
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymology of standalone elongating ketosynthases
Aochiu
Chen
 ,
Ziran
Jiang
and
Michael D.
Burkart
*
,
Ziran
Jiang
and
Michael D.
Burkart
*
Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358, USA. E-mail: mburkart@ucsd.edu
First published on 9th March 2022
Abstract
The β-ketoacyl-acyl carrier protein synthase, or ketosynthase (KS), catalyses carbon–carbon bond formation in fatty acid and polyketide biosynthesis via a decarboxylative Claisen-like condensation. In prokaryotes, standalone elongating KSs interact with the acyl carrier protein (ACP) which shuttles substrates to each partner enzyme in the elongation cycle for catalysis. Despite ongoing research for more than 50 years since KS was first identified in E. coli, the complex mechanism of KSs continues to be unravelled, including recent understanding of gating motifs, KS–ACP interactions, substrate recognition and delivery, and roles in unsaturated fatty acid biosynthesis. In this review, we summarize the latest studies, primarily conducted through structural biology and molecular probe design, that shed light on the emerging enzymology of standalone elongating KSs.
1. Introduction
The fatty acid biosynthesis (FAB) and the polyketide biosynthesis (PKB) are evolutionarily related biosynthetic pathways that synthesize primary and secondary metabolites, respectively. Utilizing the same fundamental enzymatic transformations, both FAB and PKB are of great interest for the production of valuable compounds such as biofuels, pharmaceutical leads, and agrochemicals. Fatty acid synthases (FASs) and polyketide synthases (PKSs) can be classified into type I, in which multiple domains are linked together to form megasynthases, and type II, which possesses discrete, primarily monofunctional enzymes. Despite variations of components across these assembly line pathways, the ketosynthase (KS) domain is always present, serving as the key carbon–carbon bond catalysing enzyme in the FAB and PKB elongation cycles. Another crucial protein is the acyl carrier protein (ACP), a small 4-helical bundle that shuttles substrates and intermediates throughout the pathways, interacting with each catalytic domain. A functional ACP (holo-ACP) requires post-translational installation of a 4′-phosphopantetheine moiety (PPant) onto a serine residue in which PPant provides a thiol group for substrate tethering. In this review, we aim to delineate the enzymology of elongating KSs from type II systems by focusing on three topics: the KS gating mechanism, substrate control through the KS pocket, and mechanisms involving KS–ACP interactions.The elongation mechanism of KSs
The elongating KS active site consists of a cysteine–histidine–histidine catalytic triad and an oxyanion hole formed by the backbone amides of the catalytic cysteine and a conserved phenylalanine. The ping–pong mechanism of KSs involves two half-reactions, the transacylation step and the condensation step, respectively. In the transacylation step, the acyl substrate gets transferred onto the active site cysteine, with the oxyanion hole stabilizing the tetrahedral intermediate. Subsequently, the condensation step has the two histidine residues catalysing a decarboxylative enolate attack from a malonyl group to the thioester-tethered substrate. The oxyanion hole continues to play the role of stabilizing the oxyanion formed during the reaction. The overall catalysis condenses a 2-carbon unit onto the acyl substrate to give a β-keto acyl product. Each half-reaction involves association and dissociation of ACP. Known to sequester the substrate, acyl-ACP delivers an acyl substrate to the KS pocket under a chain-flipping mechanism (Fig. 1a), and ACP is released as holo-ACP after the transacylation step. A malonyl-ACP then enters the cycle for the condensation step and leaves with the β-keto acyl product tethered (Fig. 1b).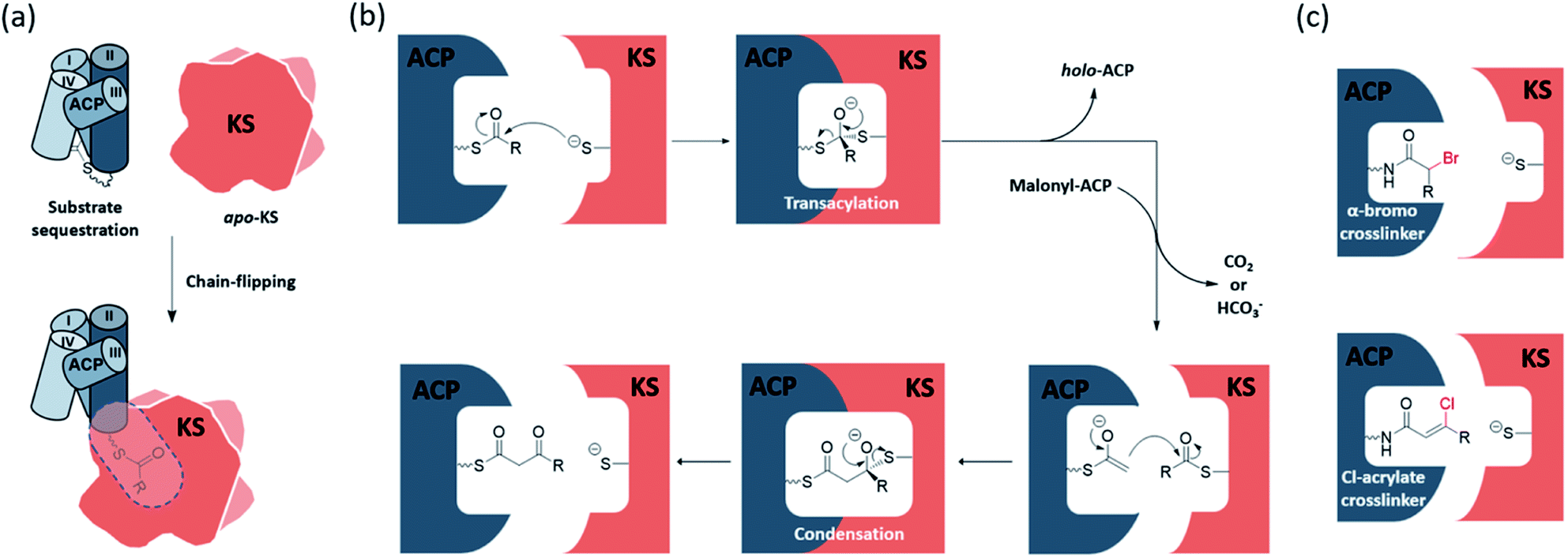 | ||
| Fig. 1 KS catalysis and mechanism-based crosslinkers: (a) transition of the substrate from the ACP pocket to the KS pocket is described as a chain-flipping event. (b) The ping–pong mechanism of KS catalysis begins with the transacylation half-reaction to generate acyl-KS followed by the condensation half-reaction that synthesizes β-ketoacyl-ACP via decarboxylative condensation. (c) The mechanism-based crosslinkers, α-bromo and Cl-acrylate, can trap KS–ACP interaction in the transacylation state and condensation state, respectively. Each leaving group is coloured by red. | ||
The detailed mechanism of the transacylation step is well understood. The active site cysteine is located at a sharp turn of the N-terminus of a long α-helix, also known as a nucleophilic elbow, which enhances the nucleophilicity and basicity of the thiol group by the α-helix dipole moment.1,2 The oxyanion hole also facilitates the reaction by accommodating the negative charge build-up on the carbonyl oxygen. The detailed mechanism of the condensation half-reaction, on the other hand, is still under debate. For type II FAS (FASII), a widely accepted version follows a sequential mechanism that starts with the decarboxylation of the malonyl group to generate an enolate, and then the enolate attacks and undergoes condensation with the KS-bound acyl substrate. Experimental data from animal FAS show that the decarboxylation by-product is bicarbonate, while releasing carbon dioxide as the by-product is still possible in FASII.3 At least two other alternative mechanisms have been proposed, and readers are directed to a review by Heil et al. for further details.4
Standalone KSs from different pathways
As the canonical and most well-studied FASII system, E. coli FAS will exemplify the following introduction of the FAS KS. Two elongating KSs, the β-ketoacyl-ACP synthase II (FabF) and the β-ketoacyl-ACP synthase I (FabB), exist in the system. FabF serves as the primary elongating KS of FAB and is also the evolutionary progenitor of PKS KSs.5 FabB specializes in the first elongation of mono-unsaturated fatty acids (UFAs) while still capable of maintaining FAB by itself.6–8 A lack of FabB in E. coli leads to UFA auxotroph. Both FabF and FabB are functional as a homodimer, forming two substrate pockets along the dimer interface. A full cycle of saturated fatty acid (SFA) elongation starts with the KS condensing the 2-carbon unit from a malonyl group to the acyl-chain followed by a ketoreductase (KR) reducing the β-keto group to a hydroxyl group, a dehydratase (DH) eliminating the hydroxyl group to generate an enoyl intermediate, and finally an enoyl-reductase (ER) to reduce the enoyl double bond (Fig. 2). In E. coli and related proteobacteria, the synthesis of UFA elongation occurs de novo through the same fashion except for the particular cycle that retains the unsaturation, in which the (2E)-enoyl intermediate is isomerized to (3Z)-enoyl by the DH that possesses isomerase activity, such as FabA in E. coli, and subsequently enters the next cycle of elongation.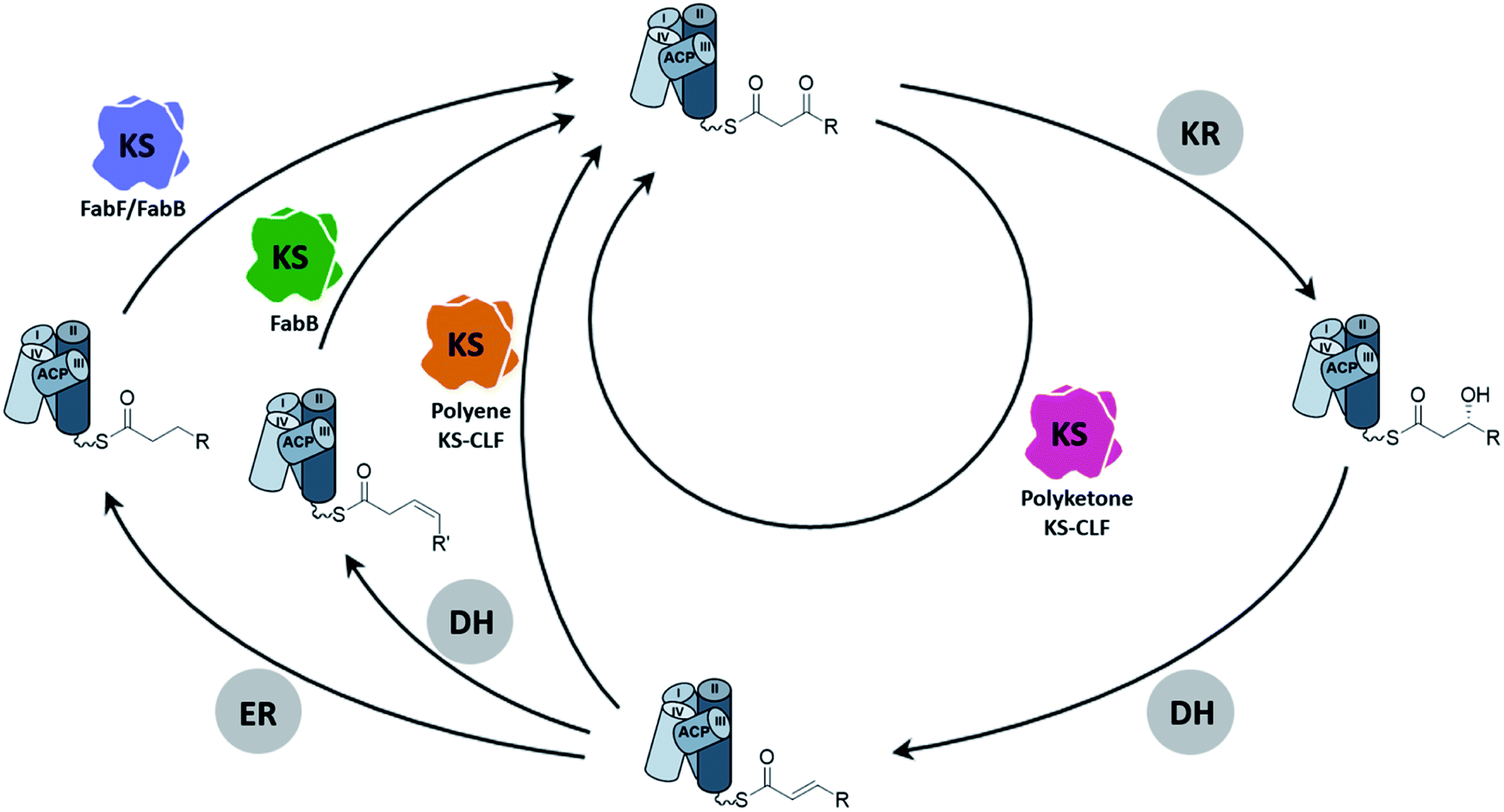 | ||
| Fig. 2 The elongation cycle of type II FAS and PKS: A full cycle of SFA elongation goes through KS-mediated elongation, ketoreduction, dehydration, and enoyl-reduction. The cis-double bond of mono-unsaturated fatty acids comes from the isomerization of (2E)-enoyl-ACP followed by the elongation of FabB (green KS) in a particular cycle. Consecutively skipping the reduction of enoyl-ACP yields polyene intermediates that are elongated by polyene KS–CLF (orange KS), and the elongation catalyzed individually by polyketone KS–CLF (magenta KS) yields chemically flexible intermediates that lead to polycyclic aromatic polyketides (KR = ketoreductase, DH = dehydratase, and ER = enoyl-reductase). | ||
The conventional type II PKB produces highly functional polycyclic aromatic compounds derived from chemically flexible polyketone intermediates which are generated from skipping all the domains but the KS in the elongation cycle (Fig. 2). Given the high reactivity of polyketone intermediates, the substrate remains protected inside the pocket during elongation until it reaches the maximum chain length.9–12 Another class of PKS, the polyene PKS that has been studied for less than a decade, has every domain from the FAB elongation cycle except the ER domain, producing polyene13–16 or aryl polyene products.17–23 In terms of substrate shuttling, the polyene PKS system is more similar to the FAS system where ACP is required to deliver substrates to multiple enzymes during elongation. Although various nomenclatures have been used to describe the two PKS systems mentioned above, for example, by their product types (aromatic vs. polyene) or the reduction level (non-reducing vs. highly reducing), in this review, we will refer to them by their elongation intermediates, polyketone and polyene, respectively, for simplicity.
The type II PKS (PKSII) KSs are heterodimers formed by KSα and KSβ subunits. The KSβ subunit has no condensation activity and lacks the canonical KS active site residues, but it plays a critical role in controlling the chain length of the resulting product and is thus often referred to as the chain-length-factor (CLF) subunit. There is one exception that the ApeR, one of the two KSs of aryl polyene biosynthesis, is instead a homodimeric KS.21,23 Historically, heterologous expression of PKSII KS–CLFs in E. coli is notorious for having the over-expressed KS–CLF resided in the insoluble fraction.24 However, recent efforts have resulted in several successful cases of individually expressing polyketone KS–CLF in E. coli, lowering the barrier for studying the enzymology of FASII KS.25–29
There are a number of organizational questions that remain to be answered. In organisms that possess both type II FAS and PKS systems, the mechanism that prevents crosstalk, or non-cognate protein catalysis, remains an important unanswered question. In addition, how FabF and FabB, structurally highly similar, differ in substrate specificity has not been solved. Growing evidence has shown that the protein–protein interactions (PPIs) of the KS and ACP and changes imparted by substrate identity, play an important role in these mechanisms, which will be covered in this review. Type III PKS pathways such as chalcone synthase are considered standalone KSs, but they are not within the scope of this review, which focuses on ACP-dependent systems.30,31
Biochemical tools to investigate KS enzymology
During the elongation of an acyl chain, many mechanistic steps must be achieved through the close communication between the KS and ACP. However, due to the conformational dynamics of the ACP and the rapid binding kinetics required to facilitate catalysis, fully understanding the protein–protein interactions (PPIs) and KS substrate recognition at the molecular level remains as an ongoing challenge. To address these issues, our lab and others have developed synthetic pantetheinamide probes to investigate the ACP–KS PPIs and substrate–protein interactions structurally and biochemically.32–35 It is noteworthy that amongst these chemical probes, mechanism-based crosslinking probes, or simply crosslinkers, have shown success in trapping KS–ACP interactions in their catalytic states.36–38 There are two major types of crosslinkers, α-bromo-pantetheinamide and chloroacrylate-pantetheinamide, respectively, for KS–ACP crosslinking. According to the carbon count and the coordination of the active site, α-bromo crosslinkers trap the KS–ACP complex in the transacylation state, while chloroacrylate crosslinkers trap KS–ACP in the condensation state (Fig. 1c). Crosslinking complexes have provided a deeper understanding of the enzymatic reaction mechanism of the KS and the PPIs between the KS and the ACP.In the study of PKS, because of the inherent chemical instability of the polyketone intermediates, different types of polyketide mimetic surrogates have been synthesized to gain more insight on the intermediate protection mechanism in the binding pocket.10,39–43 Additionally, the process of substrate sequestration in ACP and chain flipping were elucidated by vibrational spectroscopic and the solvatochromic probes.44,45 Readers can be directed to the comprehensive reviews by Chen et al. and Sulpizio et al. for more information.11,46
2. Gating mechanisms in elongating KSs
A recently discovered KS gate involving two gating loops has been proposed to regulate substrate processing and catalysis.38,47 Together with two other investigated KS gates, the KS deploys a well-controlled system to orchestrate the complex 2-step catalysis. We will discuss the mechanism, influence, and universality of each gate. If not specifically mentioned, residue numbering in this section is based on EcFabF.The front gate: double drawbridge-like gating loops
The front gate consists of two stacking loops that move co-ordinately to control the entry of substrates.48 In EcFabF, this contains loop 1 comprising G399–G402 (GFGG), and loop 2 comprising D265–N275 (DAYHMTSPPEN).38 Transition from the closed to the open form of the gate requires a large conformational change of the loops, with the Cβ of F400 moving 8.7 Å away from its original position (Fig. 3a). Such movement of F400 provides access to the substrate binding pocket and the active site cysteine, C163. In the open form, the oxyanion hole formed by the backbone amides of C163 and F400 is disrupted and therefore cannot stabilize KS reaction intermediates. Thus, the open form is believed to facilitate substrate delivery, whereas the closed form is the catalytically competent form with a well-ordered oxyanion hole.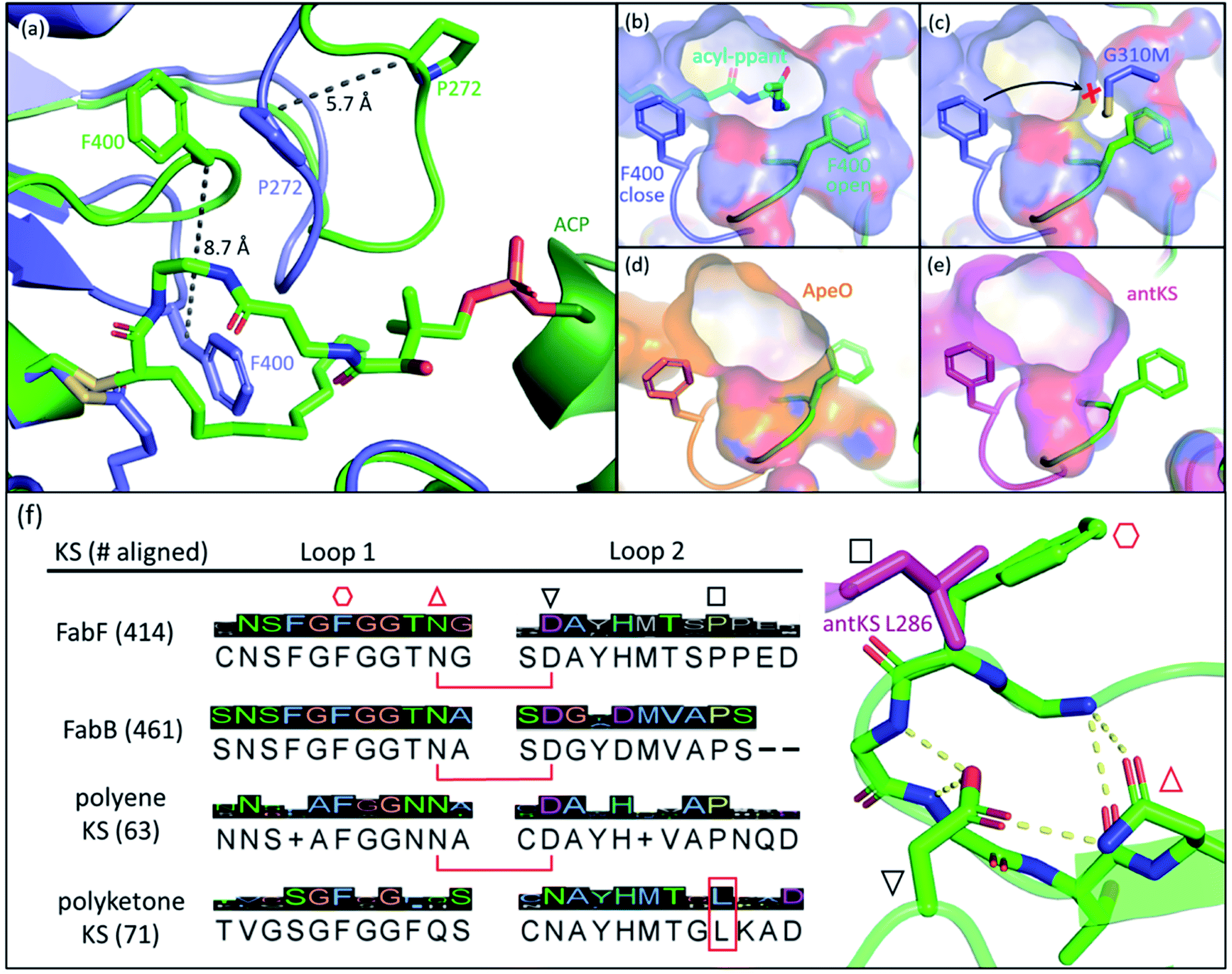 | ||
Fig. 3 The gating loops: (a) structure overlay of C12–FabF (PDB ID: 2GFY, blue) in the gate closed conformation and the KS–ACP crosslink complex (PDB ID: 6OKG, green) in the gate open conformation. Movements of F400 on loop 1 and P272 on loop 2 are measured. (b) Surface plot of gate closed FabF (blue) overlaying with the open loop 1 (green) demonstrates the side pocket that accommodates the open loop. (c) G310M mutant blocks the gate from opening. (d) Surface plot of ApeO (PDB ID: 6QSP, orange), a KS from aryl polyene biosynthesis, overlaying with the open loop 1. (e) Surface plot of a polyketone KS, antKS (PDB ID: 6SMO, magenta), overlaying with the open loop 1. The side pocket for the open gate is completely missing. (f) Over a thousand KS gating loop 1 and loop 2 sequences aligned (MUSCLE algorithm) with the consensus sequence shown (left column). The coordination of open loop 1 is depicted on the right. The conserved Asn( )–Asp( )–Asp( ) pair that is essential for the open gate is absent in the polyketone KS. The conserved Leu ( ) pair that is essential for the open gate is absent in the polyketone KS. The conserved Leu ( and L286 in antKS) of the polyketone KS clashes into the Phe ( and L286 in antKS) of the polyketone KS clashes into the Phe ( ) of open loop 1. This Leu is instead a conserved Pro in other types of KS. ) of open loop 1. This Leu is instead a conserved Pro in other types of KS. | ||
The open form of the gate was captured by crystal structures of the EcFabF–EcACP crosslink complex using the α-bromo-pantetheinamide crosslinking probes with various chain lengths of fatty acid mimetics (C12, C16, and C16:1), whereas using the C8 chloroacrylate-pantetheinamide crosslinking probe yielded a gate-closed structure. The overlay of these structures reveals a “side pocket” that accommodates the open form of loop 1 (Fig. 3b). More than a dozen EcFabF mutants designed to study the activity of the gating mechanism were developed and tested with a crosslinking gel-shift assay and two kinetic activity assays to validate the role of the gate. One particular interesting class of mutations that compromises FabF activity is the pocket block mutants, such as G310M (Fig. 3c), that fills the side pocket to prevent loop 1 from opening. The side pocket could be observed in structures of FAS and polyene KSs that release substrates for each cycle of elongation (Fig. 3b and d). However, for the polyketone KS, which is believed to keep the reactive polyketone intermediate in the pocket during elongation,11 there is no space for loop 1 in the fully open conformation (Fig. 3e). Sequence alignment over a thousand different KSs also reveals that the polyketone KS lacks a pair of crucial residues (D265–N404 of EcFabF) which stabilizes the open gate and is totally conserved in other KSs aligned. Furthermore, a conserved proline in loop 2 is instead a leucine in the polyketone KS, which blocks the gate from opening (Fig. 3f). Although the universality of the gating mechanism has yet to be confirmed, based on structure analysis and sequence alignment it seems to exist in KSs that actively accept and release substrates, an activity absent in polyketone KSs, which retain the substrate during iterative rounds of elongation.
The KS active site and gating loop arrangement during different states of the catalysis are summarized in Fig. 4. Starting from the apo-KS (1), the gating loops transition to the open conformation (2) for substrate delivery from acyl-ACP. The gate then transitions back to reform the oxyanion hole to set up the substrate in state (3) for the transacylation reaction. After holo-ACP dissociation, the acyl-KS (4) reacts with malonyl-ACP to form the decarboxylative condensation intermediate depicted in state (5). The final product, β-ketoacyl-ACP, is generated (6) and released by opening the gate (7) to bring the KS back to the starting point. In the case of aromatic PKS, rather than releasing the substrate, a transacylation reaction should tailor state (6) to position the high energy polyketone intermediate back to the KS, depicted in (PK). The reaction undergoes an iterative cycle between (4), (5), (6), and (PK) until the substrate reaches the final chain length. The gate-open states (2) and (7) may not exist in this pathway. Most of the states depicted in Fig. 4 are supported by KS or KS–ACP crosslinked crystal structures.12,36–38
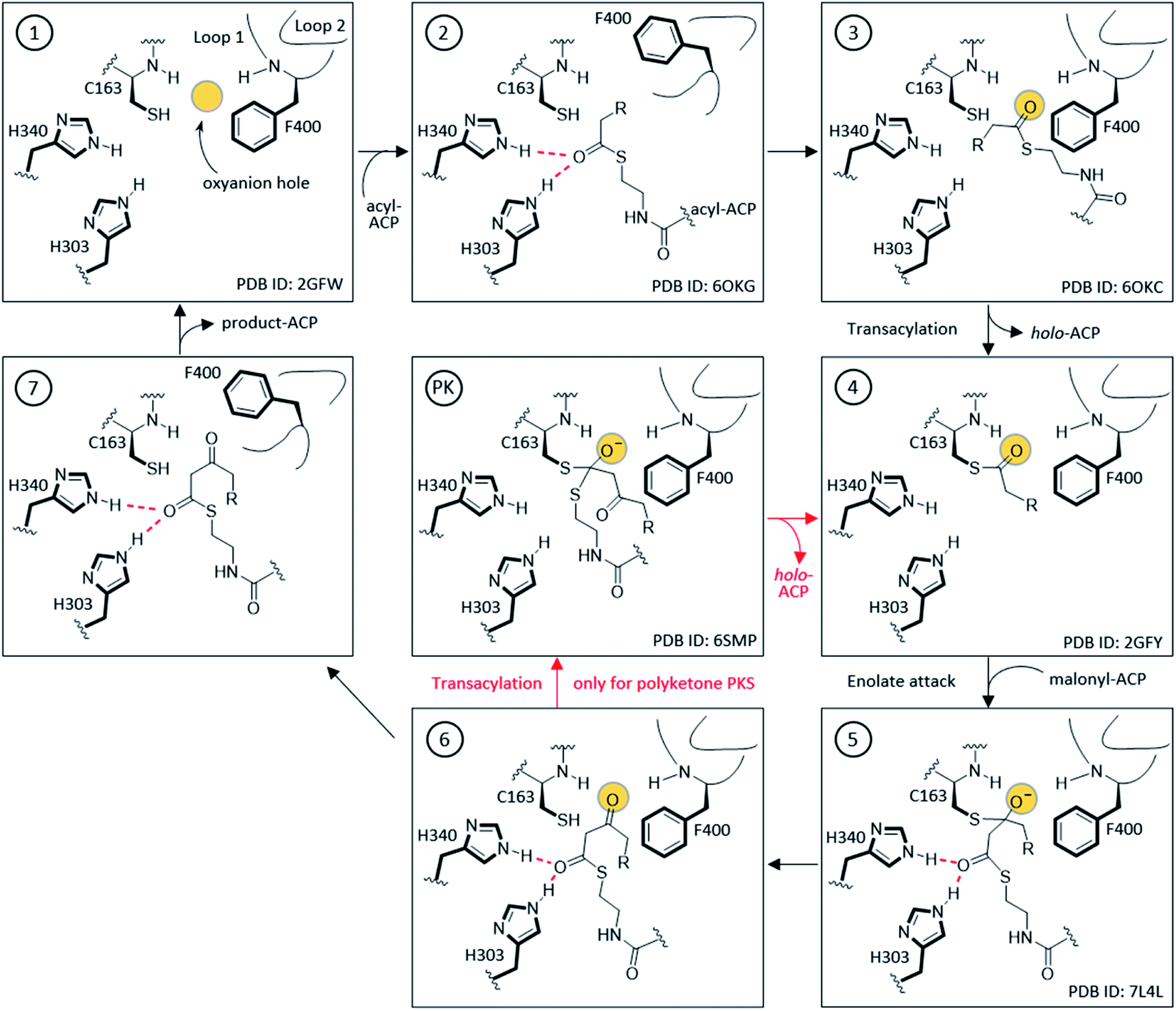 | ||
| Fig. 4 Detailed mechanism of KS elongation and the gating loops: FabF, FabB, and polyene KS go through (1)–(7) followed by the product-ACP release for one extension while the polyketone KS goes through the smaller circle (4), (5), (6), and (PK), to keep the growing chain inside the KS pocket. (2) and (7) are considered the substrate delivery and released states, respectively, with the gating loops open and the catalytic oxyanion hole absent. Most depicted states are supported by at least one crystal structure with a representative of each labelled at the bottom-right corner of the box. The amino acid numbering is from EcFabF. | ||
The gating mechanism of the gating loops provides an important piece of puzzle that connects essential elements in KS catalysis. Loop 1 contains the residue F400 (in EcFabF numbering) that plays a role in regulating the condensation half reaction. When ACP is bound, loop 2 sits in between loop 1 and ACP, bearing residues that interact with loop 1, ACP, and the phosphopantetheine moiety, indicating a potential communication role that loop 2 might play. Swapping loop 2 of EcFabF with loop 2 of EcFabB has a detrimental effect on the EcFabF function,47 suggesting that the gating loops are evolutionarily coupled in specific systems to function in a certain way. The large space on the other side of the substrate pocket, which has never before been assigned a purpose, is shown to accommodate the open form of loop 1 (Fig. 3b). Since this gate regulates substrate delivery, it could potentially play a role in KS substrate specificity. More functions of the gate have yet to be explored, but with the importance discovered so far, it is a factor that cannot be ignored in KS enzymology, particularly in the discussion of engineering efforts.
The gate for the condensation half reaction: rotamers of F400
The conserved phenylalanine residue of gating loop 1 also regulates the condensation half-reaction through its rotamers. Acylation of the active site cysteine results in a ∼60° rotation of the phenyl group along the Cβ–Cγ bond of the phenylalanine, setting up a malonyl substrate-binding pocket for the decarboxylative condensation half-reaction (Fig. 5a).49 Such a mechanism prevents KSs from taking malonyl substrates before transacylation occurs, which is critical to the timing of the ping–pong mechanism. Mutating the cysteine to a glutamine, which gives an active site arrangement similar to that of acyl-KS, is reported to convert the KS to a malonyl decarboxylase,50 while also significantly increasing KS's binding affinity towards platensimycin, a KS inhibitor that mimics the malonyl substrate.51 A phenylalanine-to-alanine mutation almost eliminates condensation activity of the KS in vitro (0.7–1.85% activity of wild type) while also decreasing the transacylation activity as well as other activities that generate shunt products.47,51 This demonstrates that simply removing the physical barrier formed by the phenylalanine gating residue does not improve the enzyme overall. These results support three functions for this residue: playing a role in the gating loops that regulate substrate delivery, controlling binding of malonyl-ACP substrates, and bearing the amide that takes part in forming the catalytic oxyanion hole, which might need the bulky side chain to fix the residue in the right position. Additionally, it has also been suggested that conformations of this phenylalanine may determine the protonation states of the active site residues in the Mycobacterium tuberculosis KS.52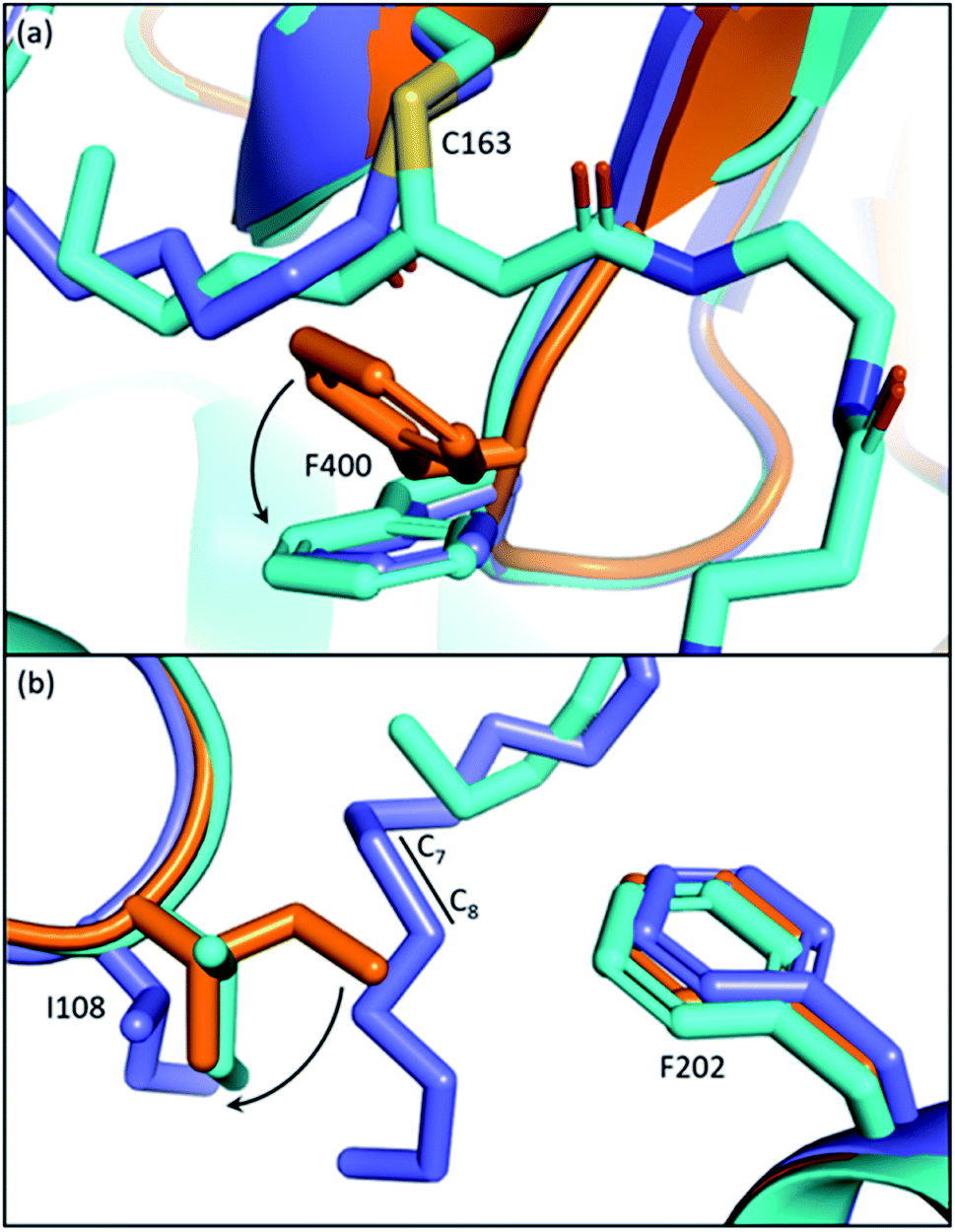 | ||
| Fig. 5 Gating residues of FabF: overlay of apoFabF (PDB ID: 2GFW, orange), C12–FabF (PDB ID: 2GFY, blue), and the FabF–ACP crosslink structure (PDB ID: 7L4L, light blue). (a) Rotation of F400 triggered by substrate binding creates the malonyl binding pocket. (b) Rotation and translation of I108 when a long acyl-chain (more than 8-carbon) is bound. | ||
The phenyl ring of F400 is positioned squarely in front of the α carbon of the acyl chain in acyl-KS and may also contribute to the selectivity towards α-branched substrates. In type I PKS, which takes branched polyketones and α-branched malonyl extender units such as methylmalonyl-CoA (mmCoA), the phenylalanine is replaced either by a valine or an isoleucine, potentially creating the space to accommodate a branching alkyl group. Interestingly, EcFabF F400V and F400I mutants, compared to the wild type (WT), suffer reduced condensation activities in vitro towards both mal-CoA and mmCoA,47 yet another indication of the complex role of F400. Considering the position of the F400 side chain, it would be interesting to test these mutants' activities towards α-branched acyl substrates.
The back gate: I108–F202
In EcFabF, I108 and F202 form a gate in the middle of the substrate pocket, approximately located at the C7–C8 position of a bound acyl chain. While FabB has the same loop 1 phenylalanine residue, I108 is highly conserved and unique to FabF and is replaced by a glycine residue in FabB. I108 has different rotamers across different EcFabF structures (Fig. 5b): in apo-FabF, the Cδ of I108 is only 3.7 Å away from the closest carbon of the F202 side chain, forming a gate-closed conformation. When a six-carbon chain is extended from the active site cysteine, like the ACP–FabF crosslinked structure 7L4L (PDB ID), I108 adopts a different rotamer that extends the closest carbon-to-carbon distance with F202 to 5.8 Å. Finally, the lauric acid bound FabF structure (PDB ID: 2GFW) has the acyl chain penetrating through the gate, with I108 moving further away to create a 7.5 Å carbon-to-carbon gap. It has been shown that the I108F mutant predominantly elongates the C6 acid to C8, resulting from a tighter gate that inhibits the loading of longer acids. A similar mutation idea has also been applied in engineering fungal FAS for short chain FA production.53 I108F mutation also prevents the binding of cerulenin, a KS inhibitor that penetrates through the back gate among binding and grants the mutant cerulenin resistance in both EcFabF and Bacillus subtilis FabF (BsFabF).54–56 I108M of BsFabF has also been reported to have a two-fold higher cerulenin resistance than the I108F mutant.57In the human mitochondrial KS (HsmtKS), a bimodal substrate preference exists that results mainly in producing the C8 fatty acid, the precursor of lipoic acid biosynthesis, and the C14–C16 fatty acids.58 Structures of mtKSs reveal that M154, the same position as I108 of EcFabF, must rearrange to allow the longer acyl chain to bind, causing the bimodal substrate profile.59EcFabF has yet not shown preference towards a certain medium chain length substrate. Whether I108 affects the substrate profile or not is still unclear. It is interesting to note that glutaryl-ACP methyl ester, equivalent to a C7 acid in length when transferred onto the active site cysteine, is elongated by the KS to generate the precursor of biotin biosynthesis.60 The methyl ester group of the dicarboxylate might be too bulky to penetrate through the back gate, thus preventing further elongation.
3. Substrate specificity and chain length control
The KS pocket has direct influence on the substrate via physical restrictions, polar contacts, and Coulomb-repulsion, thereby controlling the identity of the product. These interactions can most often be identified by analysing KS crystal structures, and on top of them are substrate bound KS structures that directly visualize substrate behaviour inside the pocket. In this section, we will discuss KS's mechanism being one of the key enzymes of product chain length control in FAS and PKS,4 and its substrate specificity realized by some key elements in the substrate pocket.The dichotomy of FabF and FabB
In the E. coli FAB, FabF and FabB have most of their SFA substrate preferences overlapped (C6–C12), with the exception of C14, which is preferentially elongated by FabF.61 In terms of UFA biosynthesis, FabB catalyses the first elongation from C10:1 to C12:1 and then both KSs can elongate C12:1 up to C16:1.6 The final elongation to C18:1 is exclusively catalysed by FabF.62 While the FabF pocket does not have an obvious end,63 as the homodimer pockets meet at the interface, FabB has a clear 12-carbon pocket depth restricted by the coordinating E200–Q113 residues located at the bottom of the pocket.64 However, both residues can adopt different conformations, dissociating the hydrogen bond between side chains, to expand the pocket depth, as seen in the C12–FabB crystal structure (PDB ID: 1EK4). This explains the existing but low activity of FabB toward the C14 substrate (Fig. 6).65 Interestingly, this longer pocket cannot coexist in both subunits of the homodimer, because such conformation of the glutamine residues (Q113), located at the center of the dimer, would clash. This set of asymmetric pockets of FabB suggests a cooperativity relationship between the subunits on long chain substrates.66 The possible cooperativity of the homodimeric type II KS has rarely been addressed, but the cooperativity effect has recently been reported in the type I murine KS domain, as well as a type I PKS.67,68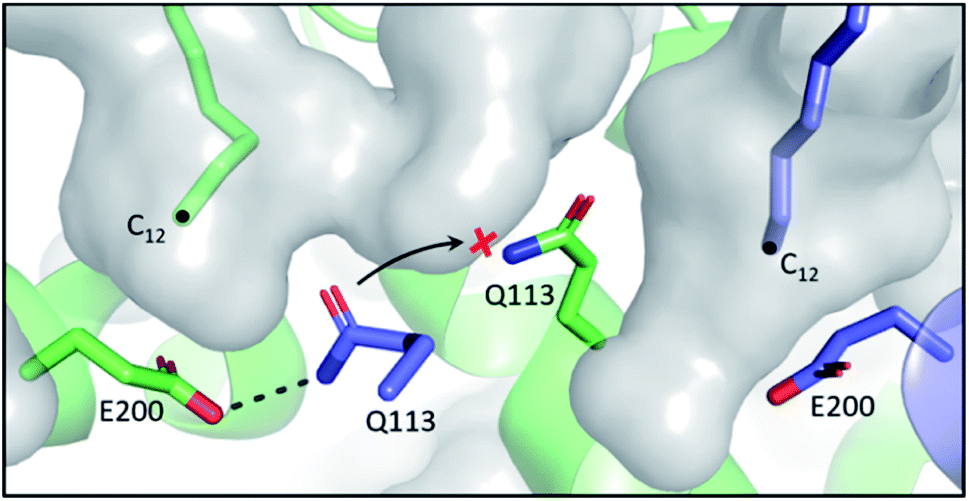 | ||
| Fig. 6 Asymmetric substrate pockets of FabB (PDB ID: 1EK4): E200–Q113 polar contact restricts the pocket size to 12-carbon on the left whereas the pocket with dissociated E200–Q113 on the right accommodates a longer substrate. Q113 from each of the subunit of the homodimer cannot co-occupy the center space, leaving the pockets asymmetric. | ||
Despite the lack of activity towards C16 SFA, EcFabF elongates C16:1 UFA, producing the C18:1 acid that could be incorporated into membrane phospholipids to increase cell membrane fluidity.69,70 It has been demonstrated in vitro and in vivo that FabF plays a key role in the thermal regulation of E. coli by its substrate preferences under different temperatures.62,71,72 It is, however, not clear how the one double bond difference between C16 and C16:1 acid can result in such distinct FabF activities. The position of the double bond, which lies between C7 and C8, is reminiscent of the FabF back gate (I108–F202) (Fig. 5b), but there has been no proof yet on if they are related.63 Interestingly, the two crosslinked structures of the FabF–ACP complex (gating loops in the open form which mimics the substrate delivery state) with C16 and C16:1 α-bromo probes, respectively, show distinct substrate conformation in which the cis double bond of C16:1 redirects the acyl chain back to the substrate pocket (Fig. 7).47 The kinked C16:1 substrate is likely more compact and provides a conformation that is more readily accommodated. This implies that the unsaturation may assist the process of C16:1 substrate delivery.
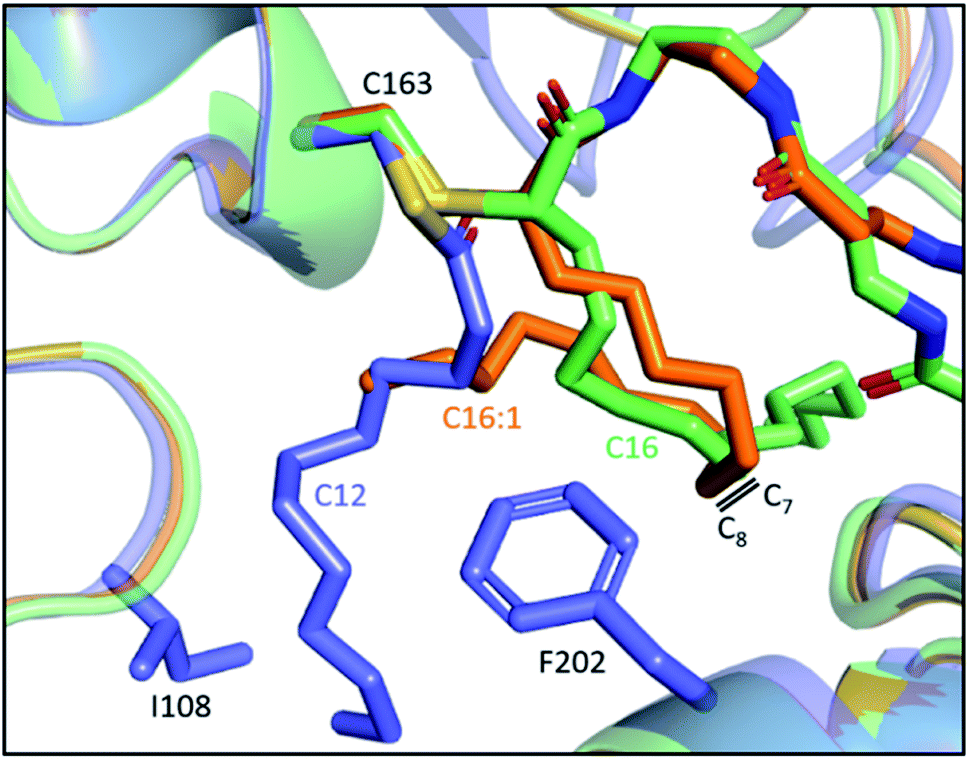 | ||
| Fig. 7 Comparison of the 16-carbon SFA and UFA at the substrate delivery state: overlay of C12–FabF (PDB ID: 2GFY, blue) and FabF–ACP complexes with crosslinkers mimicking C16 (PDB ID: 6OKG, green) and C16:1 (PDB ID: 7L4E, orange), respectively. The cis-double bond of C16:1 redirects the acyl chain to the substrate pocket. | ||
Another mysterious substrate preference is the necessity of FabB for elongating cis-3-decenoic acid (C10:1 UFA), the first UFA produced by FAB. It has been demonstrated that only FabB is capable of elongating this substrate in vivo6 and in cell extract complementation assay.73 However, an in vitro study74 shows that FabF has considerable activity on elongating the C10:1 substrate, and from comparing the pockets of the two KSs, there is no sign that FabF cannot catalyse such an elongation.63 Therefore, FabFs may have negative selectivity towards the C10:1 substrate before the substrate enters the pocket, and only under the competition of different chain length FAs. Such selectivity can potentially be achieved by allosteric regulation in combination with KS–ACP interactions. Several bacterial species do not contain a FabB orthologue, but rather have one, two, or more FabFs, with at least one of them capable of complementing FabB's function, which provides clues to how the substrate preferences arise.75–77
Comparison of KS–CLFs and homodimeric KSs
In PKSII, the genes of the KS and CLF subunits are frequently clustered, with the KS gene upstream and the CLF gene downstream, and in many cases translationally coupled (i.e. overlapping stop and start codons).28,78 The substrate pocket of KS–CLF extends along the heterodimer interface and most often ended by one or more bulky residues of the CLF.11 Examples such as F116′ (the apostrophe denotes the residue from the CLF subunit) of actinorhodin KS–CLF (PDB ID: 1TQY), W107′ of AntD–AntE (PDB ID: 6SMP), and L125′ of Iga11–Iga12 (PDB ID: 6KXF) have been demonstrated, by mutagenesis or a computational method, to control the final chain length.12,37,79 The KS–CLF of aryl polyene biosynthesis, ApeO–ApeC, has a wide pocket to accommodate the phenyl group of the substrate. Despite the difference in the width of the pocket, ApeO–ApeC still has its final product chain length determined by the depth of its substrate pocket.21,23In contrast to KS–CLFs, which normally have a well-defined pocket that leads to one single chain length final product, homodimeric KSs from the FAS have a chain length profile resulting from complex factors such as indefinite pocket size, dimer cooperativity, gating mechanisms, and acyl-ACP recognition. Such a chain length profile can often link to metabolic flux control.80,81 As previously discussed, the human mitochondrial FAB KS possesses bimodal substrate preferences that lead to two major chain length products, C8 and C14/C16. The C8 acid is the precursor of lipoic acid biosynthesis while the long chain acyl-ACP is an assembly factor for respiratory complexes as well as the iron–sulfur cluster synthesis complex.82–86 Accumulating evidence has shown the role of mitochondrial FAB as a coordinator between the acetyl-CoA level and metabolic state sensing,87,88 among other roles that have yet to be explored.
Protein–substrate interactions
The L-shape pocket of KSs consists of two segments, the PPant binding pocket and the acyl-chain binding pocket, respectively, that are roughly perpendicular to each other. The PPant binding pocket has two highly conserved threonine residues (T305 and T307 in EcFabF) that form polar contact with the oxygen of the second amide of PPant, and potentially other carbonyl groups that pass by these threonine residues when entering the active site such as those from thioester and malonate. Such interactions anchor the PPant moiety on one side of the rather spacious PPant pocket, and this orientation seems to clear out the way for the movement of gating loop 1 (Fig. 8a). One of these threonine residues is substituted by a serine in polyketone KSs, but the polar contact potentials provided by the side chains of these two residues exist among all KSs. To demonstrate their essential roles, mutation of either of these residues to alanine in both the IgaPKS (polyene) and AntPKS (polyketone) systems resulted in impaired enzyme activity.12,37 The gating loop 2 also forms polar contacts with the phosphate and the first amide of the PPant (Fig. 8b), potentially mediating between ACP and gating loop 1.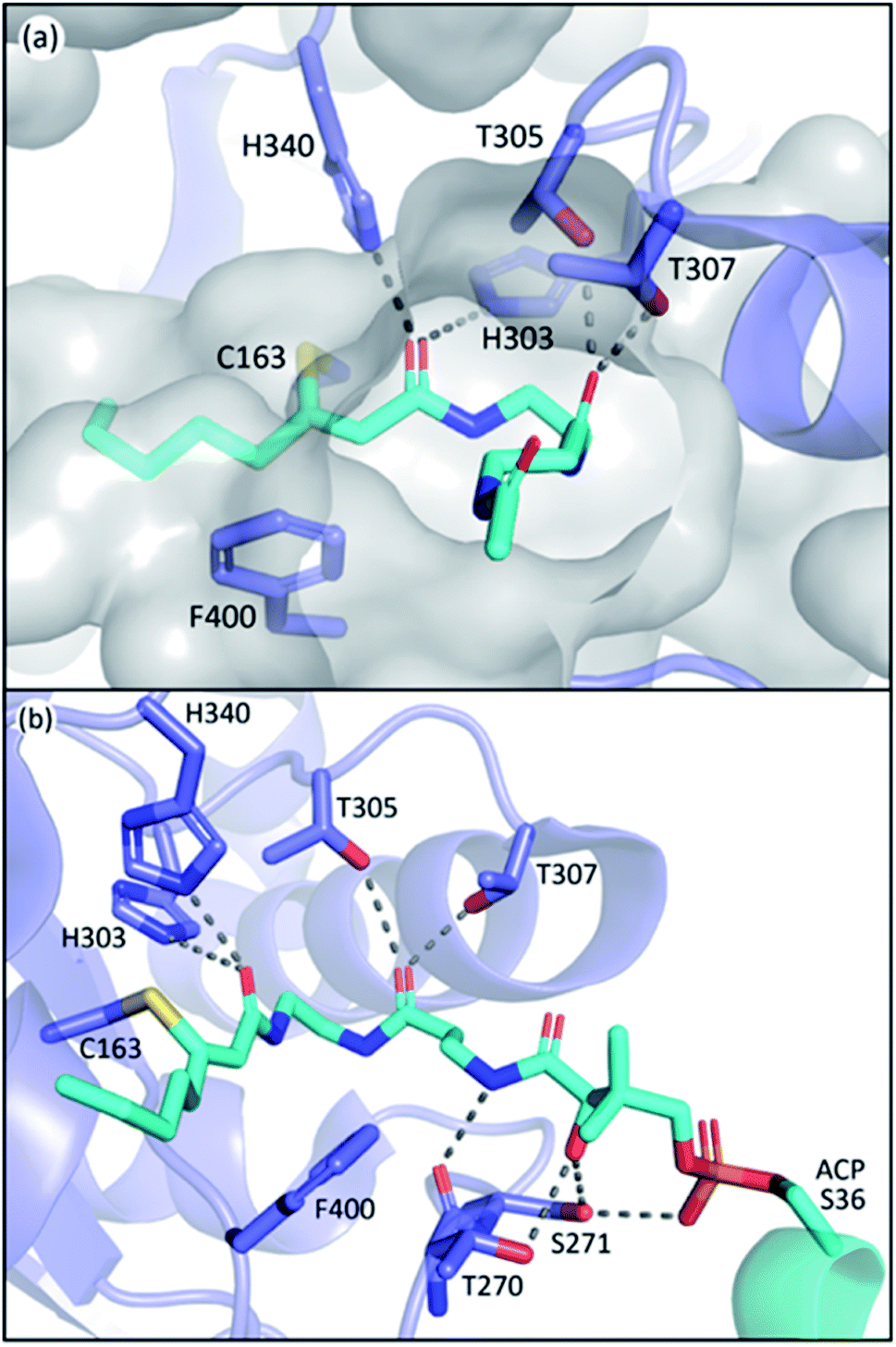 | ||
| Fig. 8 The PPant binding pocket of FabF: (a) coordination of the two threonine residues paves a hydrophilic path for PPant insertion. (b) Polar contact network of the PPant binding pocket. T270 and S271 belong to gating loop 2. | ||
In contrast to the high similarity of the PPant binding pocket, the substrate tunnels of different classes of type II KSs have distinct properties due to the diversity of substrates. The acidic side chain of the highly conserved residue D113 of IgaKS is reported to repulse the β-ketoacyl products from reloading, evidenced by the D113A mutant which produces a triketone.37 This repulsion mechanism is not observed in other kinds of KS including those of FAS as SpFabF can produce triacetic acid lactone (TAL, product of triketone) in vitro,89 and that the precursor of biotin biosynthesis, pimeloyl methyl ester, is also synthesized by FAS, indicating that a methyl ester group can fit into a KS substrate pocket.60 The aryl polyene KSs of Acinetobacter baumannii, ApeO–ApeC and ApeR, have phenylalanine and tyrosine residues in the pockets that can stack with the phenyl ring of the substrate.23 The hexaketide bound AntKS–CLF structures reveal the carbonyl orientations in the narrow pocket that lead to stable substrate binding predicted by simulations.12 These examples demonstrate the customization nature of the substrate pocket, especially for PKS, in which the precision of the product is the first priority.
4. Protein–protein interactions of KSs and ACPs
KSs possess an argenine and lysine rich surface, known as the “positive patch”, around the active site entrance that interacts with negatively charged residues on ACP. Although this is generally true for all KS–ACP interactions elucidated so far, the molecular detail of these interactions can differ to prevent crosstalk between different KS–ACP pairs in the same organism.11,90,91 This orthogonality is believed to reduce stochastic binding events and the substrate sampling time, and thus accelerate the overall kinetics of catalysis. In this section, we will delineate the molecular basis of KS–ACP interactions and the mechanisms affected by them, such as chain-flipping and allosteric regulation.Residues involved in PPIs
In the past, the interface residues involved in type II KS–ACP interactions have only been addressed in a few studies using non crystallographic methods.35,92,93 Since the first KS–ACP complex crystal structure utilizing mechanism based crosslinking probe was published in 2019, there have been in total 10 KS–ACP complex structures released on the PDB, across 4 different KSs from FAS, polyene PKS, and aromatic PKS pathways.12,36,38 By directly visualizing the protein–protein interfaces in these catalytic-relevant states, these structures allow us to assign PPIs at the atomic resolution.In Fig. 9, the interface polar contacts identified from four different KS–ACP pairs are aligned. Generally, most of the PPIs occur between the KS′ (the monomer that the catalytic site isn't involved with, or the CLF subunit in the case of PKSII) and the first two helices of ACP. To facilitate the discussion, PPIs are grouped into three regions. Region 1 involves a short helix on the KS′ (or CLF) that bears 2–3 argenine or lysine residues flanking ACP to interact with residues on the C-terminal side of helix 1 and the N-terminal side of helix 2 (Fig. 9, first row). Region 2 covers interactions of helix 2 with residues on KS′ (or CLF) as well as helix 3 with a single residue on KS (Fig. 9, second row). Lastly, region 3 features the gating loop 2 of the KS interacting with the N-terminal end of helix 2 and the PPant (Fig. 9, third row). Some differences can be observed that may lead to unique PPIs. For example, region 1 of IgaPKS has only two distal basic residues on the CLF involved, when at least three residues from each side forming a complicated polar contact network are seen in other KS–ACP pairs. AntPKS in region 2 also shows unique interactions, with the ACP bearing two interacting lysine residues, contradicting to the general thought that the ACP bears acidic residues that interact with the positive patch on the KS. EcFabB in region 3 has the least interactions, with the highly conserved residues in EcFabF, H268 and T270, substituted by D268 and V270, which are also highly conserved in FabB.
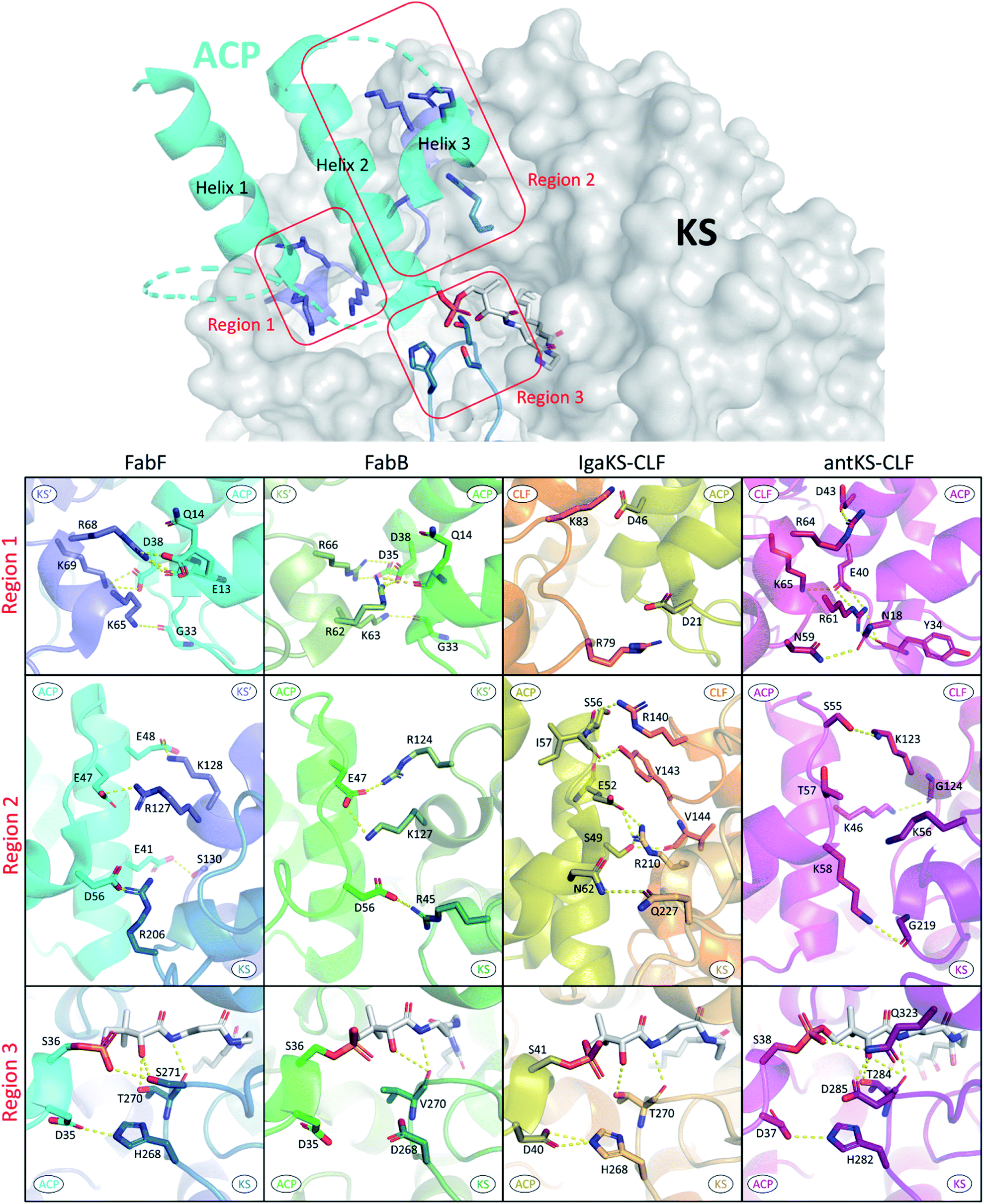 | ||
| Fig. 9 Protein–protein interactions of four different KS–ACP interfaces: PPIs are grouped into three regions as depicted in the top graph (ACP helix 4 and the inter-helices loops are not shown for simplicity). PPIs of each region from FabF (PDB ID: 7L4L, blue), FabB (PDB ID: 6OKC, green), polyene KS–CLF (PDB ID: 6KXF, yellow and orange), and polyketone KS–CLF (PDB ID: 6SMP, magenta) are aligned for comparison. | ||
Alanine scan of interacting protein–protein interface residues of FabF shows that triple mutation removing all interactions in region 1 or region 2 has a greater impact on activity than a triple mutant containing a mix of residues from both regions, indicating that anchoring ACP in both regions is essential.94 IgaKS–CLF was also subjected to the interface residue alanine scan, and all of them show some degree of decline in crosslinking activity. Among them, the R210A mutant stands out to totally abolish crosslinking activity because of the profound interactions R210 has with ACP helix 2, forming polar contacts with the side chains of S49 and E52.37 R61 of the AntCLF also plays a similar role in region 1 interacting with two ACP residues, and mutating this residue to alanine significantly reduced the enzyme activity, while the mutant leads to the loss of one single polar contact between R64 of CLF and D43 of ACP retains the same level of activity.12 Despite the ostensibly high similarity between KS–ACP interfaces, the variation of one, or a few, crucial residues among different KSs can potentially be enough to ensure the fidelity of ACP to its cognate partner.
Chain-flipping mechanism is initiated by PPIs
Carrier proteins are known to be flexible proteins that can sequester substrate intermediates in type II pathways.10,39,95–99 In EcACP, a hydrophobic cavity can be formed in between the four-alpha helical bundle to accommodate acyl substrates. Structural studies have shown that the pocket size can be altered through conformational movements of the helices with helix 3 and the two loops that connect helix 3 to helix 2 and helix 4, respectively, having the largest shift between the sequestered state and the unsequestered state.97,100 Among all the changes, the shifting of isoleucine 62, located at the loop connecting helix 3 and 4, appears to have the biggest impact on the pocket size.ACP delivers the substrate by de-sequestering and “flipping” the acyl chain into the KS pocket, a process termed chain-flipping. Structure analysis of the KS–ACP complex reveals that the de-sequestration of ACP is induced by PPIs: anchoring helix 2 and the C-terminal end of helix 1 and applying an attractive interaction to helix 3 resulted in shrinkage of the pocket size. The overlay of the FabF–ACP structure to helix 2 of the acyl-ACP structure illustrates the mechanism mentioned above (Fig. 10). With helix 2 and part of helix 1 well-anchored, the interaction of FabF–ACP (R206–D56) “flattens” ACP, tilting helix 1 and collapsing the pocket (Fig. 10a). The structure overlay also features a 2.1 Å shift of I62, which is indicative of ACP switching from the sequestered state to the unsequestered state (Fig. 10b). Mutational study on R206 reveals that mutating the residue to an alanine largely decreases the kcat value of FabF while the KM value remains the same.94 This indicates that interaction with ACP helix 3 serves more of a catalytic role rather than strictly stabilizing the protein–protein interface. Interestingly, such an interaction is quite unique across the four KSs that have the KS–ACP structure solved (Fig. 9, second row). In FabB, the corresponding position of FabF R206 is a glycine residue, and instead R45 from the neighboring loop forms polar contact with the same D56 of ACP. In IgaPKS, the interaction pair is (ACP)N62–Q227(KS), and in AntPKS, ACP becomes the side that possesses a positively charged residue, K58, to interact with the KS. The unique characteristic of this one single but important ACP(helix 3)–KS interaction may also contribute to the orthogonality of the type II pathways.
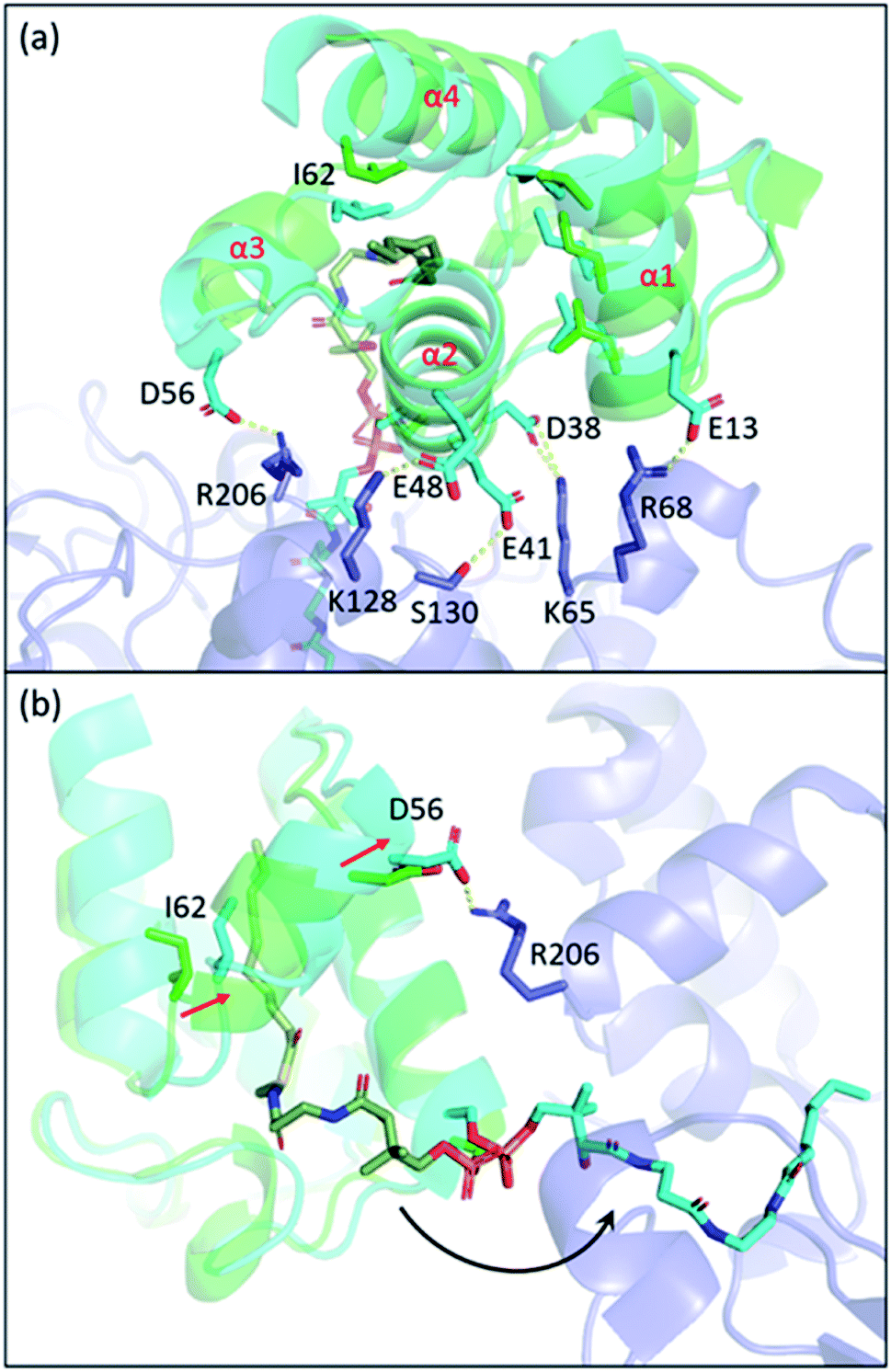 | ||
| Fig. 10 The chain-flipping mechanism: structure overlay of C10–ACP (PDB ID: 2FAE, green) to ACP helix 2 of the FabF–ACP complex (PDB ID: 7L4L, blue) shows that PPIs initiate chain-flipping. (a) PPIs anchor helix 2, E13, and D56 to twist the 4-helical bundle which shrinks the pocket. (b) Side view of the overlay. Movement of I62 shrinks the pocket and pushes the substrate out. | ||
Allosteric regulation
ACP as a substrate carrier in the type II pathways provides an extra level of regulation before sampling substrates in the pocket of the partner enzyme. Such a mechanism, termed allosteric regulation, describes indirect substrate recognition of the partner enzyme through interacting with ACP.101,102 Structural and computational studies on the substrate sequestration of acyl-ACP have long been supporting the feasibility of allosteric regulation because of the observed differences between ACPs bearing different acyl chain lengths as well as other pathway intermediates.98,100,101,103–106 NMR titration study of ACP with six E. coli FASs reveals unique ACP–partner enzyme interacting interfaces, suggesting a complex role ACP may play in the pathway.91 Recent studies on LipB, an enzyme that directs C8–ACP to lipoic acid biosynthesis, elucidates an allostery that is chain length dependent.101,107 Acyl chain terminal carbon 13C-labeled C8 and C12–ACP, whose chain-flipping event can be tracked by HSQC NMR, are titrated with active site cysteine-mutated LipB, and only the cognate C8–ACP performs chain-flipping.101 This study provides the first solid evidence of allosteric regulation of EcACP.Since structural analysis of the pocket shows no clue on FabF's negative preference towards C10:1–ACP in vivo, allosteric regulation stands out to be a plausible explanation. Furthermore, the fact that FabF can elongate C10:1–ACP in vitro indicates that the preference occurs before the substrate enters the pocket,65 and that C10:1–ACP could be competed out by other acyl-ACPs, if present, causing the in vivo result. Interestingly, the depth of the ACP pocket is approximately 7 carbons, which is exactly the carbon count of the saturated tail of C10:1 as well as other UFAs. In the sequestered state, the extra carbons in the acyl chain and the PPant moiety are exposed to the ambient environment, even forming a hairpin when the chain gets longer. This exposure may have a unique interaction with gating loop 2 (Fig. 9, region 3), potentially contributing to the allostery of ACP–KS. The cis-double bond of the C10:1 acyl chain may restrict the movement of the overhanging PPant resulting in the reduction of effective PPant conformations (those that interact with gating loop 2), while the same restriction may occur to other UFAs such as C12:1 but overcome by the longer chain length. Given that FabB has minimal polar contacts in region 3 (Fig. 9), it can be insensitive to the degree of exposure of the acyl-PPant, and thus the insensitivity towards distinguishing C10 and C10:1–ACP. While FabB has an extremely conserved gating loop 2 sequence, FabF loop 2, having a distinct sequence, is only partially conserved (Fig. 3f). This can be explained by some organisms with UFAs possessing only the FabF type of KS,7 and some of these FabFs are reported to complement FabB activity in E. coli.75–77 For example, E. faecalis has no FabB but two FabFs, FabF1 and FabF2, respectively, and only FabF1 can complement EcFabB in vivo.75 Sequence alignment shows that EfFabF2 has a more similar loop 2 sequence to EcFabF than that of EfFabF1. Gating loop 2 plays an important and complex role in the KS enzymology, interacting with both ACP and the substrate, and coordinating the movement of gating loop 1. Swapping of the whole loop 2 of FabF with FabB loop 2 results in an expressible FabF variant with no catalytic activity,47 suggesting that loop 2 likely coevolved with other elements to fine-tune KS function.
5. Conclusion
Based on a conserved mechanism, elongating KSs have evolved for distinct pathways to produce specific product intermediates. In FASII, one or more elongating KSs with complicated gating mechanisms maintain the FA profile, which provides the precursor pool for membrane phospholipids and other essential metabolites, such as biotin and lipoic acid. PKSII polyketone KSs, on the other hand, have rigid pockets that precisely synthesize a single chain length of polyketones. Between them are the PKSII polyene KSs that share some similarities with two other KS types but have some unique mechanisms to ensure product fidelity. KS–ACP interactions play an important role in substrate delivery, and these details will continue to emerge with more studies. Manipulating the KS–ACP interface has already shown success in engineering type I FAS and PKS pathways,108,109 and the recent engineering of the FASII interface of EcACP and a non-cognate thioesterase indicates that this will prove valuable in PKSII as well.110 These examples showcase the promising future of engineering FASII and PKSII pathways and would most certainly be facilitated with a thorough understanding of KS enzymology.Author contributions
A. Chen and Z. Jiang wrote the manuscript. All authors edited and finalized the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Jeffrey T. Mindrebo for his precious advice and his help in editing. We would also like to acknowledge funding from NIH R01 GM095970.Notes and references
- W. G. J. Hol, P. T. van Duijnen and H. J. C. Berendsen, Nature, 1978, 273, 443–446 CrossRef CAS PubMed.
- S. W. White, J. Zheng, Y. M. Zhang and C. O. Rock, Annu. Rev. Biochem., 2005, 74, 791–831 CrossRef CAS PubMed.
- A. Witkowski, A. K. Joshi and S. Smith, Biochemistry, 2002, 41, 10877–10887 CrossRef CAS PubMed.
- C. S. Heil, S. S. Wehrheim, K. S. Paithankar and M. Grininger, ChemBioChem, 2019, 20, 2298–2321 CrossRef CAS PubMed.
- M. E. Hillenmeyer, G. A. Vandova, E. E. Berlew and L. K. Charkoudian, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13952 CrossRef CAS PubMed.
- Y. Feng and J. E. Cronan, J. Biol. Chem., 2009, 284, 29526–29535 CrossRef CAS PubMed.
- Y. J. Lu, Y. M. Zhang and C. O. Rock, Biochem. Cell Biol., 2004, 82, 145–155 CrossRef CAS PubMed.
- K. Magnuson, S. Jackowski, C. O. Rock and J. E. Cronan Jr, Microbiol. Rev., 1993, 57, 522–542 CrossRef CAS PubMed.
- Y. Wang, G. Qian, Y. Li, Y. Wang, Y. Wang, S. Wright, Y. Li, Y. Shen, F. Liu and L. Du, PLoS One, 2013, 8, e66633 CrossRef CAS PubMed.
- G. Shakya, H. Rivera, D. J. Lee, M. J. Jaremko, J. J. La Clair, D. T. Fox, R. W. Haushalter, A. J. Schaub, J. Bruegger, J. F. Barajas, A. R. White, P. Kaur, E. R. Gwozdziowski, F. Wong, S.-C. Tsai and M. D. Burkart, J. Am. Chem. Soc., 2014, 136, 16792–16799 CrossRef CAS PubMed.
- A. Chen, R. N. Re and M. D. Burkart, Nat. Prod. Rep., 2018, 35, 1029–1045 RSC.
- A. Bräuer, Q. Zhou, G. L. C. Grammbitter, M. Schmalhofer, M. Rühl, V. R. I. Kaila, H. B. Bode and M. Groll, Nat. Chem., 2020, 12, 755–763 CrossRef PubMed.
- O. Bilyk, E. Brötz, B. Tokovenko, A. Bechthold, T. Paululat and A. Luzhetskyy, ACS Chem. Biol., 2016, 11, 241–250 CrossRef CAS PubMed.
- D. Du, Y. Katsuyama, H. Onaka, M. Fujie, N. Satoh, K. Shin-Ya and Y. Ohnishi, ChemBioChem, 2016, 17, 1464–1471 CrossRef CAS PubMed.
- D. Du, Y. Katsuyama, K. Shin-ya and Y. Ohnishi, Angew. Chem., Int. Ed., 2018, 57, 1954–1957 CrossRef CAS PubMed.
- F. F. Twigg, W. Cai, W. Huang, J. Liu, M. Sato, T. J. Perez, J. Geng, M. J. Dror, I. Montanez, T. L. Tong, H. Lee and W. Zhang, ChemBioChem, 2019, 20, 1145–1149 CrossRef CAS PubMed.
- T. A. Schöner, S. Gassel, A. Osawa, N. J. Tobias, Y. Okuno, Y. Sakakibara, K. Shindo, G. Sandmann and H. B. Bode, ChemBioChem, 2016, 17, 247–253 CrossRef PubMed.
- T. A. Schöner, S. W. Fuchs, B. Reinhold-Hurek and H. B. Bode, PLoS One, 2014, 9, e90922 CrossRef PubMed.
- T. A. Schöner, S. W. Fuchs, C. Schönau and H. B. Bode, Microb. Biotechnol., 2014, 7, 232–241 CrossRef PubMed.
- N. A. Herman, S. J. Kim, J. S. Li, W. Cai, H. Koshino and W. Zhang, Nat. Commun., 2017, 8, 1514 CrossRef PubMed.
- G. L. C. Grammbitter, M. Schmalhofer, K. Karimi, Y.-M. Shi, T. A. Schöner, N. J. Tobias, N. Morgner, M. Groll and H. B. Bode, J. Am. Chem. Soc., 2019, 141, 16615–16623 CrossRef CAS PubMed.
- W. C. Lee, S. Choi, A. Jang, K. Son and Y. Kim, Sci. Rep., 2021, 11, 7945 CrossRef CAS PubMed.
- W. C. Lee, S. Choi, A. Jang, J. Yeon, E. Hwang and Y. Kim, Sci. Rep., 2021, 11, 16340 CrossRef CAS PubMed.
- X. Gao, P. Wang and Y. Tang, Appl. Microbiol. Biotechnol., 2010, 88, 1233 CrossRef CAS PubMed.
- M. Cummings, A. D. Peters, G. F. S. Whitehead, B. R. K. Menon, J. Micklefield, S. J. Webb and E. Takano, PLoS Biol., 2019, 17, e3000347 CrossRef CAS PubMed.
- J. G. Klein, Y. Wu, B. Kokona and L. K. Charkoudian, Bioorg. Med. Chem., 2020, 28, 115686 CrossRef CAS PubMed.
- J. Zhang, S. Yuzawa, W. L. Thong, T. Shinada, M. Nishiyama and T. Kuzuyama, J. Am. Chem. Soc., 2021, 143, 2962–2969 CrossRef CAS PubMed.
- X. Liu, K. Hua, D. Liu, Z.-L. Wu, Y. Wang, H. Zhang, Z. Deng, B. A. Pfeifer and M. Jiang, ACS Chem. Biol., 2020, 15, 1177–1183 CrossRef CAS PubMed.
- D. Park, G. Swayambhu and B. A. Pfeifer, Curr. Opin. Biotechnol., 2020, 66, 123–130 CrossRef CAS PubMed.
- Y. Katsuyama and Y. Ohnishi, in Methods in Enzymology, ed. D. A. Hopwood, Academic Press, 2012, vol. 515, pp. 359–377 Search PubMed.
- D. Yu, F. Xu, J. Zeng and J. Zhan, IUBMB Life, 2012, 64, 285–295 CrossRef CAS PubMed.
- A. S. Worthington, H. Rivera, J. W. Torpey, M. D. Alexander and M. D. Burkart, ACS Chem. Biol., 2006, 1, 687–691 CrossRef CAS PubMed.
- A. S. Worthington, G. H. Hur, J. L. Meier, Q. Cheng, B. S. Moore and M. D. Burkart, ChemBioChem, 2008, 9, 2096–2103 CrossRef CAS PubMed.
- K. K. Acheampong, B. Kokona, G. A. Braun, D. R. Jacobsen, K. A. Johnson and L. K. Charkoudian, Sci. Rep., 2019, 9, 15589 CrossRef PubMed.
- Z. Ye and G. J. Williams, Biochemistry, 2014, 53, 7494–7502 CrossRef CAS PubMed.
- J. C. Milligan, D. J. Lee, D. R. Jackson, A. J. Schaub, J. Beld, J. F. Barajas, J. J. Hale, R. Luo, M. D. Burkart and S. C. Tsai, Nat. Chem. Biol., 2019, 15, 669–671 CrossRef CAS PubMed.
- D. Du, Y. Katsuyama, M. Horiuchi, S. Fushinobu, A. Chen, T. D. Davis, M. D. Burkart and Y. Ohnishi, Nat. Chem. Biol., 2020, 16, 776–782 CrossRef CAS PubMed.
- J. T. Mindrebo, A. Patel, W. E. Kim, T. D. Davis, A. Chen, T. G. Bartholow, J. J. La Clair, J. A. McCammon, J. P. Noel and M. D. Burkart, Nat. Commun., 2020, 11, 1727 CrossRef CAS PubMed.
- S. E. Evans, C. Williams, C. J. Arthur, E. Płoskoń, P. Wattana-amorn, R. J. Cox, J. Crosby, C. L. Willis, T. J. Simpson and M. P. Crump, J. Mol. Biol., 2009, 389, 511–528 CrossRef CAS PubMed.
- J. F. Barajas, G. Shakya, G. Moreno, H. Rivera, D. R. Jackson, C. L. Topper, A. L. Vagstad, J. J. La Clair, C. A. Townsend, M. D. Burkart and S.-C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E4142 CrossRef CAS PubMed.
- X. Dong, C. D. Bailey, C. Williams, J. Crosby, T. J. Simpson, C. L. Willis and M. P. Crump, Chem. Sci., 2016, 7, 1779–1785 RSC.
- A. Das and C. Khosla, Acc. Chem. Res., 2009, 42, 631–639 CrossRef CAS PubMed.
- B. D. Ellis, J. C. Milligan, A. R. White, V. Duong, P. X. Altman, L. Y. Mohammed, M. P. Crump, J. Crosby, R. Luo, C. D. Vanderwal and S.-C. Tsai, J. Am. Chem. Soc., 2018, 140, 4961–4964 CrossRef CAS PubMed.
- J. Beld, H. Cang and M. D. Burkart, Angew. Chem., Int. Ed., 2014, 53, 14456–14461 CrossRef CAS PubMed.
- K. Charov and M. D. Burkart, ACS Infect. Dis., 2019, 5, 1518–1523 CrossRef CAS PubMed.
- A. Sulpizio, C. E. W. Crawford, R. S. Koweek and L. K. Charkoudian, J. Biol. Chem., 2021, 296, 100328–100346 CrossRef CAS PubMed.
- J. T. Mindrebo, A. Chen, W. E. Kim, R. N. Re, T. D. Davis, J. P. Noel and M. D. Burkart, ACS Catal., 2021, 11, 6787–6799 CrossRef CAS.
- A. Gora, J. Brezovsky and J. Damborsky, Chem. Rev., 2013, 113, 5871–5923 CrossRef CAS PubMed.
- S. R. Luckner, C. A. Machutta, P. J. Tonge and C. Kisker, Structure, 2009, 17, 1004–1013 CrossRef CAS PubMed.
- A. Witkowski, A. K. Joshi, Y. Lindqvist and S. Smith, Biochemistry, 1999, 38, 11643–11650 CrossRef CAS PubMed.
- J. Wang, S. M. Soisson, K. Young, W. Shoop, S. Kodali, A. Galgoci, R. Painter, G. Parthasarathy, Y. S. Tang, R. Cummings, S. Ha, K. Dorso, M. Motyl, H. Jayasuriya, J. Ondeyka, K. Herath, C. Zhang, L. Hernandez, J. Allocco, A. Basilio, J. R. Tormo, O. Genilloud, F. Vicente, F. Pelaez, L. Colwell, S. H. Lee, B. Michael, T. Felcetto, C. Gill, L. L. Silver, J. D. Hermes, K. Bartizal, J. Barrett, D. Schmatz, J. W. Becker, D. Cully and S. B. Singh, Nature, 2006, 441, 358–361 CrossRef CAS PubMed.
- W. Lee and B. Engels, Biochemistry, 2014, 53, 919–931 CrossRef CAS PubMed.
- J. Gajewski, R. Pavlovic, M. Fischer, E. Boles and M. Grininger, Nat. Commun., 2017, 8, 14650 CrossRef PubMed.
- D. Val, G. Banu, K. Seshadri, Y. Lindqvist and K. Dehesh, Structure, 2000, 8, 565–566 CrossRef CAS PubMed.
- G. E. Schujman, K. H. Choi, S. Altabe, C. O. Rock and D. de Mendoza, J. Bacteriol., 2001, 183, 3032–3040 CrossRef CAS PubMed.
- M. Moche, G. Schneider, P. Edwards, K. Dehesh and Y. Lindqvist, J. Biol. Chem., 1999, 274, 6031–6034 CrossRef CAS PubMed.
- F. Trajtenberg, S. Altabe, N. Larrieux, F. Ficarra, D. de Mendoza, A. Buschiazzo and G. E. Schujman, FEBS J., 2014, 281, 2324–2338 CrossRef CAS PubMed.
- L. Zhang, A. K. Joshi, J. Hofmann, E. Schweizer and S. Smith, J. Biol. Chem., 2005, 280, 12422–12429 CrossRef CAS PubMed.
- C. E. Christensen, B. B. Kragelund, P. von Wettstein-Knowles and A. Henriksen, Protein Sci., 2007, 16, 261–272 CrossRef CAS PubMed.
- S. W. Lin, R. E. Hanson and J. E. Cronan, Nat. Chem. Biol., 2010, 6, 682–688 CrossRef CAS PubMed.
- P. Edwards, J. S. Nelsen, J. G. Metz and K. Dehesh, FEBS Lett., 1997, 402, 62–66 CrossRef CAS PubMed.
- J. L. Garwin, A. L. Klages and J. E. Cronan, J. Biol. Chem., 1980, 255, 3263–3265 CrossRef CAS.
- P. von Wettstein-Knowles, J. Olsen, K. Arnvig Mcguire and S. Larsen, Biochem. Soc. Trans., 2000, 28, 601–607 CrossRef CAS PubMed.
- J. G. Olsen, A. Kadziola, P. von Wettstein-Knowles, M. Siggaard-Andersen, Y. Lindquist and S. Larsen, FEBS Lett., 1999, 460, 46–52 CrossRef CAS PubMed.
- K. A. McGuire, M. Siggaard-Andersen, M. G. Bangera, J. G. Olsen and P. von Wettstein-Knowles, Biochemistry, 2001, 40, 9836–9845 CrossRef CAS PubMed.
- P. von Wettstein-Knowles, J. G. Olsen, K. A. McGuire and A. Henriksen, FEBS J., 2006, 273, 695–710 CrossRef CAS PubMed.
- A. Rittner, K. S. Paithankar, A. Himmler and M. Grininger, Protein Sci., 2020, 29, 589–605 CrossRef CAS PubMed.
- S. R. Bagde, I. I. Mathews, J. C. Fromme and C.-Y. Kim, Science, 2021, 374, 723–729 CrossRef CAS PubMed.
- R. Mejía, M. C. Gómez-Eichelmann and M. S. Fernández, Arch. Biochem. Biophys., 1999, 368, 156–160 CrossRef PubMed.
- E. P. Gelmann and J. E. Cronan, J. Bacteriol., 1972, 112, 381–387 CrossRef CAS PubMed.
- J. L. Garwin and J. E. Cronan Jr, J. Bacteriol., 1980, 141, 1457–1459 CrossRef CAS PubMed.
- J. L. Garwin, A. L. Klages and J. E. Cronan, J. Biol. Chem., 1980, 255, 11949–11956 CrossRef CAS.
- J. E. Cronan Jr, C. H. Birge and P. R. Vagelos, J. Bacteriol., 1969, 100, 601–604 CrossRef CAS PubMed.
- G. D'Agnolo, I. S. Rosenfeld and P. R. Vagelos, J. Biol. Chem., 1975, 250, 5289–5294 CrossRef.
- H. Wang and J. E. Cronan, J. Biol. Chem., 2004, 279, 34489–34495 CrossRef CAS PubMed.
- R. M. Morgan-Kiss and J. E. Cronan, Arch. Microbiol., 2008, 190, 427–437 CrossRef CAS PubMed.
- L. Zhu, J. Cheng, B. Luo, S. Feng, J. Lin, S. Wang, J. E. Cronan and H. Wang, BMC Microbiol., 2009, 9, 119 CrossRef PubMed.
- M. Huber, G. Faure, S. Laass, E. Kolbe, K. Seitz, C. Wehrheim, Y. I. Wolf, E. V. Koonin and J. Soppa, Nat. Commun., 2019, 10, 4006 CrossRef PubMed.
- A. T. Keatinge-Clay, D. A. Maltby, K. F. Medzihradszky, C. Khosla and R. M. Stroud, Nat. Struct. Mol. Biol., 2004, 11, 888–893 CrossRef CAS PubMed.
- B. Lowry, X. Li, T. Robbins, D. E. Cane and C. Khosla, ACS Cent. Sci., 2016, 2, 14–20 CrossRef CAS PubMed.
- S. Srivastava and C. Chan, Biotechnol. Bioeng., 2008, 99, 399–410 CrossRef CAS PubMed.
- J. Zhu, K. R. Vinothkumar and J. Hirst, Nature, 2016, 536, 354–358 CrossRef CAS PubMed.
- H. Angerer, S. Schönborn, J. Gorka, U. Bahr, M. Karas, I. Wittig, J. Heidler, J. Hoffmann, N. Morgner and V. Zickermann, Biochim. Biophys. Acta, Mol. Cell Res., 2017, 1864, 1913–1920 CrossRef CAS PubMed.
- S. A. Cory, J. G. Van Vranken, E. J. Brignole, S. Patra, D. R. Winge, C. L. Drennan, J. Rutter and D. P. Barondeau, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5325 CrossRef CAS PubMed.
- M. T. Boniecki, S. A. Freibert, U. Mühlenhoff, R. Lill and M. Cygler, Nat. Commun., 2017, 8, 1287 CrossRef PubMed.
- N. G. Fox, X. Yu, X. Feng, H. J. Bailey, A. Martelli, J. F. Nabhan, C. Strain-Damerell, C. Bulawa, W. W. Yue and S. Han, Nat. Commun., 2019, 10, 2210 CrossRef PubMed.
- A. J. Masud, A. J. Kastaniotis, M. T. Rahman, K. J. Autio and J. K. Hiltunen, Biochim. Biophys. Acta, Mol. Cell Res., 2019, 1866, 118540 CrossRef CAS PubMed.
- J. G. Van Vranken, S. M. Nowinski, K. J. Clowers, M. Y. Jeong, Y. Ouyang, J. A. Berg, J. P. Gygi, S. P. Gygi, D. R. Winge and J. Rutter, Mol. Cell, 2018, 71, 567–580 CrossRef CAS PubMed.
- Y. M. Zhang, J. Hurlbert, S. W. White and C. O. Rock, J. Biol. Chem., 2006, 281, 17390–17399 CrossRef CAS PubMed.
- Y. Tang, T. S. Lee, S. Kobayashi and C. Khosla, Biochemistry, 2003, 42, 6588–6595 CrossRef CAS PubMed.
- T. G. Bartholow, T. Sztain, A. Patel, D. J. Lee, M. A. Young, R. Abagyan and M. D. Burkart, Commun. Biol., 2021, 4, 340 CrossRef CAS PubMed.
- Y.-M. Zhang, M. S. Rao, R. J. Heath, A. C. Price, A. J. Olson, C. O. Rock and S. W. White, J. Biol. Chem., 2001, 276, 8231–8238 CrossRef CAS PubMed.
- P. Beltran-Alvarez, C. J. Arthur, R. J. Cox, J. Crosby, M. P. Crump and T. J. Simpson, Mol. BioSyst., 2009, 5, 511–518 RSC.
- J. T. Mindrebo, L. E. Misson, C. Johnson, J. P. Noel and M. D. Burkart, Biochemistry, 2020, 59, 3626–3638 CrossRef CAS PubMed.
- G. A. Zornetzer, B. G. Fox and J. L. Markley, Biochemistry, 2006, 45, 5217–5227 CrossRef CAS PubMed.
- N. M. Kosa, R. W. Haushalter, A. R. Smith and M. D. Burkart, Nat. Methods, 2012, 9, 981–984 CrossRef CAS PubMed.
- A. Roujeinikova, C. Baldock, W. J. Simon, J. Gilroy, P. J. Baker, A. R. Stuitje, D. W. Rice, A. R. Slabas and J. B. Rafferty, Structure, 2002, 10, 825–835 CrossRef CAS PubMed.
- E. Płoskoń, C. J. Arthur, A. L. P. Kanari, P. Wattana-amorn, C. Williams, J. Crosby, T. J. Simpson, C. L. Willis and M. P. Crump, Chem. Biol., 2010, 17, 776–785 CrossRef PubMed.
- R. W. Haushalter, F. V. Filipp, K. S. Ko, R. Yu, S. J. Opella and M. D. Burkart, ACS Chem. Biol., 2011, 6, 413–418 CrossRef CAS PubMed.
- A. Roujeinikova, W. J. Simon, J. Gilroy, D. W. Rice, J. B. Rafferty and A. R. Slabas, J. Mol. Biol., 2007, 365, 135–145 CrossRef CAS PubMed.
- T. Sztain, T. G. Bartholow, D. J. Lee, L. Casalino, A. Mitchell, M. A. Young, J. Wang, J. A. McCammon and M. D. Burkart, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2025597118 CrossRef CAS PubMed.
- J. Liu and R. Nussinov, PLoS Comput. Biol., 2016, 12, e1004966 CrossRef PubMed.
- G. A. Zornetzer, J. Tanem, B. G. Fox and J. L. Markley, Biochemistry, 2010, 49, 470–477 CrossRef CAS PubMed.
- D. I. Chan, T. Stockner, D. P. Tieleman and H. J. Vogel, J. Biol. Chem., 2008, 283, 33620–33629 CrossRef CAS PubMed.
- R. Farmer, C. M. Thomas and P. J. Winn, PLoS One, 2019, 14, e0219435 CrossRef CAS PubMed.
- J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29, 1111–1137 RSC.
- T. G. Bartholow, T. Sztain, M. A. Young, T. D. Davis, R. Abagyan and M. D. Burkart, RSC Chem. Biol., 2021, 2, 1466–1473 RSC.
- E. Rossini, J. Gajewski, M. Klaus, G. Hummer and M. Grininger, Chem. Commun., 2018, 54, 11606–11609 RSC.
- M. Klaus, M. P. Ostrowski, J. Austerjost, T. Robbins, B. Lowry, D. E. Cane and C. Khosla, J. Biol. Chem., 2016, 291, 16404–16415 CrossRef CAS PubMed.
- S. Sarria, T. G. Bartholow, A. Verga, M. D. Burkart and P. Peralta-Yahya, ACS Synth. Biol., 2018, 7, 1179–1187 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |