
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi-catalyzed asymmetric hydrophosphinylation of conjugated enynes and mechanistic studies†
Ya-Qian
Zhang‡
,
Xue-Yu
Han‡
,
Yue
Wu
,
Peng-Jia
Qi
,
Qing
Zhang
and
Qing-Wei
Zhang
 *
*
Department of Chemistry, University of Science and Technology of China, Hefei 230026, China. E-mail: qingweiz@ustc.edu.cn
First published on 10th March 2022
Abstract
The catalytic asymmetric synthesis of P-stereogenic phosphines is an efficient strategy to access structurally diverse chiral phosphines that could serve as organocatalysts and ligands to transition metals and motifs of antiviral drugs. Herein, we describe a Ni catalyzed highly regio and enantioselective hydrophosphinylation reaction of secondary phosphine oxides and enynes. This method afforded a plethora of alkenyl phosphine oxides which could serve as valuable precursors to bidentate ligands. A new type of mechanism was discovered by combined kinetic studies and density functional theory (DFT) calculations, which was opposed to the widely accepted Chalk–Harrod type mechanism. Notably, the alkene moiety which could serve as a directing group by coordinating with the Ni catalyst in the transition state, plays a vital role in determining the reactivity, regio and enantioselectivity.
Introduction
P-Stereogenic organophosphines have served as an important type of ligand to transition metals as well as organocatalysts in their own right (Fig. 1).1 The development of this type of ligand/catalyst has resulted in fruitful achievements in a number of highly enantioselective reactions, and culminated in industrial scale synthesis of several important chiral intermediates and medicines.2 In addition, P-stereogenic phosphines have found wide applications in organic materials science and medicinal chemistry.3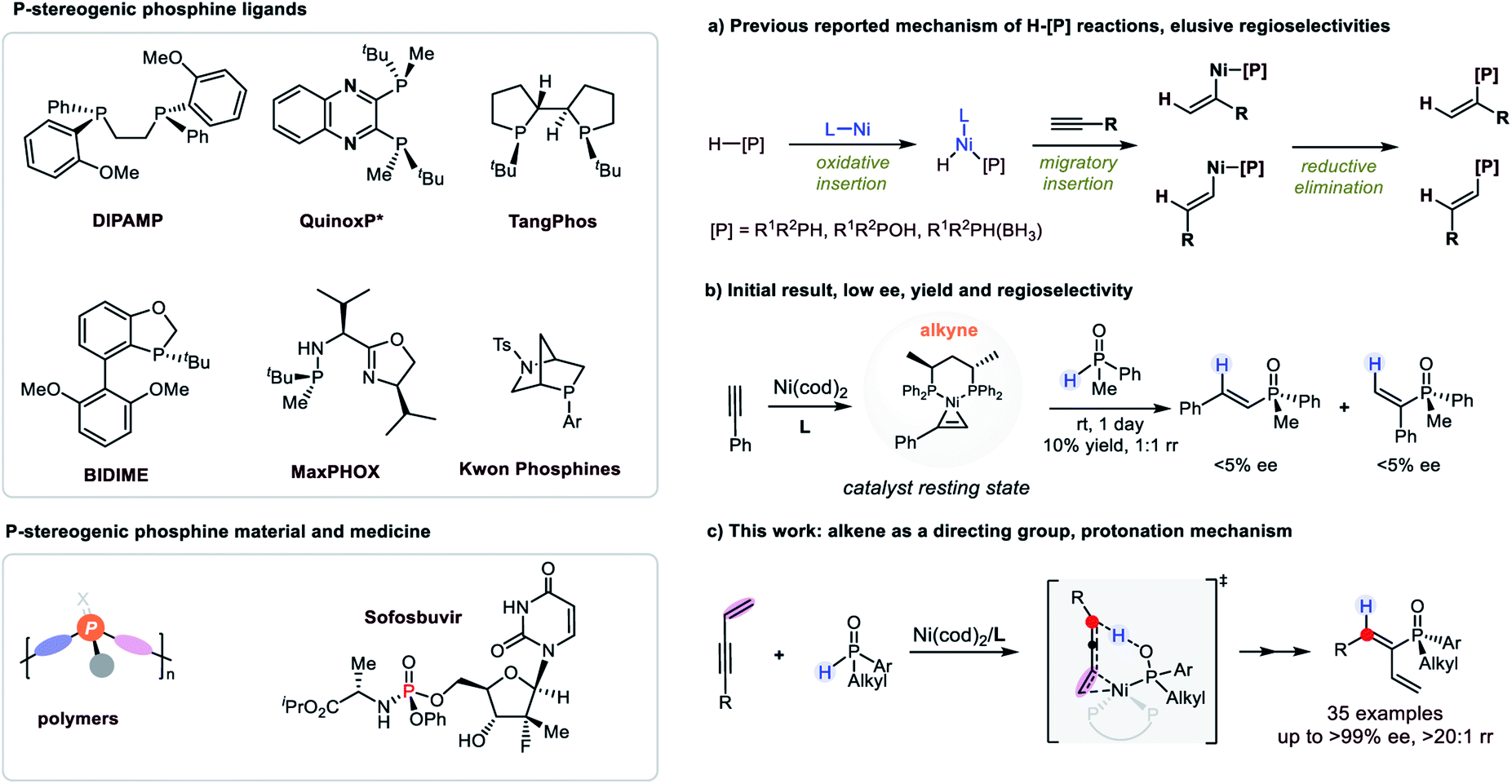 | ||
| Fig. 1 P-Stereogenic phosphines and Ni-catalyzed H–[P] reactions. (a) Chalk–Harrod type mechanism, (b) initial result, (c) this work. | ||
However, the synthesis of P-chiral compounds is challenging.4 The absence of P-stereogenic phosphine precursors in nature has impeded direct access to chiral phosphines by chiral pool syntheses. As a result, catalytic asymmetric syntheses of chiral phosphines have emerged in the last two decades. Among them, the hydrophosphination/hydrophosphinylation reactions (H–P reactions) of secondary phosphines or their derivatives with alkenes or alkynes would be the most straightforward and atom economic strategy. However, the enantioselective reactions were achieved with limited success.5 In addition, the control of P-chirality was much more challenging than carbon chiralities as exemplified by Leung’s,6 Dong’s,7 Wang’s8 and Yin's9 studies, which achieved excellent enantioselectivities of carbon stereocenters but suffered from low d.r. when racemic secondary phosphine oxides were used.
On the other hand, the control of regioselectivity of the H–P reactions of unactivated alkenes or alkynes was elusive.10 Typically, transition metals were introduced as catalysts to achieve better regioselectivities compared with uncatalyzed ones. And the mechanism of transition metal catalyzed H–P reactions was thoroughly investigated to proceed via the Chalk–Harrod type mechanism, i.e. metal catalyzed H–P oxidative insertion, hydrometallation and reductive elimination.11 Ni was also a decent catalyst for H–P reactions which were recognized to comply with the Chalk–Harrod type mechanism.12 However, during our research, phenomena that contradicted the reported mechanism were observed and herein we disclose a new finding in a Ni-catalyzed regio and enantioselective hydrophosphinylation reaction, which was explicitly supported by detailed mechanistic studies combining experimental results and DFT calculations (Fig. 1c).
Results and discussion
Initially, hydrophosphinylation was investigated with phenylacetylene and phenylmethyl phosphine oxide 1b under the catalysis of Ni. However, low ee and r.r. were obtained after extensive screening. Typically, the reaction could only give a 10% NMR yield after 1 day at r.t. with poor ee and r.r. (Fig. 1b) under the catalysis of a Ni complex. In striking contrast, when enyne 2a was used instead of simple alkynes, the desired product was obtained with high ee and regioselectivies. After extensive screening of the reaction parameters, the optimal reaction conditions were established as 1 equiv. enyne, 2 equiv. SPO, Ni(cod)2 (10 mol%), (S,S)-BDPP (12 mol%), tBuCO2K (2 equiv.) in mesitylene (1.0 mL) at −20 °C. Under these reaction conditions, 3aa was obtained in 89% yield and 91% ee (see the ESI† for details). The recovered SPO (1a) could be obtained in 76% yield along with 76% ee, indicating that the SPO was configurationally stable to a large extent under the reaction conditions.The scope examination of SPOs (Fig. 2) revealed that the reactivity and enantioselectivity of the reaction were significantly affected by the steric hindrance of the alkyl substituent attached (3ba–3ga). Generally, the bulkier the alkyl substituent, the less reactive the reaction and the higher is the ee. SPOs with phenyl primary alkyl and allyl substituents could all afford corresponding tertiary phosphine oxides 3aa–3ga with both high yields and ee (53–93% yield and 85–93% ee). The aryl or heteroaryl substituents on SPOs tested in Fig. 2 were well tolerated in this reaction, giving products 3ha–3na in moderate to high yields (53–90%) and excellent enantioselectivities (88–95% ee). Among them, the electron rich substrates were less reactive, affording 3ha and 3ia in 68% and 53% yield respectively with excellent enantioselectivities (92% and 95% ee). The reactivities of these SPOs roughly followed such a trend that the more electron donating the aryl substituent was, the less reactive the reaction was, and vice versa. This phenomenon is pivotal in elucidating the reaction mechanism and will be discussed further in the following part. In addition, the absolute configuration of product 3ka was identified by X-ray crystallography analysis (PR).
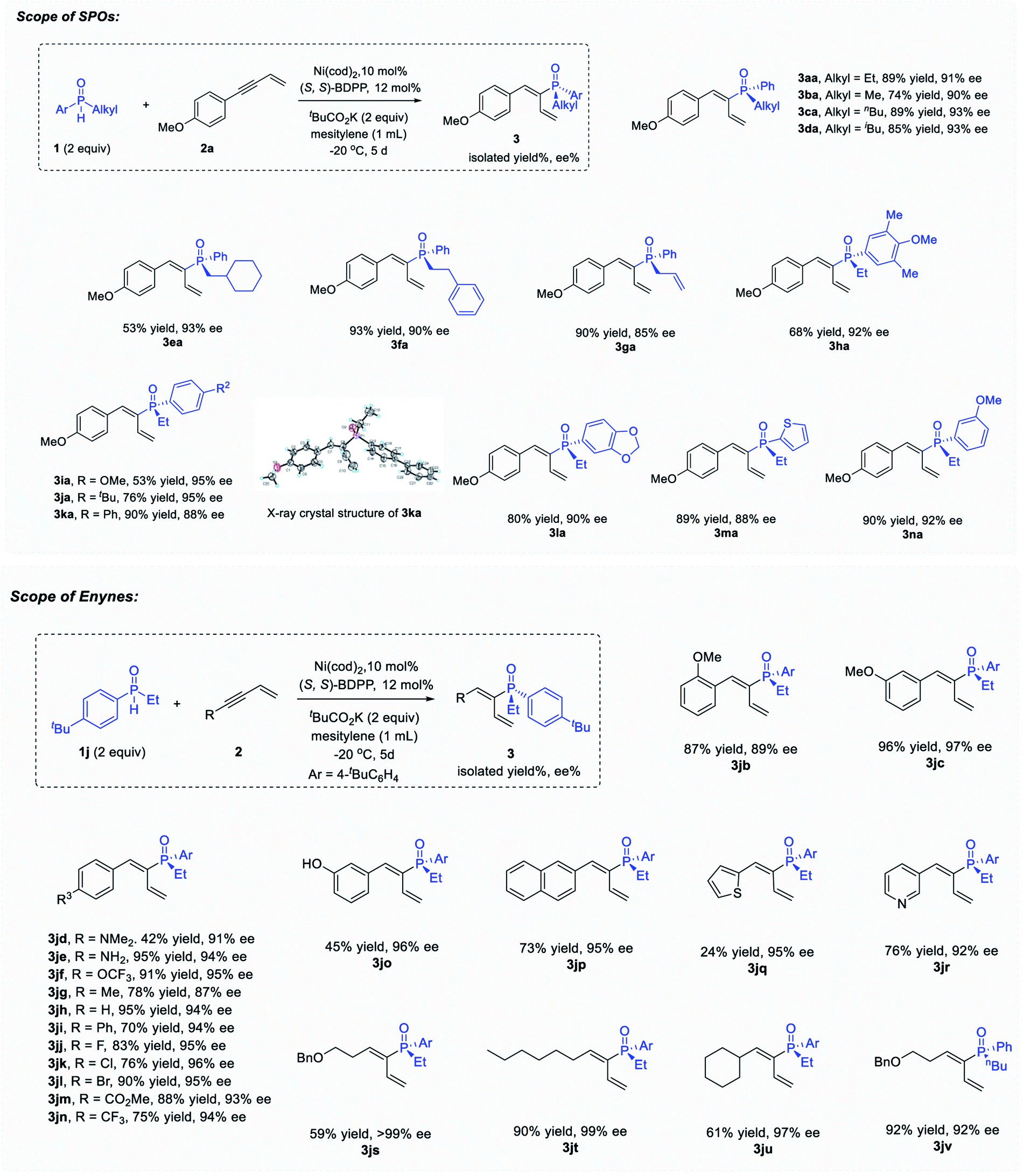 | ||
| Fig. 2 Substrate scope of SPOs and enynes. | ||
The scope of enynes was then examined with SPO 1j (Fig. 2). Aryl enynes with both electron-donating and electron-withdrawing groups, as well as alkyl enynes were all tolerated, providing products 3jb–3jv in moderate to high yields (24–96%) and excellent enantioselectivities (87–99% ee). Generally, the electron rich enynes reacted faster than the electron poor ones which was opposite to that of SPO. It is worth noting that arylbromide that could facilely undergo oxidative addition in the presence of Ni(0) survived the reaction conditions and gave product 3jl in both high yield (90%) and ee (95%). meta-Hydroxyl phenyl enyne with an acidic proton could also give the desired product 3jo during the reaction in 96% ee albeit in moderate yield (45%). Enynes with different alkyl groups could also undergo the hydrophosphinylation reaction, furnishing tertiary alkenyl phosphine oxides 3js–3jv with excellent ee (92–99%) and moderate to high yields (59–92%).
Additionally, this reaction could be accomplished at the 1 mmol-scale (Scheme 1) with comparable yield and enantioselectivity (3aa, 82% yield, and 89% ee). Further transformations of these P-chiral products were investigated (Scheme 1b and 1c). For example, reduction of 3aa was realized to afford the P-chiral phosphine–BH3 adduct 4 in 60% yield with 88% ee.13 The intramolecular olefin metathesis reaction of 3ga under the catalysis of the Hoveyda–Grubbs (II) catalyst proceeded smoothly at room temperature to afford the corresponding cyclization product 5 within 5 min without erosion of the P-chirality (92% yield and 88% ee).
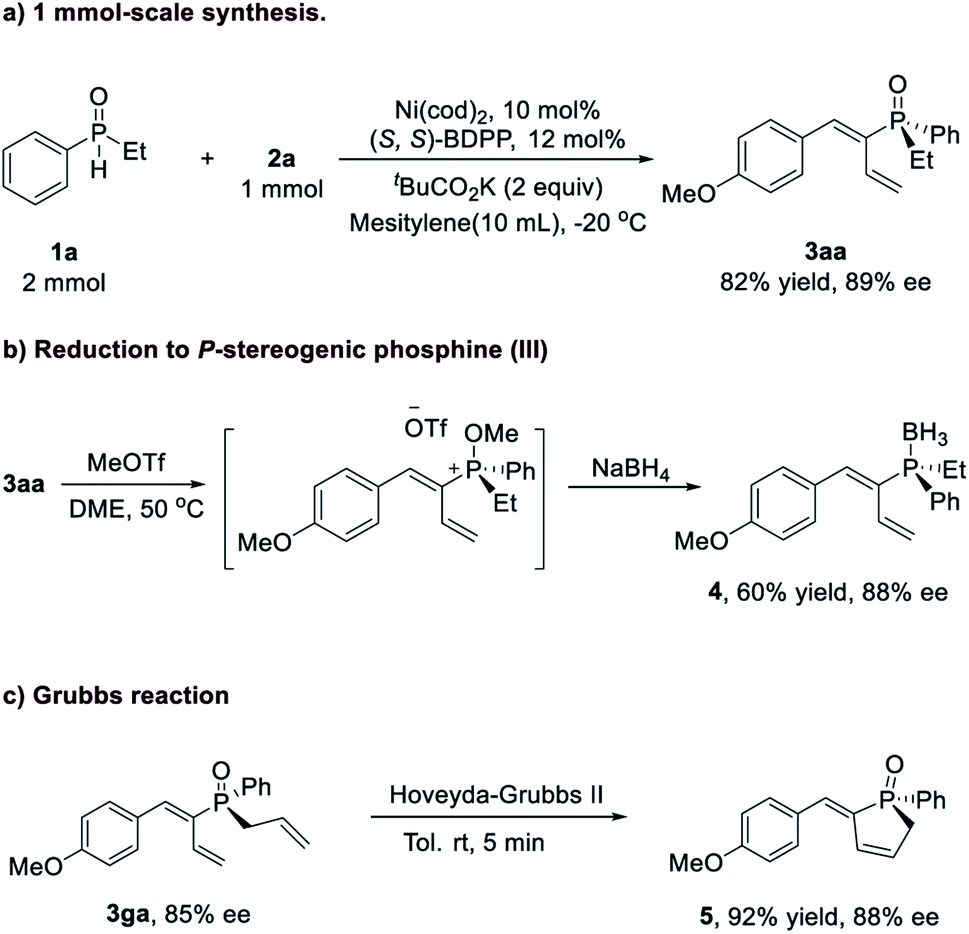 | ||
| Scheme 1 Transformations of the TPO product. (a) 1 mmol scale reaction, (b) reduction of the phosphine oxide, (c) Grubbs reaction. | ||
Identification of the catalyst resting state
In order to probe the mechanism of the reaction, we initially attempted to identify the resting state of the catalyst. Thus, the model reaction as well as the control experiments were carried out in benzene-d6 at room temperature and the aliquots of each system were determined by 31P NMR analysis. Product 3aa and substrate 1a in the presence of tBuCO2K were initially tested for comparison, and both compounds appeared as a singlet at δ 32.0 ppm and δ 25.0 ppm in the 31P NMR spectra, respectively (Fig. 3Ia and b). The aliquot of the reaction mixture of the model reaction after 10 min and 6 hours was then tested, and two new doublets appearing at 34.6 ppm (J = 20.1 Hz) and 35.6 ppm (J = 20.1 Hz) were observed (Fig. 3Ic and d). The two new doublets could probably be attributed to the resting state of the catalyst (A).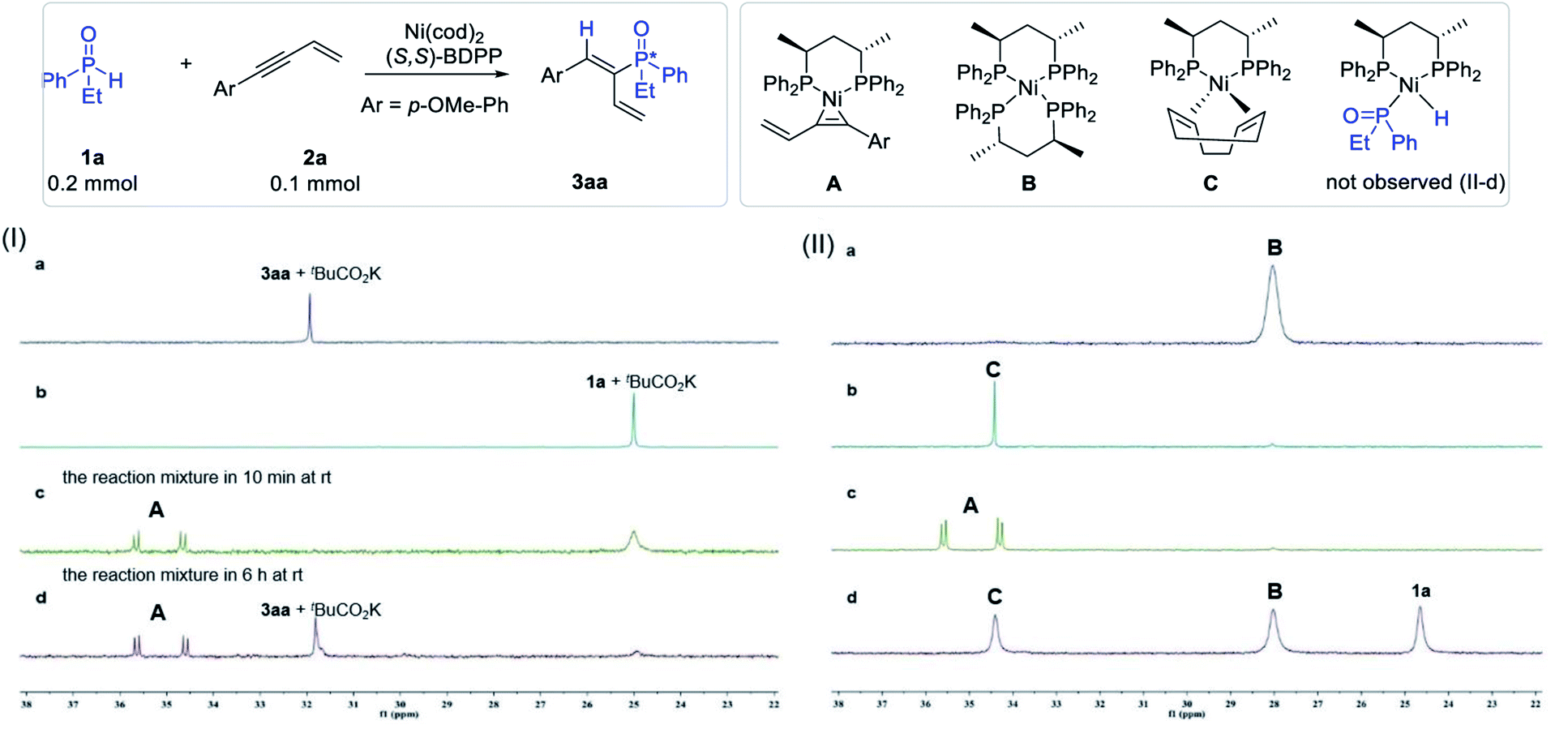 | ||
Fig. 3 Identification of possible intermediates in the reaction mixture. (I) 31P NMR spectra of (a) 3aa + tBuCO2K, (b) 1a + tBuCO2K, (c) the reaction mixture after 10 min at rt, and (d) the reaction mixture after 6 h at rt. (II) 31P NMR spectra of (a) (S,S)-BDPP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni(cod)2 = 2:1; (b) (S,S)-BDPP:Ni(cod)2 = 1:1; (c) (S,S)-BDPP:Ni(cod)2:2a = 1:1:1; (d) (S,S)-BDPP:Ni(cod)2:1a = 1:1:1. :Ni(cod)2 = 2:1; (b) (S,S)-BDPP:Ni(cod)2 = 1:1; (c) (S,S)-BDPP:Ni(cod)2:2a = 1:1:1; (d) (S,S)-BDPP:Ni(cod)2:1a = 1:1:1. | ||
To identify the structure of the catalyst resting state corresponding to the new peaks, three possible combinations that could lead to the formation of the complex were proposed, and the control experiments in benzene-d6 were carried out and monitored by 31P NMR. As shown in Fig. 3II, when the Ni(cod)2 catalyst was mixed with the (S,S)-BDPP ligand in both 1:2 and 1:1 ratios in benzene-d6, NiL2 (B, δ 28.0 ppm) and Ni(cod)L (C, δ 34.5 ppm) were observed (Fig. 3IIa and b). Delightfully, the two doublets at δ 35.6 ppm and δ 34.6 ppm corresponding to the resting state of the catalyst in the reaction mixture were observed when Ni(cod)2, (S,S)-BDPP and 2a were introduced in a 1:1:1 ratio (Fig. 3IIc). The structure of the complex was assigned as π-complex A by both 31P and 1H NMR (see the ESI†).
Since secondary phosphine oxides were reported to participate in oxidative insertion with transition metals to form a metal hydride species, we also carried out the experiment of complex C and SPO 1a to verify the validity of the mechanism in our catalytic system. Thus, Ni(cod)2, (S,S)-BDPP and 1a in a 1:1:1 ratio were stirred in benzene-d6 overnight. 31P NMR analysis revealed the presence of both B and C in the mixture (Fig. 3IId). However, no new peak that may be attributed to the coordination of 1a with Ni, or the possible oxidative insertion product P–Ni–H was observed. When enyne 2a was added to the mixture, π-complex A could be observed again accompanying the disappearance of both B and C. These experiments indicated that the formation of species B and C in the presence of cyclooctadiene (cod) was reversible and both species could be transformed to A in the presence of enyne 2a.
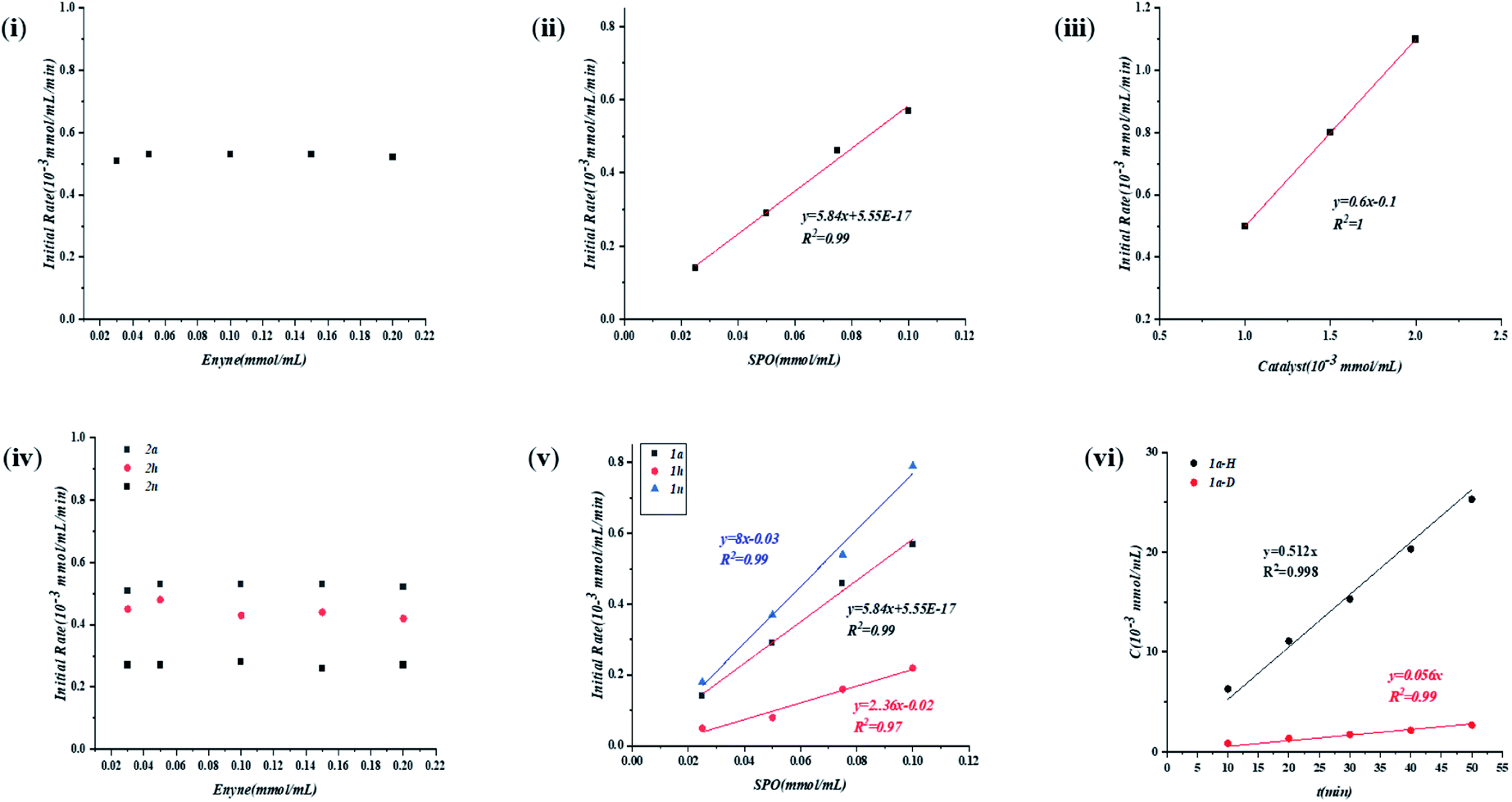 | ||
| Fig. 4 Dependence of the initial rate on (i) 2a, (ii) 1a and (iii) catalyst; the electronic effects of the substituent on enynes and SPOs: dependence of the initial rate (iv) on enynes and (v) SPOs, and (vi) deuterium experiment. | ||
Identification of the rate-limiting step of the catalytic reaction
Subsequently, to gain detailed information on the mechanism, we carried out kinetic studies to resolve the reaction order with respect to each reactant. As shown in Fig. 4, the reaction was zero-order with respect to enyne 2a (i) and first order with respect to both SPO 1a (ii) and precatalyst C (iii). The reaction should also be first order with respect to catalyst resting state A, which was formed quickly upon the addition of excess 2a.The electronic effects of the substituent on both enynes and SPOs were then examined (Fig. 4iv and v). It is interesting to find that although the reaction rate was irrelevant to the concentration of substrate 2a, there was a rate dependence on the electronic properties of enynes (iv). The more electron rich the aryl enyne, the higher the reaction rate, corresponding to a negative slope of the Hammett plot (ρ < 0). These seemingly contradictory results of both the experiments might be explained by the hypothesis that substrate 2a was transformed to an intermediate (A) which participated in the rate limiting step. The reaction also exhibited a rate dependence on the electronic properties of SPOs (v). Conversely, the more electron rich the SPO, the lower the acidity of the SPO as well as the reaction rates, corresponding to a positive slop of the Hammett plot (ρ > 0). This illustrated that both the catalyst resting state A and SPO were involved in the rate limiting step which was consistent with the above results (Fig. 4ii and iii). The inverse correlation of the electronic properties of the two substrates with the reactivity of the reaction i.e. the rate limiting protonation, suggested an acid–base neutralization type mechanism. A kinetic isotope effect experiment was then performed and the KIE from parallel reactions was determined to be 9.1 (vi, please see the ESI† for details). The large KIE was again supportive of such a mechanism, contradicting a rate limiting oxidative insertion or reduction elimination process which usually exhibits a smaller KIE.
DFT calculations
The Ni-catalyzed hydrophosphinylation reaction of phenylmethyl phosphine oxide 1b and methyl enyne 2w with (S,S)-BDPP as the ligand was selected for the density functional theory calculations (Fig. 5; see the ESI† for details). Although DFT calculations of nickel catalyzed hydrophosphinylation of the alkyne reaction were performed by Ananikov12etal. to proceed through the Chalk–Harrod type mechanism, the reaction mechanism in our study was apparently different based on the above experimental results. Herein, we present our findings in the mechanistic studies by using DFT calculations to rationalize the high reactivity, regio and enantioselectivity of the reaction (Fig. 5).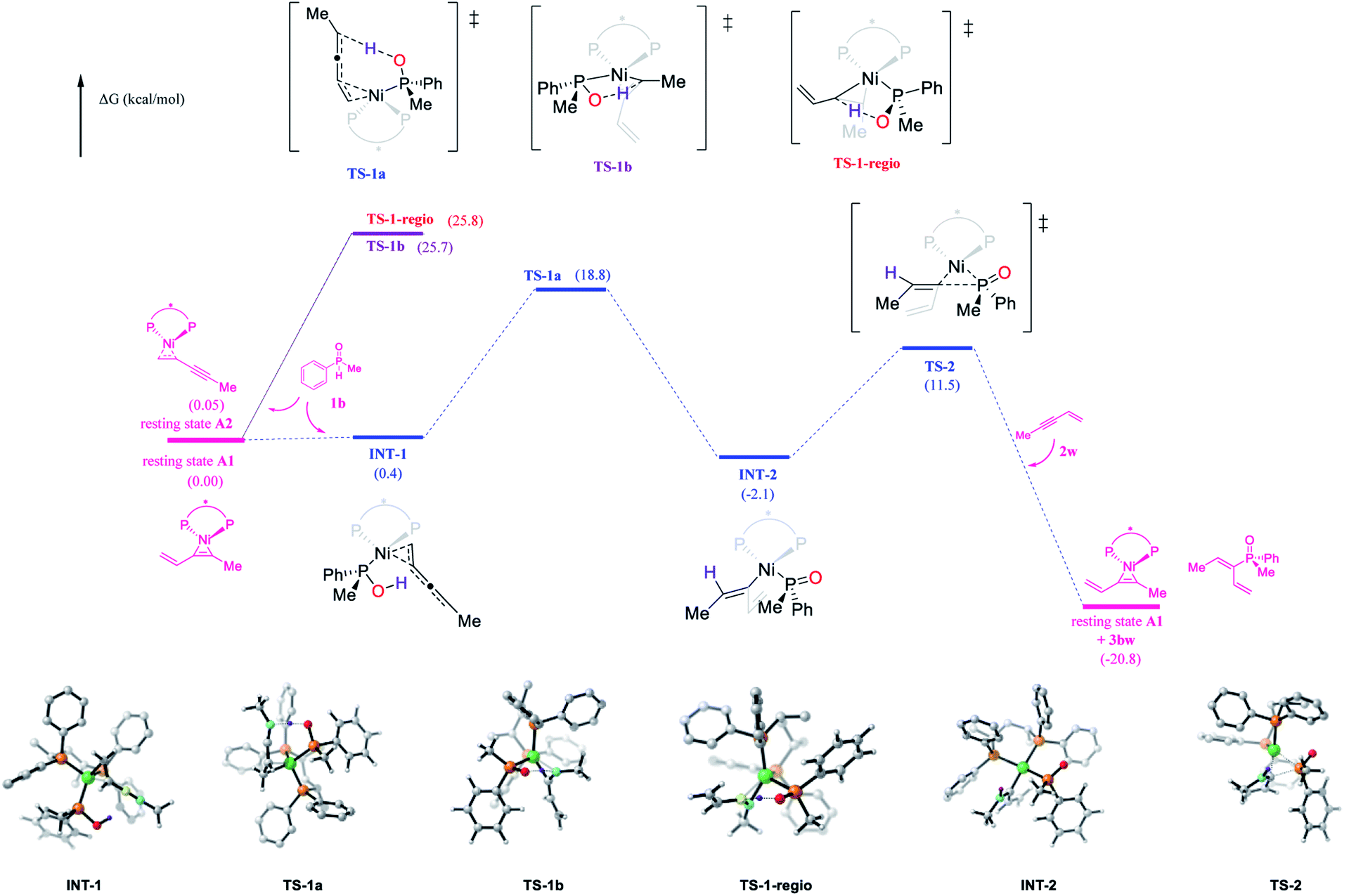 | ||
| Fig. 5 Free energy profile for the new reaction pathway. Computed at the SMD(mesitylene)/M06/6-311++G(d,p)/SDD//B3LYP-D3(BJ)/6-31G(d)/LANL2DZ level. | ||
The catalyst resting state generated from Ni(cod)2 and methyl enyne which has been identified by NMR studies was used as the starting point. To our surprise, two isomers A1 and A2 with alkene and alkyne functional groups coordinating with nickel respectively were located with almost the same Gibbs free energy (0 vs. 0.05 kcal mol−1). This result contradicted the NMR studies above in which only one conformer was observed. Further NMR studies to elucidate the new catalyst resting state were carried out using alkyl enyne 2u with Ni(cod)2 as a comparison with the result above. Gratifyingly, two pairs of doublets in a roughly 1:1 ratio which could be attributed to A1 and A2 were detected by 31P NMR (please see the ESI†), which was in perfect match with the DFT calculations.
The key transition state of the reaction of A1 or A2 with 1b was then located with the phosphorus atom of the secondary phosphine oxide coordinated with the Ni centre. Two transition states that could lead to the obtained product were found with alkene (TS-1a) and the alkyne moiety (TS-1b) respectively coordinated with Ni. The calculated energy barrier of the key intramolecular protonation step viaTS-1a (18.8 kcal mol−1) vs.TS-1b (25.7 kcal mol−1) indicated that the reaction probably proceeded through the former pathway which was favored by 6.9 kcal mol−1. This result was consistent with the experimental study that enynes were much more reactive than alkynes and could react at a very low temperature (−20 °C). As a comparison, the regioisomer could only be generated by transition state TS-1-regio in which the alkyne moiety was coordinated with Ni with an activation barrier of 25.8 kcal mol−1. This result strongly supported the excellent regioselectivity of the reaction.
In transition state TS-1a, the Ni centre and the hydroxyl group resided superficially with the enyne. As a result, simultaneous cis protonation and metalation occurred to afford Ni(II) intermediate INT-2 specifically. Reductive elimination of INT-2via transition state TS-2 (13.6 kcal mol−1) followed by ligand exchange could afford the final product and regenerate the catalyst resting state. Overall, intramolecular protonation served as the rate limiting step, which was also consistent with the kinetic studies. In addition, the oxidative insertion of SPO to Ni was also calculated to exhibit a high energy barrier (37.1 kcal mol−1), again consistent with the above experimental and calculation results (please see the ESI† for details).
The enantioselectivity could be also rationalized by the DFT calculations (please see the ESI† for details).14 The transition state of both secondary phosphine oxides (R)-1b and (S)-1b was located. The conformers with the lowest energy of TS-1a and TS-1a-S are shown in Fig. 6 with the former being more favored by 3.0 kcal mol−1 after correction. The ee was predicted to be 99% which matched with the experimental results of alkyl enynes. In summary, the cis, regio and enantioselectivity were well rationalized by DFT calculations which is fully in accordance with the experimental results. Obviously, the enyne coordinated with Ni(0) through a double bond was the key to both the high reactivity and enantioselectivity of the reaction.
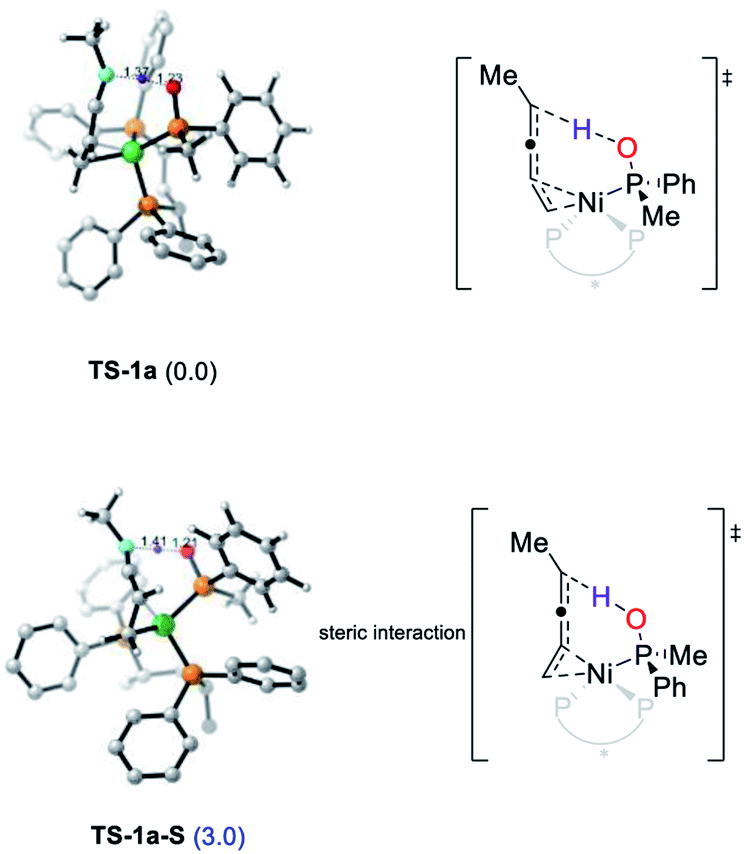 | ||
| Fig. 6 Geometries and corrected relative free energies at −20 °C of enantioselective protonation transition states TS-1a and TS-1a-S. | ||
Proposed mechanism
On the basis of the results described above, we proposed that Ni-catalyzed asymmetric hydrophosphinylation occurs by the mechanism shown in Scheme 2. Initially, the reaction of (S,S)-BDPP, Ni(cod)2 and enyne would generate the catalyst resting state A1 and A2 which also serves as the active catalyst. The phosphinous acid form of SPO which could function as a ligand for a range of transition metals, would undergo reversible coordination with A2 to form transition state D, serving as both the rate-determining and the regio-determining step. The phosphinous acid coordinated with Ni exhibiting a stronger acidity than the free one would protonate C1 of the Ni-enyne intermediate to form an allyl nickel intermediate E. Alternatively, protonation of C2 in transition state D′ is also possible to afford intermediate E′. However, this pathway is kinetically unfavorable. Reductive elimination of the generated Ni(II) intermediate via transition state F followed by ligand exchange with substrate 2w would regenerate the catalyst resting state, afford the product and complete the catalytic cycle.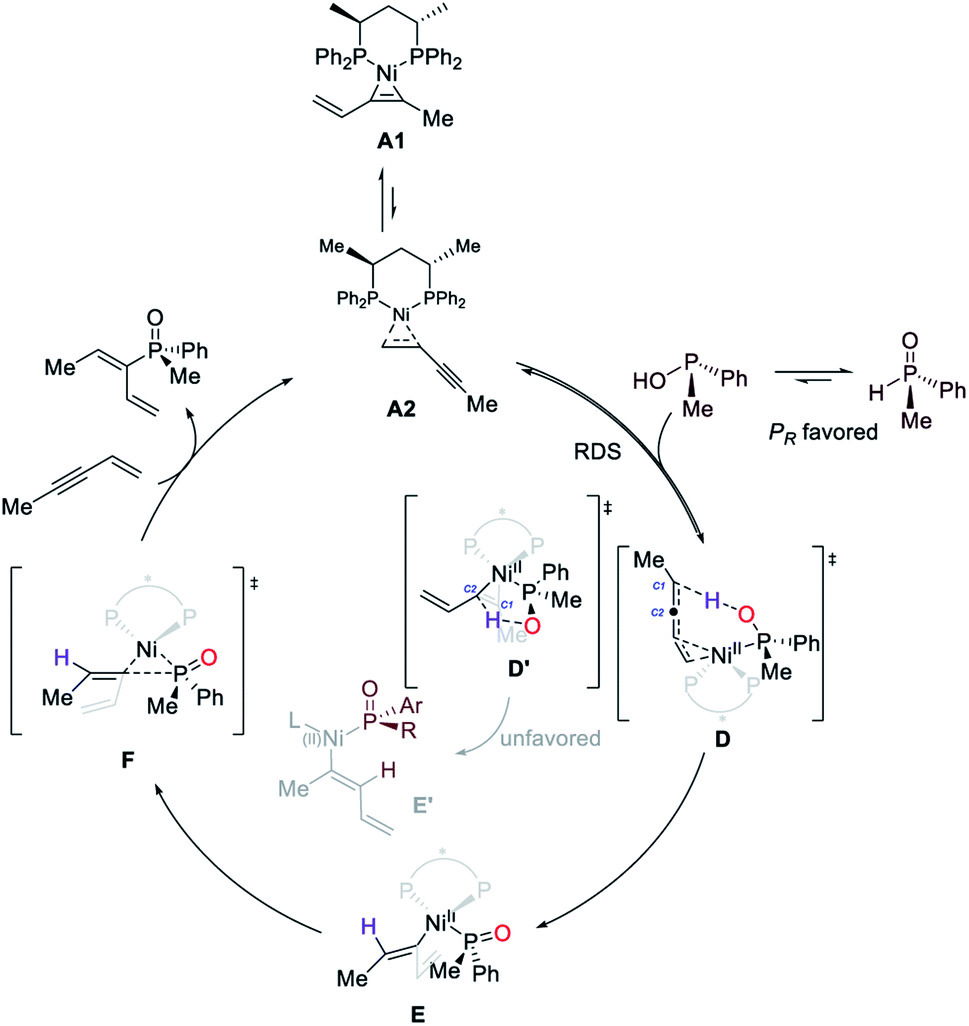 | ||
| Scheme 2 Proposed mechanism. | ||
Conclusion
In conclusion, we accomplished a highly regio and enantioselective Ni-catalyzed hydrophosphinylation reaction with excellent regio- and stereoselectivity. The mechanistic study combined with experimental and computational studies has uncovered a new pathway for the hydrophosphinylation reaction catalyzed by Ni, which is distinct from previous reports. This striking acid–base neutralization mechanism refreshes our understanding of the nickel-catalyzed hydrofunctionalization reaction. And new reactions guided by this reaction mechanism are being carried out in our group.Data availability
The data that support the findings of this study have been included in the ESI.†Author contributions
Q.-W. Z. conceived the project, designed the experiments and conducted computational studies. Y.-Q. Z., P.-J. Q. and Q. Z. performed the experimental work, X.-Y. H. performed the kinetic studies. Q.-W. Z., Y.-Q. Z., X.-Y. H., and Y. W. wrote the manuscript.Conflicts of interest
There is no conflict of interest to report.Acknowledgements
Support from NSFC (21901235 and 22071224), USTC Research Funds of the Double First-Class Initiative YD2060002010, and Anhui Provincial Natural Science Foundation (1908085MB34) is acknowledged. DFT calculations were carried out at Supercomputing Center of the University of Science and Technology of China. Prof. Liu-Zhu Gong of USTC is acknowledged for valuable discussions.Notes and references
- (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (b) H. Fernandez-Perez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119 CrossRef CAS PubMed; (c) Q.-L. Zhou, in Privileged Chiral Ligands and Catalysts, ed. W. Zhang and X. Zhang, Wiley-VCH, New York, 2011, p. 55 CrossRef; (d) M. Dutartre, J. Bayardon and S. Juge, Chem. Soc. Rev., 2016, 45, 5771 RSC; (e) T. Imamoto, Chem. Rec., 2016, 16, 2659 CrossRef PubMed; (f) H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Chem. Rev., 2018, 118, 10049 CrossRef CAS PubMed; (g) H. Ni, W.-L. Chan and Y. Lu, Chem. Rev., 2018, 118, 9344 CrossRef CAS PubMed; (h) G. Xu, C. H. Senanayake and W. Tang, Chem. Res., 2019, 52, 1101 CrossRef CAS PubMed; (i) T. Ayad, A. Gernet, J.-L. Pirat and D. Virieux, Tetrahedron, 2019, 75, 4385 CrossRef CAS; (j) A. Cabre, A. Riera and X. Verdaguer, Acc. Chem. Res., 2020, 53, 676 CrossRef CAS PubMed.
- (a) W. S. Knowles, M. J. Sabacky and B. D. Vineyard, J. Chem. Soc., Chem. Commun., 1972, 10 RSC; (b) B. D. Vineyard, W. S. Knowles and M. J. Sabacky, J. Mol. Catal., 1983, 19, 159 CrossRef CAS; (c) W. S. Knowles, J. Chem. Educ., 1986, 63, 222 CrossRef CAS; (d) C. A. Busacca and C. H. Senanayake, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Amsterdam, 2012, vol. 1, p. 167 Search PubMed.
- (a) T. Baumgartner and R. Réau, Chem. Rev., 2006, 166, 4681 CrossRef PubMed; (b) L. Clarion, C. Jacquard, O. Sainte-Catherine, S. Loiseau, D. Filippini, M. H. Hirlemann, J. N. Volle, D. Virieux, M. Lecouvey, J. L. Pirat and N. Bakalara, J. Med. Chem., 2012, 55, 2196 CrossRef CAS PubMed; (c) C. J. Schulze, G. Navarro, D. Ebert, J. DeRisi and R. G. Linington, J. Org. Chem., 2015, 80, 1312 CrossRef CAS PubMed; (d) M. P. Duffy, W. Delaunay, P. A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296 RSC; (e) N. Iwamoto, D. C. D. Butler, N. Svrzikapa, S. Mohapatra, I. Zlatev, D. W. Y. Sah, Meena, S. M. Standley, G. Lu, L. H. Apponi, M. Frank-Kamenetsky, J. J. Zhang, C. Vargeese and G. L. Verdine, Nat. Biotechnol., 2017, 35, 845 CrossRef CAS PubMed; (f) D. Siegel, H. C. Hui, E. Doerffler, M. O. Clarke, K. Chun, L. Zhang, S. Neville, E. Carra, W. Lew, B. Ross, Q. Wang, L. Wolfe, R. Jordan, V. Soloveva, J. Knox, J. Perry, M. Perron, K. M. Stray, O. Barauskas, J. Y. Feng, Y. Xu, G. Lee, A. L. Rheingold, A. S. Ray, R. Bannister, R. Strickley, S. Swaminathan, W. A. Lee, S. Bavari, T. Cihlar, M. K. Lo, T. K. Warren and R. L. Mackman, J. Med. Chem., 2017, 60, 1648 CrossRef CAS PubMed; (g) D. A. DiRocco, Y. Ji, E. C. Sherer, A. Klapars, M. Reibarkh, J. Dropinski, R. Mathew, P. Maligres, A. M. Hyde, J. Limanto, A. Brunskill, R. T. Ruck, L.-C. Campeau and I. W. Davies, Science, 2017, 356, 426 CrossRef CAS PubMed; (h) M. Wang, L. Zhang, X. Huo, Z. Zhang, Q. Yuan, P. Li, J. Chen, Y. Zou, Z. Wu and W. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20814 CrossRef CAS PubMed; (i) A. L. Featherston, Y. Kwon, M. M. Pompeo, O. D. Engl, D. K. Leahy and S. J. Miller, Science, 2021, 371, 702 CrossRef CAS PubMed; (j) M. Formica, T. Rogova, H. Shi, N. Sahara, A. J. M. Farley, K. E. Christensen, F. Duarte and D. J. Dixon, ChemRxiv, 2021 DOI:10.33774/chemrxiv-2021-5714s-v2.
- (a) K. M. Pietrusiewicz and M. Zablocka, Chem. Rev., 1994, 94, 1375 CrossRef CAS; (b) P. H. Leung, Acc. Chem. Res., 2004, 37, 169 CrossRef CAS PubMed; (c) D. S. Glueck, Chem.–Eur. J., 2008, 14, 7108 CrossRef CAS PubMed; (d) J. S. Harvey and V. Gouverneur, Chem. Commun., 2010, 46, 7477 RSC; (e) M. Oestreich, F. Tappe and V. Trepohl, Synthesis, 2010, 3037 CrossRef; (f) O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2012, 23, 1 CrossRef CAS; (g) I. Wauters, W. Debrouwer and C. V. Stevens, Beilstein J. Org. Chem., 2014, 10, 1064 CrossRef PubMed; (h) P. Bagi, V. Ujj, M. Czugler, E. Fogassy and G. Keglevich, Dalton Trans., 2016, 45, 1823 RSC; (i) R. Beaud, R. J. Phipps and M. J. Gaunt, J. Am. Chem. Soc., 2016, 138, 13183 CrossRef CAS PubMed; (j) J. Chrzanowski, D. Krasowska and J. Drabowicz, Heteroat. Chem., 2018, 29, e21476 CrossRef; (k) X.-T. Liu, Y.-Q. Zhang, X.-Y. Han, S.-P. Sun and Q.-W. Zhang, J. Am. Chem. Soc., 2019, 141, 16584 CrossRef CAS PubMed; (l) Q. Dai, W. Li, Z. Li and J. Zhang, J. Am. Chem. Soc., 2019, 141, 20556 CrossRef CAS PubMed; (m) S. Lemouzy, L. Giordano, D. Hérault and G. Buono, Eur. J. Org. Chem., 2020, 3351 CrossRef CAS; (n) Q. Dai, L. Liu and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 27247 CrossRef CAS PubMed.
- (a) I. Kovacik, D. K. Wicht, N. S. Grewal and D. S. Glueck, Organometallics, 2000, 19, 950 CrossRef CAS; (b) B. Join, D. Mimeau, O. Delacroix and A. C. Gaumont, Chem. Commun., 2006, 3249 RSC; (c) C. Li, Q.-L. Bian, S. Xu and W.-L. Duan, Org. Chem. Front., 2014, 1, 541 RSC; (d) W.-J. Yue, J.-Z. Xiao, S. Zhang and L. Yin, Angew. Chem., Int. Ed., 2020, 59, 7057 CrossRef CAS PubMed; (e) Z. Yang, X. Gu, L.-B. Han and J. Wang, Chem. Sci., 2020, 11, 7451 RSC; (f) Q. Dai, L. Liu, Y. Qian, W. Li and J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 20645 CrossRef CAS PubMed; (g) Y. B. Li, H. Tian and L. Yin, J. Am. Chem. Soc., 2020, 142, 20098 CrossRef CAS PubMed; (h) C. Wang, K. Huang, J. Ye and W. L. Duan, J. Am. Chem. Soc., 2021, 143, 5685 CrossRef CAS PubMed; (i) X.-T. Liu, X.-Y. Han, Y. Wu, Y.-Y. Sun, L. Gao, Z. Huang and Q.-W. Zhang, J. Am. Chem. Soc., 2021, 143, 11309 CrossRef CAS PubMed; (j) S. Zhang, J.-Z. Xiao, Y.-B. Li, C.-Y. Shi and L. Yin, J. Am. Chem. Soc., 2021, 143, 9912 CrossRef CAS PubMed; (k) Z.-H. Wu, A.-Q. Cheng, M. Yuan, X.-Y. Zhao, H.-L. Yang, L.-H. Wei, H.-Y. Wang, T. Wang, Z. Zhang and W.-L. Duan, Angew. Chem., Int. Ed., 2021, 60, 27241 CrossRef CAS PubMed.
- X.-Y. Yang, W. S. Tay, Y. Li, S. A. Pullarkat and P. H. Leung, Organometallics, 2015, 34, 5196 CrossRef CAS.
- S.-Z. Nie, R. T. Davison and V. M. Dong, J. Am. Chem. Soc., 2018, 140, 16450 CrossRef CAS PubMed.
- Z. Yang and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 27288 CrossRef CAS PubMed.
- J. Long, Y. Li, W. Zhao and G. Yin, Chem. Sci., 2022, 13, 1390 RSC.
- (a) C. A. Bange and R. Waterman, Chem.–Eur. J., 2016, 22, 12598 CrossRef CAS PubMed; (b) H. Hou, Org. Lett., 2021, 23, 2981 CrossRef CAS PubMed.
- (a) V. P. Ananikov, A. V. Makarov and I. P. Beletskaya, Chem.–Eur. J., 2011, 17, 12623 CrossRef CAS PubMed; (b) V. P. Ananikov and M. Tanaka, in Hydrofunctionalization, Topics in Organometallic Chemistry, ed. V. P. Ananikov and I. P. Beletskaya, Springer, Berlin, 2011, vol. 43, p. 21 Search PubMed; (c) T. Chen, C.-Q. Zhao and L.-B. Han, J. Am. Chem. Soc., 2018, 140, 3139 CrossRef CAS PubMed.
- (a) V. P. Ananikov and I. P. Beletskaya, Chem.–Asian J., 2011, 6, 1423 CrossRef CAS PubMed; (b) Y. V. Ivanova, L. L. Khemchyan, S. S. Zalesskii, V. P. Ananikov and I. P. Beletskaya, Russ. J. Org. Chem., 2013, 49, 1099 CrossRef CAS; (c) L. L. Khemchyan, J. V. Ivanova, S. S. Zalesskiy, V. P. Ananikov, I. P. Beletskaya and Z. A. Starikova, Adv. Synth. Catal., 2014, 356, 771 CrossRef CAS.
- K. V. Rajendran and D. G. Gilheany, Chem. Commun., 2012, 48, 817 RSC.
- (a) G. Luchini, J. V. Alegre-Requena, Y. Guan, I. Funes-Ardoiz and R. S. Paton, GoodVibes: GoodVibes 3.0.1, 2019, DOI: DOI:10.5281/zenodo.595246; (b) S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2056434. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00091a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |