DOI:
10.1039/D2SC00260D
(Edge Article)
Chem. Sci., 2022,
13, 7920-7932
Efficacious and sustained release of an anticancer drug mitoxantrone from new covalent organic frameworks using protein corona†
Received
15th January 2022
, Accepted 2nd June 2022
First published on 18th June 2022
Abstract
Solid porous and crystalline covalent organic frameworks (COFs) are characterized by their higher specific BET surface areas and functional pore walls, which allow the adsorption of various bioactive molecules inside the porous lattices. We have introduced a perylene-based COF, PER@PDA-COF-1, which acts as an effective porous volumetric reservoir for an anticancer drug, mitoxantrone (MXT). The drug-loaded COF (MXT–PER@PDA-COF-1) exhibited zero cellular release of MXT towards cancer cells, which can be attributed to the strong intercalation between the anthracene-dione motif of the drug and the perylene-based COF backbone. Here, we have introduced a strategy involving the serum-albumin-triggered intracellular release of mitoxantrone from MXT–PER@PDA-COF-1. The serum albumin acts as an exfoliating agent and as a colloidal stabilizer in PBS medium (pH = 7.4), rapidly forming a protein corona around the exfoliated COF crystallites and inducing the sustained release of MXT from the COF into tumorigenic cells.
Introduction
The design and synthesis of new covalent organic frameworks made from entirely sp2 hybridized, light elements (C, N, B, etc.) accommodated inside a two-dimensional lattice are of considerable scientific interest.1–5 Carbon nitride,6–8 exfoliated graphene,9 two-dimensional metal–organic frameworks (2D-MOFs),10–13 and two-dimensional covalent organic frameworks (2D-COFs)14–16 are among the most sought-after candidates in this class. In particular, COFs are an emerging class of materials in terms of gas storage17–19 and separation,20,21 catalysis,22,23 fuel cell applications,24,25 and effective drug delivery,26–28etc. COFs based on imine29 or boronate ester groups30 are promising because of their structural diversity, functional tunability, precise atomistic positions, and high porosity. Currently, attempts to load various cargos with sustained cellular delivery are being made using COFs and MOFs as porous scaffolds. However, the sustained delivery of drugs through a biocompatible and smart delivery vehicle remains an elusive task. Low dispersibility, slower release rate, leaching of toxic metal ions, etc. are some serious issues which limit their applications as biomaterials.31 Accordingly, there is a considerable scientific need for the development of an optimized smart nano-carrier by surface engineering on the porous materials with hydrophilic linear-chained or branched polymers (polyethylene glycol, polyamine, sugars, etc.).23,32–34 The stability of these materials in a physiological environment is another major concern. In the last few years, several COFs have been chosen for drug loading and release studies.35,36 The nature of the interaction of the drug molecules with the COFs may be attributed to a weak physisorption mechanism. That is why most release profiles involve an initial burst which follows by zero-order kinetics. However, often the release profile cannot be studied due to poor solubility in aqueous media, in contrast to metal oxide nanoparticles, which are easily dispersible in aqueous media due to their higher hydrophilicity. The surface modification of COF by anchoring a metal oxide is a good way of constructing a drug delivery vehicle.37 Drug delivery/therapeutic platforms are of immense significance in the scientific community for attaining higher efficiency against particular clinical manifestations, especially against cancer.38–41 Many such COFs can be used as drug delivery vectors, but due to their poor solubility and sustainability in aqueous media, it is very difficult to deliver drugs to the targeted cells using these COFs. Also, COFs may lead to the manifestation of increased toxicity and can kill normal cells, showing their inappropriateness for use as drug delivery platforms.
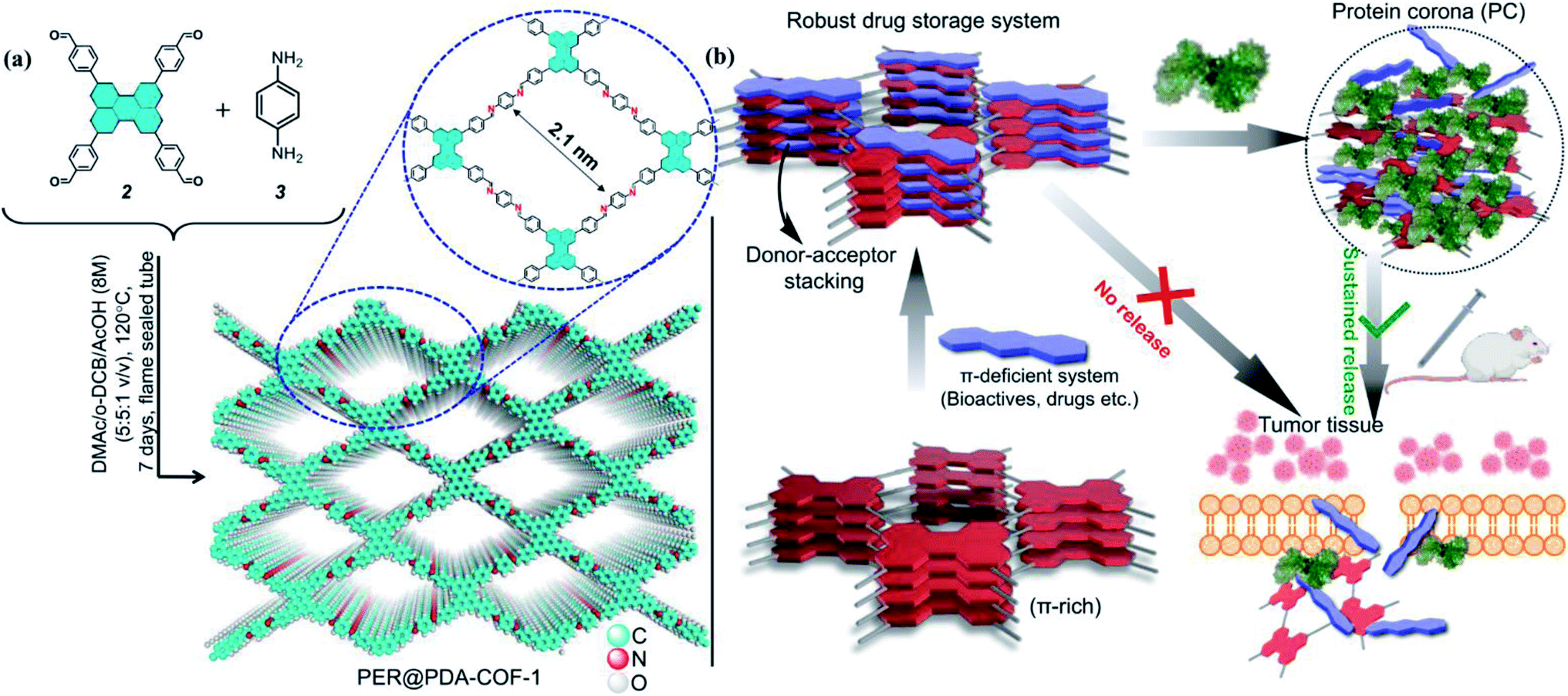
In this work, we overcome such obstacles by making COFs convenient for delivering drugs to the cells in a stimuli-responsive pathway with optimum dispersibility in aqueous and serum media. Herein, we report the synthesis of a perylene-based highly crystalline two-dimensional covalent organic framework named PER@PDA-COF-1, which has the positional precision of perylene units and offers an ordered arrangement of π-electron-rich nodes inside a rhombus net. Then the framework was utilized as a porous system for the storage of mitoxantrone (MXT), which is a type-II topoisomerase inhibitor disrupting DNA synthesis and repair by intercalating between the DNA bases within duplex strands.42,43 It has been widely used for the treatment of metastatic breast cancer, myeloid leukemia, and non-Hodgkin's lymphoma. Mitoxantrone was therefore deliberately chosen for the present studies because of the presence of π-deficient (anthracene-dione) moieties44 and its emission maximum at higher wavelength (λmax = 685 nm). Here we promulgate a new albumin corona decorated COF based nano-formulation triggered drug release strategy against cancerous cells (in vitro and in vivo) using tailor-made functional COFs with an appropriate drug molecule (Fig. 1). These not only enrich the scope and utility of the porous fluorescent materials for the uptake and albumin-triggered cellular release/monitoring of the anticancer drug in the cancer cell but also ensure the significant prospect of COF from the viewpoint of biophysical interactions between bio-actives, proteins and covalent organic frameworks.
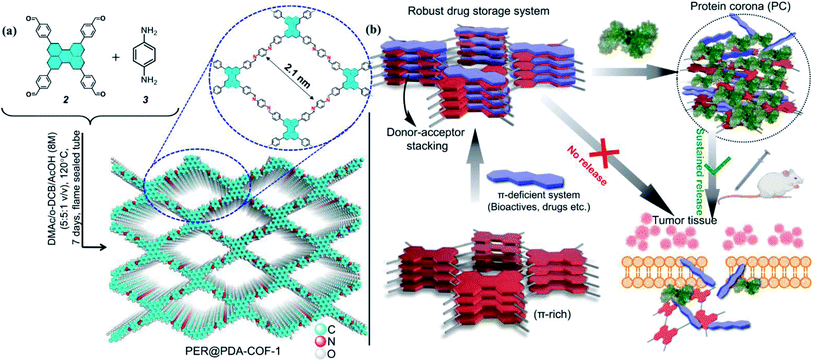 |
| | Fig. 1 (a) Schematic diagrams of the synthesis of PER@PDA-COF-1 and its structure. (b) A schematic diagram of the donor–acceptor based strategy for the loading of bio-actives inside a crystalline covalent organic framework and albumin-triggered cellular release. | |
Results and discussion
Characterization of the PER@PDA-COF-1 nano-reservoir
A stoichiometric mixture of 4,4′,4′′,4′′′-(perylene-2,5,8,11-tetrayl)tetrabenzaldehyde (2) and p-phenylenediamine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was kept under static heating conditions at 120 °C in a solvent mixture of o-dichlorobenzene/N,N-dimethyl-acetamide (v/v = 1:1, 1 mL). This afforded a powder of PER@PDA-COF-1 (Fig. 1a). The optimized reaction time was ∼7 days to obtain the imine-based COF, which was isolated as a yellowish-orange crystalline solid and found to be insoluble in common organic solvents such as ethanol, acetone, dichloromethane, and tetrahydrofuran. The powder was repeatedly washed using hot DMF and DMSO to remove any unreacted starting materials and vacuum activated (hexane) under 70 °C overnight. The perylene unit present in each vertex of the resulting crystalline lattice serves as an active site for the adsorption of a π-deficient single molecular system (Fig. 1b). Hence the perylene-decorated PER@PDA-COF-1 could be utilized as an exclusive porous host for electron-deficient guests (drugs, biomolecules etc.). The precursor perylene-based aldehyde 2 was synthesized as described in Section S2, ESI† and characterized by 1H spectroscopy (Fig. S1 and S2, ESI†).
:2) was kept under static heating conditions at 120 °C in a solvent mixture of o-dichlorobenzene/N,N-dimethyl-acetamide (v/v = 1:1, 1 mL). This afforded a powder of PER@PDA-COF-1 (Fig. 1a). The optimized reaction time was ∼7 days to obtain the imine-based COF, which was isolated as a yellowish-orange crystalline solid and found to be insoluble in common organic solvents such as ethanol, acetone, dichloromethane, and tetrahydrofuran. The powder was repeatedly washed using hot DMF and DMSO to remove any unreacted starting materials and vacuum activated (hexane) under 70 °C overnight. The perylene unit present in each vertex of the resulting crystalline lattice serves as an active site for the adsorption of a π-deficient single molecular system (Fig. 1b). Hence the perylene-decorated PER@PDA-COF-1 could be utilized as an exclusive porous host for electron-deficient guests (drugs, biomolecules etc.). The precursor perylene-based aldehyde 2 was synthesized as described in Section S2, ESI† and characterized by 1H spectroscopy (Fig. S1 and S2, ESI†).
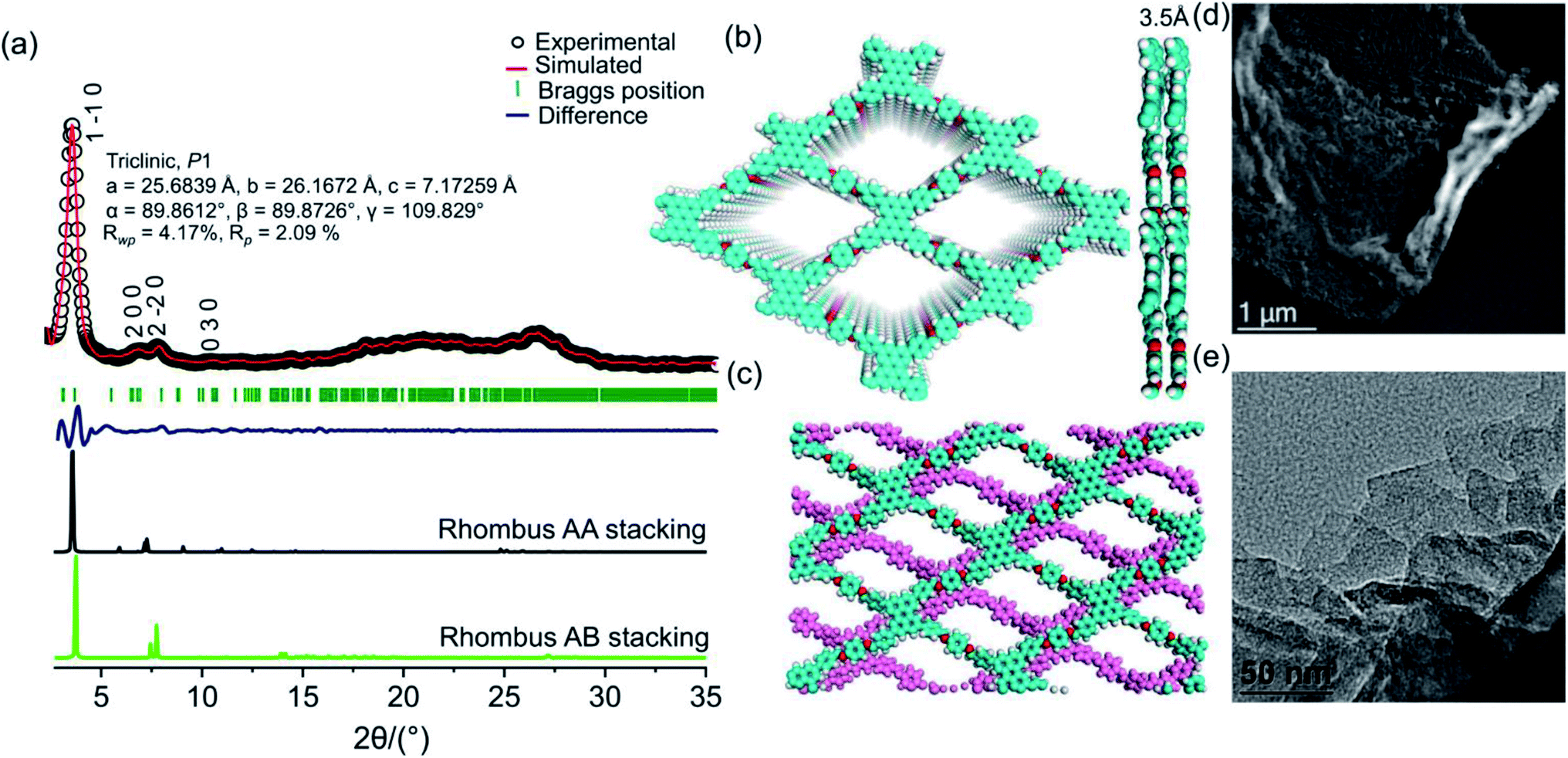
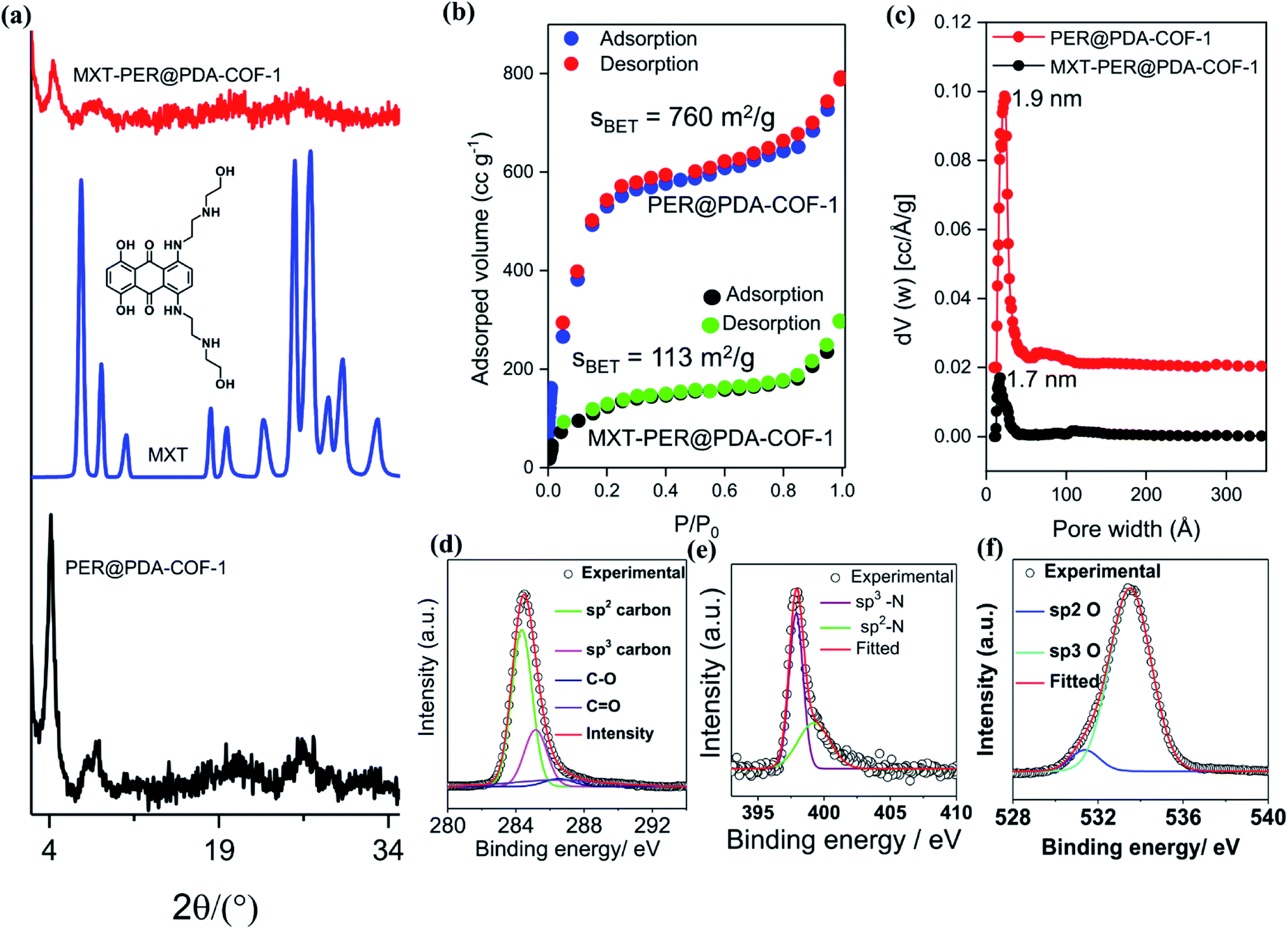
PER@PDA-COF-1 was characterized by Fourier transform infrared (FTIR) spectroscopy and 13C solid-state CP-MAS NMR spectroscopy. The peak at 1603 cm−1 may be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N functionality and that at 830 cm−1 to C–H bending (Fig. S6, ESI†). The disappearance of the CO stretching frequency (1695 cm−1) indicates the complete condensation of aldehyde and the amine unit. The 13C CP-MAS NMR resonance peak at 157 ppm indicates the presence of an imine-based functionality throughout the crystalline framework (Fig. S7, ESI†). The presence of strong resonance peaks at 122 ppm (cyan circle) and 116 ppm (red circle) may be assigned to the phenyl carbon and slightly shielded carbon atoms present in the poly-aromatic (perylene) moiety, respectively. The peak at 129 ppm (blue circle) may be assigned to the phenyl carbon belonging to the linear di-imine linker. The crystallinity and phase purity of PER@PDA-COF-1 were evaluated by powder X-ray diffraction studies and theoretical atomistic simulation. Peaks centered at 2θ = 4.1°, 7.3°, 8.3° and 10.7°, 18.2° and 26.5° were observed in the PXRD patterns (Fig. 2a, black circle plot). The peaks at 4.1°, 7.3°, 8.3°, 10.7°, 18.2° and 26.2° may be assigned to the (1 −1 0), (2 0 0), (2 −2 0), (0 3 0), (2 4 0) and (5 −6 1) facets, respectively. Pawley refinement (Fig. 2a, red solid plot) was undertaken, including crystal broadening and strain parameters using the reflex module in Accelrys material studio 7.0, which gave the minimum difference in terms of the peak intensity and width from the experimentally obtained PXRD. The R-weighted profile factor (Rwp) = 4.17% and R-pattern factor (Rp) = 2.09% could be fitted well with the AA (eclipsed) stacking mode of 2D PER@PDA-COF-1 layers (Fig. 2a, black solid line, Fig. S3, ESI†). The experimentally obtained PXRD pattern was different from the AB stacking mode (Fig. 2a, green solid line, Fig. S3, ESI†). The derived unit cell parameters of the 3D-triclinic lattice have a = 25.684 Å, b = 26.167 Å, c = 7.172 Å and α = 89.8604°, β = 89.872°, γ = 109.829°. The molecular assembly between the tetra-topic (D2h) and linear (C2) building units may crystallize either in the rhombus net or the Kagome net. These topological possibilities were investigated by molecular modeling. Neither of the AA and AB stacking modes of the Kagome lattice were adopted by the PER@PDA-COF-1 structure (Fig. S4, ESI†). The models of rhombus packing given for AA and AB stacking are shown in Fig. 2b and c. Thermogravimetric (TGA) analysis under N2 atmosphere was performed up to 800 °C to account for the thermal stability of PER@PDA-COF-1, which shows that the material is stable up to 500 °C without any weight loss (Fig. S8, ESI†). The crystalline covalent packing of 2 and the di-imine associated with the extensive π–π stacking between the perylene-decorated 2D layers make the framework purely laminar and thermally stable. The ultrahigh resolution transmission electron microscopy (UHR-TEM) images of the bulk powder material (Fig. 2d) demonstrate that the two-dimensional layered morphology had been adopted by PER@PDA-COF-1, which can be exfoliated by using a suitable solvent system (Fig. 2e) to give nanosheet of size ∼ 100–200 nm. The FFT pattern of the selected crystalline zone of the polycrystalline sample has been found, and is given in the inset to Fig. S9a and b, ESI.† The interlayer distance using GATAN digital micrography software was found to be 3.2 Å (Fig. S9c and d ESI†) which is in close agreement with the extensive van der Waals stacking parameter (3.5 Å) of the optimized AA rhombus lattice of PER@PDA-COF-1. The porosity of the PER@PDA-COF-1 was evaluated by an N2 adsorption/desorption experiment at 77 K, which shows a type-IV adsorption/desorption isotherm. The sharp rise in the low-pressure region ranging from P/P0 = 0.0001 to P/P0 = 0.15 could be attributed to the larger micropores present in PER@PDA-COF-1 (Fig. 3b, blue/red solid circle). The surface area was found to be ∼760 m2 g−1. The maximum of the pore size distribution was obtained using the NLDFT method, and was estimated to be 1.9 nm (Fig. 3c, red plot) and the pore volume was found to be 1.07 cm3 g−1. This estimated pore aperture is in agreement with the pore size expected from the structural simulation.
N functionality and that at 830 cm−1 to C–H bending (Fig. S6, ESI†). The disappearance of the CO stretching frequency (1695 cm−1) indicates the complete condensation of aldehyde and the amine unit. The 13C CP-MAS NMR resonance peak at 157 ppm indicates the presence of an imine-based functionality throughout the crystalline framework (Fig. S7, ESI†). The presence of strong resonance peaks at 122 ppm (cyan circle) and 116 ppm (red circle) may be assigned to the phenyl carbon and slightly shielded carbon atoms present in the poly-aromatic (perylene) moiety, respectively. The peak at 129 ppm (blue circle) may be assigned to the phenyl carbon belonging to the linear di-imine linker. The crystallinity and phase purity of PER@PDA-COF-1 were evaluated by powder X-ray diffraction studies and theoretical atomistic simulation. Peaks centered at 2θ = 4.1°, 7.3°, 8.3° and 10.7°, 18.2° and 26.5° were observed in the PXRD patterns (Fig. 2a, black circle plot). The peaks at 4.1°, 7.3°, 8.3°, 10.7°, 18.2° and 26.2° may be assigned to the (1 −1 0), (2 0 0), (2 −2 0), (0 3 0), (2 4 0) and (5 −6 1) facets, respectively. Pawley refinement (Fig. 2a, red solid plot) was undertaken, including crystal broadening and strain parameters using the reflex module in Accelrys material studio 7.0, which gave the minimum difference in terms of the peak intensity and width from the experimentally obtained PXRD. The R-weighted profile factor (Rwp) = 4.17% and R-pattern factor (Rp) = 2.09% could be fitted well with the AA (eclipsed) stacking mode of 2D PER@PDA-COF-1 layers (Fig. 2a, black solid line, Fig. S3, ESI†). The experimentally obtained PXRD pattern was different from the AB stacking mode (Fig. 2a, green solid line, Fig. S3, ESI†). The derived unit cell parameters of the 3D-triclinic lattice have a = 25.684 Å, b = 26.167 Å, c = 7.172 Å and α = 89.8604°, β = 89.872°, γ = 109.829°. The molecular assembly between the tetra-topic (D2h) and linear (C2) building units may crystallize either in the rhombus net or the Kagome net. These topological possibilities were investigated by molecular modeling. Neither of the AA and AB stacking modes of the Kagome lattice were adopted by the PER@PDA-COF-1 structure (Fig. S4, ESI†). The models of rhombus packing given for AA and AB stacking are shown in Fig. 2b and c. Thermogravimetric (TGA) analysis under N2 atmosphere was performed up to 800 °C to account for the thermal stability of PER@PDA-COF-1, which shows that the material is stable up to 500 °C without any weight loss (Fig. S8, ESI†). The crystalline covalent packing of 2 and the di-imine associated with the extensive π–π stacking between the perylene-decorated 2D layers make the framework purely laminar and thermally stable. The ultrahigh resolution transmission electron microscopy (UHR-TEM) images of the bulk powder material (Fig. 2d) demonstrate that the two-dimensional layered morphology had been adopted by PER@PDA-COF-1, which can be exfoliated by using a suitable solvent system (Fig. 2e) to give nanosheet of size ∼ 100–200 nm. The FFT pattern of the selected crystalline zone of the polycrystalline sample has been found, and is given in the inset to Fig. S9a and b, ESI.† The interlayer distance using GATAN digital micrography software was found to be 3.2 Å (Fig. S9c and d ESI†) which is in close agreement with the extensive van der Waals stacking parameter (3.5 Å) of the optimized AA rhombus lattice of PER@PDA-COF-1. The porosity of the PER@PDA-COF-1 was evaluated by an N2 adsorption/desorption experiment at 77 K, which shows a type-IV adsorption/desorption isotherm. The sharp rise in the low-pressure region ranging from P/P0 = 0.0001 to P/P0 = 0.15 could be attributed to the larger micropores present in PER@PDA-COF-1 (Fig. 3b, blue/red solid circle). The surface area was found to be ∼760 m2 g−1. The maximum of the pore size distribution was obtained using the NLDFT method, and was estimated to be 1.9 nm (Fig. 3c, red plot) and the pore volume was found to be 1.07 cm3 g−1. This estimated pore aperture is in agreement with the pore size expected from the structural simulation.
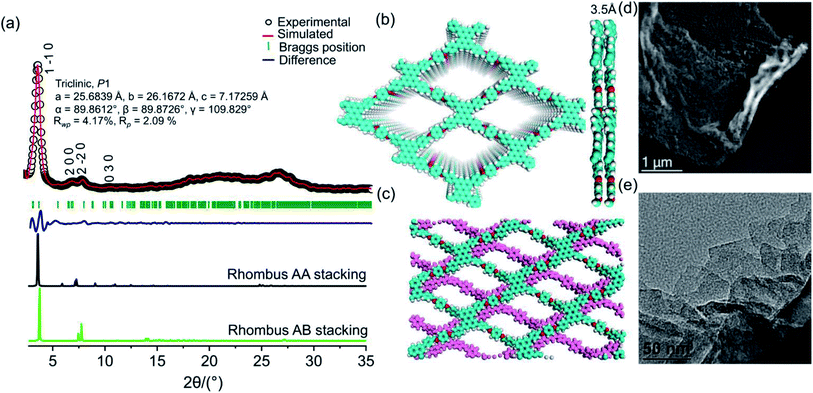 |
| | Fig. 2 (a) The powder XRD pattern of PER@PDA-COF-1 with a Cu Kα anode, where the experimental profile is shown plotted in black circles, the Pawley refinement is plotted by a red solid line (simulated), the difference is plotted in blue, the observed reflection is plotted as olive bars, the theoretical profile for rhm AA is plotted in solid black, and rhm AB stacking is plotted by a green solid line. (b) An atomistic model of rhm AA, where carbon is represented in cyan and nitrogen in red. (c) A model of rhm AB. (d) An UHRTEM image of the bulk PER@PDA-COF-1 framework (scale bar: 1 μm). (e) Solution (isopropanol) processable exfoliated PER@PDA-COF-1 nanosheets (scale bar 50 nm). | |
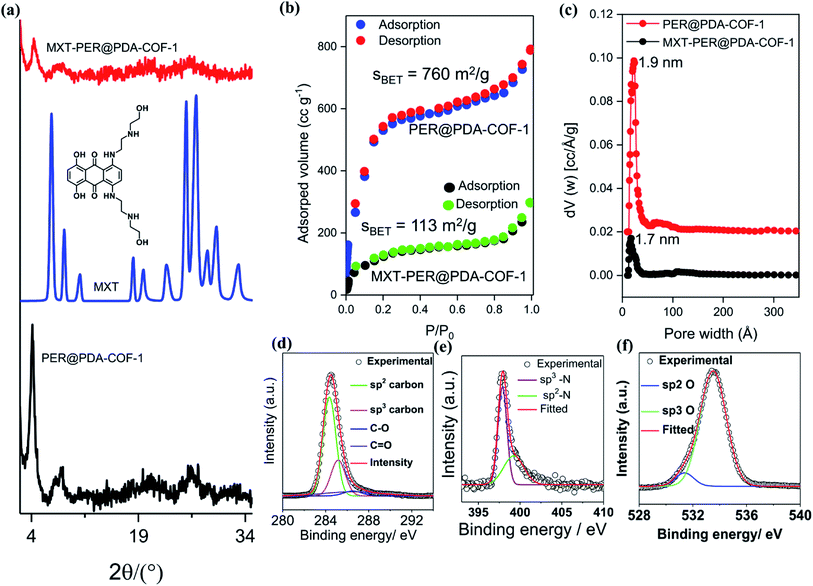 |
| | Fig. 3 (a) PXRD profiles of pristine PER@PDA-COF-1 (black), mitoxantrone crystal (blue), and drug-loaded PER@PDA-COF-1 (red). (b) N2 adsorption/desorption isotherms of PER@PDA-COF-1 before (blue and red) and after (black and green) MXT loading. (c) Pore size distributions of PER@PDA-COF-1 before (red) and after (black) MXT loading. (d) C 1s; (e) N 1s; and (f) O 1s XPS spectra of MXT–PER@PDA-COF-1. | |
Drug intercalation and storage
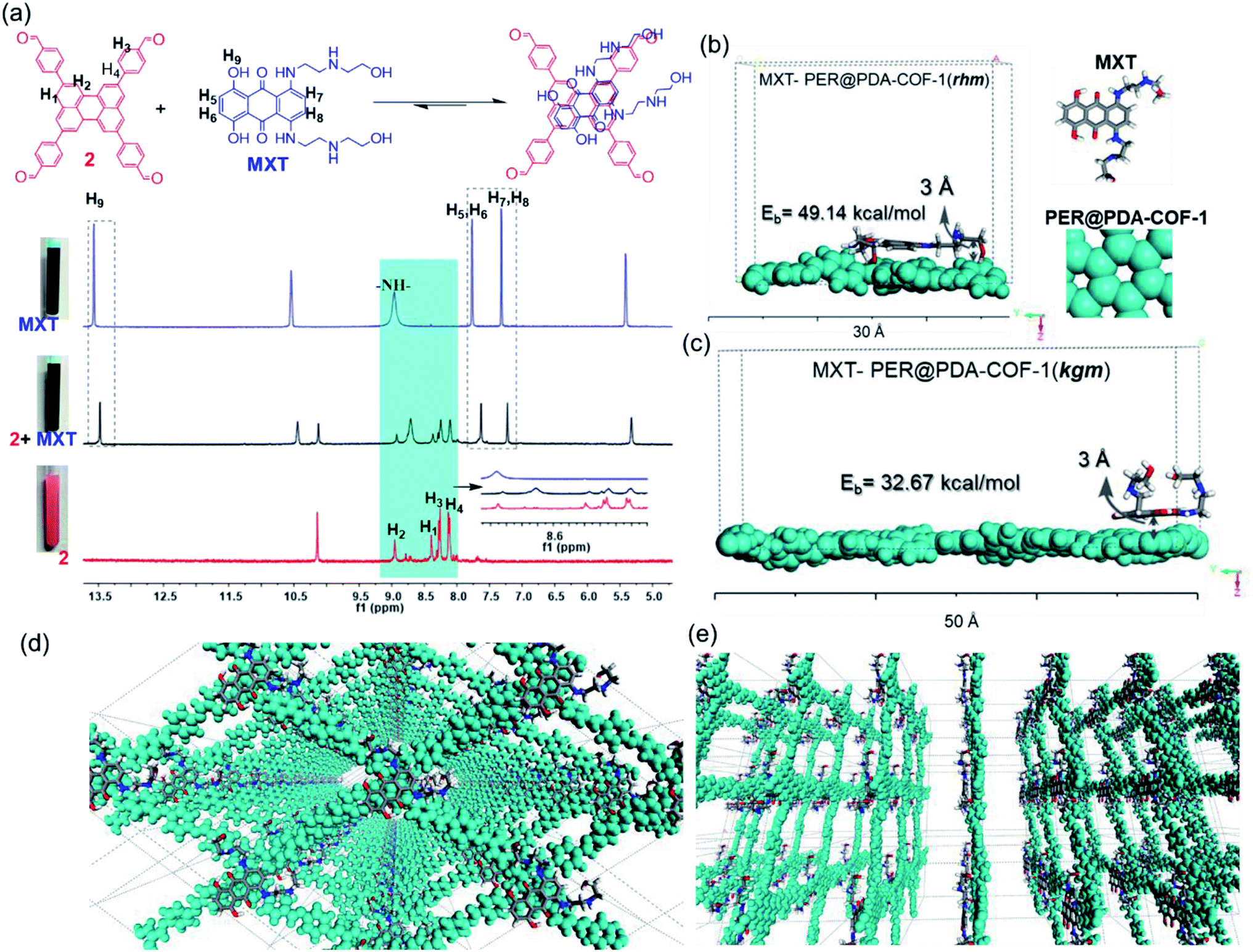
This tailor-made porous, crystalline fluorescent organic framework was used as a volumetric reservoir for loading an anticancer drug and its robustness and storage efficiency were examined. Mitoxantrone (MXT), an anticancer drug, was chosen as an electrophilic guest molecule.45 It may be demonstrated that, due to the presence of an electrophilic site in each vertex of the COF lattice, mitoxantrone can behave as an intercalant between the perylene-comprised 2D layers. Initially, PER@PDA-COF-1 (10 mg) was added in THF (20 mL) and dispersed under vigorous stirring, sonication, and vortexing. A powder X-ray diffraction study of the resulting dispersion was undertaken to investigate the effect of the MXT molecule on the PER@PDA-COF-1 lattice. Fig. 3a shows the PXRD pattern of pristine COF (black), MXT molecule (blue) and the drug-encapsulated COF matrix (red). It has been observed that a specific sharp crystalline peak due to the MXT molecule was absent in drug-loaded PER@PDA-COF-1, which confirms that the encapsulated drug molecules exist as a molecular dispersion throughout the COF lattice instead of crystalline packing of MXT itself.46 To evaluate the effect of the MXT on the porosity of PER@PDA-COF-1, we measured the BET surface area (SBET = 113 m2 g−1) of MXT-loaded PER@PDA-COF-1 (MXT–PER@PDA-COF-1) using the N2 adsorption/desorption isotherms (Fig. 3b, green and black solid circles). The abrupt reduction in SBET may be attributed to the successful loading of drug molecules into the porous lattice. A slight reduction in the pore size (1.7 nm) was observed (Fig. 3c, green plot) after drug encapsulation. On the other hand, the FTIR spectra (Fig. S10, ESI†) showed the presence of vibrational bands in MXT alone (red plot) and the drug-loaded organic framework MXT–PER@PDA-COF-1 (blue plot), which demonstrates the successful loading of drug molecules into the solid organic matrix. The elemental composition of the MXT intercalated framework was further characterized by X-ray photoelectron spectroscopy (XPS). The C 1s peak exhibits the co-existence of four secondary peaks for different carbons (Fig. 3d). The peak centered at 284.3 eV (green) may be assigned to the sp2 hybridized carbon, the peak at 285.1 eV (pink) to sp3 carbon, the peak at 286.4 eV (violet) to the trace amount of CO carbon and the peak at 286.5 eV (blue) to C–O carbon. The N 1s spectra of MXT–PER@PDA-COF-1 are given in Fig. 3e, which displays the two different peaks at 397.8 eV and 399.2 eV for the sp3 (purple) and the sp2 (green) nitrogen atoms, respectively. The O 1s XPS spectra also consist of two different peak maxima, centered at 531.3 eV and 533.5 eV for the sp2 and sp3 oxygens, respectively (Fig. 3f). The high storage capacity of MXT (∼63%) was confirmed from the reduction in absorbance of MXT in the supernatant THF solution (Fig. S11, ESI†). A fluorescence spectroscopic study was performed to examine the interaction between MXT and PER@PDA-COF-1. It was observed that there was significant quenching in the fluorescence intensity of COF with a blue shift of ∼4 nm with an increasing concentration of MXT (Fig. S12, ESI†). This indicated that the π–π stacking induced H-type aggregation between MXT and PER@PDA-COF-1.47 The Stern–Volmer constant (KSV) was estimated to be 1.348 × 108 M−1 (Fig. S13, ESI†). The donor (D)–acceptor (A) interaction between 2 and MXT was investigated by 1H NMR spectroscopy. An equimolar mixture of 2 and MXT in DMSO-d6 solution exhibits a substantial up-field shift and peak broadening of the aromatic protons of both the perylene core of 2 (H1, H2, H3, H4) and the anthracene-dione (H5, H6, H7, H8) moiety in MXT (Fig. 4a). The upfield shift of the –NH proton of mitoxantrone was observed in the mixture. The immediate color change upon adding MXT to the DMSO-d6 solution of 2 from orange to green indicates the D–A interaction between 2 and MXT. DFTB simulations from 500 random starting geometries showed that MXT preferentially undergoes π–π interactions with the perylene-based vertex of the PER@PDA-COF-1 lattice. Simulations were performed by placing an MXT molecule on a monolayer of different lattice systems (rhombus and Kagome), which results in the perylene arrangement in the rhombus lattice possessing greater binding ability towards MXT than the Kagome arrangement (Fig. 4b and c). The MXT molecule is situated at 3.0 Å above the perylene core in both systems. The theoretical binding energies (Eb) were estimated to be ∼49 kcal mol−1 and 32 kcal mol−1 for the rhombus and Kagome systems, respectively. During optimization of binding geometry, the lowest energy structure was returned for a single unit cell, confirming that PER@PDA-COF-1 can allow a single MXT molecule per perylene unit inside the system. The supercell structure of the simulated MXT–PER@PDA-COF-1 system is given in Fig. 4d and e to visualize the overall drug-intercalated nanosystem. Orientational optimization of MXT over the COF lattice, was also investigated by using a random geometry generation technique, which demonstrates that one of the two alkyl chains present in MXT aligns with the linker and the second lies in the pore, which may cause the slight reduction in the pore diameter after drug loading inside PER@PDA-COF-1 (Fig. S5a and b, ESI†). Molecular dynamic simulation was done to estimate the Eb of MXT to multilayer COF which was found to be 35 kcal mol−1 (Fig. S5e, ESI†). The as-synthesized framework acts as a porous scaffold for the storage of the anticancer drug in host–guest intercalation fashion. The high specific BET surface area of the COF-based drug carriers coupled with biocompatibility make them promising in terms of acting as an ideal host with reduced toxicity, increased bioavailability and higher molecular dispersion of the drug through the lattice space (high surface area to volume ratio). MXT–PER@PDA-COF-1 was subjected to investigation of the drug release phenomena from the COF lattice, but it did not show any significant release of MXT inside the cell. This can be attributed to the strong interaction between MXT and PER@PDA-COF-1 and makes the COF a hard storage system. To trigger MXT release, a biocompatible strategy was introduced by employing an albumin-based colloidal stabilizer. The key background for this approach is the well-known stabilizing agent-induced lowering of the interfacial energy between the material and the solvent by forming a protein corona (PC),48–50 a stealthy strategy in nanobiotechnology for drug delivery, which is probably the first report based on a COF-based nanosystem.
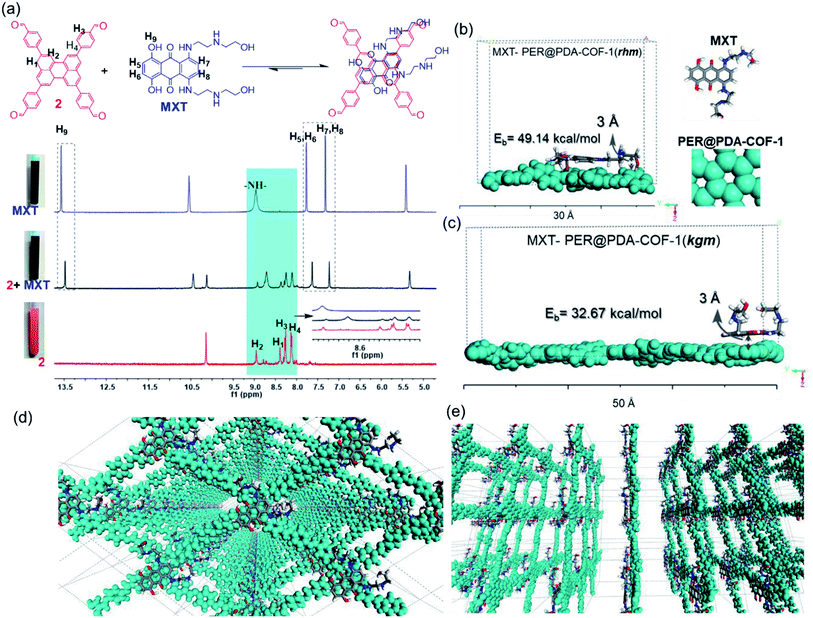 |
| | Fig. 4 (a) 1H NMR spectra (in DMSO-d6) of 2 (red), MXT (blue), and 2 + MXT (black, 1:1 v/v). (b) A simulated unit cell of an MXT-adsorbed PER@PDA-COF-1 monolayer with a rhombus (b) and Kagome (c) structure, and their binding energies. (d) A top view of the supercell structure of MXT–PER@PDA-COF-1 and (e) a side view. | |
Exfoliation and the PER@PDA-COF-1 based protein corona (PC)
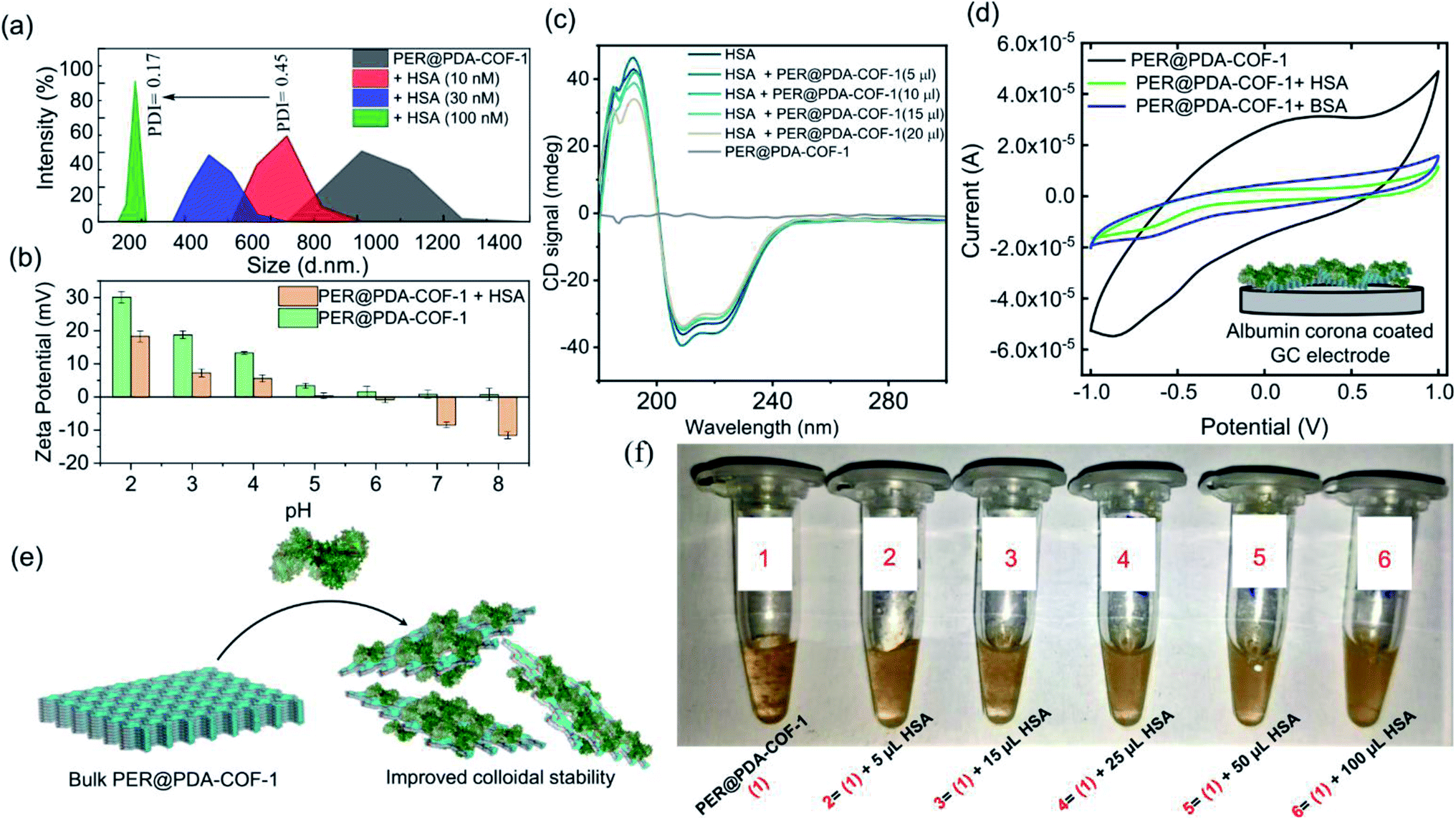
Human serum albumin (HSA) is an obvious choice for the study due to its high abundance, longer half-life and higher accumulating tendency towards tumorigenic cells51 which may form a protein corona and demonstrate efficient intracellular drug delivery. It was observed that there was a “turn-ON” type spectral response for PER@PDA-COF-1 (λmax ∼ 484 nm) upon the gradual addition of HSA into the suspension of PER@PDA-COF-1 (HSA–PER@PDA-COF-1) in aqueous media (Fig. 5a), which could be attributed to delamination of the thick lattice.52 A Job's plot (Fig. S14, ESI†) analysis indicated 1:1 complex formation between HSA and PER@PDA-COF-1 with a binding constant (logk) of 5.34. The increase in fluorescence lifetime (Fig. 5b) of the HSA–PER@PDA-COF-1 protein corona compared to the as-synthesized material alone not only proved the diffusion of the COF layers in aqueous medium but also demonstrated the rigidification of the protein-adsorbed CONs by the formation of an HSA corona.53,54 We performed the atomic force microscopy (AFM) technique (Fig. 5c). Non-aqueous exfoliation of PER@PDA-COF-1 using isopropanol, dioxane etc. led to the collection of the weakly bound small crystallites (Fig. 5ci–iii) during ultrasonication. But the aqueous exfoliation yielded nanosheets of lower aspect ratio (Fig. 5civ–vi) than when exfoliation was done in non-aqueous solvents (isopropanol), presumably due to the lower dispersibility. When HSA was added to the aqueous dispersion, delamination occurred along with the formation of an albumin corona with each de-stacked layer. At a lower concentration of HSA, a substantial decrease in sheet height was observed with a higher aspect ratio (Fig. 5cvii–ix). Upon increasing the HSA concentration, the sheet height decreased ∼5/6 fold with an abrupt increase in the aspect ratio (Fig. 5cx–xii). This clearly demonstrates the albumin-induced rapid aqueous exfoliation of PER@PDA-COF-1.55 Protein corona formation around the CONs was also confirmed from the AFM images of reduced opacity (Fig. 5di and iii) upon incubation with HSA. The outer coating of albumin (pink rectangular zone) around the PER@PDA-COF-1 could be visualized from the height-to-width profile of the HSA–PER@PDA-COF-1 complex (Fig. 5dii and iv). The ultraviolet-visible (UV-vis) spectrum of PER@PDA-COF-1 (0.2 mg mL−1) exhibited an instant hypsochromic shift (Fig. 5e) of about 7 nm compared to that of the bulk powder (462 nm vs. 455 nm) upon gradual addition of HSA (5–15 μL, 0.3 mM). The shifts (change in band gap) can be attributed to the albumin-induced exfoliation. At a lower concentration of HSA, a substantial decrease in the polydispersity index of the protein/COF colloids was investigated by dynamic light scattering (DLS) experiments. From Fig. 6a, a significant size difference was observed upon the gradual addition of HSA into the aqueous dispersion of PER@PDA-COF-1. The DLS peak shape indicates a gradual decrease in the polydispersity index (PDI) of HSA–PER@PDA-COF-1 with increasing concentration of HSA in bulk PER@PDA-COF-1. A pH-dependent zeta potential was also measured for the bare framework and its albumin corona (Fig. 6b).
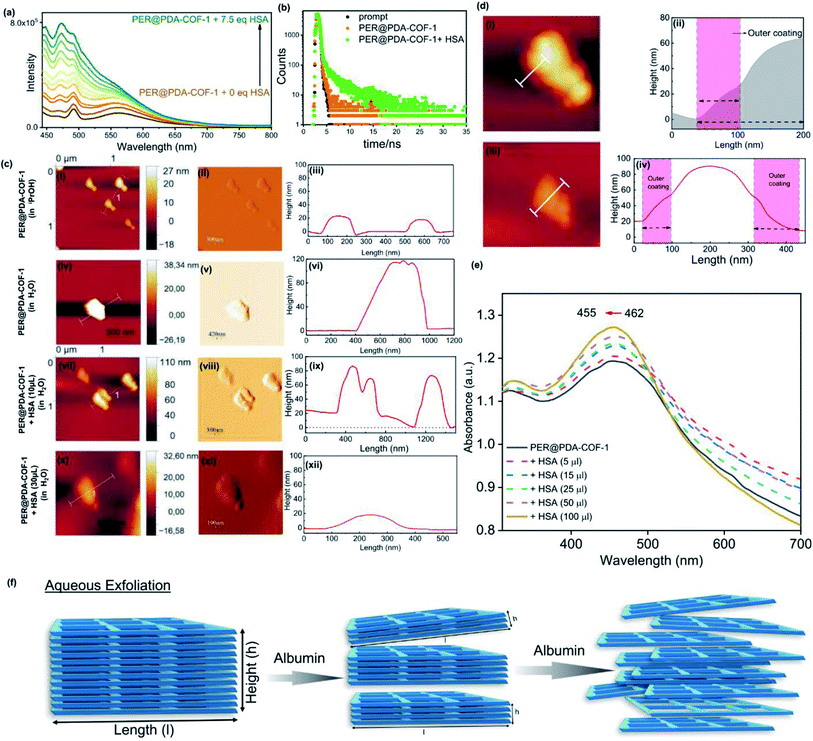 |
| | Fig. 5 (a) The exfoliation induced emission enhancement (EIEE) of PER@PDA-COF-1 (λex = 423 nm) with the gradual addition of HSA. (b) Fluorescence lifetime analysis of PER@PDA-COF-1 in the presence of HSA. (c) AFM analysis of PER@PDA-COF-1 upon non-aqueous exfoliation using isopropanol ((i–iii) represent the height image, peak force image, and height profile, respectively), aqueous exfoliation only ((iv–vi) represent the height image, peak force image, and height profile, respectively), and the effects of albumin on aqueous exfoliation [(vii and x); (viii and xi); and (ix and xii) represent height images, peak force images, and height profiles, respectively] of PER@PDA-COF-1. (d) Visualization of the protein corona based on the height–width profile of HSA–PER@PDA-COF-1 with an outer coating of serum albumin [(i and iii) and (ii and iv) represent height images and height profiles, respectively]. (e) UV-vis absorption spectra of the exfoliated PER@PDA-COF-1 nanosheets in water with different HSA concentrations (5–100 μL, 0.3 mM). The height was observed with a higher aspect ratio (cvii–ix). Upon increasing the HSA concentration, the sheet height decreased ∼5/6 fold with an abrupt increase in the aspect ratio (cx–xii). This clearly demonstrates the albumin-induced rapid aqueous exfoliation of PER@PDA-COF-1. Protein corona formation around the CONs was also confirmed from the AFM images with reduced opacity (di and iii) upon incubation with HSA. The outer coating of albumin (pink rectangular zone) around PER@PDA-COF-1 could be visualized from the height-to-width profile of the HSA–PER@PDA-COF-1 complex (dii and iv). The ultraviolet-visible (UV-vis) spectrum of PER@PDA-COF-1 (0.2 mg mL−1) exhibited a blue shift (e) of about 7 nm compared to that of the bulk powder (462 nm vs. 455 nm) instantly upon the gradual addition of HSA (5–15 μL, 0.3 mM). (f) A schematic diagram of the albumin-induced layer-by-layer exfoliation of PER@PDA-COF-1 under sonication. | |
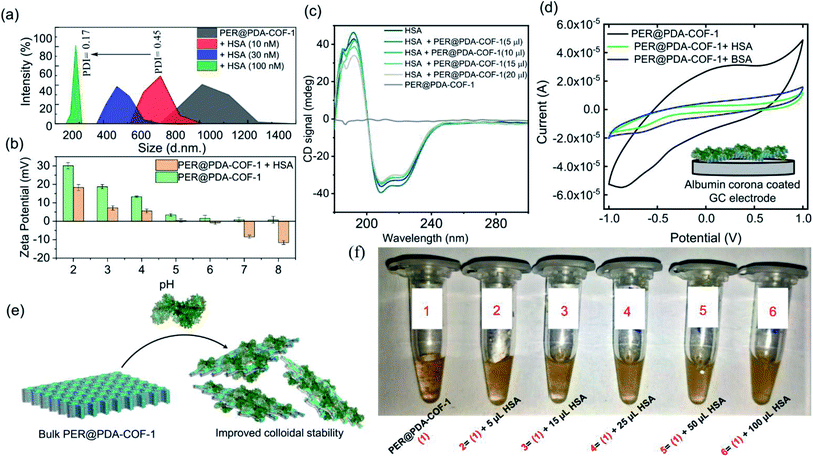 |
| | Fig. 6 (a) Average sizes of PER@PDA-COF-1 alone and exfoliated PER@PDA-COF-1 derived from DLS studies of solutions that were briefly sonicated in the presence of different concentrations of albumin (10, 30 and 100 μL; 0.3 mM) in water. (b) A pH-dependent zeta-potential study of PER@PDA-COF-1 alone and the protein corona of PER@PDA-COF-1. (c) Circular dichroism (CD) spectra of human serum albumin (HSA) upon the addition of PER@PDA-COF-1 at different concentrations (5–20 μL, 0.3 mM). (d) Cyclic voltammetry curves of bulk PER@PDA-COF-1 (black curve) and various protein coronas of the PER@PDA-COF-1 framework (green and blue curves for HSA and BSA, respectively) at a scan rate of 50 mV s−1 (e) A schematic representation of albumin-induced exfoliation and protein corona formation around PER@PDA-COF-1 nanosheets. (f) Photographs of aqueous dispersions of PER@PDA-COF-1 (0.2 mg mL−1) alone and HSA–PER@PDA-COF-1 (0.3 mM) at different albumin concentrations. (Photographs were taken after settling the aqueous dispersions overnight.) | |
The conformational variation of HSA, if any, upon adsorption onto the COF surface was investigated using circular dichroism. No significant perturbation of the secondary structure of HSA was observed in terms of peak position and spectral shape upon the gradual addition of PER@PDA-COF-1 into HSA solution (0.2 mM). PER@PDA-COF-1 showed basal ellipticity. Fig. 6c indicates the gradual loss of secondary structure with an increasing amount of COF (0.2 mg mL−1), which clearly demonstrates the surface binding of HSA on the organic backbone. Surface adsorption can be established further by a cyclic voltammetry (CV) test of PER@PDA-COF-1 and HSA-adsorbed PER@PDA-COF-1, which were coated on a glassy carbon electrode in a three-electrode cell. The significant drop in the current generated from PER@PDA-COF-1 was observed upon extensive surface entrapment of various proteins (e.g. HSA, BSA, concanavalin A, cytochrome c, trypsin) because their polyampholytic nature insulates the interfacial charge transfer between the COF matrix and the electrode.56 CVs of each sample were carried out at a scan rate of 50 mV s−1 from −1 V to +1 V using 0.4 M KCl as the electrolyte. During the experiment, the amount of protein (μM) and PER@PDA-COF-1 (μg) was kept constant for electrode preparation. From Fig. 6d and S16, ESI† it can be understood that HSA causes the maximum current drop among all the proteins tested, which substantiates the preferential highest possible adsorption affinity and higher surface coverage for HSA towards the porous organic surface of the COF backbone. This was also obvious from the fluorescence spectra where the albumin proteins like BSA and HSA showed the highest extent of interactions with PER@PDA-COF-1 among the series of proteins tested (Fig. S15, ESI†).
HSA-assisted colloidal stability for an aqueous dispersion of PER@PDA-COF-1 can be understood from the fact that albumin corona formation around individual delaminated crystallites shows a lower tendency towards re-aggregation (Fig. 6e), which can be visualized from the photographs of aqueous dispersions of PER@PDA-COF-1 in various HSA concentrations (Fig. 6f). This HSA–PER@PDA-COF-1 corona complex also showed improved urea (Fig. S17a, ESI†) and temperature-dependent stability (Fig. S17b, ESI†) compared to HSA alone. These can be attributed to the strong non-covalent surface binding onto the organic surface using the larger lipid-binding domain present in serum albumins, which bears a resemblance to the reported coordinative surface binding of proteins presumably with the metal nodes present in metal–organic frameworks.57
Serum corona triggered drug release study (in vitro)
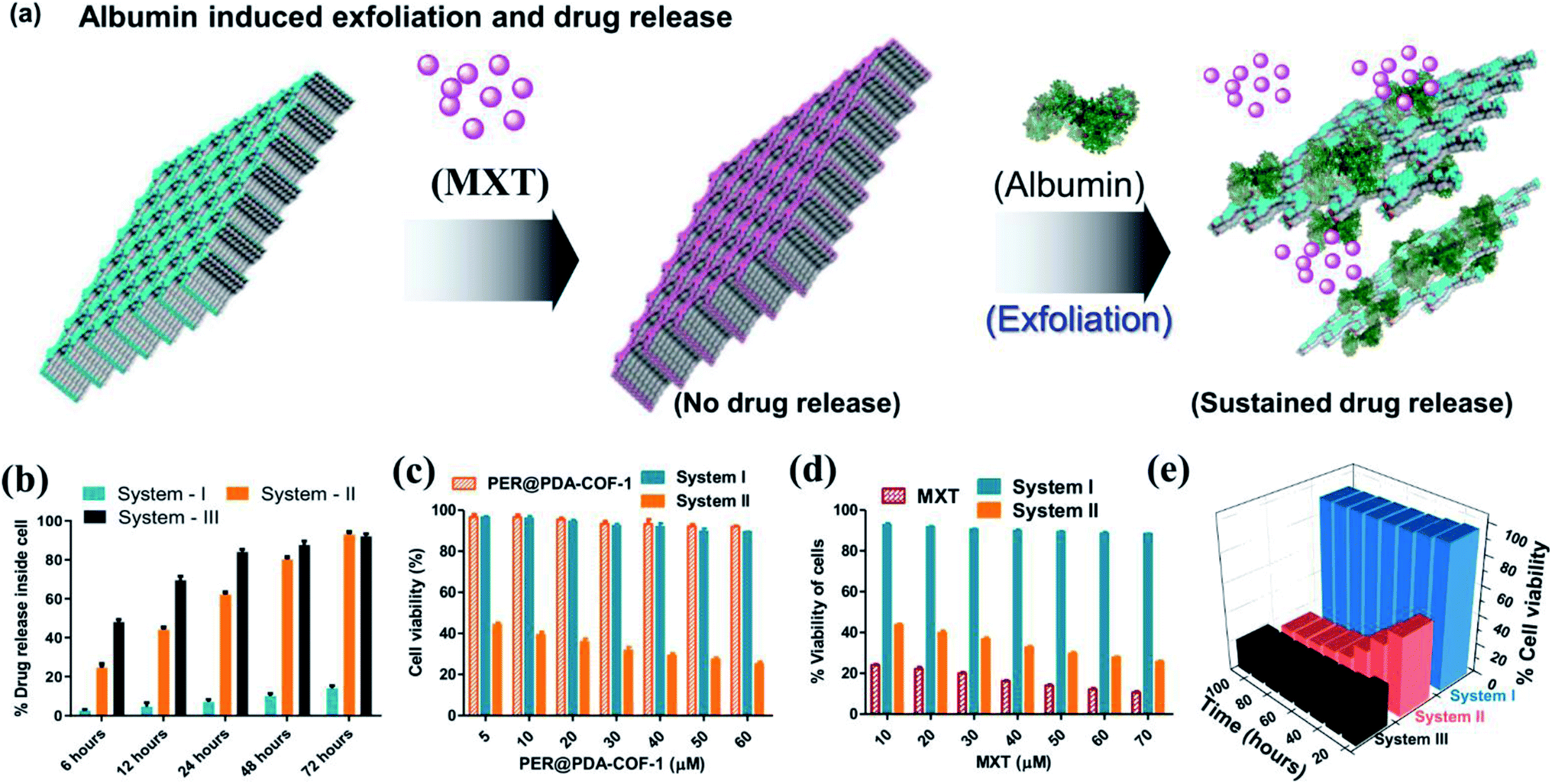
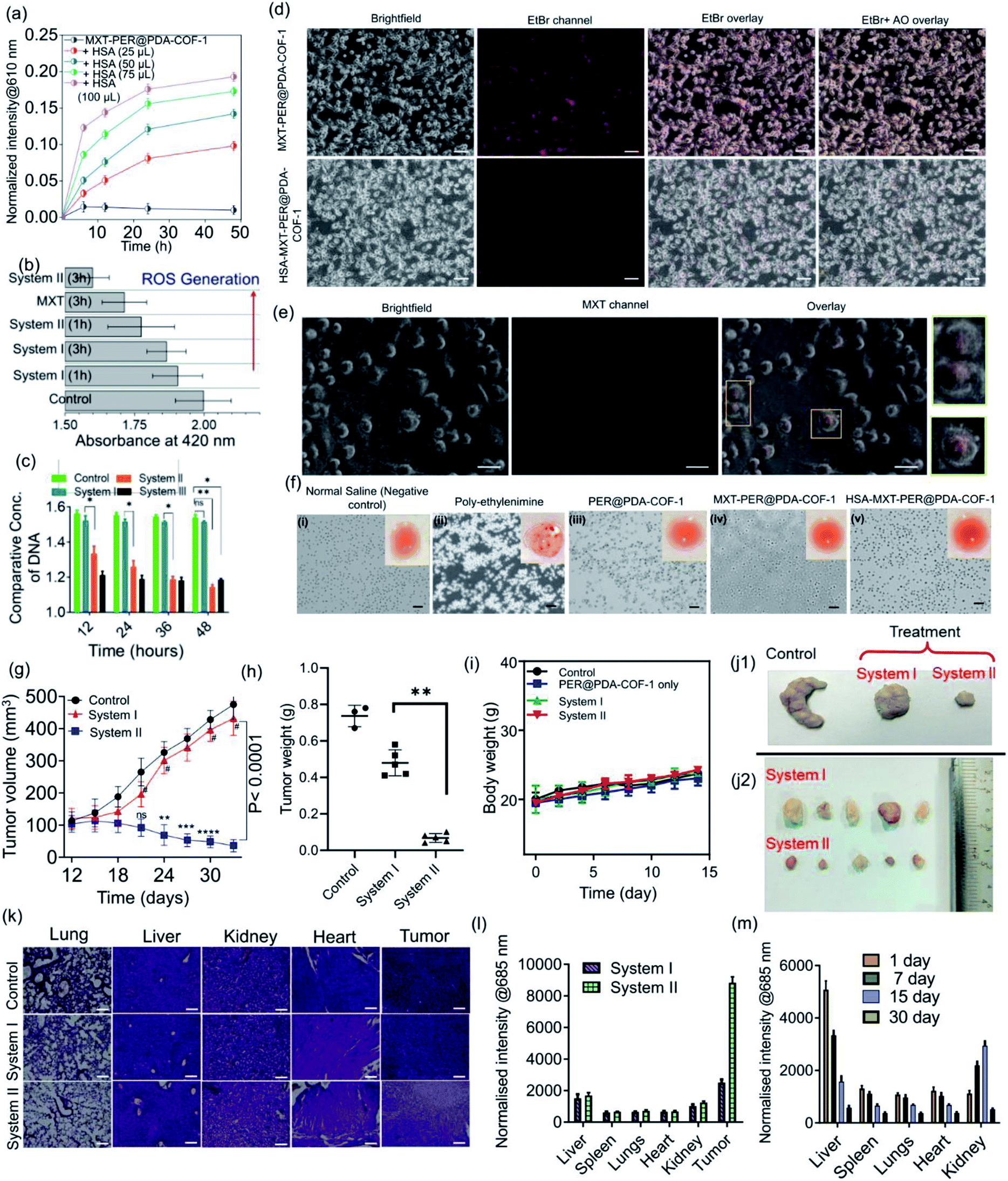
The tendency of higher surface adsorption of albumins on this covalent organic framework motivated us to evaluate the therapeutic efficacy of the HSA–MXT–PER@PDA-COF-1 complex for the cellular delivery of MXT, where HSA may act as a drug-eluting component that in turns triggers the vectorial diffusion of MXT from the fluorescent COF-based probe and the process can be detected for real-time monitoring of drug release and cell imaging.58 Various efforts have been made to develop single-component fluorescent organic nanoparticle-based nanocarriers for the drug molecule MXT and also for imaging purposes. However, a multicomponent organic protein/COF nano-formulation based passive strategy for controlled drug release has not been reported so far. Such an approach will not only increase the methodologies for therapeutic purposes but also increase the scope of applicability of COF-based system in the nanomedicine field. A protein-coated COF-based nano assembly was found to be more durable than a single macromolecular protein molecule in a biological environment (Fig. S18, ESI†). To investigate the drug binding and its release, MXT was first loaded inside the π-surface of the 2D framework (system-I, MXT–PER@PDA-COF-1) and then HSA was added to form the protein corona (system-II, HSA–MXT–PER@PDA-COF-1) to trigger the release of MXT. A small amount of premature leakage from MXT–PER@PDA-COF-1 was found to be a maximum of ∼5%, which may be attributed to the drug molecule associating with the exterior surfaces of the bulk PER@PDA-COF-1 particles.59 At first, the temperature, urea, and proteinase dependent stabilities of system-II were investigated in buffered aqueous media (1× PBS buffer, pH 7.4) by monitoring the change in the emission intensity due to MXT entrapped inside the complex. It was observed that the anticancer drug MXT was released in a sustained manner from system-II even under potentially protein-denaturing conditions like an increase in temperature (Fig. S19a, ESI†) and an increase in the urea concentration (Fig. S19b, ESI†). The proteinase K stability of the complex was not improved in the nano-formulation, probably due to the preferential protein–protein interaction (Fig. S20, ESI†). The albumin-activated release of MXT, which is schematically shown in Fig. 7a and time-dependent MXT release profiles inside the cell from each system are given in Fig. 7b (system-III; MXT with HSA) and Fig. S21, ESI.† A trace amount of premature leaching had been found from MXT–PER@PDA-COF-1, which may be attributed to the strong intercalation of the drug molecule with the π-electron rich porous host, impeding the leakage of MXT. In this study, serum albumin acts as a drug-adhesive vector, presumably because of various drug-binding domains60–62 present in the serum albumins. They interact preferentially with MXT over exfoliated CONs, which activates the release of the drug molecule from PER@PDA-COF-1. The toxicity of system-II was assayed in the HeLa63 (human cervical carcinoma) cell line. The cytotoxic effect of COF alone appeared to be insignificant (Fig. 7c). MXT-loaded COF also showed null toxicity probably due to the release of a trace amount of MXT inside the cell (Fig. 7d). The albumin coronation (system-II) triggers the release of MXT, which introduces significant drug release compared to system-I in terms of cell viability at each of the examined concentrations for the same period of incubation. The toxicity induced by system-II can be corroborated by the time-dependent cellular drug release profile from the corona complex compared to system-I. Time-dependent cell viability was also monitored, from which it was concluded that the cellular release of MXT inside the cells has been triggered by albumin and the rate of release is much more sustainable than in system-III (Fig. 7e). The time-dependent intracellular release of MXT at different stimuli (albumin) concentrations was investigated by the in situ addition of HSA to the cell plates (Fig. 8a and S22a, ESI†). The distribution of MXT across the cell wall (extracellular and intracellular) was also estimated to explore the role of the albumin carrier in intracellular delivery (Fig. S22b and c, ESI†). The increased amount of MXT in the extracellular environment for HSA–MXT–PER@PDA-COF-1 may be attributed to the higher amount of free MXT molecule than MXT–PER@PDA-COF-1. The intracellular release was further substantiated by the time-dependent increased generation of reactive oxygen species (ROS) inside the cell (Fig. 8b). The increased generation of ROS as a function of time explains the albumin stimuli-driven sustained drug release behavior. The cellular DNA was extracted by a cellular DNA extraction kit by Thermo Fisher using the manufacturer's protocol. For each cellular lysate, approximately 5 × 107 cells were seeded. The concentrations of DNA were measured using Biotek Epoch 2 nanodrop (Fig. 8c). It was seen that the concentration of DNA, as obtained from the cells treated with MXT–PER@PDA-COF-1 (system-I), was found to be more than the cells treated with system-II. It was observed that the concentration of DNA gradually decreased with an increasing incubation period of the cells with system-II. The cells were subsequently treated with ethidium bromide64 and acridine orange (AO) simultaneously to observe the DNA damage that occurred due to the treatment of system-I and system-II separately. The fluorescence images of the ethidium bromide channel for the cells treated with system-I and system-II are given in Fig. 8d. The apoptotic nature of the cells together with the cellular intake of MXT involving system-II was also observed under a fluorescence microscope (Fig. 8e, S23 and S24, ESI†). The effect of pristine COF, system-I, and system-II on hemagglutination was determined via coagulation assays (prothrombin time/activated partial thromboplastin time) via either an extrinsic pathway or an intrinsic pathway (Fig. 8f). Clot formation (Table S1, ESI†) was observed after blood incubation of PER@PDA-COF-1, system-I and system-II. The extrinsic coagulation pathway and the intrinsic pathway were not affected by the covalent framework-based drug delivery matrix; i.e., PER@PDA-COF-1, system-I, and system-II exhibited a blood clotting time of around 5–6 min (PT), whereas the positive control showed a value of around 11–16 s. The APTT values for all the samples were in the range of around 60–80 s.
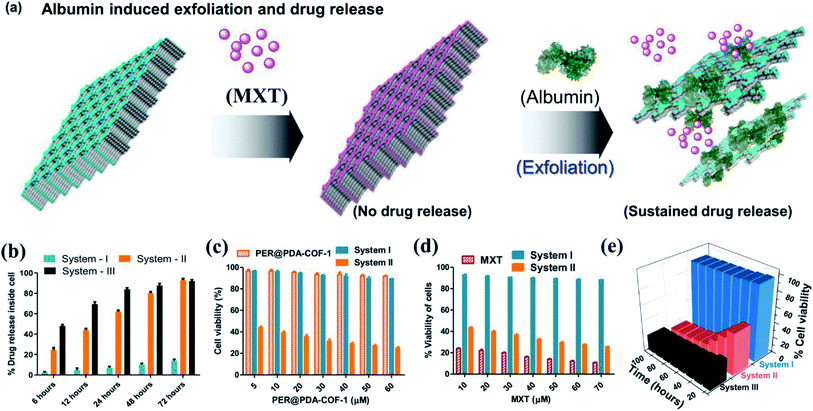 |
| | Fig. 7 (a) A schematic representation of albumin-triggered mitoxantrone (MXT) release from the porous host PER@PDA-COF-1. (b) Comparative time-dependent mitoxantrone release profiles of system-I, system-II, and system-III. (c) Comparative cellular viability (MTT assay) studies with respect to the concentration of PER@PDA-COF-1 and (d) the concentration of MXT. (e) Time-dependent cell viability studies of system-I, system-II, and system-III. | |
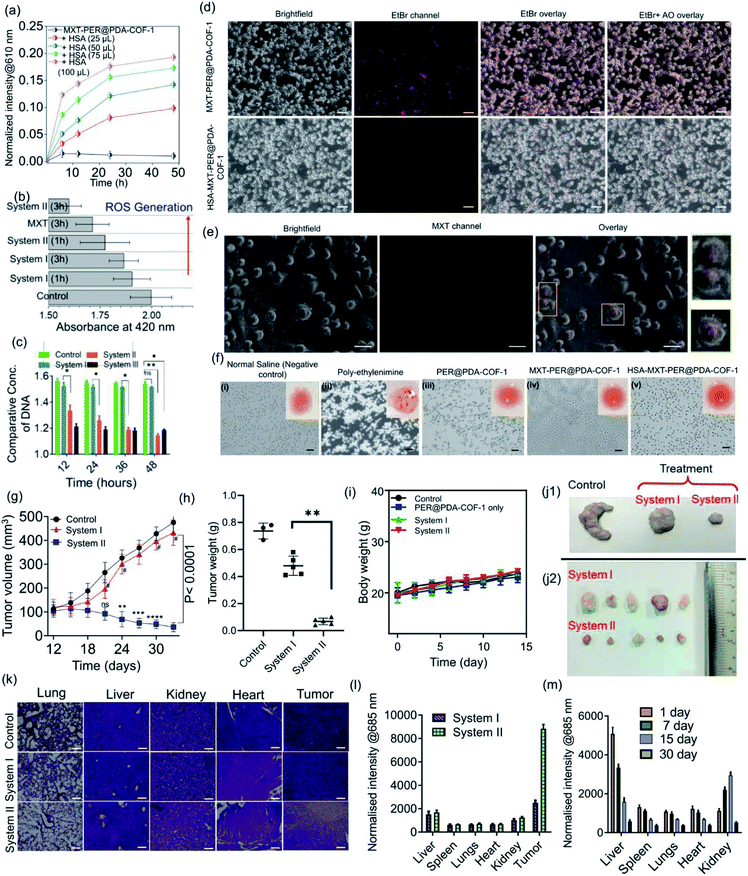 |
| | Fig. 8 (a) Time-dependent drug release profiles of MXT–PER@PDA-COF-1 alone and HSA–MXT–PER@PDA-COF-1 nano-formulations with different stimuli (albumin) concentrations. (b) The time-dependent generation of reactive oxygen species (ROS) inside the cells. (c) The time dependent quantification of DNA concentrations using system-I, system-II, and system-III; (n = 5, *p < 0.05). (d) Fluorescence intensities of ethidium bromide inside cells treated with MXT–PER@PDA-COF-1 and HSA–MXT–PER@PDA-COF-1 (scale bar: 100 μm). (e) Fluorescence microscopy images of cells treated with HSA–MXT–PER@PDA-CON-1 (scale bar: 200 μm) to visualize the apoptotic nature of the cells. (f) Micrographs of hemagglutination (scale bars = 50 μm) demonstrating the blood compatibility of PER@PDA-COF-1 (iii), MXT–PER@PDACOF-1 (iv), and HSA–PER@PDA-COF-1 (v), where normal saline is used as a negative control (i) and poly-ethylenimine is used as a positive control (ii). Evaluation of MXT storage and serum protein triggered MXT release using an in vivo mouse tumor model upon injecting normal saline (as a control), system-I, or system-II. (g) Corresponding growth curves of tumors in different groups of mice treated with normal saline solution (black plot), system-I (red plot), and system-II (blue plot, n = 5, ***p < 0.0001). (h) Average tumor weights after 33 days of treatment (n = 5, **p < 0.005). (i) Changes in body weights of the tumor-bearing mice during treatment. Representative photographs of the excised tumors from killed mice from the control and treated (system-I and system-II) groups (j1 and j2). (k) The H&E staining of major organs (lung, liver, kidney, heart) and CT-26 cells bearing tumor sections from the treated mice (scale bars: 50 μm). (l) The biodistribution of MXT upon the intra-tumoral injection of system-I and system-II in main organs and (m) the biodistribution after treatment with system-II for up to 30 days. | |
In vivo antitumor efficiency
Encouraged by the in vitro performance, serum corona triggered drug release from MXT–PER@PDA-COF-1 was further assessed using an in vivo mouse tumor model. Prior to that, the blood biochemistry was studied to assess whether the protein corona of PER@PDA-COF-1 could be injected intravenously. In this experiment, serum functional enzymes were monitored in healthy mice. Alanine aminotransferase (ALT), and aspartate aminotransferase (AST) were used to evaluate hepatotoxicity. Creatinine (Cr) and blood urea nitrogen (BUN) were used to evaluate nephrotoxicity. No significant toxicological effect was observed in healthy mice during the treatment days of intravenous administration of system-I and system-II (dose: 20 mg kg−1), as depicted by the blood parameters, including haematology, liver and kidney index (Tables S2–S5, ESI†). Then, CT-26 tumor-bearing mice were randomly divided into three groups after the 9th day from tumor implantation. Then they were subjected to intra-tumoral injection of normal saline (control), MXT–PER@PDA-COF-1 (system-I) and HSA–MXT–PER@PDA-COF-1 (system-II) where a constant concentration of mitoxantrone (1.25 mg mL−1) was maintained in both nano-formulations (system-I and system-II). The HSA:MXT ratio was maintained at 1:100 (μmol μmol−1) in system-II. After 24 days from the chemotherapeutic (system-I, system-II) injections, the mice were subjected to euthanasia and the tumors were excised. As can be seen from Fig. 8g, obvious growth of the tumor was observed for the saline-treated group. The MXT–PER@PDA-COF-1 treated group, however, exhibited much lower tumor growth suppression efficiency, which is comparable to the observation from the normal saline-treated group. In contrast, significant tumor growth suppression was observed in the system-II group. Overall, ∼10% reduction in tumor volume for the system-I and ∼92% for the system-II treated mice were observed within 24 days (9th to 33rd day). This observation confirms that PER@PDA-COF-1 can act as a safe storage system for the MXT molecule in physiological conditions and HSA significantly elutes the MXT molecule from the PER@PDA-COF-1 lattice and specifically accumulates on the tumor vasculature. The excellent antitumor efficiency of system-II can also be evidenced from the average tumor weight (Fig. 8h) which was recorded finally. The improved pharmacokinetics of MXT can be understood from the comparative growth curve of MXT-treated mice with system-II (Fig. S25, ESI†). No significant body weight loss was observed in any of the mice from each group, which suggests that this protein corona-based nano-formulation has insignificant in vivo toxic side effects (Fig. 8i). After 23 days of therapy, the final tumor sizes from the control and treated groups can be seen from Fig. 8j1 and j2. Hematoxylin and eosin (H&E) histology analyses of the lung, liver, kidney, heart, and tumor tissues, were done to further evaluate the in vivo toxicity of this nano-formulation, as shown in Fig. 8k. H&E staining of the organs confirms that no toxicity/side effect was observed even after 33 days of therapeutic treatment. In the organ histology profile, metastatic foci were detected in the livers and lungs collected from the system-I and saline-treated mice. In contrast, no metastatic foci were found in the system-II treated mice, which demonstrates the anti-metastatic property of the albumin-based nano-formulation from a drug-loaded organic framework.65 The bio-distribution of MXT inside the tissue was measured for both nano-formulations (system-I and system-II) by using tumor (Fig. 8l) and non-tumor (Fig. S26, ESI†) mouse models. MXT inside the tissue from system-II administration was found to be higher than for system-I. Moreover, in the case of intra-tumoral injections, MXT distribution in the tumor tissue was found to be selectively higher than in other different organs (liver, spleens, lungs, heart, kidney), which demonstrates the albumin corona augmented passive targeting of MXT delivery using MXT–PER@PDA-COF-1. The clearance pathway of HSA–MXT–PER@PDA-COF-1 was investigated after intra-tumoral injection. The concentrations of MXT in urine and faeces were both measured by fluorescence spectroscopy. High levels of MXT were detected in urine (Fig. S27, ESI†), strong evidence that HSA–MXT–PER@PDA-COF-1 could be degraded into small fragments due to the presence of HSA, and then eliminated from the body through the renal filtration pathway. The same can also be confirmed by long-term biodistribution of MXT in the main organs (Fig. 8m). Overall, these results show that this nano-formulation strategy could be a promising platform with an insignificant level of toxicity with an improved pharmacokinetic property.
Conclusions
In summary, we have developed a novel albumin corona-based nano-formulation strategy for therapeutic purposes where a perylene-based COF serves as an effective porous host for the anticancer drug mitoxantrone (MXT), and it did not induce any cellular release of MXT. The addition of albumin triggered drug release, which led to the elimination of cancerous cells in a controlled manner. Serum protein acts here as a drug-eluting component, which initiates vectorial diffusion of the MXT molecules from the covalent organic framework to the tumorigenic cells. The process involves a synchronized pathway of exfoliation and protein corona formation around drug-loaded COF crystallites. The methodology provides an excellent approach for passive drug delivery using a biocompatible covalent organic framework-based system, especially for administration for therapeutic purposes using COFs. This protocol was extensively investigated and found to be an excellent COF/albumin-based drug delivery nano-formulation strategy in terms of biocompatibility, cell penetrability, and sustainability. Thus, such a coronation approach utilizing COFs to induce sustained drug delivery has significant potential for developing novel nano-bioengineering strategies for cancer chemotherapy.
Author contributions
SB and PS contributed equally. Prof. SB conceived the overall idea and provided funds and infrastructure for carrying out this work. Materials and building blocks were synthesized by SB, and PS and PM conducted the experiments and analyzed the results. Dr MA conducted the theoretical analysis. SB, PS, PM, Dr MA, and Prof. SB contributed to the overall interpretation of the results and wrote the overall manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
SB and PS thank CSIR for a research fellowship. PM and other authors are thankful to the Department of Science and Technology (DST), Government of India, for financial support provided through the Technical Research Centre (TRC) at IACS. Prof. SB thanks DST for a JC Bose fellowship. We also thank Dr A. Pradhan for his assistance with the synthesis of the starting materials and preliminary discussion.
Notes and references
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS
 .
.
- B. C. Patra, S. K. Das, A. Ghosh, A. Raj K, P. Moitra, M. Addicoat, S. Mitra, A. Bhaumik, S. Bhattacharya and A. Pradhan, J. Mater. Chem. A, 2018, 6, 16655–16663 RSC .
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC .
- D. Rodríguez-San-Miguel, C. Montoro and F. Zamora, Chem. Soc. Rev., 2020, 49, 2291–2302 RSC .
- A. Schneemann, R. Dong, F. Schwotzer, H. Zhong, I. Senkovska, X. Feng and S. Kaskel, Chem. Sci., 2021, 12, 1600–1619 RSC .
- S. Y. Chong, J. T. A. Jones, Y. Z. Khimyak, A. I. Cooper, A. Thomas, M. Antonietti and M. J. Bojdys, J. Mater. Chem. A, 2013, 1, 1102–1107 RSC .
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed .
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC .
- J. N. Coleman, Acc. Chem. Res., 2013, 46, 14–22 CrossRef CAS PubMed .
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed .
- D. Sheberla, J. C. Bachman, J. S. Elias, C.-J. Sun, Y. Shao-Horn and M. Dincă, Nat. Mater., 2017, 16, 220–224 CrossRef CAS PubMed .
- W. Li, L. Sun, J. Qi, P. Jarillo-Herrero, M. Dincă and J. Li, Chem. Sci., 2017, 8, 2859–2867 RSC .
- T. Chen, J.-H. Dou, L. Yang, C. Sun, N. J. Libretto, G. Skorupskii, J. T. Miller and M. Dincă, J. Am. Chem. Soc., 2020, 142, 12367–12373 CrossRef CAS PubMed .
- S. Bhunia, K. Bhunia, B. C. Patra, S. K. Das, D. Pradhan, A. Bhaumik, A. Pradhan and S. Bhattacharya, ACS Appl. Mater. Interfaces, 2019, 11(1), 1520–1528 CrossRef CAS PubMed .
- H. Wang, Z. Zeng, P. Xu, L. Li, G. Zeng, R. Xiao, Z. Tang, D. Huang, L. Tang and C. Lai, Chem. Soc. Rev., 2019, 48, 488–516 RSC .
- S. Bhunia, N. Dey, A. Pradhan and S. Bhattacharya, Chem. Commun., 2018, 54, 7495–7498 RSC .
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef CAS PubMed .
- Q. Zhu, X. Wang, R. Clowes, P. Cui, L. Chen, M. A. Little and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 16842–16848 CrossRef CAS PubMed .
- E. Tylianakis, E. Klontzas and G. E. Froudakis, Nanoscale, 2011, 3, 856–869 RSC .
- K. Dey, M. Pal, K. C. Rout, S. H. Kunjattu, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed .
- D. B. Shinde, L. Cao, A. D. D. Wonanke, X. Li, S. Kumar, X. Liu, M. N. Hedhili, A.-H. Emwas, M. Addicoat, K.-W. Huang and Z. Lai, Chem. Sci., 2020, 11, 5434–5440 RSC .
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed .
- P. Wang, F. Zhou, K. Guan, Y. Wang, X. Fu, Y. Yang, X. Yin, G. Song, X.-B. Zhang and W. Tan, Chem. Sci., 2020, 11, 1299–1306 RSC .
- Y. Meng, G. Lin, H. Ding, H. Liao and C. Wang, J. Mater. Chem. A, 2018, 6, 17186–17191 RSC .
- D.-G. Wang, T. Qiu, W. Guo, Z. Liang, H. Tabassum, D. Xia and R. Zou, Energy Environ. Sci., 2021, 14, 688–728 RSC .
- G. Zhang, X. Li, Q. Liao, Y. Liu, K. Xi, W. Huang and X. Jia, Nat. Commun., 2018, 9, 1–11 CrossRef PubMed .
- S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. Illath, D. D. Diaz and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 4513–4520 CrossRef CAS PubMed .
- F. Benyettou, N. Kaddour, T. Prakasam, G. Das, S. K. Sharma, S. A. Thomas, F. Bekhti-Sari, J. Whelan, M. A. Alkhalifah and M. Khair, Chem. Sci., 2021, 12, 6037–6047 RSC .
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed .
- E. L. Spitler and W. R. Dichtel, Nat. Chem., 2010, 2, 672–677 CrossRef CAS PubMed .
- M. Sajid, Environ. Sci. Pollut. Res., 2016, 23, 14805–14807 CrossRef PubMed .
- B. Illes, P. Hirschle, S. Barnert, V. Cauda, S. Wuttke and H. Engelke, Chem. Mater., 2017, 29, 8042–8046 CrossRef CAS .
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette and C. Kreuz, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed .
- I. A. Lázaro, S. Haddad, S. Sacca, C. Orellana-Tavra, D. Fairen-Jimenez and R. S. Forgan, Chem, 2017, 2, 561–578 Search PubMed .
- V. S. Vyas, M. Vishwakarma, I. Moudrakovski, F. Haase, G. Savasci, C. Ochsenfeld, J. P. Spatz and B. V. Lotsch, Adv. Mater., 2016, 28, 8749–8754 CrossRef CAS PubMed .
- S. K. MIsra, P. Kondaiah, S. Bhattacharya and C. N. R. Rao, Small, 2012, 8(1), 131–143 CrossRef CAS PubMed .
- B. S. Chhikara, S. K. Misra and S. Bhattacharya, Nanotechnology, 2012, 23(6), 065101 CrossRef PubMed .
- P. Moitra, Y. Subramanian and S. Bhattacharya, J. Phys. Chem. B, 2017, 121, 815–824 CrossRef CAS PubMed .
- P. Moitra, K. Kumar, P. Kondaiah and S. Bhattacharya, Angew. Chem., 2014, 126, 1131–1135 CrossRef .
- S. K. Misra, S. Naz, P. Kondaiah and S. Bhattacharya, Biomaterials, 2014, 35, 1334–1346 CrossRef CAS PubMed .
- P. Moitra, K. Kumar, S. Sarkar, P. Kondaiah, W. Duan and S. Bhattacharya, Chem. Commun., 2017, 53, 8184–8187 RSC .
- M. D. Crespi, S. E. Ivanier, J. Genovese and A. Baldi, Biochem. Biophys. Res. Commun., 1986, 136, 521–528 CrossRef CAS PubMed .
- Y.-M. Toh and T.-K. Li, Clin. Cancer Res., 2011, 17, 5026–5037 CrossRef CAS PubMed .
- D. S. Alberts, K. S. Griffith, G. E. Goodman, T. S. Herman and E. Murray, Cancer Chemother. Pharmacol., 1980, 5, 11–15 CrossRef CAS PubMed .
- F. Shojaie and M. Dehghan, Nanomed. J., 2016, 3, 115–126 CAS .
- F. Chai, L. Sun, X. He, J. Li, Y. Liu, F. Xiong, L. Ge, T. J. Webster and C. Zheng, Int. J. Nanomedicine, 2017, 12, 1791–1802 CrossRef CAS PubMed .
- M. Más-Montoya and R. A. J. Janssen, Adv. Funct. Mater., 2017, 27, 1605779 CrossRef .
- P. C. Ke, S. Lin, W. J. Parak, T. P. Davis and F. Caruso, ACS Nano, 2017, 11, 11773–11776 CrossRef CAS PubMed .
- X. Lu, P. Xu, H.-M. Ding, Y.-S. Yu, D. Huo and Y.-Q. Ma, Nat. Commun., 2019, 10, 4520 CrossRef PubMed .
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed .
- S. Wagner, F. Rothweiler, M. G. Anhorn, D. Sauer, I. Riemann, E. C. Weiss, A. Katsen-Globa, M. Michaelis, J. Cinatl, D. Schwartz, J. Kreuter, H. von Briesen and K. Langer, Biomaterials, 2010, 31, 2388–2398 CrossRef CAS PubMed .
- P. Albacete, J. I. Martínez, X. Li, A. López-Moreno, S. a. Mena-Hernando, A. E. Platero-Prats, C. Montoro, K. P. Loh, E. M. Pérez and F. Zamora, J. Am. Chem. Soc., 2018, 140, 12922–12929 CrossRef CAS PubMed .
- A. A. Leonard, M. A. Mosquera, L. O. Jones, Z. Cai, T. J. Fauvell, M. S. Kirschner, D. J. Gosztola, G. C. Schatz, R. D. Schaller, L. Yu and L. X. Chen, Chem. Sci., 2020, 11, 7133–7143 RSC .
- L. Shang, S. Brandholt, F. Stockmar, V. Trouillet, M. Bruns and G. U. Nienhaus, Small, 2012, 8, 661–665 CrossRef CAS PubMed .
- D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef CAS PubMed .
- R. J. Toh, C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, Adv. Funct. Mater., 2017, 27, 1604923 CrossRef .
- A. Zimpel, N. Al Danaf, B. Steinborn, J. Kuhn, M. Höhn, T. Bauer, P. Hirschle, W. Schrimpf, H. Engelke, E. Wagner, M. Barz, D. C. Lamb, U. Lächelt and S. Wuttke, ACS Nano, 2019, 13, 3884–3895 CrossRef CAS PubMed .
- P. Wang, F. Zhou, C. Zhang, S.-Y. Yin, L. Teng, L. Chen, X.-X. Hu, H.-W. Liu, X. Yin and X.-B. Zhang, Chem. Sci., 2018, 9, 8402–8408 RSC .
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS PubMed .
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed .
- B. Ahmad, S. Parveen and R. H. Khan, Biomacromolecules, 2006, 7, 1350–1356 CrossRef CAS PubMed .
- F. Zsila, Mol. Pharm., 2013, 10, 1668–1682 CrossRef CAS PubMed .
- E. Callaway, Nature, 2013, 500, 132–133 CrossRef CAS PubMed .
- J.-B. LePecq and C. Paoletti, J. Mol. Biol., 1967, 27, 87–106 CrossRef CAS .
- B. Ding, P. Zheng, F. Jiang, Y. Zhao, M. Wang, M. Chang, P. a. Ma and J. Lin, Angew. Chem., 2020, 132, 16523–16526 Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bc,
Pranay
Saha‡
bc,
Pranay
Saha‡