
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRAFT-mediated polymerization-induced self-assembly (RAFT-PISA): current status and future directions
Jing
Wan
 ,
Bo
Fan
* and
San H.
Thang
*
,
Bo
Fan
* and
San H.
Thang
*
School of Chemistry, Monash University, Clayton, VIC 3800, Australia. E-mail: bo.fan@monash.edu; san.thang@monash.edu
First published on 18th March 2022
Abstract
Polymerization-induced self-assembly (PISA) combines polymerization and self-assembly in a single step with distinct efficiency that has set it apart from the conventional solution self-assembly processes. PISA holds great promise for large-scale production, not only because of its efficient process for producing nano/micro-particles with high solid content, but also thanks to the facile control over the particle size and morphology. Since its invention, many research groups around the world have developed new and creative approaches to broaden the scope of PISA initiations, morphologies and applications, etc. The growing interest in PISA is certainly reflected in the increasing number of publications over the past few years, and in this review, we aim to summarize these recent advances in the emerging aspects of RAFT-mediated PISA. These include (1) non-thermal initiation processes, such as photo-, enzyme-, redox- and ultrasound-initiation; the achievements of (2) high-order structures, (3) hybrid materials and (4) stimuli-responsive nano-objects by design and adopting new monomers and new processes; (5) the efforts in the realization of upscale production by utilization of high throughput technologies, and finally the (6) applications of current PISA nano-objects in different fields and (7) its future directions.
 Jing Wan | Jing Wan received her BEng degree from East China University of Science and Technology and completed her MSc at the University of Western Ontario (Canada). In 2018, she started her PhD studies in the group of Prof Thang at Monash University and is now pursuing her research work on RAFT-mediated polymerization-induced self-assembly. |
 Bo Fan | Dr Bo Fan is a Research Fellow at Monash University. He received his PhD in Chemical and Biochemical Engineering from the University of Western Ontario in 2018. His research interests include controlled radical polymerization, polymerization-induced self-assembly (PISA), stimuli-responsive polymers and their applications in drug delivery and mineral beneficiation. |
 San H. Thang | Prof. San H. Thang completed his PhD in chemistry at Griffith University in 1987. After a research career at CSIRO (1986–2014), he is currently a Professor of Chemistry at Monash University. His research focuses on the interface between biology and polymer chemistry. He is responsible for several key inventions in the area of controlled/living radical polymerization; significantly, he is a co-inventor of the RAFT (Reversible Addition–Fragmentation Chain Transfer) process. In June 2018, he was awarded the Companion in the General Division of the Order of Australia (AC) for his ‘eminent service to science, and to higher education, particularly in the fields of polymer chemistry and materials science, through seminal contributions as a research innovator, as a mentor, and to the community’. |
1. Introduction
Since the advent of reversible-deactivation radical polymerization (RDRP) techniques, there is no doubt that they have revolutionized the synthesis of polymers by providing a robust method for controlling the polymer compositions, architectures, molecular weights and molecular weight distributions.1 Of all the applications and development of RDRP, polymerization-induced self-assembly (PISA) is one of the research fields that has benefited most from it.2 PISA is an efficient strategy for the synthesis of diverse block copolymer nano-objects at high solid contents (up to 50% w/w), which overcomes many limitations of the conventional solution self-assembly process and is suitable for industrial scale-up. In a typical PISA process, the solvophilic stabilizer polymer (either synthetic polymers or natural polymer alternatives such as polysaccharides3–9) undergoes chain extension with a solvophobic block via RDRP, this induces the concomitant self-assembly of the amphiphilic block copolymer and the formation of nano-objects.10 PISA usually enables high or even quantitative conversion of monomers without the need for post-purification. The required reaction time is also shorter compared to solution polymerization because of the high local monomer concentration within the micellar cores.11 Over the past decade, PISA has been applied to a variety of monomers in diverse solvent systems, including water, polar solvents (e.g. alcohols), non-polar solvents12 (e.g. n-alkanes13,14) and other media including ionic liquids,15,16 supercritical CO2 (ref. 17, 18 and 19) and silicone oil.20 Up to now, many morphologies have been realized via PISA, including spheres, rods, worms, vesicles, framboidal vesicles,21,22 multilamellar vesicles,23 lamellae,24 spongosomes,25–31 hexosomes,25–27,32 cubosomes,25,26,32 Janus particles,33,34 colloidal molecules,33,34 and many others. The synthesized particles have a broad range of applications in the areas of biomedical,35–53 coating,54,55 catalysis56–60 and Pickering emulsions22,61–64 and so forth.65–70Theoretically, all RDRP techniques can produce block copolymer nano-objects via PISA. Indeed, many examples of PISA process using different RDRP techniques have been reported, including reversible addition–fragmentation chain transfer (RAFT) polymerization, nitroxide-mediated polymerization (NMP),71–73 atom transfer radical polymerization (ATRP),74–77 iodine-transfer polymerization (ITP),78 bromine–iodine transformation reversible-deactivation radical polymerization (BIT-RDRP),79,80 telluride-mediated radical polymerization (TERP),81 cobalt-mediated radical (CMR) polymerization82 and reversible complexation mediated polymerization (RCMP);83,84 other polymerization techniques such as ring-opening metathesis polymerization (ROMP)85–89 have been implemented as well. However, RAFT remains the most employed and robust technique for PISA due to its high compatibility with various monomers, solvents and reaction conditions. In recent years, there have been more than 100 research articles published on PISA every year, mainly on RAFT-mediated PISA (Fig. 1). Although PISA based on other RDRP techniques also attracted much research attention, the focus of this review is RAFT-mediated PISA.
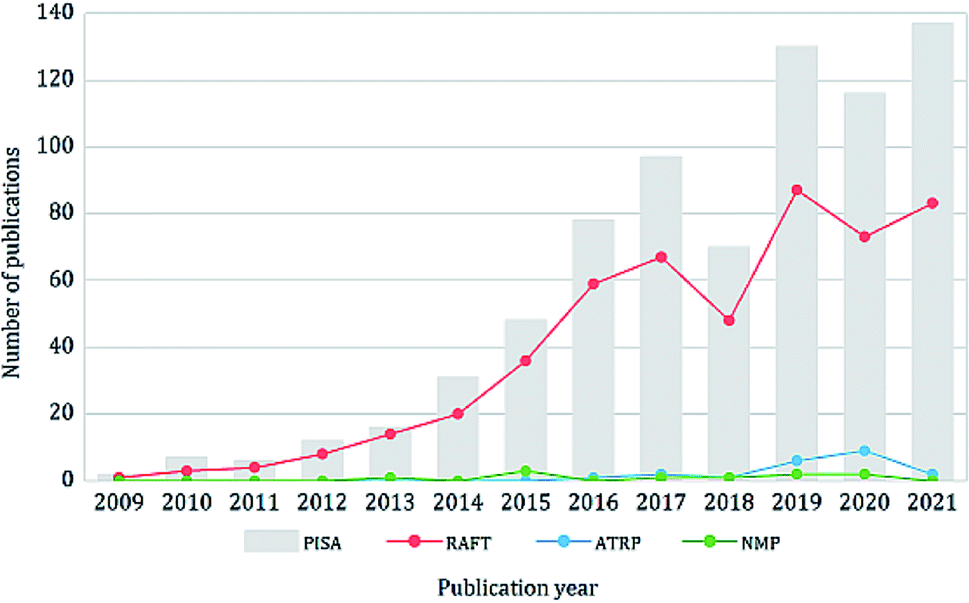 | ||
| Fig. 1 Development of PISA, RAFT-, ATRP-, and NMP-mediated PISA in 2009–2022 (source: Web of Science 02/2022, research topic: “polymerisation-induced self-assembly”, PISA: without refinement, RAFT: refined keyword “RAFT”, ATRP: refined keyword “ATRP”, NMP: refined keyword “NMP”). | ||
We note that many reviews have been published since the inception of PISA, covering some important concepts of the PISA process, such as the initiation methods,90–94 the biomedical applications,95–99 reactive and functional particles,100 controlling the morphology,96,101–103 PISA in non-aqueous dispersion,104,105 progress reports106–109 and even overviews.2,10,11,110 These previous review articles discussed about either advances in a specific topic or provided an overview of PISA within a relatively general topic. A summary of trends and future directions in PISA is essential to enable existing and new researchers of this field be keeping informed of the trends, as well as to attract attention from different disciplines. Penfold et al. summarized the emerging trends of PISA in 2019.11 However, a growing number of publications clearly reflected the interest in PISA, and many outstanding studies continue to emerge even while these lines are being read. The aim of this review is not intended to add additional concepts in RAFT-mediated PISA, rather to summarize the emerging trends in PISA and to provide an up-to-date review of this field so that the reader can better understand the current development and future direction of PISA. The particular focus of this review is on the following topics: (1) the new initiation methods for the PISA process, (2) the emerging high-order and complex morphologies by PISA, (3) the synthesis of hybrid materials via PISA, (4) the stimuli-responsive nano-objects by PISA, (5) the strategies for improved throughput and continuous flow process, (6) current and potential applications.
2. Non-thermal initiation of PISA
In contrast to other RDRP techniques such as NMP or ATRP, RAFT generally requires an external source of radicals (exogenous radicals), which are generally produced by the decomposition of organic molecules under thermal conditions.2 Most of the conventional initiators employed to implement emulsion or dispersion polymerization are azo compounds or photoactive molecules. When the dispersing phase is water, ionic azo compounds or persulfates are preferred. The reaction is usually conducted at around the 10 hours half-life temperature of the initiator. The 10 hours half-life temperature varies with the chemical nature of the initiators, usually varies between 44 to 100 °C. For RAFT-mediated PISA the most widely employed reaction temperature is 50–70 °C, as the solvent is usually water or methanol.The thermal initiation of RAFT-PISA is readily applied in diverse conditions with good compatibility with different monomers and solvent systems, and could be easily realized in the industrial upscaling process. Nevertheless, the relatively high reaction temperature (>44 °C) limits the utilization of temperature-sensitive materials, including thermo-responsive RAFT agents, temperature-sensitive polymers, DNA, RNA and enzymes. Therefore, in the past few years, there has been a surge of interest in new initiation mechanisms that unitize visible light, microwaves, enzymes, redox/oscillatory reaction, electrochemistry, ultrasound as alternative approaches for RAFT-PISA that initiated at a lower temperature. The non-thermal approaches allow the PISA to be combined with biomolecules, which may open up new scopes of research for PISA formulations.
2.1 Visible light initiated PISA (photo-PISA)
RAFT polymerization has been extensively explored in a broad range of electromagnetic spectra including gamma,111–113 ultraviolet (UV),114–117 visible,118–122 near-infrared (NIR)123 and microwave.124 It was then a spontaneous process that photo-initiated RAFT polymerizations were extended to the area of RAFT-mediated PISA. Visible light and NIR have attracted more research attention due to their low energy nature, induction of fewer side reactions, and also the potential to control the polymerizations temporally and spatially.91 In this review, the visible light photo-PISA will be mainly discussed.Initially, only photo-RAFT have been implemented in the PISA process. More recently, other RDRP techniques, such as NMP,125 ATRP,75 and bromine–iodine transformation (BIT),80 as well as some other polymerization techniques such as ring-opening polymerization126 have been explored for the photo-PISA process. Nevertheless, the photo-RAFT technique remains the most robust and widely employed technique for the photo-PISA process. The main types of photo-RAFT polymerizations can be summarized as: (1) photoinitiator process: photo-decomposition of external photoinitiators, similar to the mechanism of thermal initiation,127 (2) photoiniferter (initiator-transfer agent-terminator) process: photolytic cleavage of a C–S bond in the thiocarbonylthio RAFT agent without exogenous radical sources,128–130 and (3) photocatalyst process: a photo-induced electron transfer (PET) process using photo-redox catalysts.121,131 Thus, the photo-RAFT-PISA can also be summarized according to these three different processes (Fig. 2).
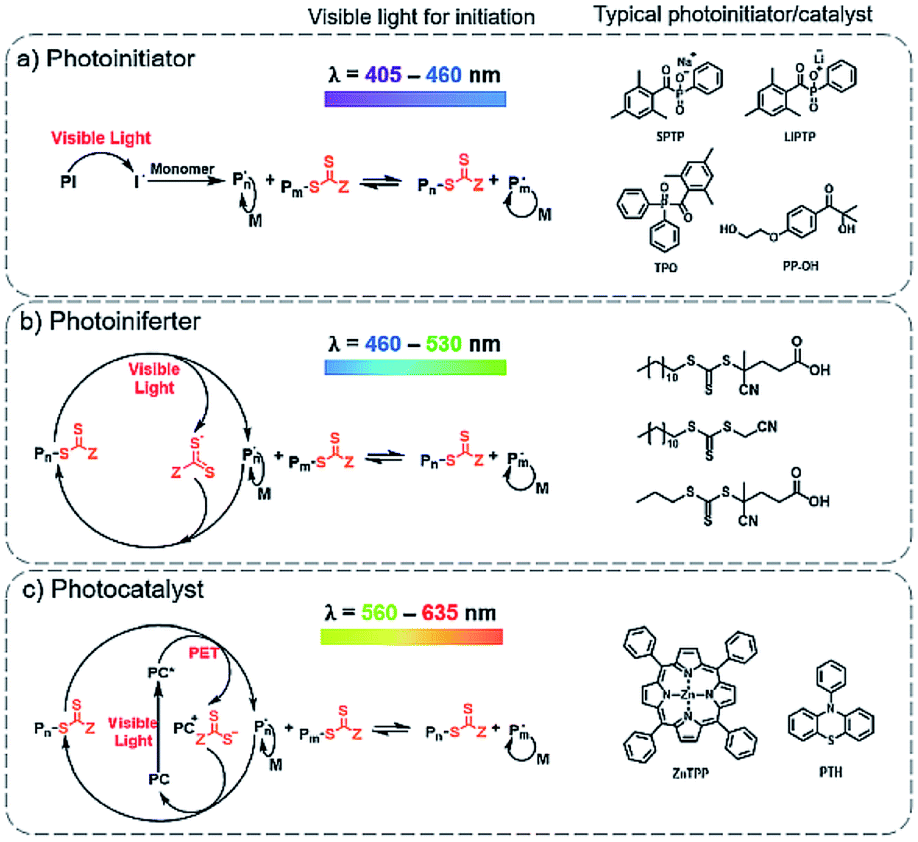 | ||
| Fig. 2 Different mechanisms for initiating PISA polymerizations under visible light: (a) photoinitiator, (b) photoiniferter and (c) photocatalyst approaches. | ||
The first example of visible light photo-PISA was reported by Cai and co-workers in 2015 using sodium phenyl-2,4,6-trimethyl-benzoylphosphinate (SPTP) as the photoinitiator.132 In this work, poly(2-hydroxypropylmethacrylamide) (PHPMAm) macro-RAFT and diacetone acrylamide (DAAM) undergo fast aqueous dispersion polymerization under visible light at 25 °C. The kinetics was found to be similar to thermally initiated PISA, both containing phase transition points and rate accelerations, however, the polymerization rates were much faster than that of traditional thermal-PISA, and the induction period was negligible. The room temperature process showed good control (Đ < 1.3) when targeting a series of PHPMAm-b-PDAAM spheres with various degree of polymerization (DP). In addition, nanoparticles synthesized with N-(2-aminoethyl)acrylamide (AEAM) as a comonomer could further functionalize with metal binding motifs via the post-polymerization process.
By applying the same photoinitiator SPTP reported in Cai's work, Zhang, Sumerlin and co-workers expanded this PISA process by employing 405 nm violet LED light to synthesize poly(ethylene glycol)–poly(2-hydroxypropyl methacrylate) (PEG–PHPMA) based classical nano-objects.133 This photo-PISA achieved a diverse set of morphologies with spheres, worms, vesicles (S, W, V) by varying the target DP of PHPMA and the solid contents. In addition, ultrafast kinetics were observed in this process, with quantitative monomer conversion achieved within 30 min at room temperature. This is a tremendous improvement compared to the conventional thermal-PISA process at 70 °C, which usually requires at least 3 hours to achieve quantitative monomer conversions. This was also the first report that the full range of morphology phase diagram was generated for photo-PISA at room temperature. The mild conditions are important for the preparation of vesicles loaded with bio-related species; as a proof of concept, silica nanoparticles and bovine serum albumin (BSA) were encapsulated in situ within the vesicles.
In addition to S, W, V, Cai et al. used the same technique to synthesize an unusual collection of morphologies such as silks, ribbons, interlinked vesicles and nanotubes (Fig. 3),134 which expands the morphologies achieved by the photo-PISA process. Recently, a similar technique was combined with electrostatic manipulation to achieve triblock copolymer multi-compartmentalized 2D nano-objects by tuning the pH of reactions.135
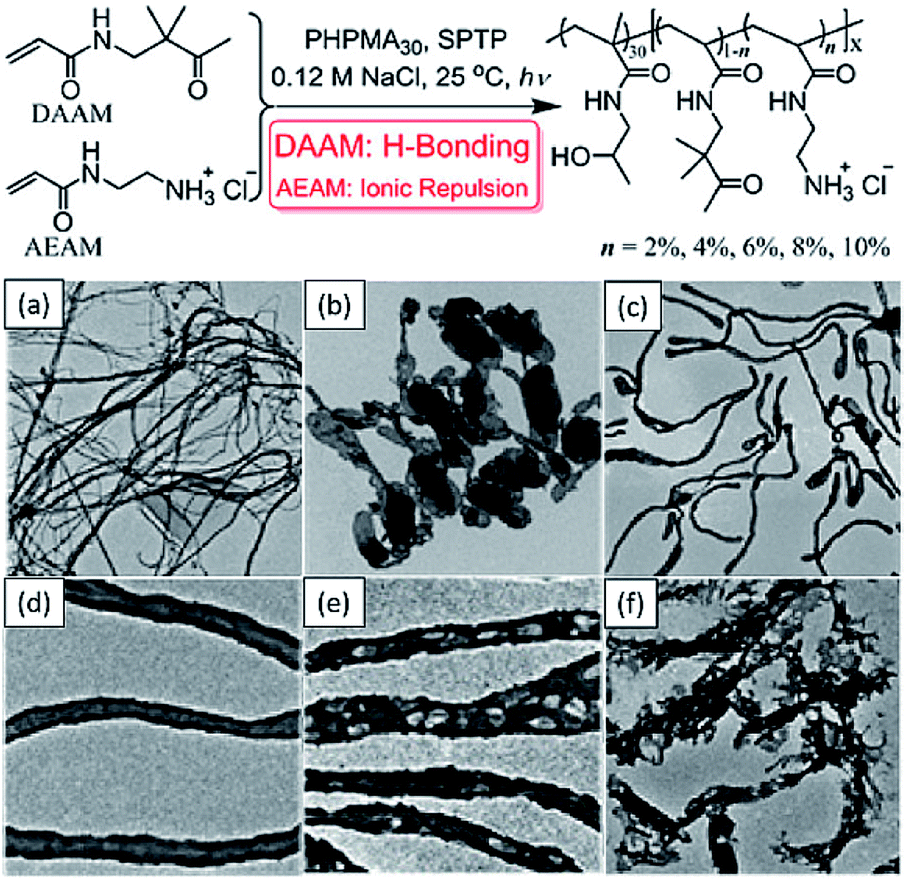 | ||
| Fig. 3 Schematic illustration of photomediated PISA at 25 °C and representative TEM images of (a) silks/films, (b) vesicles, (c and d) nanotubes and (e and f) porous nanotubes. Reprinted from ref. 134 with permission. Copyright 2016, American Chemical Society. | ||
Apart from SPTP, other photoinitiators such as 2,4,6-trimethylbenzoyl-diphenylphosphinate (TPO),136,137 2-hydroxy-4′-2-(hydroxyethoxy)-2-methylpropiophenone (PP–OH),138 and lithium phenyl(2,4,6-trimethylbenzoyl)phosphinate (LiPTP)139 were also used as photoinitiators in photo-PISA. Junkers, Zetterlund, Boyer and co-workers reported the optimized batch protocol by varying the solvents, photoinitiators as well as light intensities.140 By comparing TPO, PP–OH, BDB, camphorquinone and eosin Y (EY) disodium salt photoinitiator and photoiniferter route, TPO was selected as the most suitable photoinitiator. EtOH/DMSO (90/10 v/v%) was chosen as the optimal solvent system to achieve both high monomer conversion and high order morphologies. The optimized batch protocol was then adapted for flow processing, showing the potential of continuous tubular reactors for alcohol-based photo-PISA.
One of the trends in photoinitiator induced photo-PISA is to minimize the use of photoinitiators or to make them dual-acting. In 2020, Dai, Jung and Boyer et al. reported the dual roles of doxorubicin (DOX), which can serve as an antitumor drug and co-catalyst for a photo-PISA process under blue LED light (485 nm).141 It was found that DOX can enhance the polymerization rates of a broad range of monomers, including acrylate, acrylamide and methacrylates, however, the mechanism remains unclear. In 2021, Tan et al. developed a strategy to attach TPO to the R group of RAFT agent (TPO-CDPA), so that the type I photoinitiator could attach onto the surface of polymeric nano-objects.142 This strategy enables the photoinitiator-RAFT agent with dual-role that can both initiate and control the polymerization, thus overcoming some drawbacks of using small molecular type I photoinitiators. The yielded photoinitiator-functionalized block copolymer nanoparticles could be further used as heterogeneous photoinitiators to produce hydrogels with internally embedded nanoparticles.
In 2016, Boyer and co-workers reported the first example of photo-PISA without the addition of an external catalyst or initiator to yield nano-objects with different morphologies (S, W, V).147 A POEGMA-CDTPA macro-CTA was activated under blue (460 nm) or green (530 nm) light and chain extended with benzyl methacrylate (BzMA) in the ethanolic solvent. The in situ encapsulation of Nile red as a model hydrophobic drug was demonstrated as a proof-of-concept for drug loading. In 2017, O'Reilly compared the influence of initiation methods and light intensity on the final PISA morphology.138 The preparation of PEG-b-PHPMA nano-objects were conducted via three routes: photo-PISA with 100% intensity (405 nm, no functioning initiator), thermal-PISA, and photo-PISA with 20% (405 nm, no initiator) intensity at the same temperature. Their findings suggest both reaction kinetics and end group fidelity lead to the difference in phase diagrams generated using photo-PISA and thermal-PISA. More recently, Poly and Chemtob and co-workers reported an initiator-free photo-PISA of PHEA-b-PS spheres using 472 nm LED light at 35 °C in methanol/water co-solvent.148 The yielded nanoparticles were further demonstrated as soft templates for the synthesis of uniform nanoscale porous carbons with large mesopores size.
The advantage of the photoiniferter approach is obvious, as no external photoinitiator or catalysts need to be added, thus eliminating the potential source of toxicity. However, this visible light-mediated RAFT is strongly dependent on both the type of RAFT agent and the effective light intensity.91 While achieving high monomer conversion is not a problem, the rates of polymerization could be problematically low compared to photoinitiator or photocatalyst approaches. For example, in Poly and Chemtob's study,148 it took around 70 hours to achieve 59% conversion of styrene. Furthermore, these polymerizations require careful deoxygenation since no exogenous initiator is present, even low concentrations of oxygen can retard the polymerization.
In 2017, Pan and co-workers performed PET-RAFT dispersion polymerization using the organic dye 10-phenylphenothiazine (PTH) as an alternative to heavy metal-based photo-redox catalysts.151 Although the required concentration of PTH (1‰ to monomer) was higher than that of heavy metal-based photocatalysts (1 ppm to monomer), good control of the dispersion polymerization was achieved. Temporal control of the polymerization in this specific PET-RAFT process has also been demonstrated by adjusting the light source.
Furthermore, many other organic dyes have been exploited as photocatalysts for PET-RAFT polymerization. The order of reactivity to activate PET-RAFT was reported as eosin Y (EY) ≫ fluorescein ≫ Nile red, rhodamine 6G (R6G) and methylene blue.152 Boyer et al. reported an oxygen-tolerant ultralow volume RAFT polymerization in a 96-well microtiter plate under green light irradiation (λ = 530 nm) using EY in the presence of ascorbic acid.153 The proposed mechanism is that the photo-reduced EY is oxidized by oxygen, regenerating the original dye and converting oxygen to hydrogen peroxide. A redox reaction between the hydrogen peroxide and excess ascorbic acid then produces reactive hydroxyl radicals that initiate RAFT polymerization. This approach allows control of a range of monomer families (acrylamides, acrylates, methacrylates) for the synthesis of homo- and block copolymers, and can be applied to PISA. Recently, R6G has also been used as a photocatalyst for the PISA process by Lin and co-workers to produce micelles and other high order structures.154
Another type of widely applied catalysts for photo-PISA are porphine catalysts. In 2016, the Boyer group extended to longer visible light wavelengths such as red (λ = 635 nm) and yellow (λ = 560 nm) light by the addition of the metalloporphyrin, such as 5,10,15,20-tetraphenyl-21H,23H-porphine zinc (ZnTPP).155 In addition, encapsulation of ZnTPP into the core of nanoparticles was achieved, which makes it potentially useful in photodynamical therapy (PDT), as ZnTPP can generate singlet oxygen under visible light irradiation. This oxygen-tolerant PISA system without the traditional deoxygenation process was also demonstrated by the addition of ascorbic acid as the singlet oxygen quencher.155 Later on, the same research group explored the oxygen-tolerant PET-RAFT PISA by addition of singlet oxygen quenchers such as 9,10-dimethylanthracene, ascorbic acid and (R)-(+)-limonene.156 Conventional deoxygenation processes generally require the use of specialized equipment (Schlenk vessels, high-vacuum pump, etc.) and inert gas, the open-air PISA system allows the synthesis to perform without these restrictions at ultra-low volumes.
In 2018, a water-soluble porphine photocatalyst, zinc meso-tetra(N-methyl-4-pyridyl)porphine tetrachloride (ZnTMPyP) was investigated for aqueous PET-RAFT dispersion polymerization,157 as this water-soluble photocatalyst is more suitable for aqueous PISA systems. In this study, the photo-PISA process was conducted in 96-well microtiter plates under low energy red light (λmax = 595 nm, 10.2 mW cm−2) without deoxygenation due to the addition of biotin (vitamin B7) as the singlet oxygen quencher. A photo-responsive comonomer 7-[4-(trifluoromethyl)coumarin]methacrylamide (TCMAm) was added for the purpose of post-PISA photo-crosslinking, which was performed under a UV light source (λmax = 365 nm, 10.2 mW cm−2) to allow the core-cross-linked nanoparticles to retain their original morphologies when exposed to organic solvents.
Some PDT photo-sensitizers can be used to catalyze PET-RAFT polymerizations as well. In 2021, Sumerlin et al. reported a strategy to maximize the multifunctionality of Rose Bengal Methacrylate (RBMA) as both photocatalyst and comonomer in a photo-PISA process, also a singlet oxygen generator for PDT, and a fluorophore for imaging.158 In their work, quantitative and controlled loading of Rose Bengal (RB) was realized by converting RB to the polymerizable methacrylate that could be covalently incorporated into nanoparticles. It was further demonstrated that RB-loaded nanoparticles can be activated by visible light, and the efficiency of singlet oxygen production was higher than free RB. After post-modification of the RB-loaded nanoparticles with a DNA aptamer, in vitro study showed enhanced internalization by HCT 116 cells and significant inhibition of tumor cell proliferation under yellow light.
2.2 Enzyme-assisted/initiated PISA
In 2018, Tan and Zhang et al. reported the enzyme-initiated RAFT dispersion polymerization of 2-hydroxypropyl methacrylate (HPMA) in water at room temperature, yielding nano-objects with different morphologies.164 In their study (Fig. 4), ACAC was oxidized by H2O2 with HRP as the catalyst, generating ACAC radicals to initiate the RAFT polymerization. The enzyme-initiated RAFT-PISA enabled the rapid synthesis of nano-objects, including spheres, worms and vesicles in mild reaction conditions. The kinetics revealed that a high monomer conversion (>99%) was achieved within 20 min, indicating the fast generation of ACAC radicals and the high catalysis efficiency of HRP. SEC measurement exhibited a linear relationship between number-average molar mass (Mn) and monomer conversion, and the mPEG113–PHPMAn diblock copolymers had low dispersities (Đ < 1.20) throughout the enzyme-initiated polymerization process, indicating that good control was maintained throughout. The mild reaction conditions also allow the in situ loading of SiO2 nanoparticles and bovine serum albumin (BSA) into vesicles. In this study, they also took advantage of RAFT polymerization initiated by enzymatic cascade catalysis to develop oxygen-tolerant PISA based on GOx-HRP enzymatic cascade catalysis. As shown in Fig. 4b, GOx catalyzed the oxidation of glucose in the presence of oxygen to produce H2O2, which is subsequently used to generate ACAC radicals using HRP as the catalyst, allowing the RAFT dispersion polymerization to occur in open vessels and multiwall plates at room temperature.164 Overall, these studies demonstrated that the enzyme-initiated RAFT-PISA can serve as an efficient and facile platform for the preparation of functional nano-objects under the mild condition with oxygen tolerance.
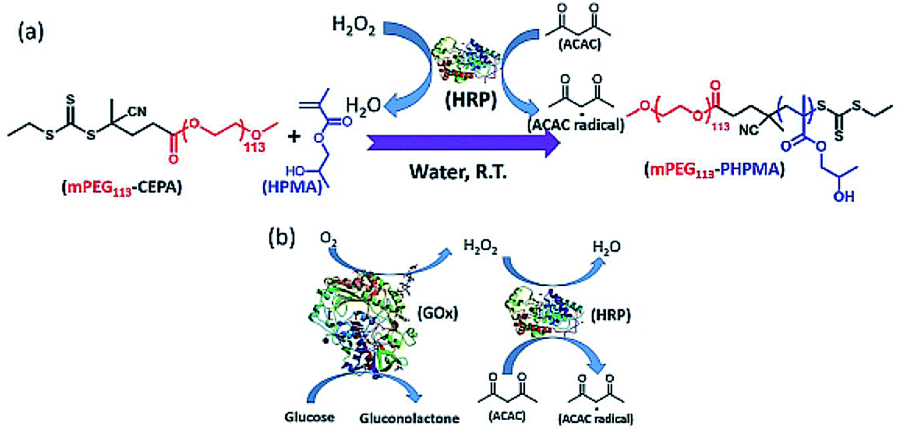 | ||
| Fig. 4 (a) Schematic illustration of enzyme-initiated aqueous RAFT dispersion polymerization of HPMA. (b) Schematic illustration of GOx-HRP cascade reaction. Reprinted from ref. 164 with permission. Copyright 2018, Wiley. | ||
Later in the same year, Tan et al. reported the synthesis of a series of epoxy-functionalized triblock copolymer vesicles via enzyme-initiated PISA.165 First, poly(glycerol monomethacrylate)-b-poly(2-hydroxypropyl methacrylate) (PGMA–PHPMA) diblock copolymer vesicles were prepared via enzyme-initiated aqueous dispersion polymerization, and then glycidyl methacrylate (GlyMA) was used for chain extension via enzyme-initiated seeded emulsion polymerization to produce epoxy-functionalized vesicles. The mild condition of enzyme-initiated RAFT polymerization at room temperature is critical to ensure the survival of epoxy groups after the polymerization. The synthesized triblock epoxy-functionalized vesicles were evaluated as a Pickering emulsifier in hexane–water emulsions. Furthermore, cross-linked vesicles were achieved by reacting epoxy groups with ethylenediamine.
In 2017, Tan and Zhang et al. expanded the range of Enz-RAFT to dispersion polymerization for high-throughput synthesis of well-defined AB diblock and ABC triblock copolymer nano-objects in open multiwell plates at room temperature.168 GOx and glucose were added for deoxygenation, and SPTP and LED (λ = 405 nm) were used to initiate the polymerization. They also demonstrated potential biologically relevant application by encapsulating HRP and BSA into vesicles without compromising protein activities. This approach facilitated high-throughput application, allowing for faster construction of the PISA phase diagram. In 2021, Tan et al. performed the same technique in a continuous flow reactor, offering the possibility of large-scale production of nano-objects in an oxygen-tolerant environment.169
Recently, the same group used a similar technique to synthesize higher-order morphologies from tert-butyl acrylate or tert-butyl methacrylate at room temperature by enzyme-assisted RAFT emulsion polymerization.170 The RAFT emulsion polymerization of hydrophobic monomers usually leads to spheres because of kinetical trapping. This may be overcome by temperature-directed morphology transformation, but the high temperature hinders the preparation of thermo-sensitive or bio-related polymer nano-objects. Therefore, their study fills the gap that higher-order morphologies can be produced by emulsion polymerization of hydrophobic methacrylic and acrylic monomers at a low temperature.
In 2020, Ng, Weil and co-authors reported a “grafting from” protocol for the preparation of DNA–polymer nanostructures from single-stranded DNA (ssDNA) under ambient conditions, aided by enzymatic degassing with glucose, glucose oxidase, and sodium pyruvate.171 In this study, they successfully produced a series of functional DNA–polymer conjugates and DNA-diblock conjugates derived from acrylamide (DMA, NAM)/acrylate (HEA, OEGA)-based monomers.
2.3 Redox-PISA/oscillatory reaction
Redox initiators have been widely used in homogeneous and heterogeneous free radical polymerizations.172–174 The low activation energy of redox initiators allow the polymerization to proceed at relatively lower temperatures, which is beneficial for the synthesis of thermal-sensitive polymer/drugs and bio-related materials. The applications of redox initiators in RAFT-mediated solution polymerization at low temperature and under even freezing conditions were explored back in 2008.175–177 Their use in RAFT-mediated dispersed system was soon reported in 2009 by Santos et al. Specifically, they reported the use of potassium persulfate (KPS)/sodium metabisulfite (SMB) as redox couple initiators for the mini-emulsion polymerization of styrene with PEO-RAFT as chain transfer agent.178 Later in 2011, An et al. reported an aqueous dispersion polymerization mediated by RAFT using KPS/L-ascorbic acid sodium salt (NaAs) as redox initiator to prepare 2-methoxyethyl acrylate (MEA)-based core-cross-linked nanogels with up to 32% w/v solid content at low temperature (30 and 40 °C).179KPS/ascorbic acid-based redox initiator is a classical redox couple that has been widely applied in low temperature polymerization.174,180,181 The redox initiator has a wide range of compatible reaction temperatures, which can be easily adjusted to study the effect of reaction temperature on polymerization and morphological evolution. For example, Tan et al. used this redox couple for RAFT emulsion polymerization at 25–50 °C to synthesize PPEGMA-b-PGlyMA and yielded higher-order morphologies.182 In this study, several factors including reaction temperature, molecular weight of macro-RAFT agent, DP of PGlyMA, and monomer concentration have been studied in detail to investigate their effects on emulsion polymerization. The results indicated that higher temperature strongly promoted the mobility of PGlyMA (Tg = ca. 45 °C), and leading to morphological evolution from spheres (25 °C) to worms (37 °C) and vesicles (50 °C). More recently in 2020, Armes et al. reported the RAFT aqueous emulsion polymerization with the incorporation of thermal and redox initiator at both 80 and 30 °C.183 In this example, poly(2-(N-acryloyloxy)ethyl pyrrolidone) (PNAEP) was used as a new non-ionic stabilizer block, and by using VA-044 as the thermal initiator at 80 °C, they achieved full conversion of styrene within 40 min while maintaining good control over dispersity (Đ < 1.30). Meanwhile, by using KPS/AscA redox initiator at pH = 3 and 30 °C, they could achieve full conversion of n-butyl acrylate (nBA) within 25 min. Despite relatively high dispersities were observed (Đ = 1.52–1.64), it is still much better control than the polymerization of nBA initiated by VA-044 at 80 °C (Đ > 3.00).
Oscillatory reactions are chemical systems in which one or more reacting compounds exhibit periodic changes in time or space. Most oscillating reactions involve redox reactions, and some of these are able to generate free radicals. Extensive studies have explored Belousov–Zhabotinsky (BZ) reaction for the initiation of radical polymerizations and investigated the roles of different free radicals in the BZ reaction.184–187 The extended study of BZ reaction to PISA has not been reported until recently. In 2017, Bastakoti and Pérez-Mercader reported a one-pot synthesis of PEG-b-PnBA giant vesicles via PISA coupled with BZ reaction.188 The authors proposed a mechanism (Fig. 5) that includes the roles of different radical species in this oscillatory reaction-initiated polymerization. The oscillations appeared after an induction time of 16 min, and the measured amplitude gradually decreased as time elapsed, which could be explained by the consumption of radicals (intermediate moieties) and the increase in viscosity during polymerization. Due to the different chemical environments inside and outside of the self-assembled vesicles, the consumption rates and osmotic pressures become different at membrane sides, resulting in the increase in vesicle size and final formation of giant vesicles. In the same year, this group repeated this technique on a different formulation composed of acrylonitrile-based PEG-b-PAN diblock copolymer and succeeded in preparing giant vesicles.189 Later on, another formulation composed of poly(ethylene glycol)-b-poly(ethyl acrylate) (PEG–PEA) was reported to form micelle with patchy voids.190
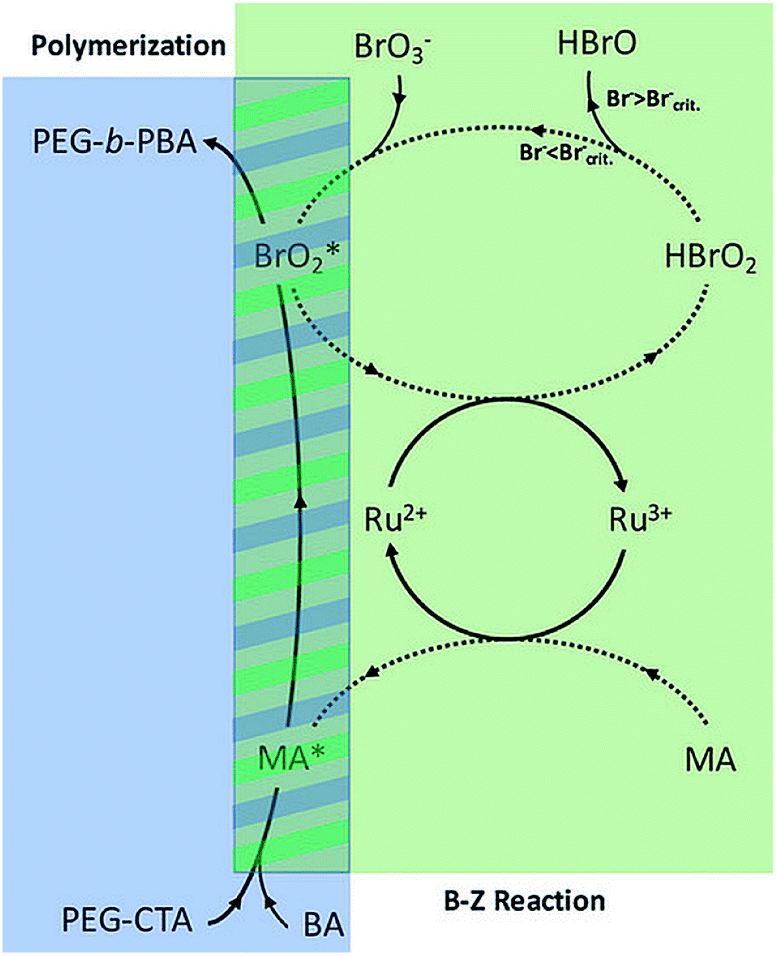 | ||
| Fig. 5 The proposed mechanism of the BZ assisted polymerization of a PEG-b-PBA block co-polymer. Reprinted from ref. 188 with permission. Copyright 2017, Wiley. | ||
In 2019, Pérez-Mercader, Dueñas-Díez and coworkers combined the oscillatory reaction-initiated PISA with a continuously stirred tank reactor (CSTR) strategy, allowing consistent control over the PISA process and chemical properties.191 In this study, different morphologies were obtained by tuning the residence time (τ), target DP and the BZ reactants. CSTR-BZ-PISA resembles the out-of-equilibrium and open-system characteristics of living systems, which may be suitable for studying the protolife scenarios.
More recently, Cheng and Pérez-Mercader presented a chemical fuel-driven PISA catalyzed by the BZ reaction.192 The amphiphilic block copolymers could self-organize into large multicompartmental structures, mimicking some dynamical aspects of living systems. These multicompartmental structures are highly dependent on the dissipate energy from BZ reaction. Overall, this study illustrated a strategy to design and synthesize complex microsystems that can be widely used for biosensors, microreactors, and molecular delivery.
The second group of oscillatory chemistry that successfully initiated RAFT-PISA was the pH oscillators. In 2019, the Pérez-Mercader's group reported a semi-batch bromate–sulfite (B–S) pH oscillator-driven PISA (pH-O-PISA), in which the two-way oxidation of the SO32− by BrO3− generated radicals and further initiated the RAFT polymerization of nBA on PEG-based macro-RAFT agent.193 The complete oxidation of SO32− to SO42− generates H+, while the partial oxidation of SO32− (1–2%) to S2O62− consumes H+. By adjusting the H+ concentration in the inflowed Na2SO3 to H2SO4 solution, this system can be universally adapted to different monomers. In 2021, the same group further investigated the effect of the oscillatory behavior on polymerization in comparison with conventional (non-oscillatory) redox initiation.194 In this study, HPMA was chosen as the monomer, which is the first time it was used in pH-O-PISA and different morphologies, including giant vesicles, were obtained. The radical formation was switched ON/OFF by the SO32−–BrO3− pH oscillator, inducing periodic polymerization. This indicates that different kinetics have a significant impact on the final conversion (%) of a particular component and the high salt concentration in the pH oscillator was found to affect the morphology and contribute to the formation of microscale structures.
2.4 Ultrasound-initiated PISA
The acoustic cavitation produced by ultrasound in water can promote several physical/chemical effects including radical formation.195 This allows the application of ultrasound in radical polymerization, providing a “green” synthesis pathway. Ultrasound has been extensively studied to initiate free radical polymerizations in both homogeneous and heterogeneous systems, as well as to accelerate the decomposition of chemical initiators to assist polymerization. In 2017, Qiao and Ashokkumar and co-workers reported the first use of ultrasound derived radicals to initiate RAFT polymerization in aqueous medium.196 In their study, a high frequency (414 kHz) was applied, at which the hydroxyl radical generation by H2O sonolysis was enhanced and the possibility of polymer degradation is minimized. This process allowed the controlled polymerization of a range of water-soluble acrylates and methacrylates. However, it was found the ability of ultrasonic irradiation to generate radicals is concentration dependent. Bulky monomers or monomers with high concentrations could suffer from no polymerization. It was also observed that sono-RAFT polymerization can be regulated on/off by simply switching on/off the sonication. The success of ultrasound-induced controlled radical polymerization provides a “green”, externally regulated and easily scalable method for a range of monomers.Later in 2018, Qiao et al. reported the first sono-RAFT-PISA using N-isopropylacrylamide (NIPAM) as the monomer to synthesize thermo-responsive spherical nanogels at 45 °C.197 Temporal control was realized by exposing the polymerization to an alternating “ON/OFF” period of ultrasound. Furthermore, N,N′-methylenebis(acrylamide) (MBA) was used as the comonomer to achieve core-cross-linked nanoparticles that would maintain the morphology below the lower critical solution temperature (LCST) of PNIPAM. The cross-linked nanogels are thermo-sensitive and can undergo reversible shrink/swell cycles in heating (45 °C)/cooling (25 °C) cycles.
To expand the scope of sono-RAFT-PISA, in 2020, our group reported the first room-temperature sono-PISA that achieved diverse morphologies, including spheres, worms, and monodispersed small vesicles (ca. 110 nm) (Fig. 6).198 Kinetic studies and GPC measurements confirmed the good control of HPMA polymerization under high-frequency (990 kHz) ultrasound irradiation. It was found that the morphology of nano-objects prepared by sono-PISA was distinct from that of thermal-PISA process, which could be the effect of reaction temperature and ultrasound wave. In addition, it has been observed that the length of pre-formed worms would decrease with time of exposure to ultrasound irradiation. This explains why no elongated worms were observed during sono-PISA. However, this was overcome by the addition of core-crosslinker ethylene glycol dimethacrylate (EGDMA) to achieve stable worms by sono-PISA. Notably, sono-PISA is easily scalable and externally tunable, does not require any exogenous initiator or catalyst, and is highly compatible with a broad range of RAFT agents and monomers. These features may open up many new prospects in the field of polymeric nano-object synthesis.
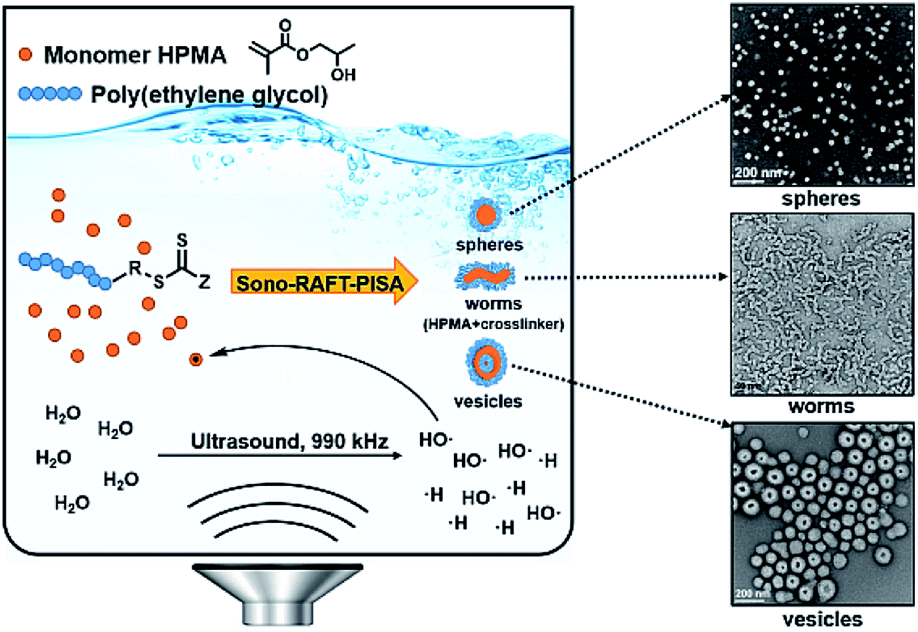 | ||
| Fig. 6 Schematic illustration of room-temperature PISA initiated by ultrasound, and TEM images of obtained nano-objects. Reprinted from ref. 198 with permission. Copyright 2020, Royal Society of Chemistry. | ||
3. Controlled and high-order morphologies by PISA
One of the main benefits of the PISA process is that the morphology of the self-assembled nano-objects can be easily tuned by adjusting the DP of the core-forming block. In a dispersed system without kinetic trapping, the evolution from spheres to worms to vesicles with the increasing DP is usually observed. Over the past decade, many efforts have been made to achieve morphological control, and interest in expanding the morphological library has continued unabatedly. In this section, we focus on some pioneering works and most recent studies that realized the control of morphology toward high-order structures. The order of morphology usually depends on the packing parameter (P, defined as P = V/a0lc, in which V and lc represent the volume and length of the hydrophobic block, respectively, and a0 stands for the effective area of the hydrophilic headgroup), high-order structures occur when P > 1. This could be achieved by adjusting several factors, including DP and solvophobicity of the hydrophobic block, solvent system, and block copolymer architecture. In the following, these factors were summarized as determinants for achieving nano-objects with high-order and complex morphologies.3.1 Varying the chain length or DP of the hydrophobic block
Tuning the chain length or DP of the hydrophobic block is the most direct way to control the morphology transformation from spheres to higher-order morphologies, such as worms, vesicles, lamella, spongosomes and cubosomes. As the molar mass/DP of the hydrophobic block increases, the packing parameter also increases simultaneously. Currently, there are a number of studies reported the morphological transformation beyond vesicles, including lamella, multilamellar vesicles,138 large compound vesicles199 or precipitates.200,201 For example, in 2010, Pan and co-authors discovered that higher-order morphology vesicles with complex internal structures; the morphologies evolved from spheres to vesicles, and to multiple morphologies including nanotubes, doughnuts, spongosomes and onion-like vesicles by tuning the chain length and thus the ratio of hydrophobic to hydrophilic blocks.202 Then in 2015, they further reported the realization of large compound vesicles (LCVs) and hexagonally packed hollow hoops (HHHs, i.e. hexasomes) with packing parameter larger than 1 by PISA.27 In addition, they probed the mechanism of the transformation from vesicles or LCVs to HHHs. Similar to the annealing of copolymers swollen by DMF in the nanoprecipitation approach,203 the high feed molar ratio of St/PDMAEMA = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 allowed PS chains to be well swollen by the residual St monomer, which has a beneficial effect on the chain mobility and the morphological transition. The good mobility of copolymer chains also allowed deformation and fusion of vesicles, eventually leading to the formation of high order structure. This is the first study to utilize the scalable PISA approach to fabricate PS-based inverse bicontinuous mesophases. However, the high feed molar ratio of St/PDMAEMA and a high monomer concentration were necessary for the fabrication of HHHs, which would result in high residual monomer content that may limit the potential applications.
000 allowed PS chains to be well swollen by the residual St monomer, which has a beneficial effect on the chain mobility and the morphological transition. The good mobility of copolymer chains also allowed deformation and fusion of vesicles, eventually leading to the formation of high order structure. This is the first study to utilize the scalable PISA approach to fabricate PS-based inverse bicontinuous mesophases. However, the high feed molar ratio of St/PDMAEMA and a high monomer concentration were necessary for the fabrication of HHHs, which would result in high residual monomer content that may limit the potential applications.
In 2019, An, Wu and Lv exploited the scalable preparation of alternating PDMA-b-P(St-alt-PFS) block copolymer particles with high order morphologies (LCVs, spongosomes, hexasomes and cubosomes).26 High monomer conversion (typically ≥90%) and high solid content (40%) were achieved simultaneously, and morphological transition from spheres to final cubosomes was observed with the growth of the core-forming block. The influence of some key parameters on the morphological evolution was investigated, it was concluded that the relatively short solvophilic block and high solid content were required to promote the production of inverse bicontinuous mesophases. In addition, the effect of different co-solvent systems was also studied, and it was found that the use of ethanol with 2% toluene afforded more ordered mesophases with improved colloidal dispersity. These mesophase structures were characterized by TEM, SEM and small-angle X-ray scattering (SAXS), revealing the Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m cubosome and p6mm hexosome structures. Later on, the successful preparation of inverse bicontinuous mesophases was also achieved by polymerization-induced cooperative assembly (PICA) using RAFT agent and PDMA-CTA as a dual controlling agent.32 In 2021, the same method has been used to obtain poly(N,N-dimethylacrylamide)-b-poly(4-tert-butoxystyrene-co-pentafluorostyrene) inverse bicontinuous mesophases via PISA.31
m cubosome and p6mm hexosome structures. Later on, the successful preparation of inverse bicontinuous mesophases was also achieved by polymerization-induced cooperative assembly (PICA) using RAFT agent and PDMA-CTA as a dual controlling agent.32 In 2021, the same method has been used to obtain poly(N,N-dimethylacrylamide)-b-poly(4-tert-butoxystyrene-co-pentafluorostyrene) inverse bicontinuous mesophases via PISA.31
In 2021, our group reported the first preparation of degradable inverse bicontinuous structures by PISA.25 First, 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl acrylate (TBA) was designed and synthesized as the core-forming monomer, which can promise a high conversion compared to styrene-based monomers. The pinacol boronic ester group in TBA is extremely sensitive to reactive oxygen species, such as H2O2, this makes the polymer and the formed nanostructures responsive to a specific stimulus. The morphological evolution from spheres to cubosomes and hexosomes with the increasing DP of PTBA was unambiguously observed and analyzed by TEM (Fig. 7b–e), SEM and SAXS. In addition, a phase diagram (Fig. 7a) illustrating the effect of DP, solid content, and stabilizer block chain length on the morphologies were also constructed. Next, the degradation study of the obtained inverse bicontinuous structures was performed by exposing the diluted cubosome suspension to H2O2. The stimuli-responsiveness and degradable feature of the inverse bicontinuous cubic and hexagonal mesophases make them potentially applicable for loading and triggering the release of payloads such as drugs and proteins.
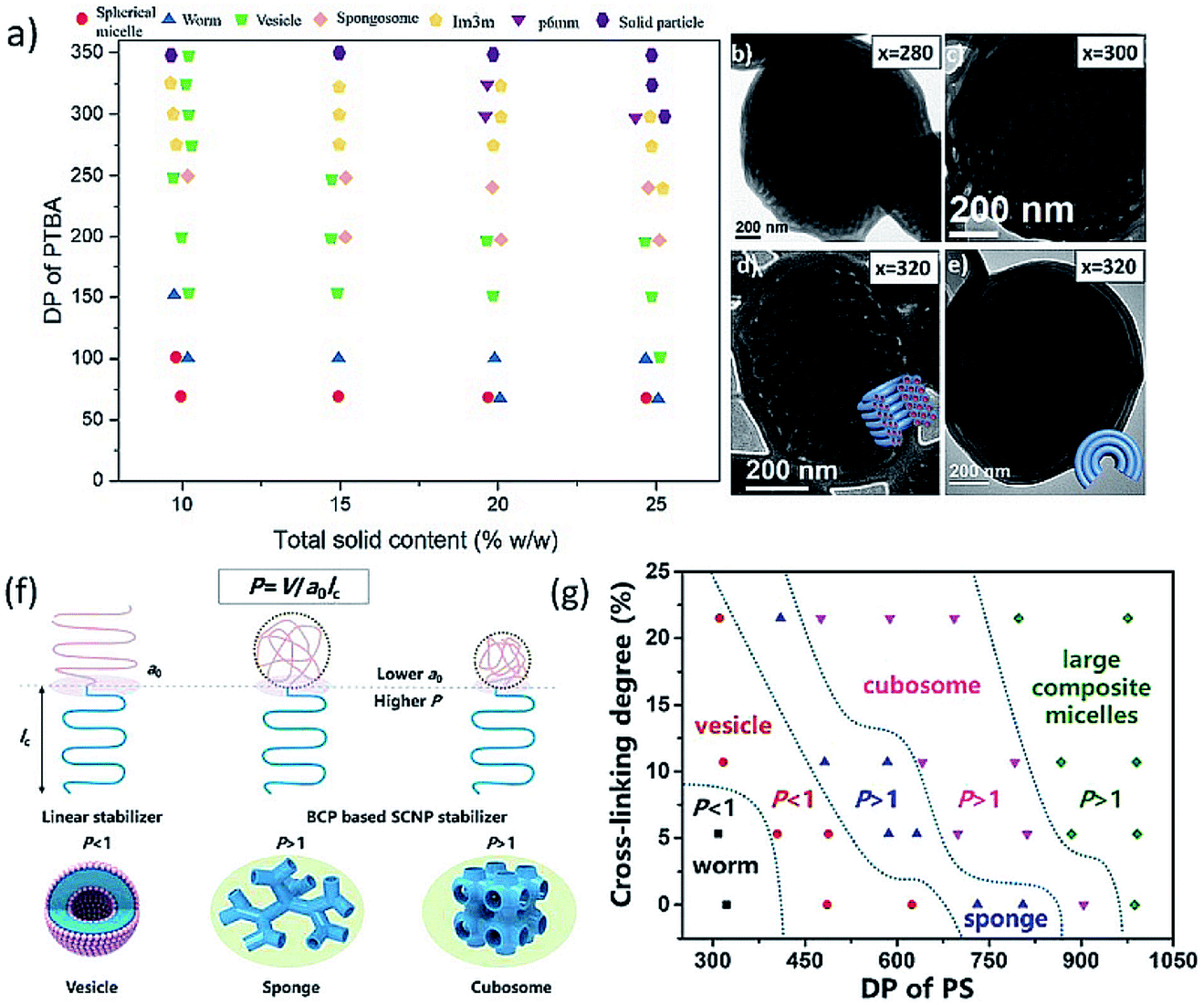 | ||
| Fig. 7 (a) Phase diagram of the PDMA45-b-PTBAx block copolymer particle during PISA with different solid contents and varying DP of PTBA. (b–e) TEM images of the particle transition from cubosome to hexosome. Reprinted from ref. 25 with permission. Copyright 2021, American Chemical Society. (f) Illustrative self-assembly of a linear block copolymer and the two SCNP-contained polymers with varied SCNP sizes and the corresponding morphologies. (g) Phase diagram of the superstructures from P4VP(SCNP)35–PSn with various cross-linking degrees of P4VP(SCNP)35. Reprinted from ref. 28 with permission. Copyright 2021, American Chemical Society. | ||
In the same year, Chen and Yang et al. demonstrated the preparation of inverse bicontinuous structures by PISA via intramolecularly folded single-chain nanoparticles (SCNPs).28 The stabilizer block (P4VP-CTA) was lightly cross-linked with 1,4-diiodobutane to obtain single-chain nanoparticles. Subsequently, PISA with styrene was performed in ethanol via these SCNPs to achieve inverse bicontinuous structures (Fig. 7f). The cross-linking of solvophilic chains leads to a smaller cross-sectional molecular area (a0), which will enlarge the packing parameter (P), and enable the preparation of polymer cubosome with well-defined structures. The morphology of structures prepared against linear P4VP-CTA and SCNPs with different degrees of cross-linking was compared. The obtained phase diagram (Fig. 7g) indicated the morphological evolution depends both on the cross-linking degree and DP of PS block, and the inverse bicontinuous structures can be easily achieved within a much broader window via this strategy.
3.2 Varying solvophobicity of core-forming block
The hydrophobicity of core-forming monomer has a great influence on the final morphology, the effect of this factor has been revealed by many studies. For example, An et al. synthesized a series of alkyl α-hydroxymethyl acrylates, including methyl (MHMA), ethyl (EHMA), isopropyl (iPrHMA), and n-butyl (nBHMA) α-hydroxymethyl acrylates, with different water solubility.139 MHMA and EHMA have higher water solubility (∼0.3 g mL−1 and ∼0.2 g mL−1 respectively) were polymerized via aqueous dispersion polymerization, and iPrHMA with moderate water solubility (∼0.06 g mL−1) was used for dispersion–emulsion polymerization. The achieved morphologies by these different monomers were compared. The TEM image and DLS analysis indicated that PEG45–PMHMA166 formed spheres, which contrasts with lamellae by PEG45–PEHMA100 and vesicles by PEG45–PEHMA200. This can be inferred that core-forming block with higher hydrophobicity provides a stronger driving force for the morphology transition, and will require lower DP to achieve higher-order morphology. For PiPrHMA with higher hydrophobicity, vesicles were obtained for PEG45–PiPrHMA100 at an even lower DP. Besides, lamellae, tubular vesicles and vesicle clusters were also observed for this polymer system, which may provide a unique opportunity to access some extraordinary morphologies.Yuan, Wei and co-workers investigated the application of three semi-fluorinated methacrylates, namely, 2-(perfluorobutyl)ethyl methacrylate (FBEMA), 2-(perfluorohexyl)ethyl methacrylate (FHEMA), and 2-(perfluorooctyl)ethyl methacrylate (FOEMA), in PISA.204 It has been shown that the dispersion polymerization of FBEMA in ethanol led to spheres, worms and vesicles, whereas FHEMA only produced kinetically frozen spheres because of the relatively strong associative interactions among the fluoro-containing side-chains. Cylindrical micelles were obtained for all PDMA-PFOEMA via PISA, due to the peculiar self-assembly behavior from the liquid crystalline ordering of PFOEMA. This additional study demonstrates the effect of core-forming monomers on morphologies.
The hydrophobicity of some monomers and polymers can be altered by external stimuli, thus affecting the PISA morphologies. For example, the hydrophobicity of PHPMA is known to increase with increasing temperature. Tan et al. then designed a temperature-programmed photo-PISA by keeping the temperature maintained or changed throughout the polymerization.199 This resulted in a diverse set of complex morphologies, including worms, vesicles and large compound vesicles, by the same mPEG113–PHPMA400 with different programmed temperature profiles.
The adjustment of polymer hydrophobicity can also be achieved by interactions with comonomers. In this regard, Zhang, Hong and co-workers explored the effect of solvophobic and aromatic interactions on PISA morphology.205 7-(2-Methacryloyloxyethoxy)-4-methylcoumarin (CMA) was selected as the core-forming monomer due to the aromaticity of the coumarin units, which allow aromatic interaction with other monomers. Three comonomers ranging from non-aromatic to strongly aromatic were selected to compare the effect of aromatic interaction. By inserting non-aromatic units into the PCMA block, both the aromatic and hydrophobic interactions were weakened, leading to the increased flexibility of the hydrophobic block. In clear contrast, the insertion of strongly aromatic comonomer enhanced both aromatic and solvophobic interactions, which lead to more rigid chains in membrane forming and vesicle fusion to form nanotubes. This is a good example demonstrating the control over the vesicular size and even steering the vesicle fusion to form the tubular structures that can be achieved by adjusting the type and aspect ratio of the comonomer.
The hydrophobicity of the particle core can also be tuned by zwitterionic copolymerization. For example, Cai et al. showed the sequence-controlled synthesis of charge-dictated alternating or gradient terpolymer via polymerization-induced electrostatic self-assembly (PIESA).206 The oppositely-charged monomers were copolymerized by photo-switched RAFT to achieve terpolymer with controlled sequence. The hydrophobicity of the polyions was adjusted, resulting in the change of the polyion complex nanostructures from water-soluble polymer to stable nano-objects, and to precipitation. By this approach, the shape of nanostructures as well as the size and thickness of micron-sized ultrathin lamellae and vesicles could be efficiently controlled.
3.3 Varying solvent
In addition to varying the monomer composition, solvent quality plays a significant role in PISA, and many researchers have investigated the role of solvent quality on the morphological evolution during PISA. Co-solvent is the most straightforward strategy to determine this effect on nanoparticle morphology, and it affects the degree of solvation or plasticization of the core-forming block.207 For instance, Armes et al. reported the preparation of inverse bicontinuous phases by alternating copolymerization of styrene with N-phenylmaleimide (NMI) in a 50:50 w/w/ethanol/methyl ethyl ketone (MEK) co-solvent system.29 The core-forming block has a relatively high Tg (219 °C), which means it is extremely rigid. However, the MEK co-solvent provided better solubilization of NMI monomer and enhanced the mobility of the growing P(St-alt-NMI) chains. Taking the advantage of high chain mobility induced by MEK, three inverse bicontinuous phases (perforated ellipsoidal lamellae, bicontinuous ellipsoids and large compound micelles) were achieved in this PISA. To further investigate the role of MEK, control experiments using 50:50 w/w ethanol/1,4-dioxane were conducted under same conditions, which only yielded kinetically trapped morphologies such as spheres, worms and worm clusters. This study also suggested that high chain mobility in an appropriate solvent system is essential for achieving high order morphologies.
An, Wu and Lv reported a rational access to poly(N,N-dimethylacrylamide)-b-poly(2,3,4,5,6-pentafluorostyrene) (PDMA-b-PPFS) via PISA in 5–20% DMF/ethanol.207 When 5% DMF/ethanol was used, a morphological transition from spheres to vesicles and finally nanotubes were observed. In 10% DMF/ethanol, morphology transformed from spheres to vesicles, then vesicles started to fuse into vesicular dimers, followed by nanotubes and multi-wall nanotubes. When 20% DMF/ethanol was used, typical sphere–worm–vesicle-large compound vesicles morphological transition was observed, with no sign of nanotubes. The authors also deduced the hypothesis that nanotubes can be formed when the polymerization temperature (Tp) is below or close to the solvated glass transition temperature (Tsg). This study revealed the effect of solvent quality on chain mobility and the subsequent morphological transition pathway. Luo also recently reported the morphological transition of PEG-b-PS by PISA using co-solvents.208 When methanol and water were used, PEG-b-PS micellar aggregates without any internal structure were observed at 30% solid content. However, when methanol/THF/water (1.15/0.45/0.4, v/v) was used as co-solvent, a morphological transition from vesicles to large compound vesicles and final inverse bicontinuous phases was observed. This study once again demonstrated the effect of co-solvent on morphology, and provided a facile method for developing inverse bicontinuous phases.
Furthermore, it was demonstrated by Fielding et al. that the nanoparticle size could be controlled by altering the co-solvent composition. In this example, anionic poly(potassium 3-sulfopropyl methacrylate) (PKSPMA) macro-CTAs were chain-extended with BzMA in alcohol/water mixtures to form PKSPMA–PBzMA nanoparticles via PISA.209 The influence of co-solvent was systematically investigated by changing the alcohol/water ratio, the alcohol type (ethanol or methanol) and relative copolymer composition. For the fixed copolymer composition, the nanoparticle diameter could be tuned from 20 to 200 nm using different ratios of ethanol/water or methanol/water with fixed copolymer composition.
Instead of co-solvents, some additives in solvent could also play a great role in PISA. For example, cyclodextrin (CD) has been used by Yuan et al. to improve the solubility of styrene (St) via the formation of a water-soluble host–guest complex. This allowed the aqueous dispersion polymerization of St, and morphologies including lamellae, nanotubes and dumbbell-like nanoparticles were achieved.210 More recently, the same group obtained new nanoflower-like morphology by using host–guest modulated PISA with increased polymerization rate.211
3.4 Varying block copolymer architecture
Varying the polymer architectures in PISA is another important strategy to achieve different morphologies and even high-order morphologies. In this regard, An et al. reported the star architecture of PEG-b-(PDAAM)2, which was synthesized by RAFT polymerization, can promote the transition to obtain higher-order morphologies at both lower solid content and lower DP.212 In another work, Zhang, Han and co-workers synthesized a series of mono- and multifunctional trithiocarbonates, which were chain extended with P4VP to achieve macro-RAFT agents with different numbers of branches.213 Linear and star block copolymer (BCP) nano-objects of [P4VP-b-PS]n with the arm number n at 1, 2, 3, and 4 were synthesized by PISA (Fig. 8). The size and morphology of the [P4VP-b-PS]n nano-objects were found to be correlative to arm number n. Nano-objects formed from star [P4VP-b-PS]n underwent the morphological transition from vesicles to lacunal spheres and porous nanospheres, presumably because star BCPs have better solubility (higher critical aggregation concentrations) than linear ones. Their research indicates that BCP architecture is a significant parameter to dedicate the size and morphology of nano-objects under PISA conditions.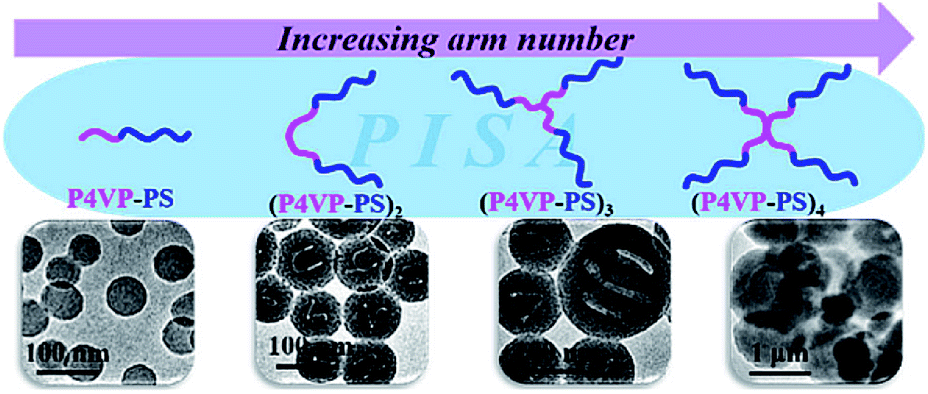 | ||
| Fig. 8 TEM images of [P4VP-b-PS]n nanoassemblies with different block copolymer architectures. Reprinted from ref. 213 with permission. Copyright 2018, American Chemical Society. | ||
Instead of star architecture, it was found that solvophobic–solvophilic–solvophobic (BAB) block copolymers allow the preparation of loop-stabilized morphologies by PISA. The Zhang group reported a series of PS-b-PEG-b-PS,214 PS-b-PNIPAM-b-PS,215 PS-b-P4VP-b-PS216 loop-stabilized nano-objects prepared by PISA, and investigated the morphology variations of nano-objects prepared from between AB and BAB copolymers. They found PS-b-PEG136-b-PS forms large-sized aggregates due to the presence of a bridging linkage between the two terminal hydrophobic PS blocks compared to their AB-type counterparts.214 In another study, it was found that the size of PS167-b-PNIPAM196-b-PS167 nanospheres (25 nm) was smaller than the PNIPAM98-b-PS328 nanospheres (50 nm).215 Furthermore, lacunal nanospheres (around 115 nm) were obtained for PS282-b-P4VP58-b-PS282, which were much different from the P4VP58-b-PS550 nanospheres (67 nm) and P4VP25-b-PS264 entrapped vesicles (143 nm).216 In addition to the work from Zhang's group, the Rieger group also studied various loop-stabilized BAB particles by PISA (Flower PISA), and they found loop solvophilic chains exhibit different stabilizing behaviors, the bridges between the B blocks lead to the formation of gel-like dynamic polymer network.217–219
Using a similar strategy, Zhang, Li and co-workers demonstrated the in situ synthesis of ingenious nano-objects by the cooperative dispersion polymerization with PEG–TTC and TTC–PEG–TTC macro-RAFT agents.220 The resulted PEG-PS/PS-PEG-PS (AB/BAB) polymer blends with various ratios could lead to vesicles (ratio 6/0), compartmentalized vesicles with different sizes and wall thickness (ratio 6/1 to 6/3), and porous nanospheres (ratio 6/4).
3.5 Multicompartment nano-objects
Multicompartment nano-objects usually were prepared from block copolymers including one solvophilic and two or more incompatible solvophobic core-forming blocks that can lead to segmented anisotropic structures or other complex nano-objects. The Zhang group have conducted extensive studies on the preparation of multicompartment block copolymer nanoparticles (MBCNs) via PISA.221–225 Nanospheres of different formulations with segregated patches on the surface were obtained by dispersion polymerization or seeded quasi-solution/dispersion/emulsion polymerization with subsequent solvent replacement or temperature change.226 The phase separation between polymer blocks was triggered by the difference in solubility of one block in the new solvent or by the LCST or upper critical solution temperature (UCST) of the polymer.Recently, in situ phase separation between incompatible core-forming blocks leading to multicompartment patchy particles has also been reported. For example, Yuan et al. reported fluoro-containing triblock terpolymer assemblies by PISA.227 In this example, PDMA-b-PBzMA-b-PFHEMA terpolymers with different DP of PBzMA and PFHEMA were prepared by seeded RAFT dispersion polymerization of FHEMA with spheres, worms and vesicles made from PDMA-b-PBzMA. Phase segregation between PBzMA and PFHEMA during polymerization led to a series of compartmentalized nanostructures including core–shell–corona, patchy-like, ribbon-shell and raspberry-like micelles as well as core–shell–corona vesicles. The same group also reported the synthesis of PDMA-b-PBzMA-b-PFMA assemblies by PISA.228 Due to the liquid crystalline alignment of PFMA, spheroids and cylinders with segregated PFMA and PBzMA nanodomains were obtained. This study demonstrated the influence of fluoro-containing mesogen on the self-assembly behavior and morphology.
Tan et al. reported the preparation of patchy cylindrical micelles by seeded photo-PISA in water using cross-linked cylindrical micelles as seeds.229 The further extension of the third block led to nanoscale phase separation within the core-forming block, which contributes to the patchy morphology. By varying the DP of the third block, the roughness of patchy cylindrical micelles could be controlled. This study indicates that the cross-linking of the second block is required for the phase separation, and non-cross-linked control only resulted in short cylindrical micelles and vesicles.
In 2020, Yuan, Yang and co-workers demonstrated the preparation of rarely achieved colloidal molecules (CMs) by seeded dispersion polymerization of HPMA with PDMA-b-PBzMA nanoparticles as seeds.33 First, the PDMA-b-PBzMA colloidal atoms (CAs) were synthesized via PISA in ethanol, it was then dialyzed against water (pH ∼ 2) to remove ethanol. In the growth of the third block, they found that phase separation between PHPMA and PBzMA occurs as the DP of PHPMA increases. When PHPMA domains reached a critical size, they combined into PHPMA central colloidal atoms and led to colloidal clusters. When the volume ratio of PHPMA and PBzMA VH/VB > 1, monovalent Janus intermediates would assemble hierarchically into ABn-type CMs, with n increased with VH/VB. When VH/VB ≤ 1, divalent intermediates lead to colloidal chains. This study achieved colloidal molecules with n from 2 to 6, and they observed the formation of “colloidal polymer”, which is a beaded chain-like architecture. However, this study involved two steps and required a solvent exchange process to meet the solubility requirements of each block. Further exploration is warrant to facilitate the application of this strategy to more block copolymer systems.
In 2021, our group provided an innovative strategy to overcome the above problems and achieved a series of triblock terpolymer-based colloidal molecules, core–shell–corona micelles and raspberry-like particles via aqueous polymerization-induced hierarchical self-assembly (PIHSA).34 As shown in Fig. 9a, in this study, dispersion polymerization of cyclodextrin (CD)–styrene water-soluble complex mediated by PEG-CDTPA was performed to obtain CAs. The remaining CD in suspension was reused to complex with second monomer tert-butyl acrylate (tBA), and the polymerization of tBA can directly lead to the formation of divalent or monovalent colloidal intermediates. These intermediates eventually formed colloidal chains or AXn-type CMs by controlling the chain length of the third block. The same method was also applied to synthesize PEG-b-PtBA-b-PS and PEG-b-PtBMA-b-PS terpolymers, instead of colloidal molecules, core–shell–corona micelles and raspberry-like particles were obtained, respectively. In this study, we proposed that for ABC triblock terpolymer nanoparticles, the distribution of B and C block domains depends on the interfacial tension between polymer block and solvent (Fig. 9b). If γCS < γBS (γiS stands for the interfacial tension between solvent and block i), block C tends to aggregate at the interface between block B and solvent interface to minimize the total interfacial energy. As a result, CM intermediates formed and hierarchically self-assemble into CMs. If γCS > γBS, block C will grow inside block B, and resulted in core–shell–corona micelles or raspberry-like particles depending on the Tg of block B. Overall, this study provided a new strategy for the synthesis of hierarchical nanoparticles and it can be readily applied to prepare nano-objects with diverse morphologies and formulations at relatively high-solid contents.
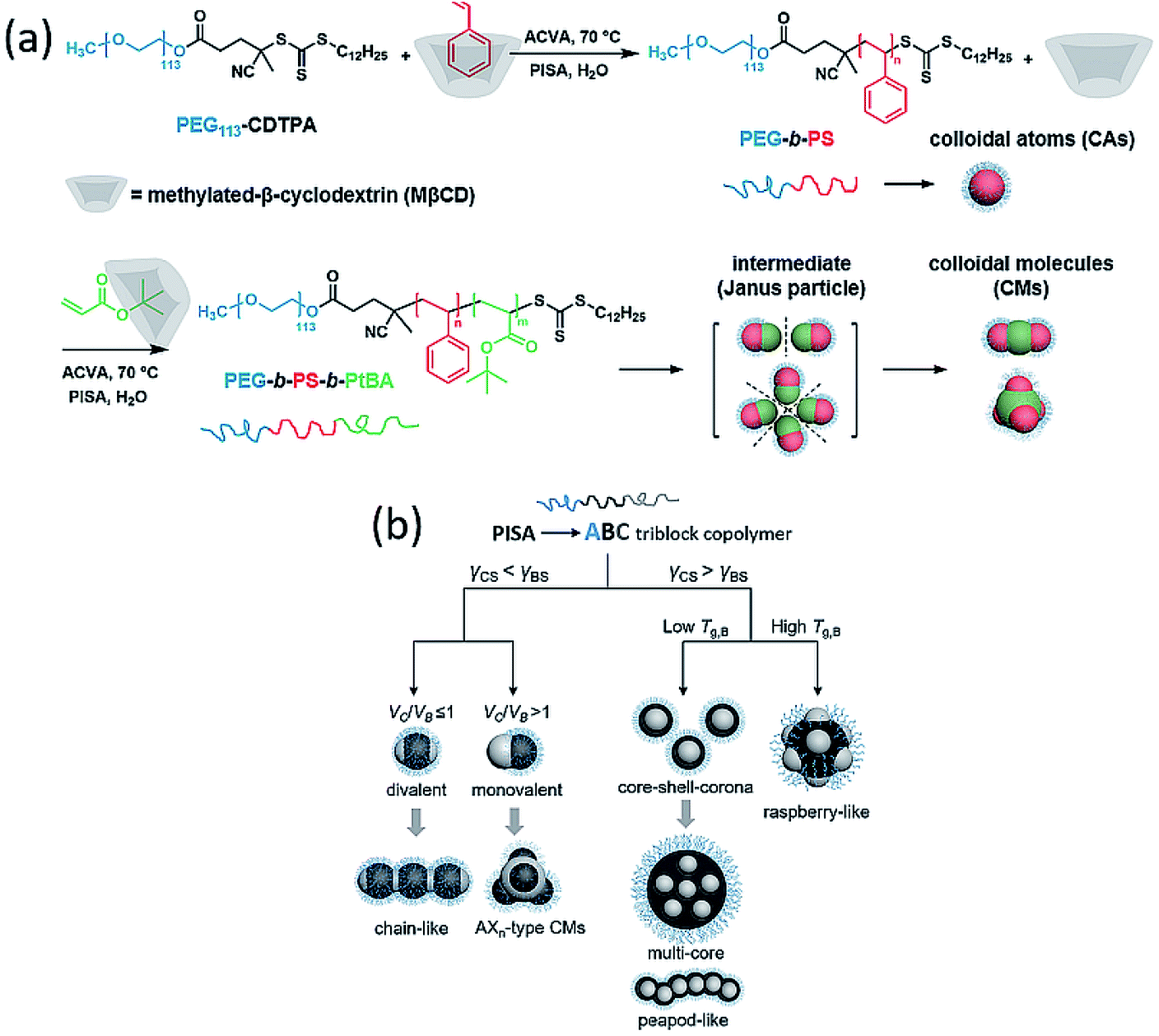 | ||
| Fig. 9 (a) Synthesis of PEG-b-PS CAs and PEG-b-PS-b-PtBA CMs using MβCD/monomer complex via PISA. (b) ABC triblock terpolymer hierarchically self-assembly mechanisms under the specific PISA conditions. Reprinted from ref. 34 with permission. Copyright 2021, American Chemical Society. | ||
4. Hybrid materials synthesized via PISA
Many efforts have been made to fabricate organic-polymer and inorganic-polymer nanocomposites with constituent materials such as proteins, (poly)peptides, metals, metal oxides, mineral oxides. The nanocomposites containing hybrid materials combine the attractive properties of both polymer and the constituents, which endows them with a wide range of synergistic effects. PISA offers a convenient and scalable strategy for the preparation of polymeric colloids, thus attracting extensive research interest in the preparation of hybrid materials with polymer colloids as scaffolds. The common connections between polymeric colloids and other materials via PISA are (1) PISA-based polymeric colloids as scaffolds with decorating materials on the surface; (2) surface-initiated PISA from the surface of other materials; (3) in situ encapsulation of cargos into PISA nano-objects during the polymerization; (4) covalently bonded hybrid polymeric colloids with other materials attaching to either the solvophilic block or the solvophobic block. This section will introduce the design of these four types of hybrid materials by discussing some relevant examples.4.1 PISA-based polymeric colloids as scaffolds for nanocomposite fabrication
One approach that allows polymeric colloids to combine with another material, such as metal nanoparticles, is to form nanocomposites by “decorating” target material on the surface of colloids. To achieve successful attaching, a common strategy is to bind on colloids by ligand exchange or complexation process; another strategy is to mix metal precursor with polymeric colloids containing docking sites, followed by in situ formation of metal nanoparticles on the colloid surface. In this approach, it is the solvophilic or interfacial block of the block copolymer colloids that usually acts as the docking sites to provide interaction with the other material, and maintain the stability of composites in the meantime. Therefore, the design of solvophilic or interfacial block becomes important for the successful fabrication of composites.An example of this approach was reported by Davis and Boyer et al. using RAFT dispersion polymerization of styrene to prepare various nano-objects and subsequent reduction of chloroauric acid to form gold nanoparticles on their surfaces.230 First, the POEGMA macro-RAFT agent was chain extended with 2-(N,N-dimethylamino)ethyl methacrylate (DMAEMA) to synthesize POEGMA-b-PDMAEMA-CTA macro-RAFT agent, which contains tertiary amine on the interfacial block. The subsequent dispersion polymerization of styrene resulted in spheres (S), worms (W) and vesicles (V) by varying the DP of PS block. Aliquots of suspension of S, W, V were mixed with chloroauric acid, then the addition NaBH4 led to the reduction of chloroauric acid and the formation of gold nanoparticles. The complexation of the tertiary amine group with chloroauric acid allowed the immobilization of gold nanoparticles between the hydrophilic and hydrophobic blocks.
The work by Zhang's group demonstrated the use of multicompartment nanoparticles (MCBNs) constructed with the brush block terpolymer as a scaffold for Au nanocatalyst.56 First, a new brush macro-RAFT agent was synthesized by polymerization of p-chloromethylstyrene (CMS) to afford PCMS21-TTC, and then the nucleophilic substitution reaction between PCMS21-TTC and pre-synthesized thiol-terminated P4VP25-SH resulted in (PCMS-g-P4VP)-TTC. MCBNs were obtained by dispersion polymerization of styrene mediated with (PCMS-g-P4VP)-TTC. Au nanoparticles were immobilized on the surface of MCBNs through the coordination between the pyridine ligand with the gold ions and the following reduction by NaBH4. The prepared Au@MCBNs were used as nanocatalyst and showed high catalytic efficiency in aerobic alcohol oxidation.
Recently, our group has also contributed to this area by providing a new and facile sonochemical pathway to prepare polymeric spheres immobilized with palladium or gold nanoparticles (Fig. 10). Ultrasound has been used to initiate the PISA process without the addition of exogenous chemical initiators.57 The versatility of ultrasound is also demonstrated by its ability to generate reducing species to form metal nanoparticles without the addition of reductant. In this study, PEG-b-PDMAEMA-TTC macro-RAFT agent was first synthesized, and then used to mediate dispersion polymerization of HPMA in water under sonication at room temperature. The synthesized spheres were then mixed with palladium or gold ion precursors and exposed to ultrasound for the in situ formation of palladium or gold nanoparticles at PDMAEMA interfacial block. Furthermore, in catalysis studies, the Pd–polymer nanocomposites showed high catalytic efficiency and good reusability as a heterogonous catalyst for the Suzuki–Miyaura cross coupling reaction, whereas the Au–polymer nanocomposites could be used to catalyze aerobic alcohol oxidation, as well as surface-enhanced Raman spectroscopy (SERS) substrates after stepwise-growth of Au nanoparticles.
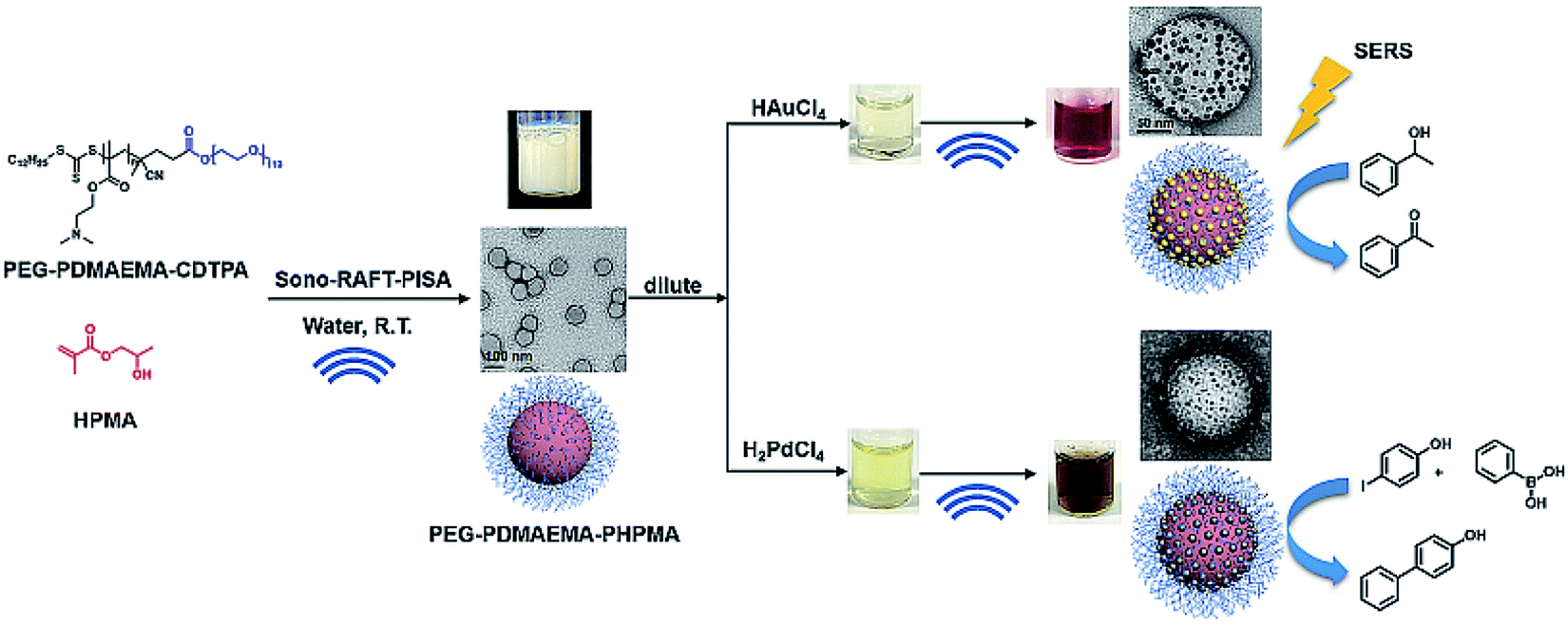 | ||
| Fig. 10 Synthesis of PEG113-b-PDMAEMA-b-PHPMA copolymers via the sono-RAFT-PISA process, and in situ formation of the Au and Pd nanocomposite by ultrasound. Reprinted from ref. 57 with permission. Copyright 2021, Royal Society of Chemistry. | ||
In addition to above work, many other studies have used this approach to synthesize a wide variety of hybrid nanocomposites. For example, in 2014, Davis and Boyer et al. reported the complexation between carboxylic acid and iron ions.231 Similar to their work of POEGMA-b-PDMAEMA-b-PS nanoparticles, POEGMA-b-MAA-b-PS triblock terpolymer nano-objects were obtained via PISA. The subsequent alkaline co-precipitation of the iron(II) and (III) salts led to formation of iron oxide nanoparticles (IONP). The complexation between MAA and iron ion (FeII/FeIII) mixture allowed the immobilization of IONP in the central block. Similarly, Matyjaszweski and Pietrasik and co-workers reported the use of PAA-b-PS nanoparticles as templates for the synthesis of Ag–polymer nanocomposites. The complexation between the PAA block and Ag+ ions and subsequent reduction resulted in AgNPs immobilized on the shell of polymeric nanoparticles. The silver–polymer nanocomposites were applied as catalyst and SERs substrate.232 An et al. employed a “multitask” monomer 2-(acetoacetoxy)ethyl methacrylate (AEMA) to produce nanospheres and vesicles bearing reactive β-ketoester groups. The β-ketoester group is multifunctional that could not only react with alkoxylamine or bisalkoxylamine to achieve oxime for cross-linking under ambient conditions, but can also complex with metal ions to produce silver nanoparticle within the nano-objects by reduction.233
Instead of forming metal nanoparticles in situ, pre-formed metal or metal oxides nanoparticles can also be immobilized with polymeric colloids via electrostatic charge interactions, ligand exchange, host–guest complexation, etc. For example, Semsarilar, Quemener and co-workers reported the PISA process to prepare PMAA-b-PMMA spheres, worms and vesicles bearing negatively charged shells.234 Magnetic iron nanoparticles coated with poly(methacrylic acid)-b-poly(quaternized 2-(dimethylamino)ethyl methacrylate) (PMAA-b-PQDMAEMA) were synthesized and incorporated with polymeric nano-objects via electrostatic interaction to form nanocomposites. The mixed matrix was spin-coated to form membranes with enhanced mechanical properties. Hawkett et al. also prepared superparamagnetic iron oxide nanoparticles (SPIONs)-decorated nanofibers by simple mixing of SPIONs with PISA-prepared nanofibers at an appropriate pH.235 A dibenzyl trithiocarbonate (DBTC) was employed as RAFT agent to synthesize DBTC-P(BA-co-AA)2 macro-RAFT agent, which was chain extended with PS block to form PS-b-P(BA-co-AA)2 nanofibers. At an appropriate pH, SPIONs were blended with nanofibers under high shear, the opposite charge interactions led to the absorption of SPIONs on the surface of nanofibers. Later on, the Semsarilar group reported a one-pot PISA synthesis of PMAA-b-PMMA block copolymer nanoparticles decorated with iron oxide nanoparticles.236 During the PISA process with the presence of oleic acid-stabilized iron oxide nanoparticles, ligand exchange occurred between oleic acid and PAA chains, resulting in the attachment of iron oxide nanoparticles on the surface of polymeric nanoparticles.
The work by Singha et al. presented a waterborne epoxy-based fluorescent adhesive ornamented with graphene quantum dots (GQDs).237 The GQDs were prepared by hydrothermal process, then mixed with the poly(1-vinyl-2-pyrrolidone) (PVP) macro-RAFT agent and glycidyl methacrylate monomer to undergo surfactant-free miniemulsion PISA. The carboxyl and hydroxyl groups on the surface of GQD nanoparticles interacted with the nitrogen of the PVP block via noncovalent interaction, resulting in a honeycomb-like structure. The prepared block copolymer/GQD emulsions were applied to adhere ceramic and glass substrates, and showed better adhesion strength than pure block copolymer adhesive.
Recently, our group exploited the “host–guest” complexation between β-cyclodextrin (β-CD) and adamantane to realize combining between AuNPs and different polymeric colloids (Fig. 11).238 First, β-CD was attached to the R group of RAFT agent via esterification. The β-CD-CDPA was chain extended with OEGMA to afford macro-RAFT agent, followed by further chain extension of HPMA via PISA to afford spheres, worms and vesicles with β-CD on the surface. Pre-formed AuNPs stabilized with polymer ligands containing adamantanyl (Ada) moiety were prepared separately. Subsequently, by incubating these polymeric nano-objects with Ada stabilized-AuNPs in different ratios, a series of different polymer/gold nanocomposites, including AuNPs decorated polymer vesicles, polymer sphere nano-flowers, polymer sphere nano-patterns, and polymer nano-worms were successfully prepared. This study provided a new and efficient strategy for the preparation of polymer–metal nanocomposites with the desired ornamentation.
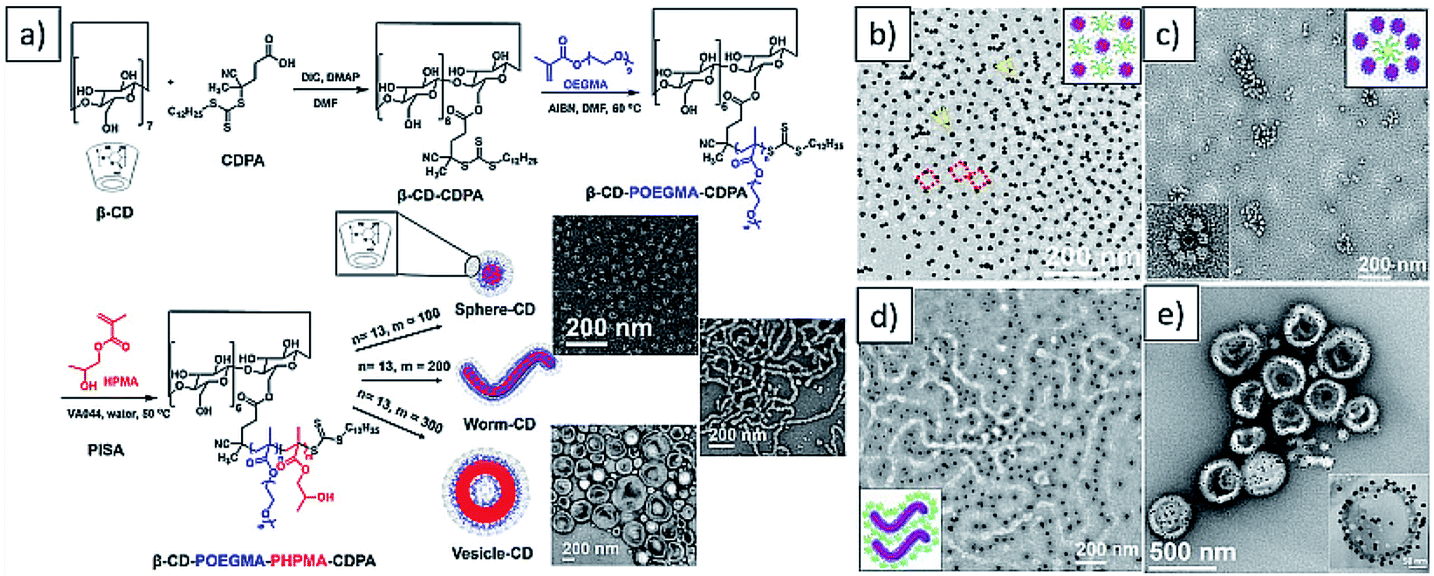 | ||
| Fig. 11 (a) Synthesis of β-CD decorated diblock copolymer nano-objects vis aqueous PISA (b–e) representative TEM images of AuNPs decorated polymer sphere nano-pattern, sphere nano-flowers, nano-worms and vesicles. Reprinted from ref. 238 with permission. Copyright 2020, American Chemical Society. | ||
4.2 Surface-initiated PISA from the particle surface
In 2016, the Benicewicz group reported the seminal work of surface-initiated PISA from SiO2 nanoparticles and obtained various 1-D structures, including short strings, branched long strings, and highly branched string networks.239 The bare silica nanoparticles of diameter ∼15 nm were attached with medium graft density (∼0.1 chain per nm2) of RAFT agent 2-cyano-2-propyl benzodithioate (CPDB) and chain extended with 2-hydroxyethyl methacrylate (HEMA). The medium graft density allowed both enough polymer chains to solubilize silica nanoparticles and enough space for the growth of second polymer population. The RAFT end of PHEMA chains were cleaved off and the silica nanoparticles were attached with the second round of RAFT agent to afford SiO2-g-(PHEMA, CPDB). It was then used to mediate the surface-initiated dispersion polymerization of BzMA in methanol. With the continuous increase of PBzMA chain length, the silica nanoparticles self-assembled into 1D silica–polymer nanocomposites (Fig. 12). Later in the same year, they extended this method by performing polymerization of BzMA from SiO2-g-(PHPMA, CPDB) nanoparticles and obtained stable 3D assemblies such as single-walled hybrid vesicles.240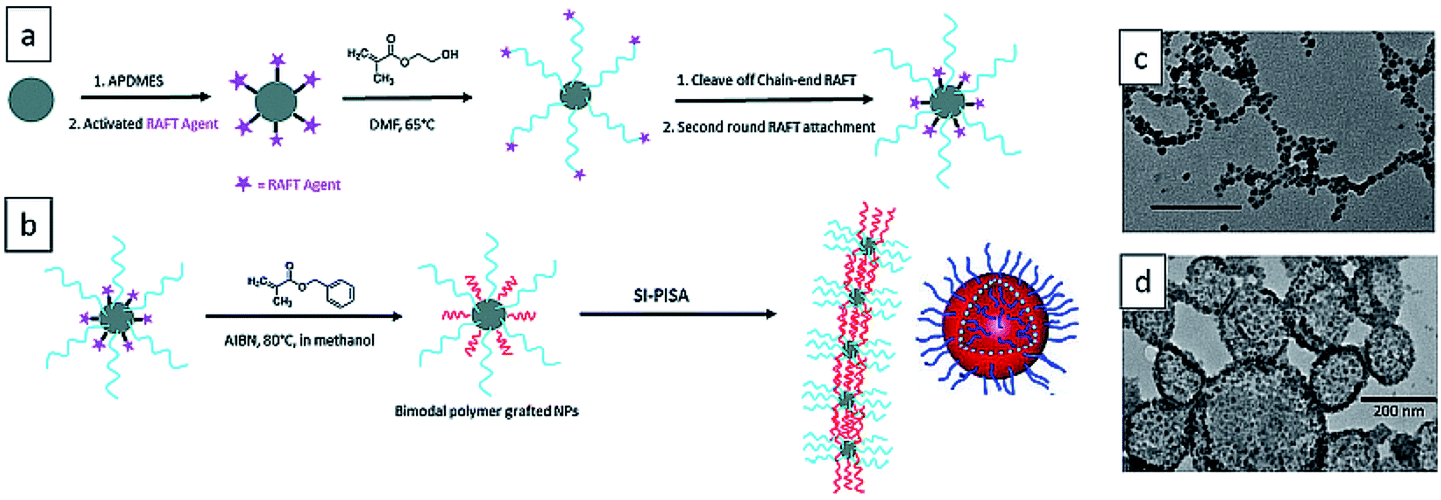 | ||
| Fig. 12 (a) Synthesis of SiO2-g-(PHPMA, CPDB) nanoparticles. (b) One-pot surface-initiated RAFT polymerization-induced self-assembly of grafted NPs into highly branched string networks and vesicles. TEM images of (c) highly branched string networks and (b) vesicles. Reprinted from ref. 239 and 240 with permission. Copyright 2016, Royal Society of Chemistry. | ||
The Bourgeat-Lami group demonstrated another approach by nitroxide-mediated emulsion polymerization of n-butyl methacrylate (BMA) and styrene to form multipod-like silica/polymer latexes.71 A PEO-based macroalkoxyamine containing a small amount of styrene (P[(OEGMA950)12-co-St1]-SG1) was synthesized and attached to the surface of acidified silica particles through hydrogen-bonding interactions. The subsequent polymerization of BMA and styrene initiated by the marcoalkoxyamine initiator led to the self-assembly of block copolymers around the central silica spheres. By varying the macroinitiator concentration or the silica particle size, dumbbell-, daisy- or raspberry-like hybrid composites were obtained. They also extended the surface PISA to achieve new hybrid morphologies. It was found that the morphology depends strongly on the size of the silica nanoparticles. When silica nanoparticles with diameter around 30 nm were used, “armoured” fibers and vesicles were formed. When ∼136 nm silica were used, the morphologies were core–shell, “half-capped” spheres, tadpole-like and “snowman”-vesicles instead. With size around 230 nm, core–shell and “half-capped” spheres were observed.241
4.3 In situ encapsulation during PISA
The encapsulation of cargos into the polymeric nanoparticles during the PISA process is one of the most common methods for preparing hybrid nanocomposites. One specific example is the encapsulation of hydrophobic drugs into the hydrophobic core of polymeric colloids. In addition to drug molecules, vesicles prepared by PISA can also serve as carriers for the encapsulation of a broad range of materials, including inorganic particles, dyes and proteins.An early example of in situ encapsulation during PISA was reported by the Armes' group in 2015.242 PGMA-b-PHPMA diblock copolymer vesicles were synthesized in the presence of varying concentrations of silica nanoparticles (∼18 nm). During the PISA, an open-ended “jellyfish” structure was formed before transforming to a vesicle structure, allowing the diffusion of silica nanoparticles into “jellyfish” before the vesicle membrane formation. The in situ encapsulation was performed with initial silica content from 0–35% w/w. Six centrifugation cycles were conducted to separate the silica–vesicle composites and non-encapsulated silica. The successful loading of silica was confirmed by TEM, cryo-TEM, thermogravimetry (TGA), disk centrifuge photosedimentometry (DCP) and SAXS. The thermo-responsive nature of the PGMA-b-PHPMA vesicles allowed the thermally induced morphological transition and the triggered release of encapsulated silica nanoparticles. This study demonstrated an encapsulation and release model system translatable to other cargoes, including globular proteins, enzymes, antibodies or other biomedical species.242
In 2015, Tan and Zhang et al. also reported the preparation of a diverse set of PEG-b-PHPMA polymeric nano-objects (spheres, worms, and vesicles) via photo-PISA at room temperature. In situ encapsulation of silica nanoparticles and bovine serum albumin (BSA) into vesicles could be achieved via photo-PISA.133 The room-temperature process makes it possible to prepare hybrid composites containing temperature-sensitive proteins and biomedical species. Later on, the same group designed and synthesized CO2-responsive PPEGMA-b-P(HPMA-co-DMAEMA) nano-objects via photo-PISA at room temperature, which is suitable for encapsulating bio-related species. In such study, BSA was encapsulated into vesicles in situ with a loading efficiency around 24%, and the CO2-triggered release was studied.243 The same methodology was also used to prepare silica–polymer hybrid nanocomposites. The CO2-responsive nature of the PDMAEMA block allowed the triggered release of silica nanoparticles under mild conditions.244
Recently, more cargo species have been encapsulated into PISA-prepared vesicles, including enzymes, dyes and drugs,35,36,245–247 and the activity of the encapsulated species were investigated. For example, Gibson and O'Reilly et al. reported the permeability of PHPMA vesicle membranes by showing the encapsulated enzyme remained active while inside the vesicles.245 Subsequently, they demonstrated the in situ encapsulation of therapeutic enzymes within permeable nanoparticles using PISA, which provided an alternative to PEGylation for the therapeutic enzymes. A one-pot synthesis approach was performed to obtain PEG-b-PHPMA vesicles loaded with L-asparaginase (ASNS) at relatively high solid content (11 wt%) under the mild condition via photo-PISA. After encapsulation, the PHPMA membrane with size-selective permeability allows the enzyme to be protected and maintain catalytic activity. The proteolytic stability of encapsulated ASNA was shown to be higher in vitro and in vivo than the native enzyme and the PEGylated conjugate, while the binding of ASNS antibodies was reduced due to being embedded inside vesicles.36 The benefits of this strategy is that it does not need chemical modification, demonstrating the potential application of PISA for in situ encapsulation and improving the stability of therapeutic enzymes. In another example, Sobotta et al. developed oxidation-sensitive poly(N-acryloylthiomorpholine) (PNAT)-based vesicles via PISA. The disintegration times of vesicles varied with the membrane thickness, and could be tuned by varying the DP of the hydrophobic PNAT block. They demonstrated the encapsulation of different cargo species including calcein (dye) and GOx enzyme. The encapsulated GOx not only retained its activity but also transformed the hybrid vesicle into glucose-responsive nanoreactors that can undergo self-degradation by converting glucose to H2O2.247
4.4 Covalently bonded biomolecule–polymer hybrid materials
Another approach to prepare hybrid materials is covalent bonding to polymer chains, especially for biomolecule–polymer hybrid materials, including (poly)peptides, proteins, nucleic acid and amino acids. Biomolecules can be linked to the chain transfer agents and employed as stabilizers for PISA; or they can be modified as polymerizable monomers and used as core-forming monomers or comonomers. The covalent bonds between biomolecules and polymers are much stronger, allowing higher loading efficiency compared to the above strategies, however, it requires additional synthesis steps.:20 ratio) from BTPA-DNA. By varying the DP of polymer, different morphologies including micelles, worms and disc-like aggregates were observed by AFM and cryo-TEM.
Gianneschi and co-workers reported the PISA using macro-CTA containing peptide moieties. In their study, KLA peptide acrylamide monomer (KLAAm) (amino acid sequence: KLAKLAKKLAKLAK) and DMA were copolymerized in pH 5 buffer to afford peptide brush macro-CTA, followed by chain extension with DAAm and DMA via photo-PISA. The one-pot process yielded nanospheres with high-density of apoptotic peptides with tunable size (ca. 36–105 nm), and tunable loading of peptides (20–48 wt%). In addition, enhanced proteolytic stability, cellular internalization, and cytotoxicity were determined for the peptide–polymer spheres in comparison with free apoptotic peptides.251
Semsarilar et al. reported the use of positively charged polylysine modified with a RAFT agent moiety as hydrophilic steric stabilizer and CTA for the dispersion polymerization of HPMA.252 First, RAFT agent 4-cyano-4-(2-phenylethanesulfanylthiocarbonyl)sulfanylpentanoic acid (PETTC) activated with NHS was used to conjugate to the amino-terminated polylysine sequence (KKK) to afford KKK-PETTC. Subsequently, it was used to conduct PISA with HPMA in water at 60 °C, resulting in the in situ formation of polylysine-decorated nano-objects (spheres, worm, vesicles) depending on DP of PHPMA. The positive charges of the nano-objects could bind to the negatively charged phospholipid head groups of bacterial membranes, allowing the antimicrobial properties of the nano-objects to be exerted. Both nanoparticle solution and spin-coated thin-film membrane made from these nano-objects showed great antibacterial activity against Gram negative (Escherichia coli) and Gram positive (Staphylococcus epidermidis) bacteria.
The same group employed a tripeptide methacrylamide derivative (MAm-Gly–Phe–Phe-NH2, denoted as MAm-GFF) to copolymerize with GMA to produce P(GMA65-stat-(MAm-GFF)7) macro-RAFT agent.253 The peptide-based macro-RAFT agent was then chain-extended with PHPMA via aqueous PISA at 70 °C. Different to commonly achieved spheres, worms and vesicles by PGMA-b-PHPMA block copolymers, the obtained P(GMA65-stat-(MAm-GFF)7)-b-PHPMA28 (cooled to ambient temperature) was in fibrous structure as observed by TEM. This confirmed the influence of the GFF interactions on self-assembly. Interestingly, the self-assemblies underwent a morphological transformation with temperature change. After annealing at 70 °C for 1 h, TEM revealed the morphology changed to worm-like structures with a diameter around 25 nm. Upon cooling to 4 °C for 1 h, the fibrous structures were replaced by large spherical objects (∼600 nm) composed of small spheres (∼25 nm). Meanwhile, the same group explored the use of MAm-GFF and MAm-FGD (MAm-Phe–Gly–Asp-NH2) as solvophobic monomers.254 Specifically, PGMA macro-CTA were chain-extended with MAm-GFF macromonomer as core-forming monomer via emulsion PISA in ethanol, yielding flake-like objects and dendritic structures composed of fibrous elements (Fig. 13a and b). PGMA-b-P((MAm-GFF)-co-HPMA) yielded a mixture of short worms and vesicles due to the insertion of a less solvophobic PHPMA block. They also investigated another peptide MAm macromonomer (MAm-FGD) as the core-forming block in aqueous PISA process, resulting in large dendritic and bow-tie shaped fibers (Fig. 13c–e).
 | ||
| Fig. 13 Representative TEM images of (a) flake-like objects, (b) branched bundles of fibers, (c–e) large dendritic and bow-tie shaped fibrous structures. Reprinted from ref. 254 with permission. Copyright 2020, Royal Society of Chemistry. | ||
5. Stimuli-responsive particles by PISA
It is well-known that for amphiphilic self-assembled nanoparticles, the morphology undergoes stimuli-responsive transformation if one or more blocks are sensitive to stimuli, such as the temperature, pH changes and reactive species. The morphological transformation includes total disassociation of the assembly and “order-to-order” transition, e.g. worm-to-sphere transition. The total disassociation has been extensively exploited in conventional solution self-assembly, i.e. nanoprecipitation, however, the latter circumstance is rarely reported. In contrast, both transitions have been explored in depth in the PISA field, especially the order-to-order transition has gained increasing research attention and even reversible responsiveness was achieved. The stimuli-responsive morphological transformation leads to change in physical properties of polymeric colloids, and allows controlled release of conjugated or encapsulated cargos, which has great potential in drug release and other biomedical applications. Here, we summarize some of the common stimuli that have been reported for stimuli-responsive nano-objects prepared by RAFT-PISA.5.1 Thermo-responsive nano-objects
Temperature-sensitive nano-objects are one of the most common stimuli-responsive polymeric nanoparticles. Typically, the design involves the incorporation of polymer blocks that exhibit either a LCST or an UCST or solubility change with temperature. Examples include thermo-responsive PNIPAM hydrogels via RAFT-mediated precipitation polymerization,258 thermal responsive spheres, worms, vesicles made from POEGMA-poly(2,3,4,5,6-pentafluorobenzyl methacrylate),259 and thermo-reversible degelation and regelation transition and thermo-responsive nanoparticles by aqueous dispersion copolymerization of N,N-diethylacrylamide (DEAAm) and N,N′-methylene bisacrylamide (MBA) using mPEG-RAFT agent.260 The LCST of PDEAAm at 32 °C led to a change in size of the resulted nanoparticles upon heating and cooling. In 2012, Blanazs et al. further extended this thermo-responsive property to PGMA-b-PHPMA worms that were synthesized via aqueous RAFT dispersion polymerization. The variable-temperature 1H NMR spectroscopy indicated the PHPMA block has a greater hydration degree at lower temperature, which can induce reversible worm-to-sphere morphological transition upon cooling from 21 to 4 °C.261 Similar cooling triggered worm-to-sphere transition was observed for poly(lauryl methacrylate)-b-poly(benzyl methacrylate) synthesized via PISA in n-dodecane.262 For PEG-b-PHPMA worms with a relatively long PEG chain (DP = 113), they exhibited thermo-responsiveness but not reversible morphological transition. Degelation of worms to spheres occurred upon cooling, but regelation failed upon return to room temperature.263 However, in a later study, the use of a binary mixture of long PEG113 and short PEG45 macro-RAFT agents in PISA process enabled the formation of nano-objects with thermo-reversible behavior.264 In this study, a systematic variation of the proportions of two PEG-RAFT agents and the DP of PHPMA were performed to obtain [x PEG45 + z PEG113]-b-PHPMAn block copolymer spheres, worms, or vesicles. Interestingly, only a single worm dispersion [0.70 PEG45 + 0.30 PEG113]-b-PHPMA115 underwent full degelation/regelation cycle.For PISA morphologies with the utilization of UCST behavior, Tran et al. synthesized hydrogen-bonding poly(N-acryloyl glycinamide) (PNAGA)-based thermo-sensitive nanogels driven by UCST via photo-PISA at 3 °C.265 POEMGA was used as macro-CTA for the polymerization of NAGA and N,N′-methylenebis(acrylamide) (MBA) cross-linker. The opalescent solution was observed, and hydrodynamic diameter decreased upon cooling to 5 °C, which was related to the collapse of the core-cross-linked P(NAGA-co-MBA). Upon heating to 55 °C, the solution became transparent with an increase in hydrodynamic diameter. This behavior was reversible and reproducible over successive shrink/swell cycles. Rieger and Stoffelbach and co-workers reported the synthesis of UCST-thermo-responsive nano-objects using PDMA as macro-CTA, acrylamide (AAm) and acrylonitrile (AN) as monomers via aqueous PISA at 45 °C.266 The molar fraction of AN (FAN) and DP of core-forming block were varied systematically. The turbidity test indicated the cloud point temperature was tunable between ∼20 to ∼80 °C by increasing the FAN. The thermo-responsiveness and morphology were greatly influenced by FAN and DP. For intermediated FAN and high DP, the formed worms transformed to spheres upon heating due to the increased hydration of the core-forming block.
While most studies reported the LCST/UCST-driven transition, Derry et al. reported the thermo-reversible crystallization-driven aggregation. A series of nanoparticles were prepared via dispersion polymerization of BzMA with poly(behenyl methacrylate)37 (PBeMA37) macro-CTA as a steric stabilizer at 90 °C in mineral oil. Turbidity and DSC studies of PBeMA37 homopolymer in mineral oil solution indicated the thermo-sensitive nature of PBeMA37 stabilizer. On cooling to 20 °C, unstable PBeMA37-b-PBzMAx spheres formed turbid pastes because of the crystallization of insoluble PBeMA block, and returned to free-flowing dispersions when heating to 50 °C. SAXS studies confirmed the strong interaction between PBeMA37-b-PBzMA100 spheres forming loose mass fractals at 20 °C.267
In most of the previous studies reported on thermo-responsive morphological transitions, a single diblock copolymer typically transforming between two morphologies (e.g., worms to spheres). Few reports have shown that a single diblock copolymer can undergo transitions between three morphologies. For example, Ratcliffe et al. reported a single copolymer composition, poly(N-(2-hydroxypropyl) methacrylamide)41-poly(2-hydroxypropyl methacrylate)180 (PHPMAC41-b-PHPMA180), can form spheres (4 °C), worms (22 °C) or vesicles (50 °C) upon temperature change. On cooling from 50 to 4 °C after dispersion polymerization, the appearance of the PHPMAC41-b-PHPMA180 exhibited three distinct states: milky-white free-flowing dispersion at 50 °C, free-standing gel at 22 °C, and slightly turbid fluid at 4 °C. TEM of the diluted dispersions equilibrated for 24 h at a specific temperature indicated the morphologies were well-defined vesicles, worms, and spheres respectively. Other characterizations such as SAXS, DLS and rheology studies all supported the observation. Theoretical analysis of the diblock copolymer system using numerical lattice computations based on a self-consistent mean field theory supported the observation as well.268
5.2 pH value as stimulus
When polyelectrolytes or polymers with ionizable functional groups are used as stabilizers, the morphological transition can be triggered by pH change. In 2014, Armes et al. reported the synthesis of PGMA-b-PHPMA nano-objects using a carboxylic acid-based RAFT agent.269 The carboxylic acid end-group on the R-group of the RAFT agent allowed the ionizable single terminal on each stabilizer chain to be exposed to the aqueous solution. The resulting HOOC-PGMA-b-PHPMA worms obtained at approximately pH 3.5 underwent worm-to-sphere morphological transition when increasing solution pH from 3.5 to 6.0 using NaOH. The pH-responsive behavior is reversible, returning the pH to its original value resulted in sphere-to-worm transition and thus regelation. Similarly, a vesicle-to-worm morphological transition upon pH change was observed; however, in this case, the transition was not reversible.Subsequently, the same group further explored the effects of polymer end-groups on nano-objects in response to external stimuli. A morpholine-functional RAFT agent (MPETTC) was synthesized and used to prepare PGMA macro-CTA containing the terminal morpholine functional group (pKa ∼ 6.3).270 Dispersion polymerization of HPMA was performed at pH 7.0–7.5. DLS, TEM and rheology studies all indicated the pH-responsiveness of the obtained worms. By lowering the solution pH from 7 to 3, the protonation of the morpholine end-group induced worm-to-sphere morphological transition and degelation. However, further reduction of the solution pH to 1 led to the reformation of worms. The increase in pH from 3 to 7 led to deprotonation of the morpholine end-group, inducing the sphere-to-worm transition. The control experiments were performed using a non-ionic RAFT agent (the carboxylic acid group was methylated), and the morphology of obtained worms remained unchanged at varied pH values.
Instead of introducing a pH-responsive end-group at the polymer chain terminal, ionic monomer could be used in stabilizer block to generate nano-objects with ionic shells. North and Armes observed size reduction of PISA-prepared PMAA-b-PHPMA nanoparticles by increasing pH from 5.5 to 10. This was because the multiple acid groups on the PMAA stabilizer were ionized and resulting in a decrease in the mean aggregation number.271 The sterically stabilized nanoparticles at pH 10 were also thermo-responsive due to the presence of PHPMA block.
Ionizable core-forming monomers could also be used to prepare pH-responsive nanoparticles via PISA. For example, Zhang and Hong et al. reported the PISA process using PEG-based macro-CTA for copolymerization of (diisopropylamino)ethyl methacrylate (DIPEMA) and BzMA. DIPEMA (pKa ∼ 6.3) with tertiary amine group enabled pH sensitivity of the formed vesicles. The immediate disaggregation of vesicles in acidic solution (pH 4.0 buffer) was observed, allowing burst release of encapsulated rhodamine B.272 In another example, Tan et al. reported the photo-PISA using HPMA and DMAEMA as core-forming monomers for preparing CO2-responsive vesicles. After treating with CO2, the solution pH decreased, a certain number of tertiary amine groups in the PDMAEMA block were protonated, inducing the increased hydrophilicity and subsequent impaired vesicle structure. After removal of CO2 by purging with N2 or Ar, the solution pH was restored, and the deprotonation of tertiary amine groups led to a decrease in conductivity and hydrodynamic diameter.273 Yuan et al. reported the fabrication of poly[oligo(ethylene glycol)methyl ether methacrylate]-b-poly(benzyl methacrylate)-b-poly[2-(diethylamino)ethyl methacrylate] (POEGMA-b-PBnMA-b-PDEA) triblock copolymer spheres, worms and vesicles via one-pot sequential PISA in ethanol, followed by replacement of the solvent with water.274 Due the presence of CO2-sensitive groups in PDEA block, upon alternative CO2/Ar treatment, PDEA underwent protonation and deprotonation cycles. Both spheres and vesicles showed reversible expansion/shrinkage, exhibiting unique “CO2-breathing” behavior.
Ampholytic diblock copolymer nanoparticles with dual pH-responsiveness were also studied. Armes et al. firstly reported the use of PISA for the synthesis of ampholytic schizophrenic nanoparticles with poly(2-(diethylamino)ethyl methacrylate) (PDEA) as stabilising block and PMAA-st-PBzMA as core-forming block. After PISA nanoparticles were prepared at pH 2.5, the ionization of PMAA and deprotonation of PDEA could induce nanoparticle inversion when solution pH was increased to 10. At pH 6–8, the dispersion was highly turbid with a large hydrodynamic diameter, which, combined with the zeta potential results, confirmed the isoelectric point at which flocculation occurred.275 Similar schizophrenic nanoparticles with dual pH-responsiveness were also reported by North and Armes. In this case, PDEA macro-CTA was chain extended with 2-carboxyethyl acrylate (CEA) via PISA in an acidic solution.276
5.3 Light-responsive nano-objects
For light/photo-responsive nano-objects, it usually involves the incorporation of some conventional photo-responsive functional groups such as azobenzene, pyrene and ortho-nitrobenzyl group. For example, Chen et al. reported the scalable synthesis of azobenzene-containing photo-responsive block copolymer nano-objects via PISA.277 PMAA macro-CTA was used as a stabilizer for the dispersion polymerization of 11-(4-(4-butylphenylazo)phenoxy)undecyl methacrylate in ethanol. During PISA, the liquid crystalline (LC) feature of azobenzene allowed internal LC ordering in the core-forming block and led to hierarchical self-assembly into worms, short belts, lamellar and rarely achieved cuboid and ellipsoidal vesicles (Fig. 14). LC behavior of azobenzene at its trans state would diminish at its cis state under UV irradiation, thus the obtained LC nanoparticles were photo-responsive. After UV irradiation, the original cuboids changed to spherical particles, and the ellipsoidal micelles changed to large compound micelles. Yuan et al. also reported the use of azobenzene-containing monomer for the synthesis of poly(N,N-dimethylaminoethyl methacrylate)-b-poly[(benzyl methacrylate)-co-(4-phenylazophenyl methacrylate)] [PDMA-b-P(BzMA-co-AzoMA)] via PISA.278 Spheres, worms and vesicles were obtained with various DP of P(BzMA-co-AzoMA). Under UV irradiation, the worms underwent worm-to-vesicle transformation due to trans-to-cis transition of azobenzene groups. During the morphological evolution, some intermediate morphologies such as “octopus” and “jellyfish”-like structures were observed before reaching vesicles.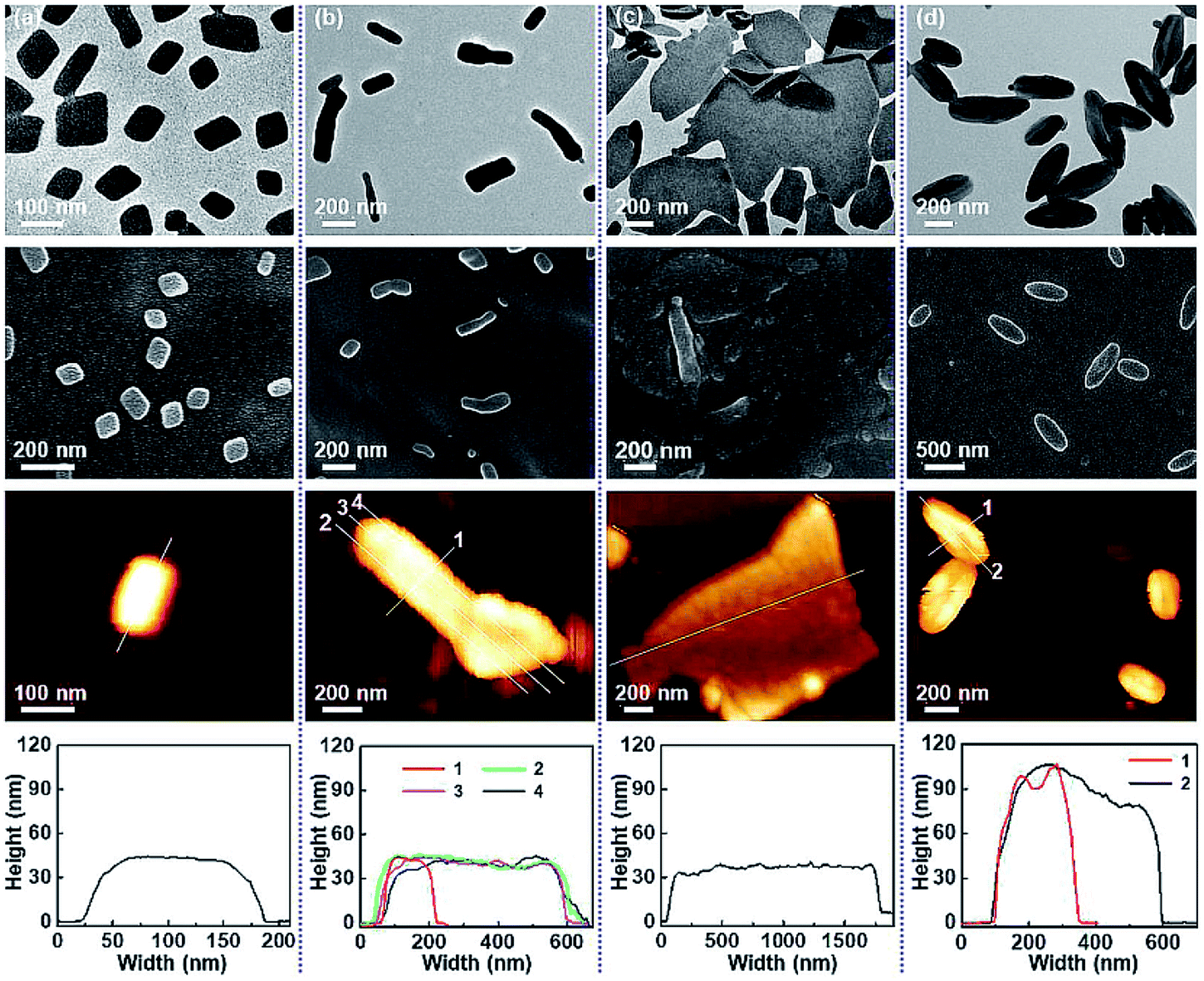 | ||
| Fig. 14 TEM, SEM, AFM images, and the corresponding height profiles of PMAA112-b-PMAAzn NPs, respectively: (a) PMAA112-b-PMAAz66 cuboids, (b) PMAA112-b-PMAAz89 short belts, (c) PMAA112-b-PMAAz115 lamellar, (d) PMAA112-b-PMAAz142 ellipsoidal vesicles. Reprinted from ref. 277 with permission. Copyright 2018, American Chemical Society. | ||
Boyer et al. demonstrated a one-pot PISA process for the preparation of light-responsive pyrene-containing nanoparticles. POEGMA macro-CTA was chain extended with 1-pyrenemethyl methacrylate (PyMA) and comonomer to yield spheres, worms and vesicles.279 The addition of comonomers such as BMA or MMA reduced the π–π stacking between pyrene moieties, this was essential for achieving relatively high PyMA conversion. The light-responsive feature of pyrene enabled light-induced gradual cleavage of pyrene moieties and gradual disassembly of nanoparticles. When exposed to UV irradiation, the POEGMA-b-PPyMA spheres and worms, POEGMA-b-P(PyMA-co-BMA) vesicles all underwent dissociation. Chemtob et al. reported the synthesis of poly(hydroxyethyl acrylate)-b-poly(o-nitrobenzyl acrylate) (PHEA-b-PNBA) to prepare photo-responsive nanoparticles via PISA.280 By exposing the PHEA-b-PNBA latex to UV irradiation, the degradation of PNBA block led to a gradual decrease in turbidity with the irradiation time. The latex also turned brown due to the decomposition of aromatic nitro products.
5.4 Reactive oxygen species as stimulus
The polymer nanoparticles with responsiveness to reactive oxygen species (ROS) have attracted many research interests, especially the ROS such as hydrogen peroxide and hypochlorite are typically found in inflammatory reactions. It is well-known that the thioether group could be oxidized by reactive oxygen species (ROS), thus the thioether functionalized vinyl monomers could be used to prepare ROS-responsive nano-objects via PISA. Yeow, Boyer and co-workers reported the use of thioether group-containing monomer, 2-(methylthio)ethyl methacrylate (MTEMA), for the preparation of vesicles via PET-RAFT-mediated PISA.281 A series of POEGMA-b-PMTEMA with different DP of PMTEMA resulted in spheres, worms and vesicles with encapsulated ZnTPP. These nano-objects can rapid disassemble after exposure to visible light in the presence of air. This was because the thioether moiety was oxidized to hydrophilic sulfoxide by the singlet oxygen generated by the encapsulated ZnTPP under visible light.Similarly, thioether-containing monomer N-acryloylthiomorpholine (NAT) was used to prepare oxidation-responsive nanoparticles via PISA.246,247 Brendel et al. synthesized poly(N-acryloylmorpholine) (PNAM)-based macro-CTA for the dispersion polymerization of NAT, and the obtained biocompatible spherical micelles exhibited different diameters. Upon exposure to H2O2, the oxidation of thioether moiety led to a gradual increase in the hydrophilicity of the core and the ultimate disassembly.246 More recently, they demonstrated unilamellar vesicles could be obtained by PISA of PNAM-b-PNAT. Calcein was encapsulated into the vesicles as a model dye, which could be released in a time-controlled manner when expose to H2O2. In addition, GOx enzyme was successfully encapsulated into vesicles as well, transforming the vesicles into glucose-responsive vesicles. With the addition of glucose, H2O2 was generated by GOx catalysis, leading to the full disassembly of vesicles.247
In 2021, our group reported the preparation of rarely achieved inverse bicontinuous mesophases using boronic ester group-containing monomer via PISA.25 Upon exposure to H2O2, the boronic ester moiety was oxidized and hydrolyzed to phenol, which further initiated the generation of quinone methide and hydrophilic PAA, thus the ultimate degradation of cubosomes and other nano-objects (Fig. 15). This allows potential application in the triggered release of payloads.
 | ||
| Fig. 15 Disassembly of the PDMA-b-PTBA-based cubosomes in response to H2O2. (a–e) TEM images of the cubosome degradation process at selected time points. (f) 1H NMR spectra evolution of the block copolymer after the addition of H2O2. (g) Digital photos of the particle suspension after the addition of H2O2. (h–j) DLS-monitored disassembly process. Reprinted from ref. 25 with permission. Copyright 2021, American Chemical Society. | ||
5.5 Reduction-responsive nano-objects
Instead of oxidising species, reducing species can work as an external stimulus for some polymers as well. For example, Zhou et al. reported using an azobenzene-derived methacrylate with a TPE (tetraphenylethylene) fluorescence probe moiety (TPE-AZO-MA) as the commoner for the dispersion copolymerization with BMA.282 A series of drug-loaded micelles and vesicles were obtained by in situ doxorubicin (DOX) loading during PISA. Azoreductase was used as the reducing agent for azo bond cleavage and resulted in micellar destruction. During enzyme hydrolysis, a slow release of DOX was observed. Meanwhile, strong fluorescence emission was detected due to the aggregation-induced emission of TPE. Goto et al. synthesized cross-linked nano-objects using a cross-linker containing disulfide group.84 Due to the stabilizing effect of the cross-linker, the morphology of the nano-objects was retained after the hydrophobic block were hydrolyzed to hydrophilic block. Subsequent addition of the reducing agent glutathione (GSH) resulted in the reduction of disulfide bonds to thiols and cleavage of cross-linking units, inducing the decomposition of nano-objects.5.6 Dual and multi-responsive nano-objects
Finally, another emerging trend in PISA for the synthesis of stimuli-responsive nanoparticles is to allow responsiveness triggered by two or more stimuli, which makes nanoparticles versatile in their applications. Lovett et al. reported pH and temperature dual-responsive HOOC-PGMA-PHPMA worms and vesicles.269,283 More specifically, the thermo-responsiveness of PHPMA block led to worm-to-sphere/vesicle-to-worm transitions on cooling; pH increase from 3.5 to 6.0 caused the ionization of the terminal carboxylic acid group located at the end of PGMA block and resulted in worm-to-sphere/vesicle-to-worm/vesicle-to-sphere transitions.Mahdavian et al. reported P(St-co-MMA) nanoparticles that could be triggered by pH change and UV irradiation. PDMAEMA macro-CTA was used for the preparation of P(St-co-MMA) along with the incorporation of spiropyranethyl acrylate (SPEA) comonomer via RAFT surfactant-free emulsion polymerization.284 The pH-responsiveness resulted in the dispersion/aggregation of the obtained latexes. The latexes were well-dispersed in acidic condition due to water-soluble PDMAEMA+Cl−, and aggregated at pH above 8 due to deprotonation. UV irradiation (λ = 365 nm) dissociated the C–O bond in the spirocarbon to form a conjugated zwitterionic merocyanine dye isomer and led to hyperchromic behavior, which could be returned by visible light.
Zhang, You and co-workers demonstrated the preparation of pH- and reduction-responsive prodrug nanoparticles with anticancer activity. First, biocompatible poly(N-(2-hydroxypropyl)methacrylamide) (PHPMAm-CPDB) was chain extended with DIPEMA and camptothecin prodrug monomer (CPTM) via RAFT solution polymerization.38 The obtained PHPMAm-b-P(DIPEMA-co-CPTM) macro-CTA was used for the dispersion polymerization of BzMA to afford prodrug nanoparticles. Upon appropriated stimulus, PDIPEMA underwent solvophobic to solvophilic transition in acidic condition, allowing faster diffusion of GSH reducing agent into the CPTM units. The reduction of the disulfide bond in the CPTM by GSH led to the release of camptothecin. The drug delivery and anticancer activity of these nanoparticles were verified by in vitro cytotoxicity study. More recently, the same group reported similar pH- and reduction-responsive prodrug nanoparticles with formulations of PHPMAm-b-PCPTM, PHPMAm/PDEAEMA-b-PCPTM, PHPMAm/PDMAEMA-b-PCPTM.39 Different to their previous study, the CPTM was used as the only core-forming monomer. HPMAm/PDEAEMA-b-PCPTM nanoparticles underwent fast hydrophobic–hydrophilic transition and charge reversal at the physiological pH value (pH = 6.8), which facilitated enhanced cell internalization. In contrast, PHPMAm-b-PCPTM without charge-reversible property showed slower cell internalization and lower anticancer activity.39
6. Improved throughput of PISA
Compared to conventional solution self-assembly, the high solid content property of PISA has dramatically improved the throughput of nanoparticle production. Based on this, many efforts have been made to further improve the throughput of PISA for the purpose of future upscale production. For instance, an automated parallel synthesizer has been employed in the RDRP polymerization285,286 and PISA287 subsequently. However, the oxygen inhibition in RDRP could be the bottleneck for high-throughput PISA. The previously reported protocols for deoxygenation are that automated parallel living polymerization performed under inert atmosphere or with automated deoxygenation process,288 which however require specialized equipment. Alternatively, as mentioned in above sections, oxygen-tolerant PISA processes have been reported using photo-PISA153,156 or enzyme-PISA,164,289 which could eliminate the time and energy needed for deoxygenation.In 2018, Cockram et al. demonstrated the first high-throughput synthesis of block copolymer nanoparticles in water via RAFT-mediated PISA.287 First, a large batch of PMAA macro-CTA was synthesized via conventional solution RAFT polymerization. Then BzMA or BMA was added as the first core-forming monomer for RAFT aqueous emulsion polymerization targeting PMMA56–PBzMA500 or PMMA56–PBMA500 nanoparticles using Chemspeed Autoplant A100. The deoxygenation protocol was performed by blowing N2 gas through all reaction vessels for 20 min (∼20 reaction vessels). Different types of stirrers and stirring speeds (propeller-type stirrer 350–650 rpm and anchor-type stirrer 150–350 rpm) were used to optimize the PISA process. The same protocol was employed to produce triblock and tetrablock copolymer nanoparticles using BMA or BzMA as the second or third water-immiscible monomer. The optimized protocols indicated that a propeller-type stirrer at stirring rates of 550–900 rpm was required to produce sufficiently small droplets of monomers. The preliminary study provided the basis for further high-throughput screening of PISA formulations. For instance, one-pot automated synthesis of both macro-CTA and block copolymer nanoparticles may be more attractive. Only spheres were achieved in this study, future study regarding the screening of different formulations to obtain various morphologies and rapid establishment of the phase diagram is foreseeable with the increasing demand from industry.
As automated synthesizers are still not readily available in most academic settings, alternative oxygen-tolerant polymerization protocols have been developed for high-throughput PISA. These protocols are more suitable for laboratory scale with even ultralow volumes, and suitable for rapid optimization especially for those with many reaction variables (monomer type, monomer concentration, targeting DP, temperature, solvent composition, solid content, etc.). In 2017, Boyer et al. demonstrated that oxygen-tolerant photo-PISA in the presence of AscA and eosin Y could be conducted in 96-well microplates.153 In the same year, Tan et al. also reported an enzyme-assisted photo-PISA in open vessels and microplates.168 The oxygen tolerance was endowed by the catalytic ability of GOx. These methods facilitate a high-throughput PISA that allows multiple parallel PISA processes to be performed simultaneously, which could realize rapid construction of phase diagrams and fast screening of reaction parameters.
On the basis of these methods, the oxygen-tolerant PISA strategy in low volumes has been extended to study the wavelength orthogonality of the [2 + 2] coumarin cycloaddition reaction and PET-RAFT PISA. Under the red light (λ = 595 nm) irradiation, exclusive activation of PET-RAFT PISA occurred, resulting in the formation of various nano-objects (spheres, worms, vesicles) with no evidence of dimerization. Rapid cross-linking of polymer chains occurred by switching to UV irradiation (λ = 365 nm), allowing the retained morphology of the nano-objects in organic solvents.157 The wavelength orthogonality of photo-induced deoxygenation and photoinitiation of PISA was reported by Tan et al. using a high-throughput multiwell strategy. Dual-wavelength photo-PISA of HPMA using PGMAn-CDPA (n = 28, 53, 69) was achieved in a fast manner using 96-well microplates.290 Furthermore, Gianneschi et al. combined UV-initiated PISA on small scales in a 96-well microplate with an automated sampling of every well and automated TEM and image analysis for the rapid generation of phase diagrams.291 Selected compositions were repeated by scaled-up experiments, which showed identical morphologies and sizes with the low volume version. This approach could be readily applied for handling fast screening of a large number of samples. In summary, high throughput PISA and characterization could serve as a robust method for rapid discovery of target materials and subsequent optimization of formulations and experimental conditions, which promise great potential in future industrial mass production.
It is generally accepted that PISA under continuous flow is a more favourable method for scaling up PISA over batch synthesis, as it provides better heat/mass transfer, increased reaction rates and the ability to integrate into feedback control loops. In 2017, Zhu and Zhang et al. firstly reported the thermal-initiated PISA performed under continuous flow conditions, where POEGMA-CTA and MMA were used as a steric block and core-forming monomer in water/ethanol co-solvent system in a lab-scale two-stage continuous tubular reactor.292 The concentration and flow rate of each feedstock were kept constant, and the target DP was achieved by varying the concentration of POPEMGA macro-CTA. Only spheres were obtained due to the hindered morphological transition resulting from the high amount of water (56% v/v). Later on, Parkinson et al. reported an all-aqueous synthesis of both PDMA macro-CTA and PDMA-PDAAm diblock copolymer spheres, worms and vesicles by thermal-initiated RAFT polymerizations in coil continuous flow reactors.293
Recently, researchers in Junkers, Boyer and Zetterlund groups attempted to convert light-initiated RAFT polymerization and PISA from batch to continuous flow systems.140,294–296 In their report in 2018, different morphologies including spheres worms and vesicles were obtained via photoiniferter RAFT PISA in a continuous flow reactor under blue LED light. A rapid polymerization was found, which reached full monomer conversion in only 75 min, compared to 6 h in batch process. Their study also optimized the protocol by varying the experimental conditions including light intensity, solid content, and residence time, allowing the production of 60 g polymeric nanoparticles per day in the lab.294 Warren et al. reported the ultrafast continuous-flow RAFT dispersion polymerization which achieved conversions >90% within 8 minutes for target PDAAM DPs of 50 and 100, and achieved 79% conversion of DP of 200 within 20 minutes.297 In the further study (Fig. 16), PET-RAFT dispersion polymerization with oxygen tolerance employing eosin Y/triethanol amine as catalytic system was exploited for the synthesis of poly(dimethyl acrylamide)-b-(poly(diacetone acrylamide)-co-poly(dimethyl acrylamide)) (PDMAA-b-(PDAAm-co-PDMAA)) in one-step continuous-flow process without intermediate purification.295 The photo-PISA in less polar solvents via continuous flow process was investigated as well.140 Several parameters including solvents, photoinitiators, and light intensities were optimized in the batch process. The optimized protocol was then performed in continuous flow reactors, which realized non-aqueous photo-PISA in flow with high order morphologies.
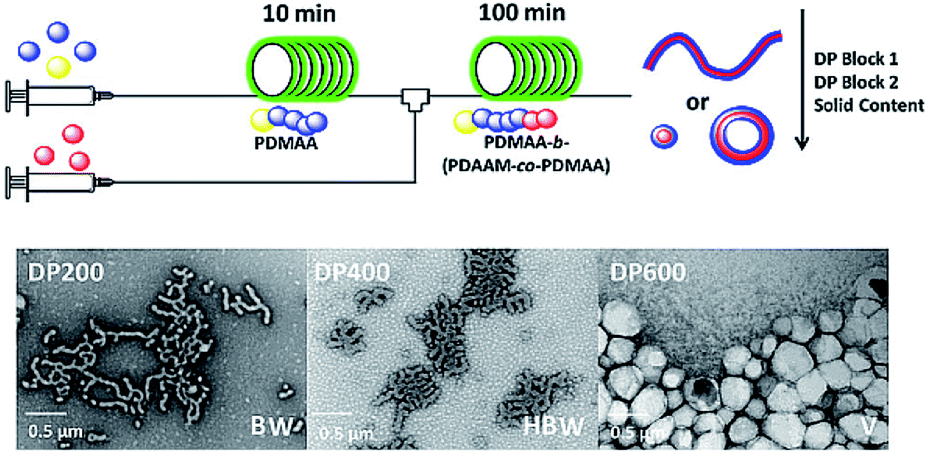 | ||
| Fig. 16 Schematic diagram of the flow reactor setup coupling two reactors, and TEM micrographs of PDMAA-b-(PDAAm-stat-PDMAA) nano-objects with DP 200–600 solvophobic block. Reprinted from ref. 295 with permission. Copyright 2019, American Chemical Society. | ||
More recently, the enzyme-assisted photo-PISA using GOx and glucose catalytic system was converted from batch to flow process as well. Tan et al. reported the oxygen-tolerant process conducted in a flow reactor, which constructed a PEG-b-PHPMA morphological phase diagram by systematically varying the DP of PHPMA.169 Another oxygen-tolerant in-flow PISA system was reported by Hou et al. based on the oscillatory BZ redox reaction. Dispersion polymerization of DAAM from PEG macro-CTA was implemented in a CSTR yielded spheres, worms, vesicles and giant vesicles, depending on the factors including residence time, BZ oscillation and DP of PDAAM.191
7. Applications
PISA allows the production of nanoparticles in various morphologies in relatively high solid contents, which opens up the applications of PISA in a broad range of areas. The most common applications for polymeric nanoparticles including those prepared by the PISA process, is drug delivery and other biomedical applications. Besides, PISA-prepared nano-objects have been explored as catalysts,56–60 Pickering emulsifiers,22,61–64 imaging agents,65,66 lubricants,67 templating agents,68–70 pigments,54,55etc. The following section summarizes some applications of PISA-prepared nano-objects.Due to the versatility of PISA, nanoparticles with biocompatible formulations could be efficiently synthesized, which have drawn increasing interest in exploring their applications in the biomedical field. As early as in 2012, Armes group reported that PISA-derived worms could form free-standing hydrogels with thermo-reversible behavior, which allowed the facile preparation of sterile gels with potential biomedical applications.261 Later on, the loading of guest compounds into PISA-derived nano-objects during polymerization were investigated by Boyer and Davis using Nile red as a guest molecule,298 as well as Mable et al. by loading silica nanoparticles into vesicles via in situ encapsulation.242 The successful loading of guest compounds make it possible to load drug molecules during PISA as well. O'Reilly et al. demonstrated efficient loading of L-asparaginase (ASNS) to vesicles during PISA. After encapsulation, the morphology of vesicles remained, and the enzyme remained catalytically active. In addition, the encapsulated enzyme showed higher proteolytic stability than the free enzyme in vitro and in vivo.36 Recently, Malmström et al. reported the in situ physical encapsulation of hydrophobic drug (DOX) during aqueous PISA. The hydrophobic core of PDMAPMA-b-PMMA nanoparticles allowed the migration of DOX into the core. The DOX-loaded nanoparticles showed higher cell toxicity than free DOX toward macrophages cell line.35 Another approach for drug loading is to use prodrug monomers as a sole core-forming monomer or comonomer.37–41 Drug molecules are covalently linked to polymer chains, leading to high loading efficiencies, even up to 100% when full monomer conversion is achieved. For example, Zhang et al. synthesized PEG-b-P(MEO2MA-co-CPTM) prodrug macro-CTA, which contains camptothecin linked to the polymer chains via reductive-sensitive linkage. The prodrug macro-CTA was then extended with BzMA as core-forming monomer and N,N-cystaminebismethacrylamide (CBMA) as cross-linker, obtaining core-cross-linked nanoparticles with enhanced structural stability. The presence of disulfide bond allowed the controlled release of camptothecin under the trigger of reductive species, such as GSH in the cytosol (Fig. 17a). The prodrug nanoparticles also exhibited excellent anticancer efficiency against HeLa cells.40 Recently, Zhang et al. used camptothecin prodrug monomer synthesized anticancer nanoparticles with both pH- and reductive-regulated drug release,38 and pH-responsive charge-reversible property.39 In more recent reports by Stenzel et al., the anticancer drug 4-(N-(S-penicillaminylacetyl)amino)phenylarsenonous acid (PENAO) methacrylate prodrug was copolymerized with OEGMA or zwitterionic 2-methacryloyloxyethyl phosphorylcholine (MPC) to synthesize macro-CTAs for PISA.42,43 The prepared nanoparticles with drug covalently linked to shell block were compared in terms of cytotoxicities, cellular uptakes, spheroid penetration, and cell localization profiles.
 | ||
| Fig. 17 (a) Proposed mechanism of reductive-responsive release of CPT from the prodrug nanoparticles. Reprinted from ref. 40 with permission. Copyright 2016, American Chemical Society. (b) Synthesis of PDMAEMA-b-PMAEBA diblock copolymer nano-objects via PISA, and conjugation between MAEBA and DOX. Reprinted from ref. 48 with permission. Copyright 2016, American Chemical Society. | ||
Apart from the above mentioned loading before or during polymerization, many other studies explored the post-polymerization loading, including post-encapsulation and modification.44,45 Zhang et al. exhibited the fabrication of intelligent vesicles with tunable size-selective membrane permeability.46 In this example, PHPMAm macro-CTA was chain extended with 2-(diisopropylamino)ethyl methacrylate (DIPEMA) and 7-(2-methacryloyloxyethoxy)-4-methylcoumarin (CMA) to afford vesicles. The post-polymerization dimerization of coumarin groups was achieved under UV irradiation, leading to cross-linking of the vesicular membrane. The pore size range of the transmembrane traffic could be tuned by cross-linking density via changing the UV irradiation time. Post-loading of gold nanoparticles with different sizes (5, 10, 15 nm) into vesicles in acidic solution was performed, indicating vesicles with different cross-linking densities exhibited size-selective permeability.46 In 2013, Boyer, Davis and co-workers conducted post-modification of PISA-generated nanoparticles for the synthesis of DOX-loaded nanoparticles with the therapeutic application.47 First, POEMGA macro-CTA was extended with styrene and vinyl benzaldehyde (VBA) via dispersion polymerization to obtain spheres, worms and vesicles. The aldehyde groups of VBA were conjugated with DOX via pH-sensitive bonds (drug loading 5 wt%). Cell viability study using MCF-7 breast cancer cells indicated the cytotoxicity of the DOX-loaded nano-objects was significantly affected by morphology.47 The conjugation between aldehyde group and primary amine group in DOX for drug-loading was also reported by Hong and Pan et al.48 Dispersion polymerization of p-(methacryloxyethoxy)benzaldehyde (MAEBA) using PDMAEMA macro-CTA obtained four different nano-objects with aldehyde-based polymer as core, followed by conjugation between MAEBA and DOX (Fig. 17b). The DOX-loaded nano-objects displayed cytotoxicity in the order of nanorods-DOX > vesicles-DOX > spheres-DOX > nanowires-DOX, which also indicated the correlation between cytotoxicity and morphology. Recently, Mable et al. demonstrated the design and synthesis of dengue virus-mimicking framboidal vesicles containing rhodamine B piperazine (Rh) for targeting triple-negative breast cancer cells.49 Two macro-CTAs PGMA and poly(2-(methacryloyloxy)ethyl phosphorylcholine) (PMPC) (pure PGMA and 97:3 binary mixture) were chain-extended via copolymerization of HPMA and GlyMA, obtaining triblock copolymer vesicles. The epoxy groups of GlyMA were reacted with Rh, this allowed post-PISA loading in the vesicular membrane. Further extension with 2-(diisopropylamino)ethyl methacrylate (DPA) led to the formation of pH-responsive framboidal vesicles. In vitro studies indicated that the introducing of phosphorylcholine-based targeting ligand is essential for intracellular uptake by MDA-MB-231 breast cancer cells.49
In addition to drug delivery applications, PISA-prepared vesicles have drawn interest as mimicking systems for living cells.50,51 Researchers have modulated vesicles with controllable permeability,52,245 and used vesicles as enzymatic nanoreactors.36,53 Other biomedical applications of PISA include cellular imaging, where nanoparticles with fluorescent property and biocompatibility were prepared via PISA.65,66
Catalysis is another application of nanoparticles prepared by PISA. Polymeric nanoparticles derived by PISA are usually not catalytically active but can serve as scaffolds for catalytic species, allowing better dispersion in solvents, higher stability, higher recyclability and less leaching. A series of studies investigated the synthesis of triphenylphosphine299–302 or nixantphos303 ligand-containing polymer nanoparticles via PISA. The catalytic composite produced by complexation between rhodium and triphenylphosphine proved to be useful for the aqueous biphasic hydroformylation of 1-octane. Oble and Rieger et al. synthesized core-cross-linked nanogels via PISA to stabilize and support Pd nanoparticles. The hybrid polymer–Pd nanomaterial was applied as quasi-homogeneous catalyst for the Mizoroki–Heck reaction between n-butyl acrylate and bromo- or iodoarenes.58 The complexations between metal nanoparticles and functional groups in nanoparticles derived by PISA were also used to prepare polymer–Ag and polymer–Au nanocomposites with catalytic activities.56,59,60 Recently, our group used ultrasound for the synthesis of both polymeric nanoparticles and the in situ formation of Au and Pd nanoparticles.57 The metal nanoparticles were immobilized on the PDMAEMA block, which was in between the hydrophilic shell and hydrophobic core, resulting in a larger exposed area of metal nanoparticles and more accessible active sites. The synthesized polymer–Au and polymer–Pd showed good catalytic activity and recyclability.
The merits of PISA, including high solid contents, tuneable morphology and functionality, also make nanoparticles suitable candidates for Pickering emulsifiers. In this regard, Armes and co-workers conducted some seminal work of Pickering emulsifiers based on block copolymer nano-objects prepared by PISA.22,61–64 Since then, block copolymer nano-objects including spheres,62,63,304 worms,61,62,305 vesicles64 and even framboidal particles22 have been used to prepare oil-in-water,22,304,306 water-in-oil,305,307 and double61,308 emulsions. Armes et al. reported that the oil-in-water-in-oil transparent Pickering double emulsions could be obtained by using refractive index-matched nanoparticles with appropriate core-forming block.308 Recently, Yuan et al. studied the effect of solvophilic chain length of nanoparticles on Pickering emulsion, and found a positive correlation between the stability of emulsion and the chain length.306 For more details on Pickering emulsifier application of PISA-prepared nanoparticles, the reader is referred to a recent review for good insight.309
Other applications of PISA-derived nano-objects are emerging in different fields as well. For example, Derry, Armes and co-workers synthesized core-cross-linked poly(stearyl methacrylate)–poly(benzyl methacrylate)–poly(ethylene glycol dimethacrylate) nanoparticles via PISA, which offer excellent boundary lubrication performance and ultralow-viscosity.67 PISA-derived nano-objects can also serve as templating agents for the fabrication of silica nanotubes.68 Nanoparticle occlusion of silica/dye-loaded nano-objects offers a “Trojan horse” strategy to incorporate payloads into host inorganic crystals such as calcite.69,70 PISA also plays an important role in coating industry, such as producing surfactant-free latexes to solve free surfactants issue, and preparing hollow particles54 or encapsulating pigments55 for opacity enhancement.
8. Summary and future perspective
This review summarizes the new aspects in PISA including non-thermal initiation methods, high order morphologies, hybrid materials, stimuli-responsive nano-objects, improved throughput and applications of PISA. As elaborated in the previous sections, PISA has proven to be an efficient process for the preparation of polymeric nano-objects. To date, many morphologies observed in conventional solution self-assembly have been realized by PISA in a more efficient way, however, there are still some complex structures that have yet to be realized, for example, Janus cylinder, ellipsoidal particle stacked internal lamellae and oblate ellipsoids. More efforts to achieve complex structures by PISA are expected, where hierarchical self-assembly will attract more attention to achieve 2D and 3D hierarchical structures, and eventually leading to higher-level architecture by PISA. Meanwhile, aqueous formulations will be the focus of future PISA, as the “green” solvent benefits most in industrial applications. The non-thermal initiation methods include photo-, enzyme- and redox-initiation, providing oxygen-tolerant PISA in open vessels, which allow for high throughput PISA and eliminate the requirement for specialised equipment. This may lead to a platform technology for rapid screening of formulations and the establishment of phase diagrams. In the meantime, PISA performed in a continuous-flow fashion has provided a possible route for mass production. Another emerging trend in the PISA process is the concept of “multifunctionality” of nano-objects, for example, where the core-forming monomer containing a therapeutic drug is also stimuli-responsive, or where the synthesised nanoparticles are responsive to multiple triggers. With the addition of multifunctional components, fewer building blocks will be required and the process will become more user-friendly, thus, the versatility of PISA can be further leveraged. Automated or even artificial intelligence-controlled PISA will be another future direction in this field, by which the hands of experienced chemists can be freed. PISA has evolved into a more powerful tool than it was a decade ago, and it is foreseeable that it will perform better and attract more attention in the future, gaining enhanced performance in existing applications or being applied to new fields.Author contributions
J. W., B. F. and S. H. T. devised the main conceptual ideas. J. W. drafted the manuscript and designed the figures. B. F. and S. H. T. revised and edited the draft. All authors participated in the discussion of the draft and carefully revised the draft before the final submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. W. thanks Monash University for the MGS and MITS scholarships and the Monash University Postgraduate Publications Award. B. F. and S. H. T. would like to acknowledge the support by ARC Centre of Excellence for Enabling Eco-Efficient Beneficiation of Minerals (Grant No. CE200100009).References
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 Search PubMed.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 Search PubMed.
- F. L. Hatton, M. Ruda, M. Lansalot, F. D'Agosto, E. Malmström and A. Carlmark, Biomacromolecules, 2016, 17, 1414–1424 Search PubMed.
- D. Ikkene, A. A. Arteni, M. Ouldali, J.-L. Six and K. Ferji, Polym. Chem., 2020, 11, 4729–4740 Search PubMed.
- J. Bernard, M. Save, B. Arathoon and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2845–2857 Search PubMed.
- V. Kapishon, R. A. Whitney, P. Champagne, M. F. Cunningham and R. J. Neufeld, Biomacromolecules, 2015, 16, 2040–2048 Search PubMed.
- K. Ferji, P. Venturini, F. Cleymand, C. Chassenieux and J.-L. Six, Polym. Chem., 2018, 9, 2868–2872 Search PubMed.
- D. Ikkene, A. A. Arteni, M. Ouldali, G. Francius, A. Brûlet, J.-L. Six and K. Ferji, Biomacromolecules, 2021, 22, 3128–3137 Search PubMed.
- V. L. Romero Castro, B. Nomeir, A. A. Arteni, M. Ouldali, J.-L. Six and K. Ferji, Polymers, 2021, 13, 4064 Search PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 Search PubMed.
- N. J. W. Penfold, J. Yeow, C. Boyer and S. P. Armes, ACS Macro Lett., 2019, 8, 1029–1054 Search PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 Search PubMed.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 Search PubMed.
- B. Darmau, M. J. Rymaruk, N. J. Warren, R. Bening and S. P. Armes, Polym. Chem., 2020, 11, 7533–7541 Search PubMed.
- Q. Zhang and S. Zhu, ACS Macro Lett., 2015, 4, 755–758 Search PubMed.
- H. Zhou, C. Liu, C. Gao, Y. Qu, K. Shi and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1517–1525 Search PubMed.
- X. Wang, L. Shen and Z. An, Prog. Polym. Sci., 2018, 83, 1–27 Search PubMed.
- A. Xu, Q. Lu, Z. Huo, J. Ma, B. Geng, U. Azhar, L. Zhang and S. Zhang, RSC Adv., 2017, 7, 51612–51620 Search PubMed.
- J. Jennings, M. Beija, A. P. Richez, S. D. Cooper, P. E. Mignot, K. J. Thurecht, K. S. Jack and S. M. Howdle, J. Am. Chem. Soc., 2012, 134, 4772–4781 Search PubMed.
- M. J. Rymaruk, S. J. Hunter, C. T. O'Brien, S. L. Brown, C. N. Williams and S. P. Armes, Macromolecules, 2019, 52, 2822–2832 Search PubMed.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 Search PubMed.
- C. J. Mable, N. J. Warren, K. Thompson, O. Mykhaylyk and S. P. Armes, Chem. Sci., 2015, 6, 6179–6188 Search PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromolecules, 2014, 47, 1664–1671 Search PubMed.
- P. Yang, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 8545–8556 Search PubMed.
- B. Fan, J. Wan, J. Zhai, X. Chen and S. H. Thang, ACS Nano, 2021, 15, 4688–4698 Search PubMed.
- F. Lv, Z. An and P. Wu, Nat. Commun., 2019, 10, 1397 Search PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2015, 36, 1428–1436 Search PubMed.
- W. Wen, S. Guan, Z. Yang and A. Chen, ACS Macro Lett., 2021, 10, 603–608 Search PubMed.
- P. Yang, Y. Ning, T. J. Neal, E. R. Jones, B. R. Parker and S. P. Armes, Chem. Sci., 2019, 10, 4200–4208 Search PubMed.
- X. Luo and Z. An, Macromol. Rapid Commun., 2020, 41, 2000209 Search PubMed.
- X. Luo and Z. An, Chin. J. Chem., 2021, 39, 1819–1824 Search PubMed.
- F. Lv, Z. An and P. Wu, CCS Chem., 2021, 3, 2211–2222 Search PubMed.
- D. Li, X. Chen, M. Zeng, J. Ji, Y. Wang, Z. Yang and J. Yuan, Chem. Sci., 2020, 11, 2855–2860 Search PubMed.
- J. Wan, B. Fan, K. Putera, J. Kim, M. M. Banaszak Holl and S. H. Thang, ACS Nano, 2021, 15, 13721–13731 Search PubMed.
- J. Engström, H. Asem, H. Brismar, Y. Zhang, M. Malkoch and E. Malmström, Macromol. Chem. Phys., 2020, 221, 1900443 Search PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O'Reilly, ACS Cent. Sci., 2018, 4, 718–723 Search PubMed.
- D. Das, D. Gerboth, A. Postma, S. Srinivasan, H. Kern, J. Chen, D. M. Ratner, P. S. Stayton and A. J. Convertine, Polym. Chem., 2016, 7, 6133–6143 Search PubMed.
- M. Chen, W.-G. Zhang, J.-W. Li, C.-Y. Hong, W.-J. Zhang and Y.-Z. You, Sci. China: Chem., 2018, 61, 1159–1166 Search PubMed.
- X. Zhao, M. Chen, W.-G. Zhang, C.-H. Wang, F. Wang, Y.-Z. You, W.-J. Zhang and C.-Y. Hong, Macromol. Rapid Commun., 2020, 41, 2000260 Search PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Biomacromolecules, 2016, 17, 2992–2999 Search PubMed.
- C. Tian, J. Niu, X. Wei, Y. Xu, L. Zhang, Z. Cheng and X. Zhu, Nanoscale, 2018, 10, 10277–10287 Search PubMed.
- J.-M. Noy, F. Chen and M. Stenzel, Beilstein J. Org. Chem., 2021, 17, 2302–2314 Search PubMed.
- J.-M. Noy, F. Chen, D. T. Akhter, Z. H. Houston, N. L. Fletcher, K. J. Thurecht and M. H. Stenzel, Biomacromolecules, 2020, 21, 2320–2333 Search PubMed.
- H. Asem, W. Zheng, F. Nilsson, Y. Zhang, M. S. Hedenqvist, M. Hassan and E. Malmström, ACS Appl. Bio Mater., 2021, 4, 1045–1056 Search PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Biomacromolecules, 2017, 18, 1210–1217 Search PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 15086–15095 Search PubMed.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 Search PubMed.
- L. Qiu, C.-R. Xu, F. Zhong, C.-Y. Hong and C.-Y. Pan, ACS Appl. Mater. Interfaces, 2016, 8, 18347–18359 Search PubMed.
- C. J. Mable, I. Canton, O. O. Mykhaylyk, B. Ustbas Gul, P. Chambon, E. Themistou and S. P. Armes, Chem. Sci., 2019, 10, 4811–4821 Search PubMed.
- S. Varlas, L. D. Blackman, H. E. Findlay, E. Reading, P. J. Booth, M. I. Gibson and R. K. O'Reilly, Macromolecules, 2018, 51, 6190–6201 Search PubMed.
- M. Garni, R. Wehr, S. Y. Avsar, C. John, C. Palivan and W. Meier, Eur. Polym. J., 2019, 112, 346–364 Search PubMed.
- S. Varlas, J. C. Foster, P. G. Georgiou, R. Keogh, J. T. Husband, D. S. Williams and R. K. O'Reilly, Nanoscale, 2019, 11, 12643–12654 Search PubMed.
- J. He, J. Cao, Y. Chen, L. Zhang and J. Tan, ACS Macro Lett., 2020, 9, 533–539 Search PubMed.
- B. T. T. Pham, D. Nguyen, V. T. Huynh, E. H. Pan, B. Shirodkar-Robinson, M. Carey, A. K. Serelis, G. G. Warr, T. Davey, C. H. Such and B. S. Hawkett, Langmuir, 2018, 34, 4255–4263 Search PubMed.
- D. Nguyen, V. Huynh, M. Lam, A. Serelis, T. Davey, O. Paravagna, C. Such and B. Hawkett, Macromol. Rapid Commun., 2021, 42, 2100008 Search PubMed.
- P. Shi, C. Gao, X. He, P. Sun and W. Zhang, Macromolecules, 2015, 48, 1380–1389 Search PubMed.
- J. Wan, B. Fan and S. H. Thang, Nanoscale Adv., 2021, 3, 3306–3315 Search PubMed.
- A. Pontes da Costa, D. R. Nunes, M. Tharaud, J. Oble, G. Poli and J. Rieger, ChemCatChem, 2017, 9, 2167–2175 Search PubMed.
- M. Tan, Y. Shi, Z. Fu and W. Yang, Polym. Chem., 2018, 9, 1082–1094 Search PubMed.
- Y. Zhang, Z. Wang, K. Matyjaszewski and J. Pietrasik, Eur. Polym. J., 2019, 110, 49–55 Search PubMed.
- K. L. Thompson, C. J. Mable, J. A. Lane, M. J. Derry, L. A. Fielding and S. P. Armes, Langmuir, 2015, 31, 4137–4144 Search PubMed.
- K. L. Thompson, C. J. Mable, A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Soft Matter, 2014, 10, 8615–8626 Search PubMed.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Macromolecules, 2014, 47, 5613–5623 Search PubMed.
- K. L. Thompson, P. Chambon, R. Verber and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 12450–12453 Search PubMed.
- J. Wang, Y. Zhu, S. Zhou, W. Wu, Q. Tong, J. Hu, S. Min, G. Hou, B. Dong and B. Song, Dyes Pigm., 2021, 190, 109353 Search PubMed.
- Y. Wang, D. Yang, Y. Hu, Y. Wang, W. J. Yang and L. Wang, Talanta, 2021, 232, 122182 Search PubMed.
- M. J. Derry, T. Smith, P. S. O'Hora and S. P. Armes, ACS Appl. Mater. Interfaces, 2019, 11, 33364–33369 Search PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, J. Mater. Chem. A, 2014, 2, 7819–7828 Search PubMed.
- Y. Ning, D. J. Whitaker, C. J. Mable, M. J. Derry, N. J. W. Penfold, A. N. Kulak, D. C. Green, F. C. Meldrum and S. P. Armes, Chem. Sci., 2018, 9, 8396–8401 Search PubMed.
- Y. Ning, L. Han, M. J. Derry, F. C. Meldrum and S. P. Armes, J. Am. Chem. Soc., 2019, 141, 2557–2567 Search PubMed.
- X. G. Qiao, P. Y. Dugas, B. Charleux, M. Lansalot and E. Bourgeat-Lami, Macromolecules, 2015, 48, 545–556 Search PubMed.
- X. G. Qiao, M. Lansalot, E. Bourgeat-Lami and B. Charleux, Macromolecules, 2013, 46, 4285–4295 Search PubMed.
- G. Delaittre, M. Save and B. Charleux, Macromol. Rapid Commun., 2007, 28, 1528–1533 Search PubMed.
- S. Sugihara, K. Sugihara, S. P. Armes, H. Ahmad and A. L. Lewis, Macromolecules, 2010, 43, 6321–6329 Search PubMed.
- A. Shahrokhinia, R. A. Scanga, P. Biswas and J. F. Reuther, Macromolecules, 2021, 54, 1441–1451 Search PubMed.
- X. Liu and W. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 2023–2028 Search PubMed.
- W.-M. Wan and C.-Y. Pan, Macromolecules, 2007, 40, 8897–8905 Search PubMed.
- S. Sue-eng, T. Boonchuwong, P. Chaiyasat, M. Okubo and A. Chaiyasat, Polymer, 2017, 110, 124–130 Search PubMed.
- Q. Xu, C. Tian, L. Zhang, Z. Cheng and X. Zhu, Macromol. Rapid Commun., 2019, 40, 1800327 Search PubMed.
- H. Li, Q. Xu, X. Xu, L. Zhang, Z. Cheng and X. Zhu, Polymers, 2020, 12, 150 Search PubMed.
- Y. Kitayama, H. Moribe, K. Kishida and M. Okubo, Polym. Chem., 2012, 3, 1555–1559 Search PubMed.
- D. Cordella, A. Debuigne, C. Jérôme, Z. Kochovski, D. Taton and C. Detrembleur, Macromol. Rapid Commun., 2016, 37, 1181–1187 Search PubMed.
- J. Sarkar, L. Xiao, A. W. Jackson, A. M. van Herk and A. Goto, Polym. Chem., 2018, 9, 4900–4907 Search PubMed.
- J. Sarkar, K. B. J. Chan and A. Goto, Polym. Chem., 2021, 12, 1060–1067 Search PubMed.
- D. B. Wright, M. A. Touve, L. Adamiak and N. C. Gianneschi, ACS Macro Lett., 2017, 6, 925–929 Search PubMed.
- S. Varlas, J. C. Foster and R. K. O'Reilly, Chem. Commun., 2019, 55, 9066–9071 Search PubMed.
- L. Zhang, C. Song, J. Yu, D. Yang and M. Xie, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5231–5238 Search PubMed.
- K.-Y. Yoon, I.-H. Lee, K. O. Kim, J. Jang, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2012, 134, 14291–14294 Search PubMed.
- I.-H. Lee, P. Amaladass, K.-Y. Yoon, S. Shin, Y.-J. Kim, I. Kim, E. Lee and T.-L. Choi, J. Am. Chem. Soc., 2013, 135, 17695–17698 Search PubMed.
- N. An, X. Chen and J. Yuan, Polym. Chem., 2021, 12, 3220–3232 Search PubMed.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 Search PubMed.
- K. M. Burridge, T. A. Wright, R. C. Page and D. Konkolewicz, Macromol. Rapid Commun., 2018, 39, 1800093 Search PubMed.
- W. Xie, L. Zhao, Y. Wei and J. Yuan, Cell Rep. Phys. Sci., 2021, 2, 100487 Search PubMed.
- Q. Xiong, X. Zhang, W. Wei, G. Wei and Z. Su, Polym. Chem., 2020, 11, 1673–1690 Search PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2019, 40, 1800279 Search PubMed.
- S. Y. Khor, J. F. Quinn, M. R. Whittaker, N. P. Truong and T. P. Davis, Macromol. Rapid Commun., 2019, 40, 1800438 Search PubMed.
- G. Cheng and J. Pérez-Mercader, Macromol. Rapid Commun., 2019, 40, 1800513 Search PubMed.
- H. Phan, V. Taresco, J. Penelle and B. Couturaud, Biomater. Sci., 2021, 9, 38–50 Search PubMed.
- S. Varlas, G. L. Maitland and M. J. Derry, Polymers, 2021, 13, 2603 Search PubMed.
- D. Le, D. Keller and G. Delaittre, Macromol. Rapid Commun., 2019, 40, 1800551 Search PubMed.
- J.-L. Six and K. Ferji, Polym. Chem., 2019, 10, 45–53 Search PubMed.
- S. Li, G. Han and W. Zhang, Polym. Chem., 2020, 11, 4681–4692 Search PubMed.
- S. Pearce and J. Perez-Mercader, Polym. Chem., 2021, 12, 29–49 Search PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Prog. Polym. Sci., 2016, 52, 1–18 Search PubMed.
- A. B. Lowe, Polymer, 2016, 106, 161–181 Search PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873–881 Search PubMed.
- S. C. Thickett and G. H. Teo, Polym. Chem., 2019, 10, 2906–2924 Search PubMed.
- M. Lansalot, J. Rieger and F. D'Agosto, in Macromolecular Self-Assembly, ed. L. Billon and O. Borisov, John Wiley & Sons, Inc., 2016, ch. 2, pp. 33–82 Search PubMed.
- J. Cao, Y. Tan, Y. Chen, L. Zhang and J. Tan, Macromol. Rapid Commun., 2021, 42, 2100498 Search PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 Search PubMed.
- P.-E. Millard, L. Barner, M. H. Stenzel, T. P. Davis, C. Barner-Kowollik and A. H. E. Müller, Macromol. Rapid Commun., 2006, 27, 821–828 Search PubMed.
- J. F. Quinn, L. Barner, E. Rizzardo and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 19–25 Search PubMed.
- R.-K. Bai, Y.-Z. You and C.-Y. Pan, Macromol. Rapid Commun., 2001, 22, 315–319 Search PubMed.
- M. Y. Khan, M.-S. Cho and Y.-J. Kwark, Macromolecules, 2014, 47, 1929–1934 Search PubMed.
- M. A. Tasdelen, Y. Y. Durmaz, B. Karagoz, N. Bicak and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3387–3395 Search PubMed.
- S. Muthukrishnan, E. H. Pan, M. H. Stenzel, C. Barner-Kowollik, T. P. Davis, D. Lewis and L. Barner, Macromolecules, 2007, 40, 2978–2980 Search PubMed.
- L. Lu, N. Yang and Y. Cai, Chem. Commun., 2005, 5287–5288 Search PubMed.
- P. Maximiano, P. V. Mendonça, J. R. C. Costa, N. L. Haworth, A. C. Serra, T. Guliashvili, M. L. Coote and J. F. J. Coelho, Macromolecules, 2016, 49, 1597–1604 Search PubMed.
- S. Shanmugam, J. Xu and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9174–9185 Search PubMed.
- M. Chen, M. J. MacLeod and J. A. Johnson, ACS Macro Lett., 2015, 4, 566–569 Search PubMed.
- J. Xu, K. Jung and C. Boyer, Macromolecules, 2014, 47, 4217–4229 Search PubMed.
- G. Liu, H. Shi, Y. Cui, J. Tong, Y. Zhao, D. Wang and Y. Cai, Polym. Chem., 2013, 4, 1176–1182 Search PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 Search PubMed.
- S. L. Brown, C. M. Rayner, S. Graham, A. Cooper, S. Rannard and S. Perrier, Chem. Commun., 2007, 2145–2147 Search PubMed.
- E. Yoshida, J. Dispersion Sci. Technol., 2020, 41, 763–770 Search PubMed.
- S. Varlas, P. G. Georgiou, P. Bilalis, J. R. Jones, N. Hadjichristidis and R. K. O'Reilly, Biomacromolecules, 2018, 19, 4453–4462 Search PubMed.
- J.-P. Fouassier and J. Lalevée, Photoinitiators for polymer synthesis: scope, reactivity, and efficiency, John Wiley & Sons, 2012 Search PubMed.
- J. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, in Controlled Radical Polymerization: Mechanisms, ed. K. Matyjaszewski, B. S. Sumerlin, N. V. Tsarevsky and J. Chiefari, American Chemical Society, 2015, ch. 13, vol. 1187, pp. 247–267 Search PubMed.
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 Search PubMed.
- C. Ding, C. Fan, G. Jiang, X. Pan, Z. Zhang, J. Zhu and X. Zhu, Macromol. Rapid Commun., 2015, 36, 2181–2185 Search PubMed.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 Search PubMed.
- Y. Jiang, N. Xu, J. Han, Q. Yu, L. Guo, P. Gao, X. Lu and Y. Cai, Polym. Chem., 2015, 6, 4955–4965 Search PubMed.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 Search PubMed.
- P. Gao, H. Cao, Y. Ding, M. Cai, Z. Cui, X. Lu and Y. Cai, ACS Macro Lett., 2016, 5, 1327–1331 Search PubMed.
- L. Cao, Q. Zhao, Q. Liu, L. Ma, C. Li, X. Wang and Y. Cai, Macromolecules, 2020, 53, 2220–2227 Search PubMed.
- J. He, Q. Xu, J. Tan and L. Zhang, Macromol. Rapid Commun., 2019, 40, 1800296 Search PubMed.
- J. Tan, J. He, X. Li, Q. Xu, C. Huang, D. Liu and L. Zhang, Polym. Chem., 2017, 8, 6853–6864 Search PubMed.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O'Reilly, Polym. Chem., 2017, 8, 2860–2871 Search PubMed.
- X. Wang, S. Man, J. Zheng and Z. An, ACS Macro Lett., 2018, 7, 1461–1467 Search PubMed.
- N. Zaquen, W. A. A. W. Azizi, J. Yeow, R. P. Kuchel, T. Junkers, P. B. Zetterlund and C. Boyer, Polym. Chem., 2019, 10, 2406–2414 Search PubMed.
- L. Zhang, L. Xie, S. Xu, R. P. Kuchel, Y. Dai, K. Jung and C. Boyer, Biomacromolecules, 2020, 21, 3887–3897 Search PubMed.
- Y. Du, S. Jia, Y. Chen, L. Zhang and J. Tan, ACS Macro Lett., 2021, 10, 297–306 Search PubMed.
- H. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 Search PubMed.
- Y.-Z. You, C.-Y. Hong, R.-K. Bai, C.-Y. Pan and J. Wang, Macromol. Chem. Phys., 2002, 203, 477–483 Search PubMed.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 Search PubMed.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 Search PubMed.
- J. Yeow, O. R. Sugita and C. Boyer, ACS Macro Lett., 2016, 5, 558–564 Search PubMed.
- V. Tkachenko, C. Matei Ghimbeu, C. Vaulot, L. Vidal, J. Poly and A. Chemtob, Polym. Chem., 2019, 10, 2316–2326 Search PubMed.
- J. Yeow, J. Xu and C. Boyer, ACS Macro Lett., 2015, 4, 984–990 Search PubMed.
- S. Han, Y. Gu, M. Ma and M. Chen, Chem. Sci., 2020, 11, 10431–10436 Search PubMed.
- J. Zhou, C. Hong and C. Pan, Mater. Chem. Front., 2017, 1, 1200–1206 Search PubMed.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 Search PubMed.
- J. Yeow, R. Chapman, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 5012–5022 Search PubMed.
- C. Lin, S. K. Katla and J. Perez-Mercader, J. Photochem. Photobiol., A, 2021, 406, 112992 Search PubMed.
- J. Yeow, S. Shanmugam, N. Corrigan, R. P. Kuchel, J. Xu and C. Boyer, Macromolecules, 2016, 49, 7277–7285 Search PubMed.
- G. Ng, J. Yeow, J. Xu and C. Boyer, Polym. Chem., 2017, 8, 2841–2851 Search PubMed.
- S. Xu, J. Yeow and C. Boyer, ACS Macro Lett., 2018, 7, 1376–1382 Search PubMed.
- A. B. Korpusik, Y. Tan, J. B. Garrison, W. Tan and B. S. Sumerlin, Macromolecules, 2021, 54, 7354–7363 Search PubMed.
- Y.-H. Ng, F. di Lena and C. L. L. Chai, Chem. Commun., 2011, 47, 6464–6466 Search PubMed.
- Y.-H. Ng, F. di Lena and C. L. L. Chai, Polym. Chem., 2011, 2, 589–594 Search PubMed.
- B. Zhang, X. Wang, A. Zhu, K. Ma, Y. Lv, X. Wang and Z. An, Macromolecules, 2015, 48, 7792–7802 Search PubMed.
- Y. Lv, Z. Liu, A. Zhu and Z. An, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 164–174 Search PubMed.
- Z. Liu, Y. Lv and Z. An, Angew. Chem., Int. Ed., 2017, 56, 13852–13856 Search PubMed.
- J. Tan, Q. Xu, X. Li, J. He, Y. Zhang, X. Dai, L. Yu, R. Zeng and L. Zhang, Macromol. Rapid Commun., 2018, 39, 1700871 Search PubMed.
- Q. Xu, Y. Zhang, X. Li, J. He, J. Tan and L. Zhang, Polym. Chem., 2018, 9, 4908–4916 Search PubMed.
- R. Chapman, A. J. Gormley, K.-L. Herpoldt and M. M. Stevens, Macromolecules, 2014, 47, 8541–8547 Search PubMed.
- R. Chapman, A. J. Gormley, M. H. Stenzel and M. M. Stevens, Angew. Chem., Int. Ed., 2016, 55, 4500–4503 Search PubMed.
- J. Tan, D. Liu, Y. Bai, C. Huang, X. Li, J. He, Q. Xu and L. Zhang, Macromolecules, 2017, 50, 5798–5806 Search PubMed.
- W.-B. Cai, D.-D. Liu, Y. Chen, L. Zhang and J.-B. Tan, Chin. J. Polym. Sci., 2021, 39, 1127–1137 Search PubMed.
- J. Tan, X. Dai, Y. Zhang, L. Yu, H. Sun and L. Zhang, ACS Macro Lett., 2019, 8, 205–212 Search PubMed.
- T. Lückerath, K. Koynov, S. Loescher, C. J. Whitfield, L. Nuhn, A. Walther, C. Barner-Kowollik, D. Y. W. Ng and T. Weil, Angew. Chem., Int. Ed., 2020, 59, 15474–15479 Search PubMed.
- N. Kohut-Svelko, R. Pirri, J. M. Asua and J. R. Leiza, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2917–2927 Search PubMed.
- T.-P. Chiu and T.-M. Don, J. Appl. Polym. Sci., 2008, 109, 3622–3630 Search PubMed.
- G. S. Misra and C. V. Gupta, Makromol. Chem., 1973, 165, 205–216 Search PubMed.
- X.-L. Sun, W.-D. He, T.-T. Pan, Z.-L. Ding and Y.-J. Zhang, Polymer, 2010, 51, 110–114 Search PubMed.
- H. Zheng, W. Bai, K. Hu, R. Bai and C. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2575–2580 Search PubMed.
- X.-L. Sun, W.-D. He, J. Li, L.-Y. Li, B.-Y. Zhang and T.-T. Pan, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6863–6872 Search PubMed.
- A. M. dos Santos, T. Le Bris, C. Graillat, F. D'Agosto and M. Lansalot, Macromolecules, 2009, 42, 946–956 Search PubMed.
- G. Liu, Q. Qiu, W. Shen and Z. An, Macromolecules, 2011, 44, 5237–5245 Search PubMed.
- H. Narain, S. M. Jagadale and N. D. Ghatge, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 1225–1238 Search PubMed.
- D. E. Cabelli and B. H. J. Bielski, J. Phys. Chem., 1983, 87, 1809–1812 Search PubMed.
- X. Dai, L. Yu, Y. Zhang, L. Zhang and J. Tan, Macromolecules, 2019, 52, 7468–7476 Search PubMed.
- O. J. Deane, O. M. Musa, A. Fernyhough and S. P. Armes, Macromolecules, 2020, 53, 1422–1434 Search PubMed.
- J. A. Pojman, D. C. Leard and W. West, J. Am. Chem. Soc., 1992, 114, 8298–8299 Search PubMed.
- R. P. Washington, W. W. West, G. P. Misra and J. A. Pojman, J. Am. Chem. Soc., 1999, 121, 7373–7380 Search PubMed.
- B. Venkataraman and P. G. Soerensen, J. Phys. Chem., 1991, 95, 5707–5712 Search PubMed.
- H. D. Foersterling, S. Muranyi and Z. Noszticzius, J. Phys. Chem., 1990, 94, 2915–2921 Search PubMed.
- B. P. Bastakoti and J. Pérez-Mercader, Angew. Chem., Int. Ed., 2017, 56, 12086–12091 Search PubMed.
- B. P. Bastakoti and J. Pérez-Mercader, Adv. Mater., 2017, 29, 1704368 Search PubMed.
- B. P. Bastakoti, S. Guragain and J. Pérez-Mercader, Chem.–Eur. J., 2018, 24, 10621–10624 Search PubMed.
- L. Hou, M. Dueñas-Díez, R. Srivastava and J. Pérez-Mercader, Commun. Chem., 2019, 2, 139 Search PubMed.
- G. Cheng and J. Pérez-Mercader, Chem, 2020, 6, 1160–1171 Search PubMed.
- J. Guo, E. Poros-Tarcali and J. Pérez-Mercader, Chem. Commun., 2019, 55, 9383–9386 Search PubMed.
- J. Guo, E. Poros-Tarcali and J. Pérez-Mercader, Front. Chem., 2021, 9, 576349 Search PubMed.
- T. J. Mason, Chem. Soc. Rev., 1997, 26, 443–451 Search PubMed.
- T. G. McKenzie, E. Colombo, Q. Fu, M. Ashokkumar and G. G. Qiao, Angew. Chem., Int. Ed., 2017, 56, 12302–12306 Search PubMed.
- S. Piogé, T. N. Tran, T. G. McKenzie, S. Pascual, M. Ashokkumar, L. Fontaine and G. Qiao, Macromolecules, 2018, 51, 8862–8869 Search PubMed.
- J. Wan, B. Fan, Y. Liu, T. Hsia, K. Qin, T. Junkers, B. M. Teo and S. H. Thang, Polym. Chem., 2020, 11, 3564–3572 Search PubMed.
- Y. Zhang, J. He, X. Dai, L. Yu, J. Tan and L. Zhang, Polym. Chem., 2019, 10, 3902–3911 Search PubMed.
- M. Sponchioni, C. T. O'Brien, C. Borchers, E. Wang, M. N. Rivolta, N. J. W. Penfold, I. Canton and S. P. Armes, Chem. Sci., 2020, 11, 232–240 Search PubMed.
- D. Le, F. Wagner, M. Takamiya, I. L. Hsiao, G. Gil Alvaradejo, U. Strähle, C. Weiss and G. Delaittre, Chem. Commun., 2019, 55, 3741–3744 Search PubMed.
- W.-M. Wan and C.-Y. Pan, Polym. Chem., 2010, 1, 1475–1484 Search PubMed.
- K. Yu, C. Bartels and A. Eisenberg, Macromolecules, 1998, 31, 9399–9402 Search PubMed.
- M. Huo, D. Li, G. Song, J. Zhang, D. Wu, Y. Wei and J. Yuan, Macromol. Rapid Commun., 2018, 39, 1700840 Search PubMed.
- X.-F. Xu, R.-M. Zhu, C.-Y. Pan, Y.-Z. You, W.-J. Zhang and C.-Y. Hong, Macromolecules, 2021, 54, 2729–2739 Search PubMed.
- L. Wang, Y. Ding, Q. Liu, Q. Zhao, X. Dai, X. Lu and Y. Cai, ACS Macro Lett., 2019, 8, 623–628 Search PubMed.
- F. Lv, Z. An and P. Wu, Macromolecules, 2020, 53, 367–373 Search PubMed.
- X. Luo, Eur. Polym. J., 2021, 158, 110639 Search PubMed.
- S.-P. Wen, J. G. Saunders and L. A. Fielding, Polym. Chem., 2020, 11, 3416–3426 Search PubMed.
- X. Chen, L. Liu, M. Huo, M. Zeng, L. Peng, A. Feng, X. Wang and J. Yuan, Angew. Chem., Int. Ed., 2017, 56, 16541–16545 Search PubMed.
- X. Chen, N. An, M. Zeng and J. Yuan, Chem. Commun., 2021, 57, 13720–13723 Search PubMed.
- X. Wang, C. A. Figg, X. Lv, Y. Yang, B. S. Sumerlin and Z. An, ACS Macro Lett., 2017, 6, 337–342 Search PubMed.
- Y. Zhang, M. Cao, G. Han, T. Guo, T. Ying and W. Zhang, Macromolecules, 2018, 51, 5440–5449 Search PubMed.
- C. Gao, S. Li, Q. Li, P. Shi, S. A. Shah and W. Zhang, Polym. Chem., 2014, 5, 6957–6966 Search PubMed.
- W. Wang, C. Gao, Y. Qu, Z. Song and W. Zhang, Macromolecules, 2016, 49, 2772–2781 Search PubMed.
- Y. Qu, S. Wang, H. Khan, C. Gao, H. Zhou and W. Zhang, Polym. Chem., 2016, 7, 1953–1962 Search PubMed.
- P. Biais, M. Engel, O. Colombani, T. Nicolai, F. Stoffelbach and J. Rieger, Polym. Chem., 2021, 12, 1040–1049 Search PubMed.
- P. Biais, O. Colombani, L. Bouteiller, F. Stoffelbach and J. Rieger, Polym. Chem., 2020, 11, 4568–4578 Search PubMed.
- P. Biais, P. Beaunier, F. Stoffelbach and J. Rieger, Polym. Chem., 2018, 9, 4483–4491 Search PubMed.
- C. Gao, J. Wu, H. Zhou, Y. Qu, B. Li and W. Zhang, Macromolecules, 2016, 49, 4490–4500 Search PubMed.
- P. Shi, Q. Li, X. He, S. Li, P. Sun and W. Zhang, Macromolecules, 2014, 47, 7442–7452 Search PubMed.
- X. He, Q. Li, P. Shi, Y. Cui, S. Li and W. Zhang, Polym. Chem., 2014, 5, 7090–7099 Search PubMed.
- Z. Song, X. He, C. Gao, H. Khan, P. Shi and W. Zhang, Polym. Chem., 2015, 6, 6563–6572 Search PubMed.
- X. Shen, F. Huo, H. Kang, S. Zhang, J. Li and W. Zhang, Polym. Chem., 2015, 6, 3407–3414 Search PubMed.
- F. Huo, S. Li, Q. Li, Y. Qu and W. Zhang, Macromolecules, 2014, 47, 2340–2349 Search PubMed.
- J. Huang, Y. Guo, S. Gu, G. Han, W. Duan, C. Gao and W. Zhang, Polym. Chem., 2019, 10, 3426–3435 Search PubMed.
- M. Huo, M. Zeng, D. Li, L. Liu, Y. Wei and J. Yuan, Macromolecules, 2017, 50, 8212–8220 Search PubMed.
- M. Huo, Y. Zhang, M. Zeng, L. Liu, Y. Wei and J. Yuan, Macromolecules, 2017, 50, 8192–8201 Search PubMed.
- X. Dai, Y. Zhang, L. Yu, X. Li, L. Zhang and J. Tan, ACS Macro Lett., 2019, 8, 955–961 Search PubMed.
- R. Bleach, B. Karagoz, S. M. Prakash, T. P. Davis and C. Boyer, ACS Macro Lett., 2014, 3, 591–596 Search PubMed.
- B. Karagoz, J. Yeow, L. Esser, S. M. Prakash, R. P. Kuchel, T. P. Davis and C. Boyer, Langmuir, 2014, 30, 10493–10502 Search PubMed.
- Y. Zhang, P. Filipczak, G. He, G. Nowaczyk, L. Witczak, W. Raj, M. Kozanecki, K. Matyjaszewski and J. Pietrasik, Polymer, 2017, 129, 144–150 Search PubMed.
- W. Zhou, Q. Qu, W. Yu and Z. An, ACS Macro Lett., 2014, 3, 1220–1224 Search PubMed.
- L. Upadhyaya, M. Semsarilar, S. Nehache, D. Cot, R. Fernández-Pacheco, G. Martinez, R. Mallada, A. Deratani and D. Quemener, Macromolecules, 2016, 49, 7908–7916 Search PubMed.
- D. Nguyen, V. Huynh, N. Pham, B. Pham, A. Serelis, T. Davey, C. Such and B. Hawkett, Macromol. Rapid Commun., 2019, 40, 1800402 Search PubMed.
- L. Upadhyaya, C. Egbosimba, X. Qian, R. Wickramasinghe, R. Fernández-Pacheco, I. M. Coelhoso, C. A. M. Portugal, J. G. Crespo, D. Quemener and M. Semsarilar, Macromol. Rapid Commun., 2019, 40, 1800333 Search PubMed.
- S. Samanta, S. L. Banerjee, K. Bhattacharya and N. K. Singha, ACS Appl. Mater. Interfaces, 2021, 13, 36307–36319 Search PubMed.
- B. Fan, Y. Liu, J. Wan, S. Crawford and S. H. Thang, ACS Mater. Lett., 2020, 2, 492–498 Search PubMed.
- Y. Zheng, Y. Huang, Z. M. Abbas and B. C. Benicewicz, Polym. Chem., 2016, 7, 5347–5350 Search PubMed.
- Y. Zheng, Y. Huang, Z. M. Abbas and B. C. Benicewicz, Polym. Chem., 2017, 8, 370–374 Search PubMed.
- X. G. Qiao, O. Lambert, J. C. Taveau, P. Y. Dugas, B. Charleux, M. Lansalot and E. Bourgeat-Lami, Macromolecules, 2017, 50, 3796–3806 Search PubMed.
- C. J. Mable, R. R. Gibson, S. Prevost, B. E. McKenzie, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 16098–16108 Search PubMed.
- J. Tan, X. Zhang, D. Liu, Y. Bai, C. Huang, X. Li and L. Zhang, Macromol. Rapid Commun., 2017, 38, 1600508 Search PubMed.
- J. Tan, D. Liu, X. Zhang, C. Huang, J. He, Q. Xu, X. Li and L. Zhang, RSC Adv., 2017, 7, 23114–23121 Search PubMed.
- L. D. Blackman, S. Varlas, M. C. Arno, A. Fayter, M. I. Gibson and R. K. O'Reilly, ACS Macro Lett., 2017, 6, 1263–1267 Search PubMed.
- F. H. Sobotta, F. Hausig, D. O. Harz, S. Hoeppener, U. S. Schubert and J. C. Brendel, Polym. Chem., 2018, 9, 1593–1602 Search PubMed.
- F. H. Sobotta, M. T. Kuchenbrod, F. V. Gruschwitz, G. Festag, P. Bellstedt, S. Hoeppener and J. C. Brendel, Angew. Chem., Int. Ed., 2021, 60, 24716–24723 Search PubMed.
- B. Le Droumaguet and K. Velonia, Angew. Chem., Int. Ed., 2008, 47, 6263–6266 Search PubMed.
- C. Ma, X. Liu, G. Wu, P. Zhou, Y. Zhou, L. Wang and X. Huang, ACS Macro Lett., 2017, 6, 689–694 Search PubMed.
- T. Lueckerath, T. Strauch, K. Koynov, C. Barner-Kowollik, D. Y. W. Ng and T. Weil, Biomacromolecules, 2019, 20, 212–221 Search PubMed.
- H. Sun, W. Cao, N. Zang, T. D. Clemons, G. M. Scheutz, Z. Hu, M. P. Thompson, Y. Liang, M. Vratsanos, X. Zhou, W. Choi, B. S. Sumerlin, S. I. Stupp and N. C. Gianneschi, Angew. Chem., Int. Ed., 2020, 59, 19136–19142 Search PubMed.
- L. Luppi, T. Babut, E. Petit, M. Rolland, D. Quemener, L. Soussan, M. A. Moradi and M. Semsarilar, Polym. Chem., 2019, 10, 336–344 Search PubMed.
- T. P. T. Dao, L. Vezenkov, G. Subra, M. Amblard, M. In, J.-F. Le Meins, F. Aubrit, M.-A. Moradi, V. Ladmiral and M. Semsarilar, Macromolecules, 2020, 53, 7034–7043 Search PubMed.
- T. P. T. Dao, L. Vezenkov, G. Subra, V. Ladmiral and M. Semsarilar, Polym. Chem., 2021, 12, 113–121 Search PubMed.
- V. Ladmiral, A. Charlot, M. Semsarilar and S. P. Armes, Polym. Chem., 2015, 6, 1805–1816 Search PubMed.
- K. Bauri, A. Narayanan, U. Haldar and P. De, Polym. Chem., 2015, 6, 6152–6162 Search PubMed.
- K. Bauri, B. Maiti and P. De, Macromol. Symp., 2016, 369, 101–107 Search PubMed.
- Z. An, Q. Shi, W. Tang, C.-K. Tsung, C. J. Hawker and G. D. Stucky, J. Am. Chem. Soc., 2007, 129, 14493–14499 Search PubMed.
- N. Busatto, V. Stolojan, M. Shaw, J. L. Keddie and P. J. Roth, Macromol. Rapid Commun., 2019, 40, 1800346 Search PubMed.
- J. Rieger, C. Grazon, B. Charleux, D. Alaimo and C. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2373–2390 Search PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 Search PubMed.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 Search PubMed.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 Search PubMed.
- N. J. W. Penfold, J. R. Whatley and S. P. Armes, Macromolecules, 2019, 52, 1653–1662 Search PubMed.
- T. N. Tran, S. Piogé, L. Fontaine and S. Pascual, Macromol. Rapid Commun., 2020, 41, 2000203 Search PubMed.
- N. Audureau, F. Coumes, J.-M. Guigner, T. P. T. Nguyen, C. Ménager, F. Stoffelbach and J. Rieger, Polym. Chem., 2020, 11, 5998–6008 Search PubMed.
- M. J. Derry, O. O. Mykhaylyk, A. J. Ryan and S. P. Armes, Chem. Sci., 2018, 9, 4071–4082 Search PubMed.
- L. P. D. Ratcliffe, M. J. Derry, A. Ianiro, R. Tuinier and S. P. Armes, Angew. Chem., Int. Ed., 2019, 58, 18964–18970 Search PubMed.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2015, 54, 1279–1283 Search PubMed.
- N. J. W. Penfold, J. R. Lovett, N. J. Warren, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2016, 7, 79–88 Search PubMed.
- S. M. North and S. P. Armes, Polym. Chem., 2020, 11, 2147–2156 Search PubMed.
- X.-F. Xu, C.-Y. Pan, W.-J. Zhang and C.-Y. Hong, Macromolecules, 2019, 52, 1965–1975 Search PubMed.
- L. Yu, Y. Zhang, X. Dai, Q. Xu, L. Zhang and J. Tan, Chem. Commun., 2019, 55, 11920–11923 Search PubMed.
- M. Zeng, M. Huo, Y. Feng and J. Yuan, Macromol. Rapid Commun., 2018, 39, 1800291 Search PubMed.
- S. L. Canning, T. J. Neal and S. P. Armes, Macromolecules, 2017, 50, 6108–6116 Search PubMed.
- S. M. North and S. P. Armes, Polym. Chem., 2021, 12, 5842–5850 Search PubMed.
- S. Guan, C. Zhang, W. Wen, T. Qu, X. Zheng, Y. Zhao and A. Chen, ACS Macro Lett., 2018, 7, 358–363 Search PubMed.
- Q. Ye, M. Huo, M. Zeng, L. Liu, L. Peng, X. Wang and J. Yuan, Macromolecules, 2018, 51, 3308–3314 Search PubMed.
- A. Bagheri, C. Boyer and M. Lim, Macromol. Rapid Commun., 2019, 40, 1800510 Search PubMed.
- E. Chaabouni, V. Tkachenko, L. Vidal, N. Allouche and A. Chemtob, Eur. Polym. J., 2021, 156, 110633 Search PubMed.
- S. Xu, G. Ng, J. Xu, R. P. Kuchel, J. Yeow and C. Boyer, ACS Macro Lett., 2017, 6, 1237–1244 Search PubMed.
- Y. Zhou, Z. Wang, Y. Wang, L. Li, N. Zhou, Y. Cai, Z. Zhang and X. Zhu, Polym. Chem., 2020, 11, 5619–5629 Search PubMed.
- J. R. Lovett, N. J. Warren, S. P. Armes, M. J. Smallridge and R. B. Cracknell, Macromolecules, 2016, 49, 1016–1025 Search PubMed.
- F. Khakzad, A. R. Mahdavian, H. Salehi-Mobarakeh and M. H. Sharifian, J. Colloid Interface Sci., 2018, 515, 58–69 Search PubMed.
- C. Chen, F. Richter, J. Zhang, C. Guerrero-Sanchez, A. Traeger, U. S. Schubert, A. Feng and S. H. Thang, Eur. Polym. J., 2021, 160, 110777 Search PubMed.
- C. Guerrero-Sanchez, R. M. Paulus, M. W. M. Fijten, M. J. de la Mar, R. Hoogenboom and U. S. Schubert, Appl. Surf. Sci., 2006, 252, 2555–2561 Search PubMed.
- A. A. Cockram, R. D. Bradley, S. A. Lynch, P. C. D. Fleming, N. S. J. Williams, M. W. Murray, S. N. Emmett and S. P. Armes, React. Chem. Eng., 2018, 3, 645–657 Search PubMed.
- C. Guerrero-Sanchez, D. J. Keddie, S. Saubern and J. Chiefari, ACS Comb. Sci., 2012, 14, 389–394 Search PubMed.
- M. Wang, J. Zhang, C. Guerrero-Sanchez, U. S. Schubert, A. Feng and S. H. Thang, ACS Comb. Sci., 2019, 21, 643–649 Search PubMed.
- D. Liu, W. Cai, L. Zhang, C. Boyer and J. Tan, Macromolecules, 2020, 53, 1212–1223 Search PubMed.
- M. A. Touve, D. B. Wright, C. Mu, H. Sun, C. Park and N. C. Gianneschi, Macromolecules, 2019, 52, 5529–5537 Search PubMed.
- J. Peng, C. Tian, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2017, 8, 1495–1506 Search PubMed.
- S. Parkinson, N. S. Hondow, J. S. Conteh, R. A. Bourne and N. J. Warren, React. Chem. Eng., 2019, 4, 852–861 Search PubMed.
- N. Zaquen, J. Yeow, T. Junkers, C. Boyer and P. B. Zetterlund, Macromolecules, 2018, 51, 5165–5172 Search PubMed.
- N. Zaquen, H. Zu, A. M. N. B. P. H. A. Kadir, T. Junkers, P. B. Zetterlund and C. Boyer, ACS Appl. Polym. Mater., 2019, 1, 1251–1256 Search PubMed.
- N. Zaquen, A. M. N. B. P. H. A. Kadir, A. Iasa, N. Corrigan, T. Junkers, P. B. Zetterlund and C. Boyer, Macromolecules, 2019, 52, 1609–1619 Search PubMed.
- S. Parkinson, S. T. Knox, R. A. Bourne and N. J. Warren, Polym. Chem., 2020, 11, 3465–3474 Search PubMed.
- B. Karagoz, C. Boyer and T. P. Davis, Macromol. Rapid Commun., 2014, 35, 417–421 Search PubMed.
- E. Lobry, A. F. Cardozo, L. Barthe, J.-F. Blanco, H. Delmas, S. Chen, F. Gayet, X. Zhang, M. Lansalot, F. D'Agosto, R. Poli, E. Manoury and C. Julcour, J. Catal., 2016, 342, 164–172 Search PubMed.
- A. F. Cardozo, C. Julcour, L. Barthe, J.-F. Blanco, S. Chen, F. Gayet, E. Manoury, X. Zhang, M. Lansalot, B. Charleux, F. D'Agosto, R. Poli and H. Delmas, J. Catal., 2015, 324, 1–8 Search PubMed.
- S. Chen, A. F. Cardozo, C. Julcour, J.-F. Blanco, L. Barthe, F. Gayet, M. Lansalot, F. D'Agosto, H. Delmas, E. Manoury and R. Poli, Polymer, 2015, 72, 327–335 Search PubMed.
- X. Zhang, A. F. Cardozo, S. Chen, W. Zhang, C. Julcour, M. Lansalot, J.-F. Blanco, F. Gayet, H. Delmas, B. Charleux, E. Manoury, F. D'Agosto and R. Poli, Chem.–Eur. J., 2014, 20, 15505–15517 Search PubMed.
- A. Joumaa, F. Gayet, E. J. Garcia-Suarez, J. Himmelstrup, A. Riisager, R. Poli and E. Manoury, Polymers, 2020, 12, 1107 Search PubMed.
- M. Zeng, X. Li, Y. Zhang, X. Chen, X. Sui and J. Yuan, Polymer, 2020, 206, 122853 Search PubMed.
- K. L. Thompson, J. A. Lane, M. J. Derry and S. P. Armes, Langmuir, 2015, 31, 4373–4376 Search PubMed.
- M. Zeng, S. Zhou, X. Sui and J. Yuan, Chin. J. Chem., 2021, 39, 3448–3454 Search PubMed.
- K. L. Thompson, L. A. Fielding, O. O. Mykhaylyk, J. A. Lane, M. J. Derry and S. P. Armes, Chem. Sci., 2015, 6, 4207–4214 Search PubMed.
- M. J. Rymaruk, K. L. Thompson, M. J. Derry, N. J. Warren, L. P. D. Ratcliffe, C. N. Williams, S. L. Brown and S. P. Armes, Nanoscale, 2016, 8, 14497–14506 Search PubMed.
- S. J. Hunter and S. P. Armes, Langmuir, 2020, 36, 15463–15484 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |