
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-mediated outer-sphere CO2 electro-reduction mechanism over the Ag111 surface†
Vivek
Sinha
 *,
Elena
Khramenkova
and
Evgeny A.
Pidko
*
*,
Elena
Khramenkova
and
Evgeny A.
Pidko
*
Inorganic Systems Engineering, Department of Chemical Engineering, Faculty of Applied Sciences, Delft University of Technology, Delft, The Netherlands. E-mail: v.sinha@tudelft.nl; e.a.pidko@tudelft.nl
First published on 24th February 2022
Abstract
The electrocatalytic CO2 reduction reaction (CO2RR) is one of the key technologies of the clean energy economy. Molecular-level understanding of the CO2RR process is instrumental for the better design of electrodes operable at low overpotentials with high current density. The catalytic mechanism underlying the turnover and selectivity of the CO2RR is modulated by the nature of the electrocatalyst, as well as the electrolyte liquid, and its ionic components that form the electrical double layer (EDL). Herein we demonstrate the critical non-innocent role of the EDL for the activation and conversion of CO2 at a high cathodic bias for electrocatalytic conversion over a silver surface as a representative low-cost model cathode. By using a multiscale modeling approach we demonstrate that under such conditions a dense EDL is formed, which hinders the diffusion of CO2 towards the Ag111 electrocatalyst surface. By combining DFT calculations and ab initio molecular dynamics simulations we identify favorable pathways for CO2 reduction directly over the EDL without the need for adsorption to the catalyst surface. The dense EDL promotes homogeneous phase reduction of CO2via electron transfer from the surface to the electrolyte. Such an outer-sphere mechanism favors the formation of formate as the CO2RR product. The formate can undergo dehydration to CO via a transition state stabilized by solvated alkali cations in the EDL.
Introduction
Electrochemical conversion of CO2 holds promise to help mitigate the carbon footprint of the production of fuels and chemicals.1 The abundant CO2 green-house waste gas is an attractive substrate to stabilize excess “electrons” generated from renewable energy via the CO2 reduction reaction (CO2RR).2 A wide range of electrocatalysts have been described so far for the CO2RR.3,4 Depending on the catalyst employed the primary CO2RR product can be either formate or CO resulting from a 2e− reduction, or multi-electron transfer products such as alcohols and hydrocarbons. The electrocatalytic reduction of CO2 to CO opens a path for carbon recycling within the established syngas chemistry infrastructure to produce fuels and chemicals.1In an electrocatalytic cell, the CO2RR proceeds on the cathode side. The cathode material, its morphology and electrolyte properties collectively influence the electrocatalytic activity and selectivity at the solid–liquid interface.5,6 Gold-based electrocatalysts have been reported to reduce CO2 to CO with high activity and selectivity. Hori and co-workers used bulk Au to reduce CO2 to CO with 87.1% faradaic efficiency (FE) at −1.14 V (NHE) with a partial CO current of 5 mA cm−2.7 At the same partial current, Ag showed a FE of 81.5% towards CO at −1.37 V (NHE).7 The lower cost, and comparable selectivity and activity to Au make Ag an attractive electrocatalyst for the CO2RR.
The selectivity and activity of an electrocatalyst for the CO2RR are strongly influenced by the electrolyte and the local environment close to the cathode.5,6,8–11 At potentials (ΦM) below the potential of zero charge (pzc) (Φpzc) the negative charge density on the cathode surface increases, attracting more cations and resulting in the formation of an electrical double layer (EDL). The EDL influences the local electrochemical environment close to the cathode surface such as the interfacial pH and the structure of water at the interface.11–13 The cations in the EDL also interact with the surface intermediates and tune the stabilization of transition states and adsorbates on the electrocatalyst surface.14,15 However, at a higher cathodic bias, the EDL becomes very dense and compact, thus strongly hampering the mass transport of CO2 to the electrocatalyst surface.16–18
The electron transfer (ET) from the cathode to the reagent is the key mechanistic step of any electrocatalytic conversion. ET can in principle proceed via two alternative mechanisms: inner- and outer-sphere ET (Fig. 1). The inner-sphere mechanism starts with the chemisorption of the reagent (CO2) to the catalyst surface that enables direct ET via overlapping orbitals (Fig. 1a). In the outer-sphere mechanism indirect ET from the catalyst surface to CO2 takes place through the electrolyte without the direct chemical interaction between the reagent and the electrocatalyst (Fig. 1b).
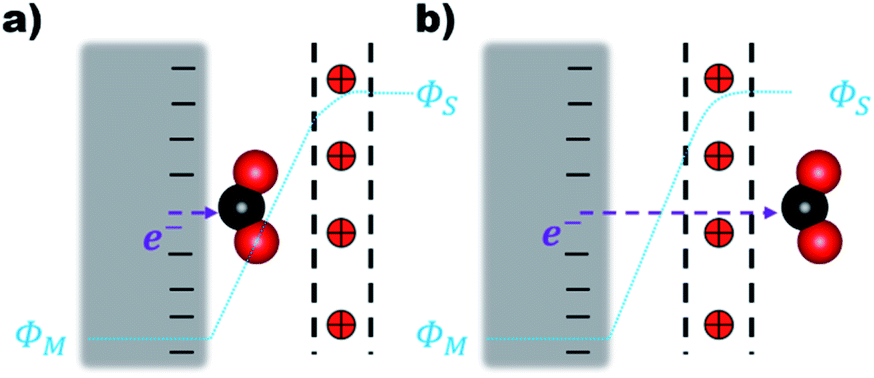 | ||
| Fig. 1 (a) Inner-sphere and (b) outer-sphere electron transfer from the cathode to CO2. The red positive charges within the black dotted lines denote the EDL. (ΦM) and (Φs) are the surface and bulk solution phase potentials respectively. | ||
The inner-sphere mechanisms describing the electrocatalytic conversions in the framework of surface adsorbed species dominate the current literature.5,19–25 Investigating the selectivity of the CO2RR on various Ag facets, Bohra and co-workers proposed that the formation of formate species is self-inhibited on Ag surfaces resulting in improved selectivity to CO at low to moderate potentials, and to H2 at higher potentials.26 Their work did not consider the effects from the EDL and the electrolyte explicitly. A realistic description of the reaction medium and conditions in modelling studies has been currently emphasized across the field of catalysis.27–40 The importance of including an explicit representation of the EDL and accounting for the reaction conditions in mechanistic studies of the electrocatalytic CO2RR has been emphasized in recent literature.14,15,41,42
However, most mechanistic studies assume facile mass transport of CO2 from the bulk phase to the surface via the EDL. Such an assumption is reasonable for hydrodynamic transport through a low concentration electrolyte but under the operando CO2RR potentials, the EDL can get more condensed and strongly impact the mass transport of CO2.16,17 Under such conditions an outer-sphere ET in the homogeneous phase over the EDL is a plausible mechanism for the CO2RR.
Herein, we have taken a multiscale operando modelling approach to investigate the possibility and the impact of homogeneous ET on the CO2RR under realistic electrocatalytic conditions. The combination of classical molecular dynamics (CMD) and ab initio molecular dynamics (AIMD) simulations shows that CO2 can be favourably reduced to formate anions via outer-sphere ET over the dense EDL. The formate species can then convert to CO via a thermally activated dehydration reaction facilitated by the solvated cations within the EDL.
Results and discussion
Molecular structure of the EDL
To rationally construct an atomistic operando model of the cathode-electrolyte interface under the reaction conditions, the formation and structure of the EDL at the Ag111 surface was first investigated by classical molecular dynamics (CMD) simulations. The electrocatalytic system was modelled as an aqueous electrolyte containing 0.86 M KCl and 0.06 M CO2, confined between two Ag111 slabs (the model cathode and the anode) in a super cell of dimensions 33.1 × 37.2 × 265.5 Å3 periodic in the x and y directions (see Fig. 2a). Complete details of the CMD model and simulations are presented in the ESI.† These simulations aimed to probe the formation of the EDL at the electrodes under different polarization conditions. The polarization conditions were mimicked by placing uniform distributions of point charges behind the Ag111 slabs resulting in negative (cathode) and positive (anode) surface charge densities on the electrolyte facing surfaces. | ||
| Fig. 2 (a) A snapshot of the model of the KCl electrolyte with CO2 confined between two silver slabs simulated at 0.5 V nm−1. The surface on the left represents the anode, with positive ghost charges imposed behind the wall, while the right surface represents the cathode, with negative ghost charges imposed behind the wall. The color code is as follows: silver is grey, oxygen is red, hydrogen is white, potassium is violet, chlorine is green, carbon of CO2 is yellow, and the ghost atoms are light brown. (b–e) Density profile of H2O (b), CO2 (c), Cl− (d), and K+ (e), under increasing polarization conditions represented by the electric field at the surface of the electrode. At low or zero polarization most of the ions are present in the bulk phase. With increasing polarization, the respective densities of K+ at the cathode and Cl− at the anode show a sharp increase leading to the formation of compact EDLs at the respective electrodes. | ||
The CMD simulations of the extended electrocatalyst system representing the electrochemical cell revealed the formation of a dense EDL at the cathode as the polarization was increased (Fig. 2). The density of water oscillates within 1 nm of the cathode surface indicating the formation of ordered layers of solvation, while it was found to be constant at 1 kg m−3 in the bulk phase. The simulations show a deeper penetration of water molecules into the outer Helmholtz plane (OHP; indicated by the dashed line at 1 Å from the electrode) of the cathode compared to the anode. K+ ions accumulated near the cathode while Cl− anions accumulated at the anode and their respective concentrations in the EDL region increased with increasing surface polarization. This resulted in the compaction of the EDL and associated depletion of the CO2 near the electrocatalyst surface. The latter is fully covered by solvated alkali cations, which can be further considered as the reactive sites for CO2 activation instead of the bare metal surface.
Outer-sphere CO2RR
The outer-sphere reduction of CO2 over the solvated Ag111 surface was next investigated using periodic density functional theory (DFT) calculations (see the ESI for details†). The reactive events were simulated using a smaller molecular model representing the reaction environment near the Ag111 cathode (4 × 4 × 5 slab model). The initial static DFT calculations on the simplified models revealed the critical role of the EDL in the outer-sphere charge transfer eliciting indirect reduction of CO2. Indeed, the interaction of CO2 with an aqueous solvation layer on Ag111 is very weak and does not lead to notable perturbations of the adsorbed molecules (Ag111–(H2O)24–CO2, Fig. 3a). The situation drastically changes upon the introduction of sodium ions and the formation of the EDL (Ag111–(Na+)4-(H2O)24–CO2, Fig. 3b), which facilitates the reduction of CO2. The CO2 molecule in this case adopts a bent configuration due to the partial charge transfer from the silver slab. The bent anionic CO2 moiety is stabilized by hydrogen bonding with the neighbouring H2O molecules.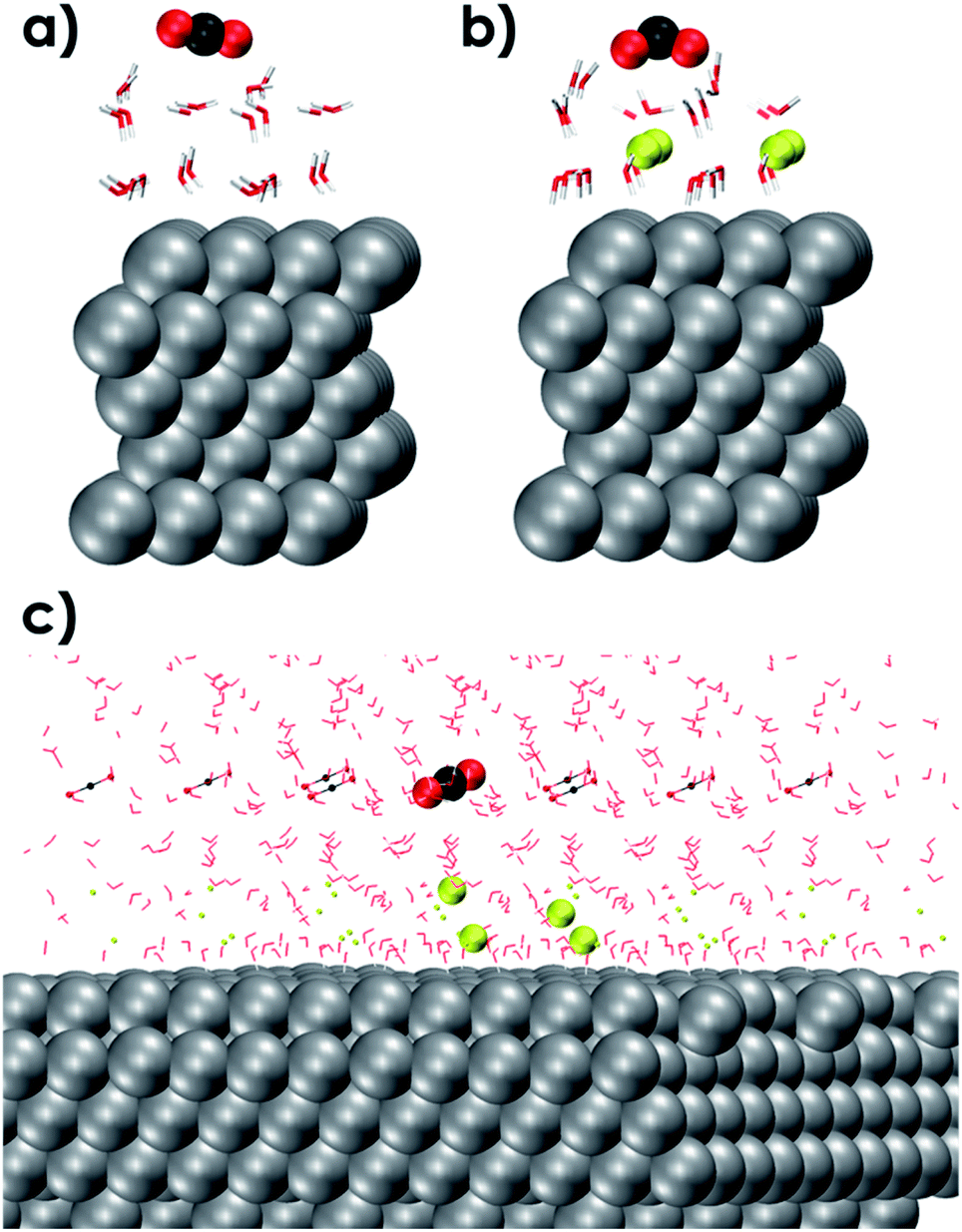 | ||
| Fig. 3 DFT optimized molecular models of a 4 × 4 × 5 Ag111 slab with one CO2 molecule in vacuum over the (a) Ag111-water interface (b) Ag111-EDL interface EDL = (Na+)4(H2O)24 and (c) fully solvated and periodic (in x, y and z directions) system used for AIMD simulations composed of a 4 × 4 × 5 Ag111 slab, 4Na+ cations and 61 water molecules. Periodic images have also been shown. The CO2 and Na+ species in the original simulation cell are shown as larger VdW spheres. Color code: C (black), Ag (silver), O (red), Na (Yellow), and H (white). | ||
To better investigate the outer-sphere ET and the subsequent conversions of CO2 over the EDL, an extended fully solvated model was employed containing (Ag111–4Na+–(H2O)61–CO2, Fig. 2c) in combination with AIMD simulations (BLYP-PW-400 eV, VASP 5.5.4, for further details see the ESI†). The reactive environment was simulated with a 19 ps long AIMD simulation of CO2 in the solvated phase over the Ag111-EDL interface. CO2 was found to preferentially stay in the 3rd and 4th water layers of solvation at about 10 Å from the Ag111 surface. The average O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO angle was ∼172° during the runs. Two of the four Na+ cations forming the EDL were found at about ∼3 Å from the surface while the other two Na+ cations were located further away at ∼5 Å on the Ag111 surface (Fig. 3c).43 The water molecules within the EDL close to the surface facet were found to show a limited mobility. They pointed their protons towards the metal surface during the simulations. In the absence of the EDL, the water molecules preferentially oriented with O moieties pointing towards the Ag111 surface (Fig. 2a). Next, constrained AIMD simulations were carried out on 11 intermediate states representing different stages of the outer-sphere ET CO2RR. The OCO angle was chosen as the reaction coordinate (Q), and it was varied from 172° to 125°. The resulting Gibbs free energy profile along with the representative snapshots of the relevant reactant and product configurations are presented in Fig. 4.
CO angle was ∼172° during the runs. Two of the four Na+ cations forming the EDL were found at about ∼3 Å from the surface while the other two Na+ cations were located further away at ∼5 Å on the Ag111 surface (Fig. 3c).43 The water molecules within the EDL close to the surface facet were found to show a limited mobility. They pointed their protons towards the metal surface during the simulations. In the absence of the EDL, the water molecules preferentially oriented with O moieties pointing towards the Ag111 surface (Fig. 2a). Next, constrained AIMD simulations were carried out on 11 intermediate states representing different stages of the outer-sphere ET CO2RR. The OCO angle was chosen as the reaction coordinate (Q), and it was varied from 172° to 125°. The resulting Gibbs free energy profile along with the representative snapshots of the relevant reactant and product configurations are presented in Fig. 4.
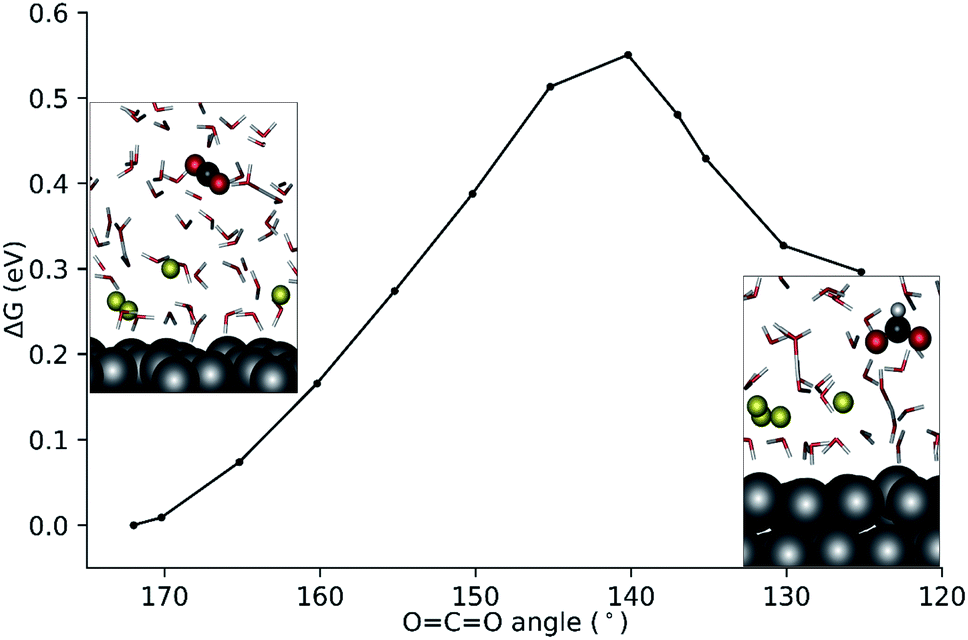 | ||
| Fig. 4 Computed Gibbs free energy profile for the homogeneous reduction of CO2 to formate using the ∠OCO angle as the reaction coordinate. | ||
The constrained AIMD simulations revealed that upon bending, the CO2 moiety diffused closer to the EDL. The transition state was located between OCO angles of 140°–145° (Fig. 4). Releasing the constraint at ∠OCO = 140° directly results in the formation of the formate product. The activation free energy barrier for the outer-sphere CO2RR is 0.55 eV with reference to the linear CO2 molecule.
Bader charge analysis at Q = 140° (equilibrated supercell at ∼18 ps) revealed a net atomic charge of −0.80 on the CO2 moiety. This is comparable to the Bader net atomic charge of −0.74 units computed for the 1e− reduced CO2 radical in water (see the ESI†). Therefore, the bent CO2 moiety at 140° represents a 1e− reduced CO2 radical. Upon further bending the transient radical species accepts an H+ from the solvent simultaneously with the second ET to yield the formate product. A snapshot of the proton transfer at ∠OCO = 137° is shown in the ESI.† Three water molecules coordinated to a Na+ in the EDL are actively involved during the proton transfer via H-bonding interactions. The bent CO2 moiety is strongly solvated forming 5 H-bonds.
The interaction with the EDL is critical for the reduction of CO2 and can be compared with cation mediated outer-sphere ET among species in the homogeneous phase (Fig. 5a).44 In the homogeneous phase solvated alkali cations have been reported to mediate outer-sphere ET between two species.44 AIMD simulations show that the solvated cations in the EDL can facilitate a similar outer-sphere ET between the cathode surface and CO2 (Fig. 5b and c). The critical role of the EDL in facilitating the CO2RR was further highlighted by the slow-growth approach (SGA) simulations that slowly bent the CO2 moiety when it was located far away from the EDL. In the absence of interaction of CO2 with the EDL, bending the CO2 moiety resulted in the formation of the HCO3− species by the nucleophilic attack of water suggesting the importance of the EDL in facilitating electroreduction (see the ESI†).
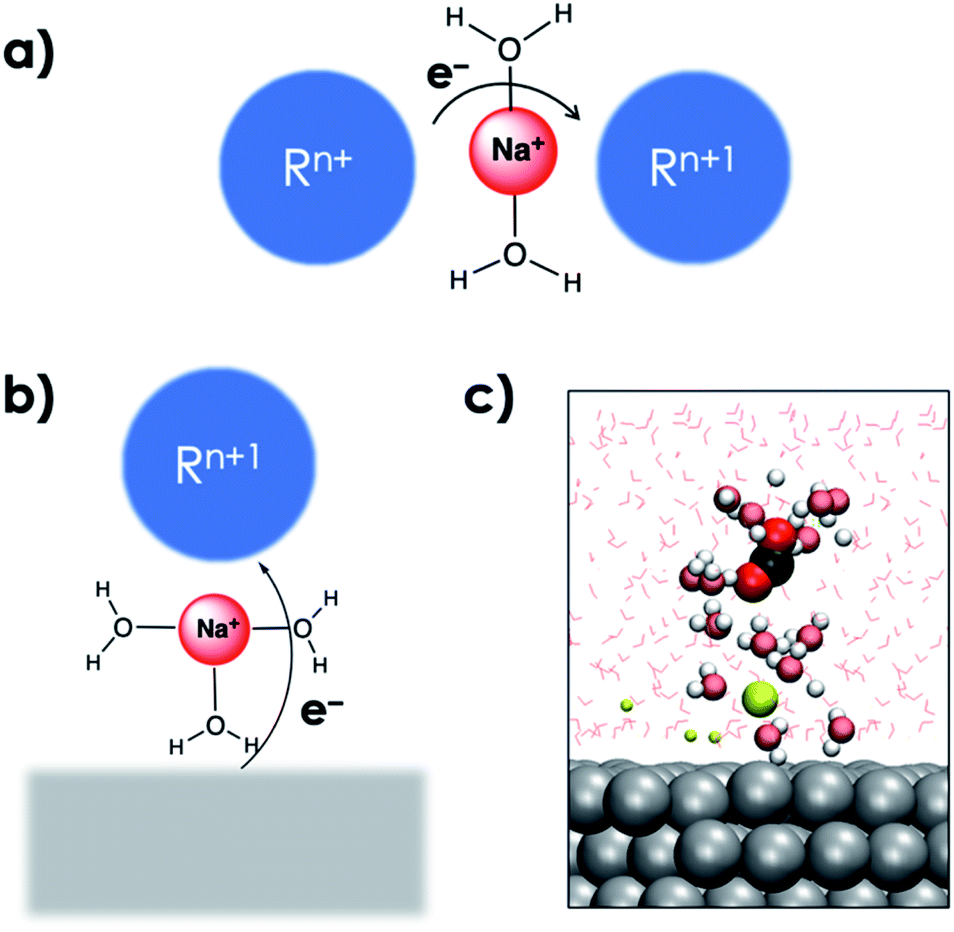 | ||
| Fig. 5 (a) Schematic representation of alkali cation promoted outer-sphere ET between two species in the homogenous phase. (b) Proposed schematic representation of alkali cation mediated outer-sphere ET from a cathode surface to a species in the homogeneous phase. (c) Snapshot of an AIMD trajectory with the OCO angle constrained at 140° showing the interaction between a solvated cation in the EDL and the solvated CO2 moiety analogous to the schematic depiction in (b). VdW representation is used for the CO2 moiety interacting with a solvated Na+ cation along with its first solvation shells which are shown in brushed metal colors. Other Na+ cations are shown as smaller spheres, and water molecules are shown via line representation. Periodic images have also been included. | ||
The formate species formed via the outer sphere ET mechanism can undergo dehydration to form CO, which is expected to be favoured by low pH conditions near the EDL.45 Dehydration of formic acid/formate in acidic conditions is well-established chemistry. Consistently, AIMD simulations revealed similar free energy barriers for the dehydration of formate to CO in the solvated phase in the presence of the EDL (Ag111–4Na+–(H2O)61–CO2 system; 1.19 eV) and without the EDL (Ag111–(H2O)61–CO2 system; 1.26 eV) (see the ESI†). The presence of the EDL provides a small stabilization (0.07 eV) to the dehydration TS. The dehydration of formate to CO is expected to be more favourable with larger cations such as K+ and Cs+ where the pH near the EDL is lower.45
Experimental results for the CO2RR over Ag show that at high cathodic bias the hydrogen evolution reaction outcompetes the formation of CO. Jaramillo and co-workers showed that the partial current density for the formation of H2 exceeds that for CO at cathodic potentials below −1.3 V (RHE).46 The rate of formation of CO peaks around −1.1 V (RHE) and then decreases as the potential is lowered. Further analysis revealed that the decrease in CO formation was due to mass transport limitations. Both formate and CO require CO2 to reach the cathode surface for the CO2RR to proceed via an inner-sphere ET mechanism. Therefore, the rate of formate production is also expected to decrease around the same potential where CO production dips due to mass transport limitations. Contrastingly, the partial current density of formate, although always lower than CO and H2 kept growing as the cathodic bias was decreased.
An outer-sphere ET mechanism, which does not require mass transport of CO2 to the surface, can explain the increasing partial formate current density. At moderate to low cathodic bias, CO2 can reach the surface, and its adsorption is stabilized by the EDL14,15 leading to the production of CO (kinetically favoured) and HCOO− (less favoured). Thus, the current density for CO and HCOO− both increase as the applied voltage is lowered. At high cathodic bias the current density switches from kinetic control to mass transport limitations leading to decreased CO production. We suggest that the mass transport limitations (at least partially) result from a condensed EDL rather than only solubility and diffusion of CO2 in the electrolyte. CO2 is therefore available in the region close to the EDL and gets reduced to formate via an outer-sphere ET mechanism, which explains the increasing formate current density.
To gain further insight into HCOO−versus CO production we compare the free energy barriers for CO formation (inner-sphere ET) reported in the literature versus HCOO− formation (outer-sphere ET) as computed by us. Based on the results reported by Chen and co-workers for the CO2RR over the Ag111 surface in the presence of a model EDL, a free energy barrier of 0.52 eV can be estimated for surface-mediated CO formation at an applied external potential of −1 V (SHE) at pH = 7.14 The current AIMD-computed free energy barrier of 0.55 eV at −1.45 V (SHE) (∼1 V versus the PZC)47 reflects a relatively higher barrier for the CO2RR via the outer sphere ET, explaining the lower partial current density for formate.
Therefore, we propose that the mechanism of 2e− reduction of CO2 is dependent on the applied bias. Scheme 1 summarizes the outer- and inner-sphere 2e− mechanisms to produce CO and formate via the CO2RR. Jaramillo and co-workers also demonstrated the formation of >2e− reduction products of the CO2RR over Ag at high overpotentials46 and an outer-sphere ET could potentially be involved in those mechanistic steps as well.
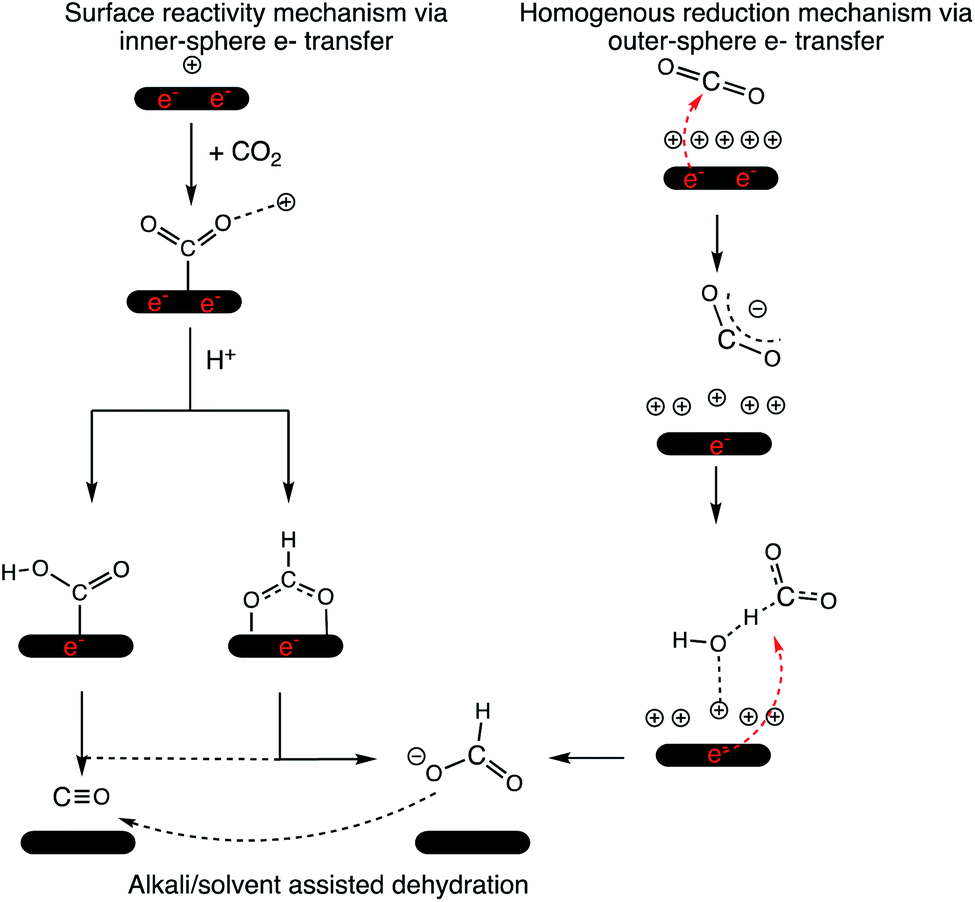 | ||
| Scheme 1 Mechanism of the CO2RR over a cathode surface via inner- and outer-sphere electron transfer. 2e− that originate from the cathode surface and reduce the CO2 moiety are shown in red for representative purposes. The cathode surface is maintained at a constant potential in the electrolyzer. | ||
Conclusions
We have explored the CO2RR over the Ag111 surface via an outer-sphere ET mechanism. Following a multiscale operando modelling strategy, we first simulated the multi-component electrolyte–cathode interface under various applied potentials. Investigation of density profiles of water, ions and CO2 revealed the formation of a condensed EDL within 1 nm of the cathode surface composed of cations and ordered layers of solvation at high overpotentials. This finding motivated the development of a smaller periodic model of the cathode-electrolyte interface which was used to investigate the reactive events during the CO2RR at high cathodic potentials via AIMD simulations. AIMD simulations showed that an outer-sphere ET mechanism resulted in the formation of formate species over the EDL. The formate species was further shown to undergo alkali promoted dehydration to CO with a moderate free energy barrier of 1.19 eV. The presence of the EDL was found to be the key to promote an outer-sphere ET CO2RR mechanism.The outer-sphere ET CO2RR is a plausible mechanism to produce formate and CO under high cathodic bias. A surface-based, alkali promoted CO2RR is likely still the dominant mechanism for the formation of CO. Our calculations show that an alternative reaction channel to reduce CO2 is accessible in the presence of a dense EDL, and the reaction mechanism is a complex network of voltage dependent inner- and outer-sphere ET steps. Outer-sphere mechanisms should be further explored for heterogeneous electrocatalytic systems and can be especially relevant for electrocatalytic reduction of organic substrates which occur under high voltage conditions.
Data availability
Data related to this publication is available via the 4TU database under the DOI: http://10.4121/19142303.Author contributions
EAP conceived and supervised the project. VS carried out DFT and AIMD simulations. EK performed CMD simulations. All the authors discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. S. acknowledges the ARC-CBBC project 2016.008 for funding. E. A. P. acknowledges the financial support from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement no. 725686). E. K. thanks Dr R. M. Hartkamp and Mr M. F. Döpke for providing help with CMD simulations. This work was sponsored by NWO Domain Science for the use of the national computer facilities. We acknowledge that the results of this research have been partially achieved using the DECI resource, Kay, based in Ireland at ICHEC with support from PRACE under the DECI.References
- S. van Bavel, S. Verma, E. Negro and M. Bracht, ACS Energy Lett., 2020, 5, 2597–2601 CrossRef CAS
.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS PubMed
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed
- Y. Quan, J. Zhu and G. Zheng, Small Sci., 2021, 1, 2100043 CrossRef
- Y. Pei, H. Zhong and F. Jin, Energy Sci. Eng., 2021, 9, 1012–1032 CrossRef CAS
- S. A. Mahyoub, F. A. Qaraah, C. Chen, F. Zhang, S. Yan and Z. Cheng, Sustainable Energy Fuels, 2020, 4, 50–67 RSC
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS
- C. Xi, F. Zheng, G. Gao, M. Ye, C. Dong, X. W. Du and L. W. Wang, J. Mater. Chem. A, 2020, 8, 24428–24437 RSC
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed
-
J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Modulating Electric Field Distribution by Alkali Cations for CO2 Electroreduction in Strongly Acidic Medium. ChemRxiv, Cambridge, Cambridge Open Engage, 2021, DOI, https://doi.org/ DOI:10.33774/chemrxiv-2021-zgq9k
- M. M. Waegele, C. M. Gunathunge, J. Li and X. Li, J. Chem. Phys., 2019, 151, 160902 CrossRef PubMed
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC
- V. Briega-Martos, F. J. Sarabia, V. Climent, E. Herrero and J. M. Feliu, ACS Meas. Sci. Au, 2021, 1, 48–55 CrossRef CAS
- L. D. Chen, M. Urushihara, K. Chan and J. K. Nørskov, ACS Catal., 2016, 6, 7133–7139 CrossRef CAS
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, ACS Catal., 2021, 4, 654–662 CAS
- H. Zhong, K. Fujii and Y. Nakano, J. Electrochem. Soc., 2017, 164, F923–F927 CrossRef CAS
- D. Bohra, J. H. Chaudhry, T. Burdyny, E. A. Pidko and W. A. Smith, Energy Environ. Sci., 2019, 12, 3380–3389 RSC
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed
- D. Ješić, D. Lašič Jurković, A. Pohar, L. Suhadolnik and B. Likozar, Chem. Eng. J., 2021, 407, 126799 CrossRef
- Y. Wang, J. Liu, Y. Wang, A. M. Al-Enizi and G. Zheng, Small, 2017, 13, 1701809 CrossRef PubMed
- E. R. Cave, J. H. Montoya, K. P. Kuhl, D. N. Abram, T. Hatsukade, C. Shi, C. Hahn, J. K. Nørskov and T. F. Jaramillo, Phys. Chem. Chem. Phys., 2017, 19, 15856–15863 RSC
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS
- S. Saeidi, S. Najari, F. Fazlollahi, M. K. Nikoo, F. Sefidkon, J. J. Klemeš and L. L. Baxter, Renewable Sustainable Energy Rev., 2017, 80, 1292–1311 CrossRef CAS
- S. Xu and E. A. Carter, Chem. Rev., 2019, 119, 6631–6669 CrossRef CAS PubMed
- D. Bohra, I. Ledezma-Yanez, G. Li, W. de Jong, E. A. Pidko and W. A. Smith, Angew. Chem., Int. Ed., 2019, 58, 1345–1349 CrossRef CAS PubMed
- N. Fey and J. M. Lynam, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2021, e1590, DOI:10.1002/wcms.1590
- J. A. Gauthier, S. Ringe, C. F. Dickens, A. J. Garza, A. T. Bell, M. Head-Gordon, J. K. Nørskov and K. Chan, ACS Catal., 2019, 9, 920–931 CrossRef CAS
- J. J. Varghese and S. H. Mushrif, React. Chem. Eng., 2019, 4, 165–206 RSC
- M. Saleheen and A. Heyden, ACS Catal., 2018, 8, 2188–2194 CrossRef CAS
- J. N. Harvey, F. Himo, F. Maseras and L. Perrin, ACS Catal., 2019, 9, 6803–6813 CrossRef CAS
- V. Sinha, N. Govindarajan, B. de Bruin and E. J. Meijer, ACS Catal., 2018, 8, 6908–6913 CrossRef CAS PubMed
- N. Govindarajan, V. Sinha, M. Trincado, H. Grützmacher, E. J. Meijer and B. de Bruin, ChemCatChem, 2020, 12, 2610–2621 CrossRef CAS
- V. Sinha, D. Sun, E. J. Meijer, T. J. H. Vlugt and A. Bieberle-Hütter, Faraday Discuss., 2021, 229, 89–107 RSC
- S. E. Weitzner, S. A. Akhade, J. B. Varley, B. C. Wood, M. Otani, S. E. Baker and E. B. Duoss, J. Phys. Chem. Lett., 2020, 11, 4113–4118 CrossRef CAS PubMed
- A. M. Krieger, P. Kuliaev, F. Q. Armstrong Hall, D. Sun and E. A. Pidko, J. Phys. Chem. C, 2020, 124, 26990–26998 CrossRef CAS PubMed
- J. Meeprasert, G. Li and E. A. Pidko, Chem. Commun., 2021, 57, 7890–7893 RSC
- A. M. Krieger and E. A. Pidko, ChemCatChem, 2021, 13, 3517–3524 CrossRef CAS PubMed
- R. K. A. van Schendel, W. Yang, E. A. Uslamin and E. A. Pidko, ChemCatChem, 2021, 13, 4886–4896 CrossRef CAS
- A. Kovalenko and V. Neburchilov, J. Mol. Model., 2020, 26, 267 CrossRef CAS PubMed
- J. A. Gauthier, M. Fields, M. Bajdich, L. D. Chen, R. B. Sandberg, K. Chan and J. K. Nørskov, J. Phys. Chem. C, 2019, 123, 29278–29283 CrossRef CAS
- N. J. Firet and W. A. Smith, ACS Catal., 2017, 7, 606–612 CrossRef CAS
- M. M. Waegele, C. M. Gunathunge, J. Li and X. Li, J. Chem. Phys., 2019, 151, 160902 CrossRef PubMed
- P. D. Metelski and T. W. Swaddle, Inorg. Chem., 1999, 38, 301–307 CrossRef CAS
- F. Zhang and A. C. Co, Angew. Chem., Int. Ed., 2020, 59, 1674–1681 CrossRef CAS PubMed
- T. Hatsukade, K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Phys. Chem. Chem. Phys., 2014, 16, 13814–13819 RSC
- G. Valette, J. Electroanal. Chem. Interfacial Electrochem., 1989, 269, 191–203 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Full datasets are separately available via the 4TU database. See DOI: 10.1039/d1sc07119j |
| This journal is © The Royal Society of Chemistry 2022 |