCT) to a higher excited state. Then, the excited molecules in the 3MLCT state are likely to relax to the lowest excited state, the 3LC state, through fast internal conversion (IC). Indeed, the IC from 3MLCT to 3LC in Ir complexes has been reported.46 Based on these considerations, we conclude that the 10.8 ps dynamics for I1 → I2 correspond to the 3MLCT → 3LC transition.
According to previous studies, PCET in Ir complexes can occur through one of the following processes: (i) stepwise ET-PT, (ii) stepwise PT-ET, and (iii) concerted PCET.11,37 If CT precedes ESIPT (process (i)), 1LCE, which is generated upon excitation, rapidly transits to the 1MLCTE state, and then the 3MLCTE state should be formed through ultrafast ISC. Therefore, in process (i), 1LCK is not formed during PCET dynamics, resulting in the absence of fluorescence from the 1LCK state. If the transition from 1LCE to 3MLCTK occurs directly through a concerted PCET (process (iii)), fluorescence from 1LCK will not be observed either. Thus, in processes (i) and (iii), fluorescence from 1LCK cannot be observed because the 1LCK state is not formed. In contrast, if ESIPT precedes CT (process (ii)), the 1LCK state will be rapidly formed through ESIPT from the 1LCE state, resulting in fluorescence from the 1LCK state. For Ir-PIPN in DCM, fluorescence from the 1LCK state is observed around 450 nm (see the previous section). This indicates that PCET in Ir-PIPN occurs via a stepwise PT-ET mechanism (Fig. 3f). In this mechanism, the 1LCE state rapidly transits into the 1LCK state via ESIPT. The resulting 1LCK state is converted to 3MLCTK through ultrafast CT and ISC. If this were the only pathway, the 3MLCTE state would not be formed, and emission from the 3MLCTE state should not be observed. The emission spectrum of Ir-PIPN exhibits emission from the 3MLCTE state as well as that from the 3MLCTK state (Fig. 1d). This observation suggests that, in addition to the stepwise PT-ET process, a part of the excited molecules in the 1LCE state transits to the 3MLCTE state through the 1MLCTE state; thereafter, the excited molecules in the 3MLCTE state are deactivated to the ground state via phosphorescence.
Charge density difference
To visualize the charge distribution related to MLCT, we calculated the difference between the charge densities of the highest occupied natural transition orbital and the lowest unoccupied natural transition orbital using TDDFT calculations. The calculated charge density difference (CDD) shows that the positive and negative charge densities are confined to the Ir atom and imidazo[4,5-f]-1,10-phenanthroline moiety of the ESIPT ligand, respectively, as shown in Fig. S15.† The calculated CDD supports that the MLCT from the Ir atom to the ESIPT ligand occurs in Ir-PIPN. If the ESIPT process alone occurs, the proton-accepting part, which is imidazo[4,5-f]-1,10-phenanthroline shown in red in Fig. 1a and b, of the ESIPT ligand would have a positive charge due to ESIPT. Contrary to this expectation, the CDD shows that the proton-accepting part has a negative charge density (Fig. S15†). A possible explanation for the CDD is as follows: once ESIPT occurs, the proton-accepting part, which tentatively has a positive charge, may become a strong electron-accepting part. This charge rearrangement induced by ESIPT likely facilitates CT from the Ir atom to the imidazo[4,5-f]-1,10-phenanthroline moiety, which corresponds to MLCT. Indeed, the emission intensity from MLCT is greatly increased as the ratio of EtOH, which can suppress ESIPT, decreases (Fig. 2). Thus, we suggest that the ESIPT process improves the MLCT of the Ir complexes.Solvent-modulated PCET
As discussed in the section on Absorption/emission spectra and emission lifetimes, the PT and CT processes of the Ir complexes can be disturbed by EtOH. It is also known that the PT process in a protic solvent is much slower than that in an aprotic solvent.20 Furthermore, the steady-state spectroscopic data show that the enol form in the excited state is dominant in a protic solvent, DCM/EtOH (20/80) (Fig. 2). Thus, we can expect that the PCET dynamics in DCM/EtOH (20/80) can be affected by the external hydrogen-bonding network with the solvent. To confirm this hypothesis, we investigated the effect of a protic solvent on the PCET mechanism in Ir-PIPN using fs-TA spectroscopy. The same excitation wavelength (350 nm) used for Ir-PIPN in DCM was used. In comparison with the ESA observed in DCM (∼620 nm, Fig. 3a), the absorption band corresponding to the ESA in DCM/EtOH (20/80) is observed at a longer wavelength (∼640 nm, Fig. 4a), and the TA spectra are blue-shifted with time. The time profiles monitored at specific wavelengths are well fitted by a bi-exponential function with two relaxation times of 1.2 ± 0.1 and 6.6 ± 0.5 ps (Fig. 4b), unlike those in DCM fitted well with a single time constant (6.4 ps) (Fig. 3b). These results imply that the excited-state relaxation dynamics occurring in DCM/EtOH (20/80) are different from those in DCM, and the stepwise PT-ET process in DCM is altered by external hydrogen bonding upon the addition of EtOH.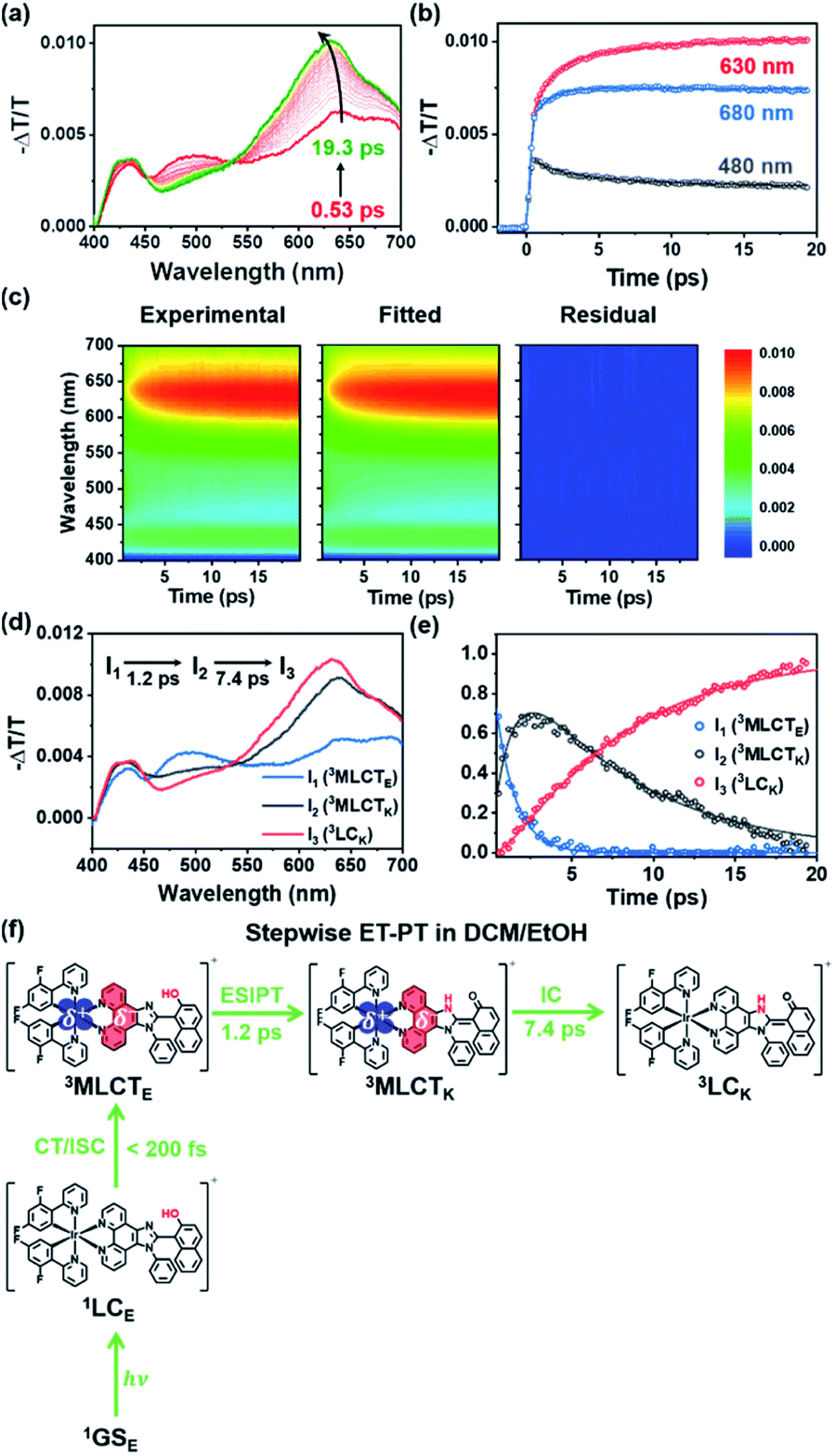 | ||
| Fig. 4 Time-resolved TA spectra and kinetic analysis of Ir-PIPN in DCM/EtOH (20/80) with a sequential kinetic model of I1 → I2 → I3. (a) TA spectra of Ir-PIPN in DCM/EtOH (20/80). The excitation wavelength is 350 nm. (b) Time profiles monitored at three different wavelengths of Ir-PIPN in DCM/EtOH (20/80). The solid lines are the fits by the sum of two exponential functions. (c) Experimental TA spectra (left), fitted spectra (middle) by a linear combination of the three species-associated difference spectra (SADS), and residuals (right), which are obtained by subtracting the fitted data from the experimental data. All plots share a color scale representing −ΔT/T. (d) SADSs determined from the kinetic analysis of TA spectra based on the sequential kinetic model. (e) Time-dependent population changes. The solid lines are the concentrations obtained from the kinetic analysis. The open circles correspond to the optimized populations obtained by fitting the experimental curve at each time point with a linear combination of SADSs for I1, I2, and I3. (f) Stepwise ET-PT mechanism of Ir-PIPN in DCM/EtOH (20/80). 1GSE indicates the ground state, which is singlet and the enol form. | ||
To extract information on excited-state dynamics more systematically, we performed a global kinetic analysis of the time-resolved TA spectra based on SVD, as in the data in DCM. SVD analysis identified three significant singular components, suggesting that the three components mainly contribute to the excited-state dynamics in DCM/EtOH (20/80). The three significant rSVs from SVD can be satisfactorily fitted by bi-exponential functions with shared relaxation times of 1.2 ± 0.1 and 7.4 ± 0.5 ps, which are similar to the time constants (1.2 and 6.6 ps) obtained from the fitting of time profiles at specific wavelengths (Fig. 4b and S17b†). Using the result of SVD analysis, we used a sequential kinetic model of I1 → I2 → I3 involving three components (I1, I2, and I3) and two time constants (1.2 ps for I1 → I2 and 7.4 ps for I2 → I3) in the global kinetic analysis. The TA spectra calculated with linear combinations of the SADSs of three components give a satisfactory agreement with the measured TA spectra (Fig. 4c).
The three species (I1, I2, and I3) that correspond to the three SADSs were assigned to 3MLCTE, 3MLCTK, and 3LCK, respectively, for the following reasons. First, I3 is a long-lived species whose TA intensity does not decay to zero, as in DCM. Similar to the spectra of the long-lived species in DCM, the third SADS is similar to the previously reported TA feature of the ESA band corresponding to the T1 → Tn transition of the keto form of a free PIPN ligand (Fig. 4d).38 Because a triplet excited state confined to the ligand with the keto form must be a triplet ligand-centered state of the keto form (3LCK), I3 can be assigned to the 3LCK state. The time constant of 7.4 ps for I2 → I3 is close to 6.4 ps observed in DCM, which is assigned to the 3MLCTK → 3LCK transition, implying that I2 is 3MLCTK. Therefore, I2 → I3 corresponds to the 3MLCTK → 3LCK transition.
We further checked whether these assignments of the chemical species are consistent with the SADSs. Because there is the 3MLCTK → 3LCK transition in both DCM and DCM/EtOH (20/80), the spectra for 3MLCTK and 3LCK and their relative changes corresponding to the 3MLCTK → 3LCK transition in both solvents would be similar if the assignment is correct. As shown in Fig. S18,† the shapes of the spectra of 3MLCTK and 3LCK and their relative differences are similar throughout the entire wavelength range. For instance, the major peak of 3LCK has a higher intensity than that of 3MLCTK in both solvents. In addition, the peak position of 3LCK is blue-shifted compared to that of 3MLCTK in both solvents. These similarities provide additional support for species assignment.
Now, we turn to I1, which is assigned to 3MLCTE for the following reason. Upon excitation, 1LCE is generated, as in the case of DCM because Ir-PIPN in DCM/EtOH exists as the enol form in the ground state. In contrast, unlike in DCM, Ir-PIPN in DCM/EtOH (20/80) in the emitting state exists mainly as the enol form, as discussed in the section on Absorption/emission spectra and emission lifetimes. Because the transition of the keto form (3MLCTK → 3LCK) occurs with the slower one (7.4 ps) of the two observed time constants, we can infer that the earlier time constant (1.2 ps) is likely to account for the transition from the enol form (I1) to 3MLCTK. Because CT and ISC occur within 100 fs owing to the strong SOC of the Ir atom,29,49 I1 should be a triplet enol form with MLCT character, that is, 3MLCTE. In this scenario, the 1.2 ps time constant corresponds to the 3MLCTE → 3MLCTK transition, implying that the ESIPT process (3MLCTE → 3MLCTK) in DCM/EtOH (20/80) is decelerated compared to the ESIPT process (1LCE → 1LCK) in DCM occurring within 200 fs. This scenario is consistent with a previous report that the PT process in a protic solvent such as EtOH is generally much slower than that in an aprotic solvent because of external hydrogen bonding upon the addition of EtOH.20 In other words, the ESIPT process (3MLCTE → 3MLCTK), which rapidly occurs in DCM, can become slower than CT (1LCE → 3MLCTE) in DCM/EtOH (20/80) owing to the addition of EtOH. Thus, we suggest that PCET in Ir-PIPN in DCM/EtOH (20/80) occurs via a stepwise ET-PT mechanism, as shown in Fig. 4f.
A comparison of the kinetic frameworks in DCM and in DCM/EtOH (20/80) reveals the solvent-dependent pathways of excited-state relaxation dynamics: the CT process precedes ESIPT in a protic solvent, whereas the ESIPT precedes the CT process in an aprotic solvent (Fig. 5). Considering that a protic solvent is known to disturb the PT process from the enol form to the keto form because of hydrogen bonding between the solute and the solvent,44,45 we suggest that the change in the temporal order of PT and CT is caused by the retarded ESIPT owing to the external hydrogen bonding effect of a protic solvent, EtOH.
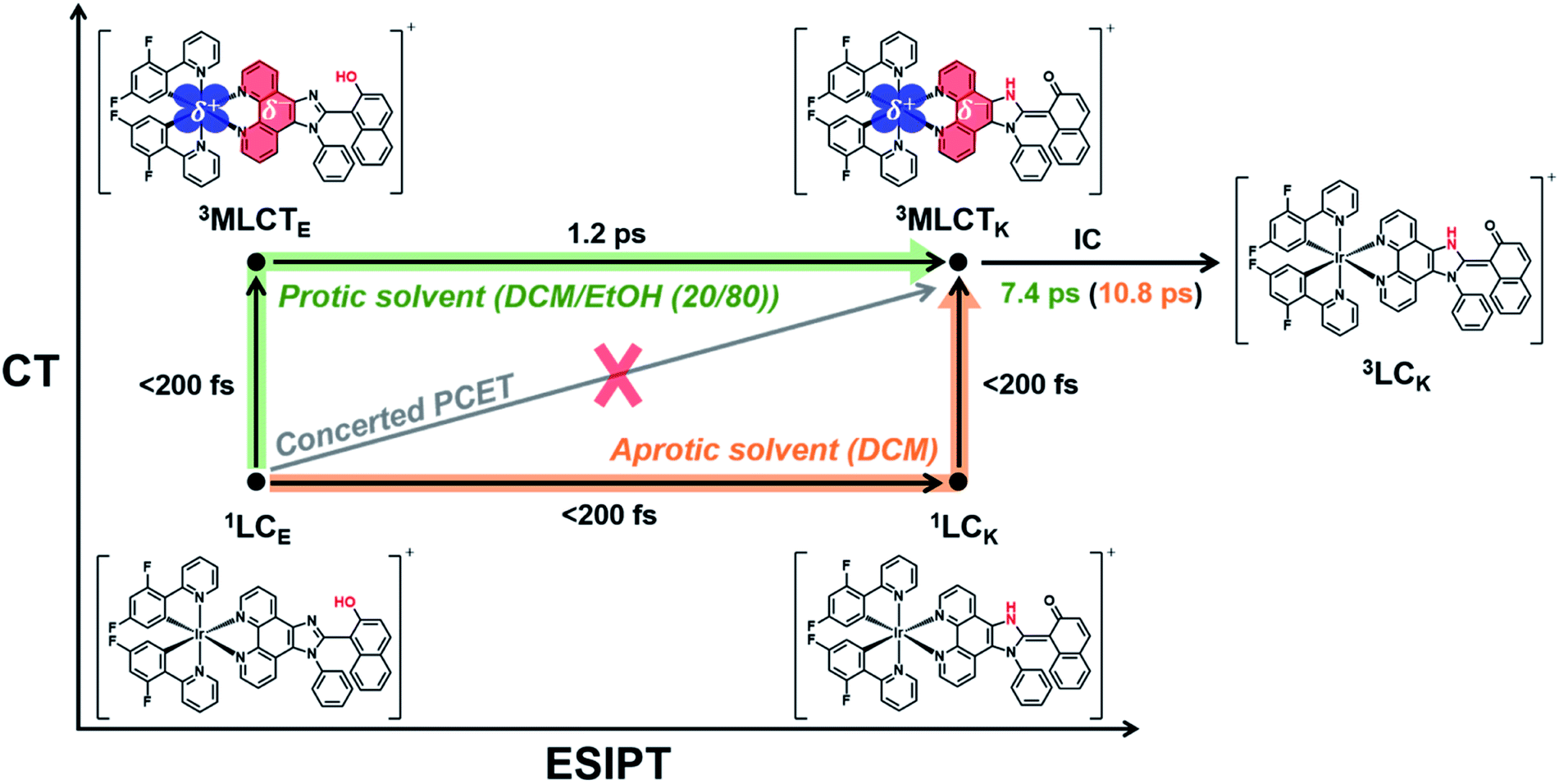 | ||
| Fig. 5 Excited-state relaxation dynamics for PCET in Ir-PIPN in an aprotic solvent and in a protic solvent in the axes of the CT and ESIPT. Reaction pathways in an aprotic solvent and a protic solvent are indicated by orange and green arrows, respectively. The subscripts E and K refer to the enol and keto forms, respectively. In the CT axis, the charge density difference shows the change in the charge distribution induced by the MLCT transition. The blue- and red-marked regions indicate the decrease (δ+) and increase (δ−) in negative charge densities, respectively. | ||
At this point, we also examined the possibility of an alternative scenario in which the solvent-modulated PCET is derived from the increased dielectric constant of the solvent instead of the effect of hydrogen bonding. As it is generally known that CT in organic compounds is accelerated by the increase in the dielectric constant of the solvent,53,54 we examined the possibility that the change in the temporal order of PT and CT is caused solely by accelerated CT owing to the increased dielectric constant of the solvent. This scenario to explain the observed temporal ordering of CT and PT requires that the following two conditions are satisfied: (i) CT is accelerated by the high dielectric constant of the solvent, and (ii) PT is not significantly affected by the solvent. Regarding condition (i), it is known that, unlike an organic compound, MLCT in inorganic compounds is less affected by the dielectric constant of the solvent compared to CT in organic compounds.55 The solvatochromic shift of MLCT in Ir-PIPN was not observed, as shown in Fig. 2, suggesting that the effect of the dielectric constant on the time scale of CT in the investigated Ir complex is not significant. This implies that condition (i) is not satisfied. Even if condition (i) were satisfied, our data support that PT itself exhibits an outstanding dependence on the solvent, indicating that condition (ii) is not satisfied. On the one hand, it is known that the ESIPT generally occurs within 100 fs.56 If the reversal of the temporal order of PT and CT in DCM/EtOH (20/80) is caused solely by the CT accelerated owing to the increased dielectric constant of the solvent, the time scale of PT would remain unchanged, with PT occurring within 100 fs as in DCM. In this case, no additional time constant should have been observed in DCM/EtOH (20/80). In reality, a time constant of 1.2 ps corresponding to ESIPT was observed in DCM/EtOH (20/80), unlike in DCM. Based on these points, we propose that the reversion of the temporal order of PT and ET is due to the slowed PT rather than the accelerated CT owing to the high dielectric constant of EtOH. Notably, the MLCT efficiency was improved in DCM, as discussed in Absorption/emission spectra and emission lifetimes section. Linking the improved MLCT efficiency in DCM with the fact that PT precedes CT in DCM, unlike in DCM/EtOH (20/80), we suggest that the MLCT efficiency can be improved when PT precedes CT. In brief, the observed solvent-modulated PCET dynamics present the possibility that the reaction pathway and CT efficiency in PCET-based molecular systems can be easily regulated by an external environment, such as hydrogen bonding with the solvent.
Ir-PIPP-H in comparison with Ir-PIPN
It was reported that the ESIPT process from the enol form to the keto form occurs more efficiently in PIPP-H than in PIPN, and the dynamics of PIPP-H and PIPN are significantly different owing to the structural difference between the two molecules.38 Based on this result, we investigated whether the dynamics of Ir-PIPP-H differ from those of Ir-PIPN. When the ESIPT ligand is changed from PIPN to PIPP-H, the general spectral features of Ir-PIPP-H exhibit a similar trend to those of Ir-PIPN. First, compared with the free PIPP-H ligand, the absorption spectrum of Ir-PIPP-H in DCM is broader in all wavelength ranges (Fig. S19†). Ir-PIPP-H exhibits an absorption band up to 500 nm, whereas the free dfppy and PIPP-H ligands exhibit no absorption at wavelengths longer than 310 and 380 nm, respectively.41 As in Ir-PIPN, these spectral differences between the free ligands and Ir-PIPP-H indicate the interaction between Ir and the ligand, and we assign the newly formed absorption band in Ir-PIPP-H to MLCT.The steady-state emission spectrum of Ir-PIPP-H in DCM exhibits a single broad emission band from 400 to 700 nm, and the emission decay profile is well fitted by a single-exponential function (Fig. S5 and Table S1.†) These results indicate the existence of a single emissive state (Fig. S19c†). A comparison of the emission spectra of the free PIPP-H ligand and Ir-PIPP-H indicates that the emission from the excited singlet state of the free PIPP-H ligand (1LC) is not clearly observed in Ir-PIPP-H (Fig. S19c†), unlike in Ir-PIPN, which exhibits fluorescence from 1LC. This difference is consistent with the low quantum yield of Ir-PIPP-H (0.004), which is five times lower than that of Ir-PIPN (0.021). As in Ir-PIPN, the emission lifetime of Ir-PIPP-H is strongly influenced by the presence of oxygen, indicating that the emission is from a triplet state (Fig. S5 and Table S1†). As in Ir-PIPN, we suggest that the emission band of Ir-PIPP-H is phosphorescence from the 3MLCT state (see the section on Absorption/emission spectra and emission lifetimes). Unlike PIPN, PIPP-H exists mainly as a keto form in the excited states, according to the previous study.38 Therefore, in the case of Ir-PIPP-H, the observed single emission band is attributed to the 3MLCTK state.
For further comparison with Ir-PIPN, we performed fs-TA measurements of Ir-PIPP-H in DCM. The fs-TA experiments were conducted under the same conditions as those used for Ir-PIPN. The TA spectra of Ir-PIPP-H in DCM exhibit a broad positive signal corresponding to the ESA from 400 to 700 nm (Fig. S20†). The time profiles of Ir-PIPP-H are well fitted with time constants of 3.7 ± 0.7 ps (Fig. S20b and Table S2†). The TA signal intensities after the dynamics of 3.7 ps exhibit a constant value, suggesting the existence of a long-lived component (Fig. S20b†). The TA spectra of the long-lived component measured at late time delays (>10 ps) are similar to the features of the ESA band corresponding to the transition from the T1 state of the keto form of a free PIPP-H ligand to Tn.38 Considering the experimental and theoretical calculation results, we can expect that the lowest triplet state of Ir-PIPP-H is the 3LC state, as in Ir-PIPN (Fig. S13 and S14†). As discussed in the section on Excited-state dynamics, in Ir complexes, the transition from 1LC to 3MLCT state is ultrafast (<100 fs) through the 1MLCT state. Thus, we can infer that because the 1MLCT → 3MLCT transition proceeds via ISC occurring within 100 fs, the 3MLCT state of the excited molecules is likely to convert to the lowest excited state, 3LC, through fast IC. After photoexcitation, ESIPT can efficiently occur in Ir-PIPP-H, resulting in the keto form of Ir-PIPP-H, as in Ir-PIPN in DCM, and the emission band suggests that the excited molecules exist as the keto form. Based on these considerations, we conclude that the 3.7 ps dynamics correspond to the 3MLCTK → 3LCK transition. The observed dynamics of Ir-PIPP-H are not significantly different from those of Ir-PIPN except the time constant value (3.7 ps vs. 10.8 ps) of the 3MLCTK → 3LCK transition. We note that the dynamics of 3.7 ps in Ir-PIPP-H are not accompanied by large spectral changes (Fig. S20†), whereas the dynamics of 10.8 ps in Ir-PIPN were accompanied by a spectral blue shift (Fig. 3a). This indicates that the energy gap between the two states (3MLCTK and 3LCK) of Ir-PIPP-H is smaller than that of Ir-PIPN. The faster dynamics of Ir-PIPP-H (3.7 ps) than those of Ir-PIPN (10.8 ps) may originate from the smaller energy gap of Ir-PIPP-H.
Conclusions
In this study, Ir-PIPN and Ir-PIPP-H, where an ESIPT ligand was used as the ancillary ligand to avoid the complexity of intermolecular PCET, were designed and synthesized as model systems to understand the PCET mechanism in Ir complexes. To elucidate the effect of external hydrogen bonding on the PCET dynamics of Ir complexes with the ESIPT ligand, Ir-PIPN in DCM and DCM/EtOH (20/80) and Ir-PIPP-H in DCM were investigated by combining fs-TA spectroscopy and theoretical calculations. In particular, the experimental results provide evidence that the temporal order of ESIPT and CT is reversed by the solvent environment in Ir-PIPN. This work is the first experimental observation of the reversion of the temporal order of PT and CT induced by the solvent in metal complexes. In an aprotic solvent (DCM), ESIPT from the enol form to the keto form (1LCE → LCK) precedes CT (1LCK → 3MLCTK), resulting in an improvement in the efficiency of MLCT. In contrast, CT (1LCE → 3MLCTE) in a protic solvent (EtOH) precedes ESIPT (3MLCTE → 3MLCTK) because ESIPT becomes slower by external hydrogen bonding with the solvent. In both aprotic and protic solvents, the 3MLCTK state decays to the 3LCK state, which is the lowest excited state of both Ir-PIPP-H and Ir-PIPN because of the largely expanded π-conjugation of the ESIPT ligands, whereas the lowest excited state of a metal complex is generally 3MLCT. These results provide a better understanding of the PCET mechanism in Ir complexes with an ESIPT ligand, which may provide fundamental insights into the design and application of PCET-based Ir complexes. In particular, this study demonstrates the possibility of controlling PCET dynamics in the desired direction and improving the efficiency of CT by simply perturbing the external hydrogen-bonding network with the solvent. Moreover, our experimental results suggest that the efficiency of CT increases when PT precedes CT. We envision that the findings of our study may provide insight into the improvement of photocatalysts that harness PCET involving ESIPT. For instance, a study reported that an Ir complex capable of PCET involving ESIPT exhibited improved photocatalytic performance compared to an Ir complex without PCET.23 It is worth exploring the possibility of solvent-modulated photocatalytic activities and studying the link between the solvent-modulated PCET found in our study to photocatalytic properties. In addition, it has been reported that organic chromophores with PCET involving ESIPT can exhibit dual emission and that the ratiometric response of emission can vary depending on the surrounding hydration environment. Based on this feature, the chromophore tagged to the protein has been applied as a molecular probe to quantify the hydration environment of a biomolecule.20,57 In this regard, Ir complexes with PCET involving ESIPT, which have high photostabilities and long emission lifetimes of Ir complexes compared with organic chromophores, have the potential to be better molecular probes for biological studies because their ratios of emission intensities are affected by external hydrogen bonding.Data availability
All relevant data is presented in the main text and ESI.†Author contributions
K.-R. W., and H. I. designed research. S. K., J. C., D. W. C., M. A., S. E., J. K., K.-R. W., and H. I. performed research. S. K., J. C., D. W. C., M. A., S. E., J. K., K.-R. W., and H. I. contributed to the interpretation of the results and S. K., J. C., K.-R. W., and H. I. wrote the paper.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the Institute for Basic Science (IBS-R033). This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF-2020R1C1C1009007 and NRF-2020R1I1A3054352).Notes and references
- C. Tommos and G. T. Babcock, Acc. Chem. Res., 1998, 31, 18–25 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- S. Y. Reece and D. G. Nocera, Annu. Rev. Biochem., 2009, 78, 673–699 CrossRef CAS PubMed.
- T. J. Meyer, M. H. V. Huynh and H. H. Thorp, Angew. Chem., Int. Ed., 2007, 46, 5284–5304 CrossRef CAS PubMed.
- J. M. Mayer, Annu. Rev. Phys. Chem., 2004, 55, 363–390 CrossRef CAS PubMed.
- J. C. Lennox, D. A. Kurtz, T. Huang and J. L. Dempsey, ACS Energy Lett., 2017, 2, 1246–1256 CrossRef CAS.
- M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed.
- C. C. Hsieh, C. M. Jiang and P. T. Chou, Acc. Chem. Res., 2010, 43, 1364–1374 CrossRef CAS PubMed.
- S. Hammes-Schiffer, Acc. Chem. Res., 2009, 42, 1881–1889 CrossRef CAS PubMed.
- C. J. Gagliardi, B. C. Westlake, C. A. Kent, J. J. Paul, J. M. Papanikolas and T. J. Meyer, Coord. Chem. Rev., 2010, 254, 2459–2471 CrossRef CAS.
- O. S. Wenger, Acc. Chem. Res., 2013, 46, 1517–1526 CrossRef CAS PubMed.
- N. Berg, S. Bergwinkl, P. Nuernberger, D. Horinek and R. M. Gschwind, J. Am. Chem. Soc., 2021, 143, 724–735 CrossRef CAS PubMed.
- Y. Yoneda, S. J. Mora, J. Shee, B. L. Wadsworth, E. A. Arsenault, D. Hait, G. Kodis, D. Gust, G. F. Moore, A. L. Moore, M. Head-Gordon, T. A. Moore and G. R. Fleming, J. Am. Chem. Soc., 2021, 143, 3104–3112 CrossRef CAS PubMed.
- S. J. Mora, E. Odella, G. F. Moore, D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2018, 51, 445–453 CrossRef CAS PubMed.
- G. F. Moore, M. Hambourger, M. Gervaldo, O. G. Poluektov, T. Rajh, D. Gust, T. A. Moore and A. L. Moore, J. Am. Chem. Soc., 2008, 130, 10466–10467 CrossRef CAS PubMed.
- S. K. Thiyagarajan, R. Suresh, V. Ramanan and P. Ramamurthy, Chem. Sci., 2018, 9, 910–921 RSC.
- H. Kotani, H. Shimomura, M. Horimoto, T. Ishizuka, Y. Shiota, K. Yoshizawa, S. Yanagisawa, Y. Kawanara-Nakagawa, M. Kubo and T. Kojima, Dalton Trans., 2019, 48, 13154–13161 RSC.
- T. Kojima, Dalton Trans., 2020, 49, 7284–7293 RSC.
- S. Miyazaki, T. Kojima, J. M. Mayer and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 11615–11624 CrossRef CAS PubMed.
- A. P. Demchenko, K. C. Tang and P. T. Chou, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC.
- W. B. Swords, G. J. Meyer and L. Hammarstrom, Chem. Sci., 2020, 11, 3460–3473 RSC.
- M. Bourrez, R. Steinmetz, S. Ott, F. Gloaguen and L. Hammarstrom, Nat. Chem., 2015, 7, 140–145 CrossRef CAS PubMed.
- H. Sayre, H. H. Ripberger, E. Odella, A. Zieleniewska, D. A. Heredia, G. Rumbles, G. D. Scholes, T. A. Moore, A. L. Moore and R. R. Knowles, J. Am. Chem. Soc., 2021, 143, 13034–13043 CrossRef PubMed.
- G. F. Manbeck, E. Fujita and J. J. Concepcion, J. Am. Chem. Soc., 2016, 138, 11536–11549 CrossRef CAS PubMed.
- T. Kojima, Bull. Chem. Soc. Jpn., 2020, 93, 1571–1582 CrossRef CAS.
- J. I. Goldsmith, W. R. Hudson, M. S. Lowry, T. H. Anderson and S. Bernhard, J. Am. Chem. Soc., 2005, 127, 7502–7510 CrossRef CAS PubMed.
- S. Tschierlei, A. Neubauer, N. Rockstroh, M. Karnahl, P. Schwarzbach, H. Junge, M. Beller and S. Lochbrunner, Phys. Chem. Chem. Phys., 2016, 18, 10682–10687 RSC.
- R. Bevernaegie, L. Marcelis, A. Moreno-Betancourt, B. Laramee-Milette, G. S. Hanan, F. Loiseau, M. Sliwa and B. Elias, Phys. Chem. Chem. Phys., 2018, 20, 27256–27260 RSC.
- F. Spaenig, J. H. Olivier, V. Prusakova, P. Retailleau, R. Ziessel and F. N. Castellano, Inorg. Chem., 2011, 50, 10859–10871 CrossRef CAS PubMed.
- T. Devi, Y. M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2018, 140, 8372–8375 CrossRef CAS PubMed.
- A. Dey, J. Dana, S. Aute, P. Maity, A. Das and H. N. Ghosh, Chem.–Eur. J., 2017, 23, 3455–3465 CrossRef CAS PubMed.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC.
- M. S. Lowry and S. Bernhard, Chem.–Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- J. C. Freys, G. Bernardinelli and O. S. Wenger, Chem. Commun., 2008, 4267–4269 RSC.
- A. R. B. Yusoff, A. J. Huckaba and M. K. Nazeeruddin, Top. Curr. Chem., 2017, 375, 39 CrossRef PubMed.
- Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061–7084 RSC.
- O. S. Wenger, Coord. Chem. Rev., 2015, 282, 150–158 CrossRef.
- J. Choi, D. S. Ahn, S. Y. Gal, D. W. Cho, C. Yang, K. R. Wee and H. Ihee, J. Phys. Chem. C, 2019, 123, 29116–29125 CrossRef CAS.
- V. Coropceanu, X.-K. Chen, T. Wang, Z. Zheng and J.-L. Brédas, Nat. Rev. Mater., 2019, 4, 689–707 CrossRef.
- Y.-J. Cho, A.-R. Lee, S.-Y. Kim, M. Cho, W.-S. Han, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 18, 22921–22928 RSC.
- Y. J. Cho, S. Y. Kim, H. J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2017, 19, 8778–8786 RSC.
- W. T. Wu, W. H. Wu, S. M. Ji, H. M. Guo and J. Z. Zhao, Dalton Trans., 2011, 40, 5953–5963 RSC.
- S.-H. Wu, J.-W. Ling, S.-H. Lai, M.-J. Huang, C. H. Cheng and I. C. Chen, J. Phys. Chem. A, 2010, 114, 10339–10344 CrossRef CAS PubMed.
- D. LeGourrierec, V. Kharlanov, R. G. Brown and W. Rettig, J. Photochem. Photobiol., A, 1998, 117, 209–216 CrossRef CAS.
- S. Lazzaroni, D. Dondi, A. Mezzetti and S. Protti, Photochem. Photobiol. Sci., 2018, 17, 923–933 CrossRef CAS PubMed.
- J. M. Favale, C. E. Hauke, E. O. Danilov, J. E. Yarnell and F. N. Castellano, Dalton Trans., 2020, 49, 9995–10002 RSC.
- J. Choi, M. Ahn, J. H. Lee, D. S. Ahn, H. Ki, I. Oh, C. W. Ahn, E. H. Choi, Y. Lee, S. Lee, J. Kim, D. W. Cho, K. R. Wee and H. Ihee, Inorg. Chem. Front., 2021, 8, 2987–2998 RSC.
- A. J. Howarth, D. L. Davies, F. Lelj, M. O. Wolf and B. O. Patrick, Inorg. Chem., 2014, 53, 11882–11889 CrossRef CAS PubMed.
- Y. J. Cho, S. Y. Kim, M. Cho, K. R. Wee, H. J. Son, W. S. Han, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2016, 18, 15162–15169 RSC.
- G. J. Hedley, A. Ruseckas and I. D. W. Samuel, J. Phys. Chem. A, 2009, 113, 2–4 CrossRef CAS PubMed.
- E. Pomarico, M. Silatani, F. Messina, O. Braem, A. Cannizzo, E. Barranoff, J. H. Klein, C. Lambert and M. Chergui, J. Phys. Chem. C, 2016, 120, 16459–16469 CrossRef CAS.
- Y.-J. Cho, S.-Y. Kim, H.-J. Son, W.-S. Han, D. W. Cho and S. O. Kang, J. Photochem. Photobiol., A, 2018, 356, 673–680 CrossRef CAS.
- S. Kim, D.-S. Ahn, M. Ahn, K.-R. Wee, J. Choi and H. Ihee, Phys. Chem. Chem. Phys., 2020, 22, 28440–28447 RSC.
- C. Kim, T. W. Kim, S. Kim, I. Oh, H. Wonneberger, K. Yoon, M. Kwak, J. Kim, J. Kim, C. Li, K. Müllen and H. Ihee, J. Phys. Chem. C, 2020, 124, 22993–23003 CrossRef CAS.
- B.-S. Yun, J.-H. Kim, S.-Y. Kim, H.-J. Son, D. W. Cho and S. O. Kang, Phys. Chem. Chem. Phys., 2019, 21, 7155–7164 RSC.
- C. Schriever, M. Barbatti, K. Stock, A. J. A. Aquino, D. Tunega, S. Lochbrunner, E. Riedle, R. de Vivie-Riedle and H. Lischka, Chem. Phys., 2008, 347, 446–461 CrossRef CAS.
- J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc07250a |
| This journal is © The Royal Society of Chemistry 2022 |