
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAggregation caused quenching to aggregation induced emission transformation: a precise tuning based on BN-doped polycyclic aromatic hydrocarbons toward subcellular organelle specific imaging†
Huanan
Huang
 *a,
Lingxiu
Liu
b,
Jianguo
Wang
*b,
Ying
Zhou
a,
Huanan
Hu
a,
Xinglin
Ye
a,
Guochang
Liu
a,
Zhixiong
Xu
a,
Han
Xu
*a,
Wen
Yang
a,
Yawei
Wang
a,
You
Peng
a,
Pinghua
Yang
a,
Jianqi
Sun
a,
Ping
Yan
a,
Xiaohua
Cao
*a and
Ben Zhong
Tang
*c
*a,
Lingxiu
Liu
b,
Jianguo
Wang
*b,
Ying
Zhou
a,
Huanan
Hu
a,
Xinglin
Ye
a,
Guochang
Liu
a,
Zhixiong
Xu
a,
Han
Xu
*a,
Wen
Yang
a,
Yawei
Wang
a,
You
Peng
a,
Pinghua
Yang
a,
Jianqi
Sun
a,
Ping
Yan
a,
Xiaohua
Cao
*a and
Ben Zhong
Tang
*c
aCollege of Chemistry and Chemical Engineering, Jiangxi Province Engineering Research Center of Ecological Chemical Industry, Jiujiang Key Laboratory of Organosilicon Chemistry and Application, Xinghuo Organosilicon Industry Research Center, Jiujiang University, Jiujiang 332005, China. E-mail: huanan200890@163.com; tmxuhan@163.com; 910039498@qq.com
bCollege of Chemistry and Chemical Engineering, Inner Mongolia Key Laboratory of Fine Organic Synthesis, Inner Mongolia University, Hohhot 010021, China. E-mail: wangjg@iccas.ac.cn
cShenzhen Institute of Molecular Aggregate Science and Engineering, School of Science and Engineering, The Chinese University of Hong Kong, Shenzhen 518172, China. E-mail: tangbenz@ust.hk
First published on 11th February 2022
Abstract
Polycyclic aromatic hydrocarbons (PAHs) with boron–nitrogen (BN) moieties have attracted tremendous interest due to their intriguing electronic and optoelectronic properties. However, most of the BN-fused π-systems reported to date are difficult to modify and exhibit traditional aggregation-caused quenching (ACQ) characteristics. This phenomenon greatly limits their scope of application. Thus, continuing efforts to seek novel, structurally distinct and functionally diverse structures are highly desirable. Herein, we proposed a one-stone-two-birds strategy including simultaneous exploration of reactivity and tuning of the optical and electronic properties for BN-containing π-skeletons through flexible regioselective functionalization engineering. In this way, three novel functionalized BN luminogens (DPA-BN-BFT, MeO-DPA-BN-BFT and DMA-DPA-BN-BFT) with similar structures were obtained. Intriguingly, DPA-BN-BFT, MeO-DPA-BN-BFT and DMA-DPA-BN-BFT exhibit completely different emission behaviors. Fluorogens DPA-BN-BFT and MeO-DPA-BN-BFT exhibit a typical ACQ effect; in sharp contrast, DMA-DPA-BN-BFT possesses a prominent aggregation induced emission (AIE) effect. To the best of our knowledge, this is the first report to integrate ACQ and AIE properties into one BN aromatic backbone with subtle modified structures. Comprehensive analysis of the crystal structure and theoretical calculations reveal that relatively large twisting angles, multiple intermolecular interactions and tight crystal packing modes endow DMA-DPA-BN-BFT with strong AIE behavior. More importantly, cell imaging demonstrated that luminescent materials DPA-BN-BFT and DMA-DPA-BN-BFT can highly selectively and sensitively detect lipid droplets (LDs) in living MCF-7 cells. Overall, this work provides a new viewpoint of the rational design and synthesis of advanced BN–polycyclic aromatics with AIE features and triggers the discovery of new functions and properties of azaborine chemistry.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are among the most widely studied organic molecules for organic photoelectric materials.1 Thus, many efforts to modify their structures and properties have been undertaken in the past several decades. The incorporation of main-group elements such as N, S, Si, P, or B into π-systems has emerged as a viable strategy for the development of new materials with unique properties and functions.2 Among these various possible dopants, boron and nitrogen have attracted tremendous attention due to their distinct electronic and optical characteristics. One appealing strategy for doping both boron and nitrogen into PAHs is substituting one or more C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C units by boron–nitrogen (BN) units.3 The replacement of the CC unit by the diatomic BN not only perturbs the electronic structure and distribution but also changes the chemical reactivities while maintaining the aromaticity and basic structural features of a PAH.4 These unique characteristics provide a great opportunity for the design and production of new materials. In fact, over the last few decades, considerable progress has been made in BN-doped PAHs, and some efficient entries to new types of BN-aromatic compounds with unique electronic properties have been developed.5 Although considerable progress has been achieved in BN-doped π-systems, most of the currently available materials usually present traditional luminous behavior,6 that is, luminogens fluoresce strongly in dilute solution but emit weak fluorescence in the aggregate state. Since the concept of aggregation-induced emission (AIE), an exciting and intriguing phenomenon coined by the Tang group in 2001,7 great attention has been attracted to the development of efficient luminogens with AIE feature. However, as an important class of luminogens, boron nitrogen-containing π-systems featuring AIE, especially three-coordinate boron species, have been rarely developed.5f Therefore, extending BN-doped heteroaromatics to the AIE luminogen (AIEgen) family and constructing BN-modified AIE molecular systems remain challenging.
C units by boron–nitrogen (BN) units.3 The replacement of the CC unit by the diatomic BN not only perturbs the electronic structure and distribution but also changes the chemical reactivities while maintaining the aromaticity and basic structural features of a PAH.4 These unique characteristics provide a great opportunity for the design and production of new materials. In fact, over the last few decades, considerable progress has been made in BN-doped PAHs, and some efficient entries to new types of BN-aromatic compounds with unique electronic properties have been developed.5 Although considerable progress has been achieved in BN-doped π-systems, most of the currently available materials usually present traditional luminous behavior,6 that is, luminogens fluoresce strongly in dilute solution but emit weak fluorescence in the aggregate state. Since the concept of aggregation-induced emission (AIE), an exciting and intriguing phenomenon coined by the Tang group in 2001,7 great attention has been attracted to the development of efficient luminogens with AIE feature. However, as an important class of luminogens, boron nitrogen-containing π-systems featuring AIE, especially three-coordinate boron species, have been rarely developed.5f Therefore, extending BN-doped heteroaromatics to the AIE luminogen (AIEgen) family and constructing BN-modified AIE molecular systems remain challenging.
Over the last 20 years, the luminescence mechanism and application of AIEgens have been scientifically and systematically studied.8 Several reasonable principles concerning molecular design have been proposed to construct AIEgens.9 The efficient emission of AIEgens in the solid state is ascribed to their twisted conformations, which hamper detrimental π–π stacking and the restriction of intramolecular motion by multiple noncovalent interactions, such as hydrogen bonding and CH–π interactions between molecules. In particular, intermolecular interactions and molecular packing modes are two crucial factors in regulating molecular luminescence properties. The rational balance of different intermolecular interactions and effective adjustment of the packing modes are thus approached as crucial issues in the design of current AIE materials. As stated earlier, benefitting from the significant polarization of the B–N bond, the additional intermolecular dipole–dipole interaction resulting from the BN unit could effectively tailor the intermolecular interactions and molecular packing modes in the solid state. Thus, we believe that the BN-substituted aromatic structure with appropriate structural modifications may serve as a novel building block to achieve the possible AIE effect. In addition, due to the unique properties in terms of high emission efficiency in aggregates, large Stokes shift and signal “turn on” features, AIE-based turn on probes have recently been developed at a tremendous pace and involved an array of applications ranging from optoelectronic devices to biological imaging. Thus, we speculate that BN-aromatic compounds with AIE feature may also possess special biosensing properties and excellent bioimaging capability. To date, BN-substituted PAHs have mainly been applied for materials in organic optoelectronic devices,5a–c and few results have been reported in the biomedical field,10 both experimentally and theoretically. Therefore, regardless of fundamental research or material application, it is fascinating to probe and verify the possible AIE characteristics and bioimaging application of BN-embedded PAHs.
Herein, to realize and understand the possibility of the AIE effect in BN PAH system, we selected the BN-embedded benzo[f]tetraphene (BN-BFT) as the precursor core (Fig. 1). Molecular skeleton BN-BFT contains a proper conjugate size, ensuring that the molecule presents crystalline state, while avoiding strong π–π stacking interactions, which is an essential condition to achieve the AIE performance of organic materials in aggregates. In addition, the existence of N–H bond may prompt the formation of hydrogen bonds and restrain molecular rotation in aggregated states,11 enhancing the possibility of AIE behavior. On the other hand, although molecule BN-BFT had no AIE effect, the excellent chemical stability of BN-BFT and multiple modifiable sites may allow for us to implement further structural modifications and finally realize AIEgen. Indeed, BN-BFT could undergo highly selective bromination at a specific position, and subsequent cross coupling with the C–Br bond was accomplished. Through the regioselective functionalization strategy, we obtain three expectant BN PAH molecules DPA-BN-BFT, MeO-DPA-BN-BFT and DMA-DPA-BN-BFT. Interestingly, the derived structures exhibit completely different emission behaviors in solution and the solid state. DPA-BN-BFT, MeO-DPA-BN-BFT and DMA-DPA-BN-BFT possess ACQ and AIE features, respectively. To the best of our knowledge, this is the first example of a simple BN aromatic backbone with subtle modification that achieves the regulation of luminescence performance from ACQ to AIE. More importantly, further investigations demonstrated that DPA-BN-BFT and DMA-DPA-BN-BFT can highly selectively and sensitively detect lipid droplets (LDs) in living MCF-7 cells, indicating that azaborine compounds may be promising candidates for targeting specific organelles. These meaningful results provide a new opportunity for BN aromatic systems to discover new properties and functions in a variety of disciplines ranging from materials science to biomedical research.
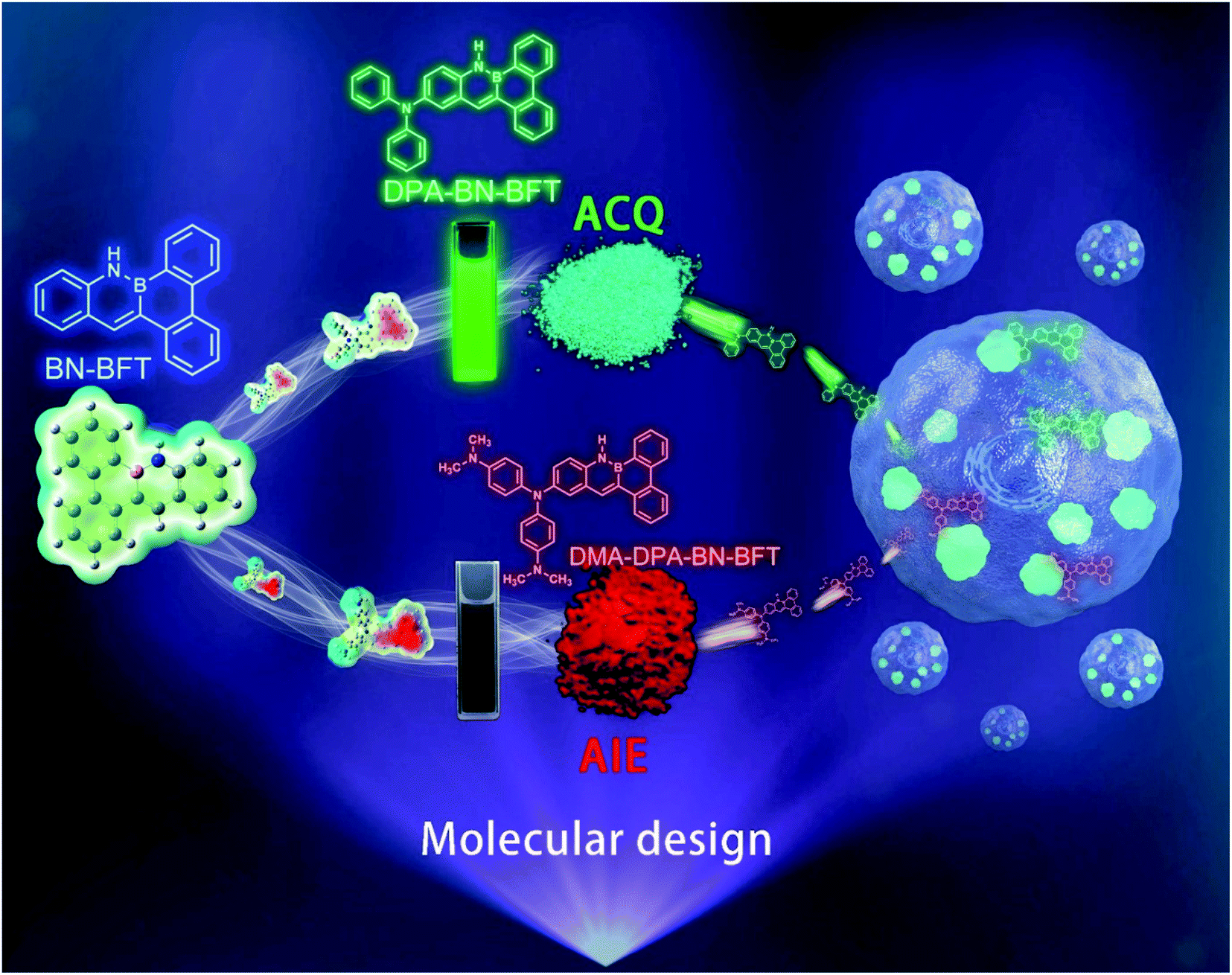 | ||
| Fig. 1 The design idea and the different emission forms of BN-functionalized molecules as well as their applications in subcellular organelle specific imaging. | ||
Results and discussion
BN-BFT is readily producible according to the metal-catalysed cyclization reaction.5e This compound is air- and moisture-stable in both the solid state and solution. To highlight the importance of BN incorporation into PAHs, its hydrocarbon analog BFT serves as a counterpart for comparison. The spectral properties of compounds BN-BFT and BFT were well investigated via UV-vis spectroscopy (Fig. S1†). The UV-vis spectrum of BFT in CH2Cl2 exhibited the lowest-energy absorption at 289 nm, but the lowest-energy absorption of BN-BFT shifted to 380 nm, and the bathochromic was nearly 100 nm. To understand the differences, the conformation and electronic structure of BN-BFT and BFT were observed. Their electronic distribution of HOMO and LUMO were studied theoretically by density functional theory (DFT) calculations at the level of B3LYP/6-31G* (Fig. S2†). The optimized structures of BN-BFT and BFT showed a fully planar skeleton within the fused polycyclic moiety, and noticeable differences between the frontier orbitals of the two compounds can be seen due to the BN substitution in BN-BFT. Although the electrons are delocalized over the whole fused rings in the HOMO and LUMO, the HOMO has no distribution at the nitrogen atom, which is consistent with previously reported heteroatom-substituted PAHs containing separated boron and nitrogen, where the charge can facilely transfer from the nitrogen to the boron center. We predicted that the localized charge characteristic should have some effect on the aromaticity of BN-BFT, and this assumption was further supported by nucleus-independent chemical shift (NICS) calculations. The NICS values for each of the rings are given in Fig. S2.† As predicted, the aromaticity of the BC5 and BNC4 rings in BN-BFT is significantly reduced in comparison to the corresponding rings in BFT. One plausible explanation is the partially localized charge on the B atom due to the polarization of the B–N bond, which reduces the effective diatropic ring current and leads to lowered aromaticity. These results indicate that BN unit incorporation led to significantly distinct electronic structures, which determine their varying reactivity and photophysical properties (Fig. 2).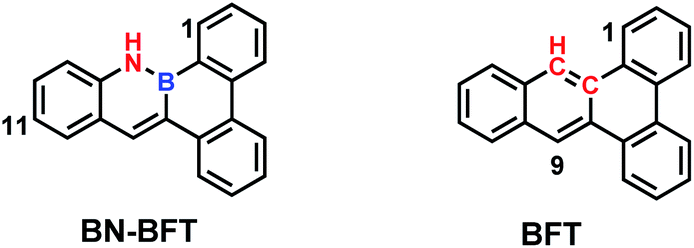 | ||
| Fig. 2 The molecular structures of BN-BFT and BFT. | ||
Evaluation of the optical and aromaticity properties of BN-BFT and BFT confirmed that the BN unit indeed plays an irreplaceable role in regulating luminescence behaviors. Thus, to gain a more in-depth understanding of the properties of this new family of BN-BFT, we turned our attention to introducing representative functional groups to the skeleton of BN-BFT and to extend the possibilities for obtaining typical AIEgens. Transition metal-catalyzed direct functionalization of C–X bonds (X = Cl, Br, I) has emerged as one of the most efficient and straightforward methods for achieving diverse materials with tunable properties.12 In particular, the selective activation of C–Br bonds has proven to be very useful for late-stage functionalization by cross-coupling reactions. Gratifyingly, preliminary exploration confirmed that BN-BFT could undergo highly selective bromination reaction (Scheme 1). Here, we mainly discuss the regioselective bromination of BN-BFT. The impact of site-selective bromination can be demonstrated by comparison with the bromination of its hydrocarbon analogue BFT, which naturally undergoes electrophilic aromatic substitution selectively at the 9-position.13 While the BN-BFT occur at the 11-position. This reaction provided an opportunity to understand the different reactivities of BN-containing aromatic systems and their analogous hydrocarbons. Thus, DFT calculations were undertaken to explain the highly regioselective bromination of BN-BFT. We selected one of the most reliable dual descriptors for further study.14 The first procedure is based on the typical definition of the dual descriptor:
| ΔfA = 2qAN − qAN+1 − qAN−1 |
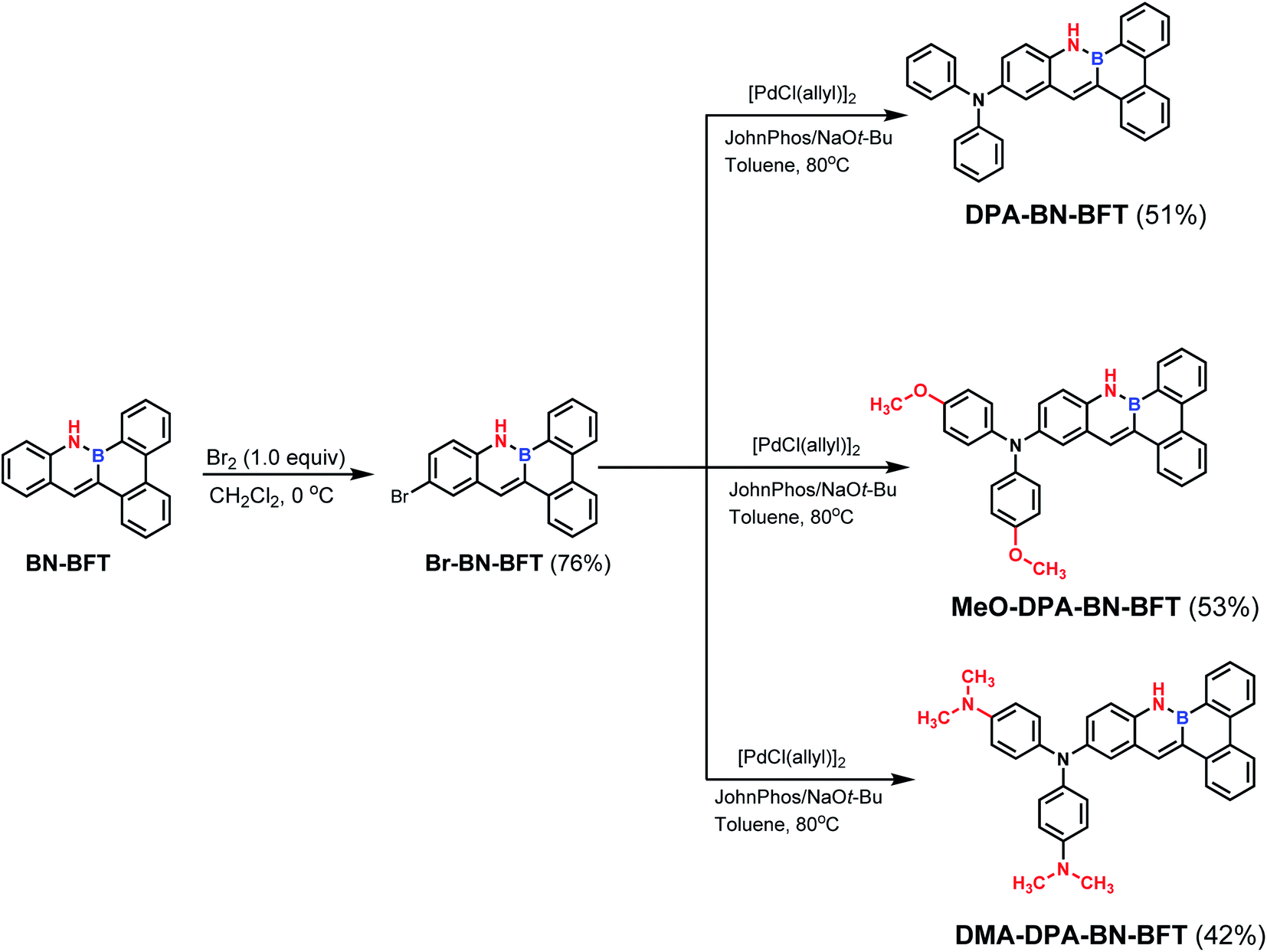 | ||
| Scheme 1 Regioselective bromination and late-stage functionalization of BN-BFT. | ||
Based on the above analysis of the NICS (valid ring current distribution), BN-BFT exhibited prominent electron deficiency, which indicated that BN-BFT has the potential to be an electron acceptor block. In fact, the optoelectronic properties of organic functional materials are often tuned using a combination of electron-rich (donor, D) and electron-poor (acceptor, A) substituents.15 However, the potential of this strategy in extending π conjugation and modulating the optical and electronic properties of BN aromatic systems has not yet been fully investigated. Therefore, systematically designed suitable BN aromatic systems with electron-donating and electron-accepting substituents are highly desired to probe the structure–property relationships, which may trigger the discovery of new functions and properties of azaborine chemistry. Considering all this information, we designed three novel BN-benzo[f]tetraphene-based “push–pull” chromophores (Scheme 1). First, we introduced a widely used diphenylamino moiety in the BN-BFT tail-region as the donor unit, forming the final D–A chromophore DPA-BN-BFT. The photophysical properties of DPA-BN-BFT were well investigated by UV-vis absorption and fluorescence emission spectroscopies (Fig. 3A and B). The UV-vis spectra of DPA-BN-BFT in dichloromethane exhibited the lowest energy absorption at 420 nm, and a redshift of 40 nm was observed compared to that of BN-BFT. Moreover, the photoluminescence (PL) spectra of DPA-BN-BFT in dichloromethane were also investigated, exhibiting emission ranging from 405 to 520 nm compared with BN-BFT (Fig. 3B), and the quantum yields (QY) of DPA-BN-BFT in dichloromethane and solid state were 38.5%, 15.2%, respectively (Table 1). Push–pull systems with donor and acceptor units may exhibit intramolecular charge transfer (ICT) characteristics due to dipole–dipole interactions.16 It is worth mentioning that the ICT interactions of BN-BFT were ignorable (Fig. S3†). Therefore, the solvatochromic effect of DPA-BN-BFT in various solvents was investigated. The emission spectrum of DPA-BN-BFT in highly polar solvent is broadened and exhibits a significant redshift from 456 to 520 nm (Fig. 3C), which can be attributed to a highly polarized ICT excited state, an important phenomenon for donor–acceptor (D–A) compounds. To further explain this phenomenon, DFT calculations were performed at the B3LYP/6-31G(d) level to explore the electronic distribution of molecule DPA-BN-BFT. As shown in Fig. S4,† the charge separation in DPA-BN-BFT is considerable in the ground state, resulting in strong charge transfer interactions in this system, which is highly consistent with the experimental results. Thus far, we have realized the regulation of the ICT effect from scratch. In addition, the methoxy substituted diphenylamine as a new donor unit was introduced to the BN-BFT backbone, constructing a novel D–A structure MeO-DPA-BN-BFT. Compared with DPA-BN-BFT, the emission spectrum of MeO-DPA-BN-BFT is further redshifted in dichloromethane, exhibiting emission ranging from chartreuse to yellow, with λem shifting from 520 to 562 nm (Fig. 3B). Meanwhile, its quantum yields (43.29% in dichloromethane solution, 27.51% in solid state, Table 1) are also higher than DPA-BN-BFT (38.5% in dichloromethane solution, 15.2% in solid state, Table 1). It is worth noting that MeO-DPA-BN-BFT also possess significant ICT effect (Fig. S5†).
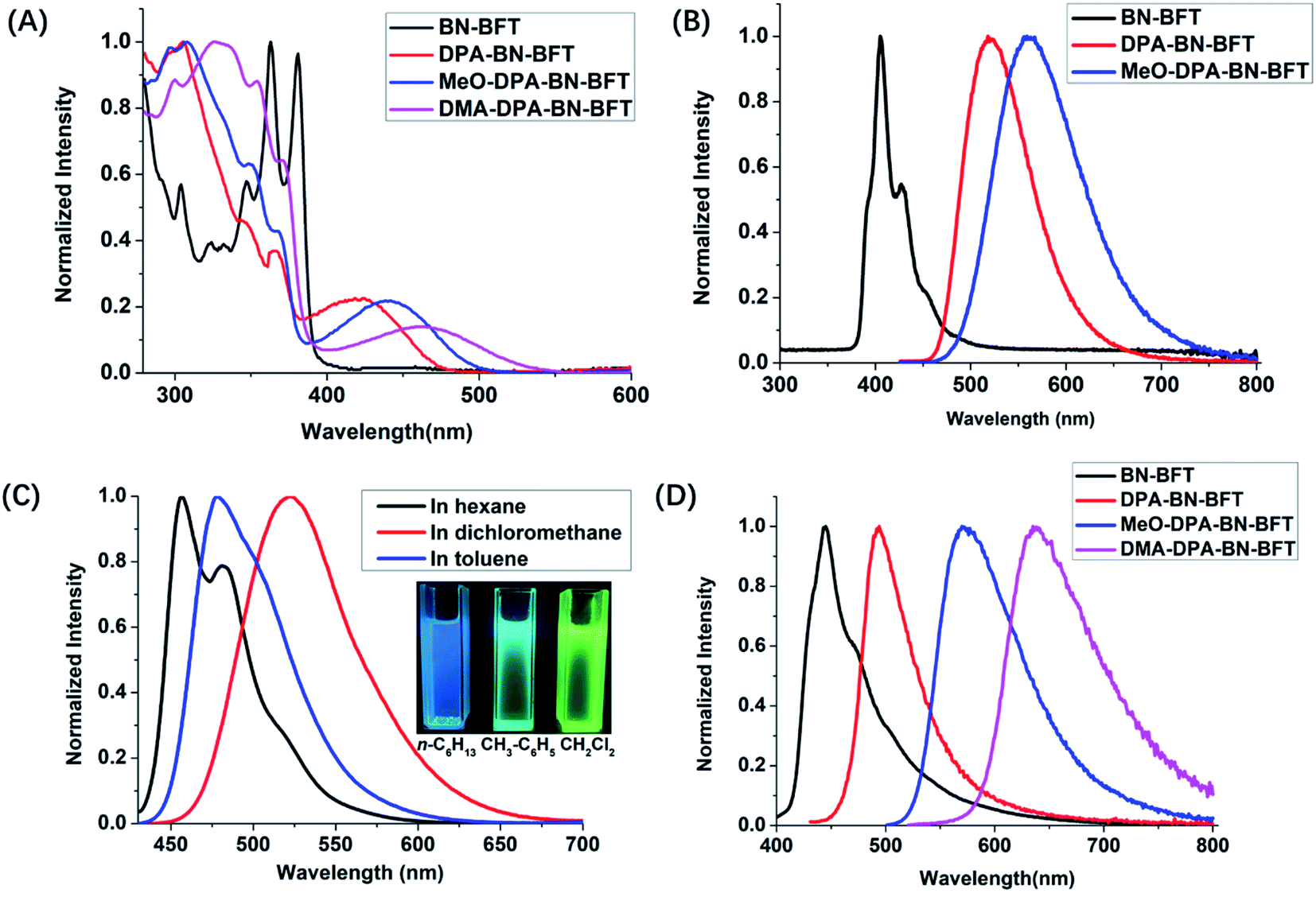 | ||
| Fig. 3 (A) The UV-vis spectra of BN-BFT, DPA-BN-BFT, MeO-DPA-BN-BFT and DMA-DPA-BN-BFT in CH2Cl2 (concentration = 10−5 M); (B) fluorescence spectras of BN-BFT, DPA-BN-BFT and MeO-DPA-BN-BFT in CH2Cl2, excitation wavelength: 380 nm (for BN-BFT) and 420 nm (for DPA-BN-BFT and MeO-DPA-BN-BFT); (C) normalized solvent-dependent fluorescence spectra of DPA-BN-BFT in n-hexane (black), toluene (blue), dichloromethane (red) solutions at 10−6 M (inset: fluorescent photographs of DPA-BN-BFT in different solvents); (D) solid fluorescence spectras of BN-BFT, DPA-BN-BFT, MeO-DPA-BN-BFT and DMA-DPA-BN-BFT, excitation wavelength: 365 nm (for BN-BFT) and 420 nm (for DPA-BN-BFT, MeO-DPA-BN-BFT and DMA-DPA-BN-BFT). | ||
| Compound | In CH2Cl2 | Solid state | ||||
|---|---|---|---|---|---|---|
| λ emmax (nm) | τ (ns) | Φ F (%) | λ emmax (nm) | τ (ns) | Φ F (%) | |
| a All experiments were performed in CH2Cl2 solution at 10−5 M. b Absolute quantum yield determined by a calibrated integrating sphere system; the absolute quantum yield was measured in dichloromethane and solid state at room temperature. | ||||||
| DPA-BN-BFT | 520 | 6.95 | 38.50 | 493 | 5.44 | 15.2 |
| MeO-DPA-BN-BFT | 564 | 11.26 | 43.29 | 570 | 12.45 | 27.51 |
| DMA-DPA-BN-BFT | — | 6.97 | 0.2 | 637 | 5.36 | 3.0 |
The above results indicated that electron donor moieties such as diphenylamine and methoxy substituted diphenylamine can dramatically change the optical character. We assume that the introduction of a relatively strong electron capable group may impart BN-BFT better photoelectric performance. Thus, an N,N′-dimethylamino substituted diphenylamine was introduced to the skeleton of BN-BFT, forming D–A molecule DMA-DPA-BN-BFT. Excitedly, the as-prepared DMA-DPA-BN-BFT exhibited quite distinct photoluminescent properties. The molecule presented negligible emission in solution but high emission in the aggregation state, namely, manifesting a representative AIE effect. Encouraged by this result, the AIE behavior of DMA-DPA-BN-BFT was investigated in detail. Its quantitative AIE activity was measured by adding H2O (as a poor solvent) to the solution of DMA-DPA-BN-BFT in THF (as a good solvent). When the H2O content is less than 70%, the solution of DMA-DPA-BN-BFT is transparent with poor emission (Fig. 4A and B). As the water content increases gradually from 60% to 70%, however, DMA-DPA-BN-BFT becomes insoluble in the THF/H2O mixture of solvents and begins to aggregate, resulting in a gradual increase in luminescent intensities at the emission maximum at 620 nm (Fig. 4B), with a significant increase in the photoluminescent quantum yield (QY) from 0.67% to 24.22% (Fig. 4A-up). This AIE behavior can also be observed with the naked eye and is accompanied by a gradual increase in the turbidity and luminescence of the solution. The AIE behavior of DMA-DPA-BN-BFT was further verified by the luminescent changes during the evaporation of a drop of the solution of DMA-DPA-BN-BFT on a thin layer chromatography plate (Fig. 4C and Video S1†). As the solvent evaporates, DMA-DPA-BN-BFT aggregates on the thin-layer chromatography plate, resulting in an increase in the luminescence under UV lamp irradiation. In sharp contrast, DPA-BN-BFT and MeO-DPA-BN-BFT exhibit the significant ACQ effect, and their emissions were weakened in water–tetrahydrofuran solutions with a high water proportion (Fig. 4A, D, and S6†), with a significant decrease in the QY from 37.56% to 8.82%, 37.32% to 6.32%, respectively (Fig. 4A-down, and S6†). The controlled results are attributed to the introduction of a donor unit, indicating that the concept of D–A substitution was suitable for tuning the optical and electronic properties of the BN aromatic system, confirming that our original design philosophy is accessible. In addition, these discrepancies in the photoluminescent properties piqued our interest in further exploring their underlying mechanisms.
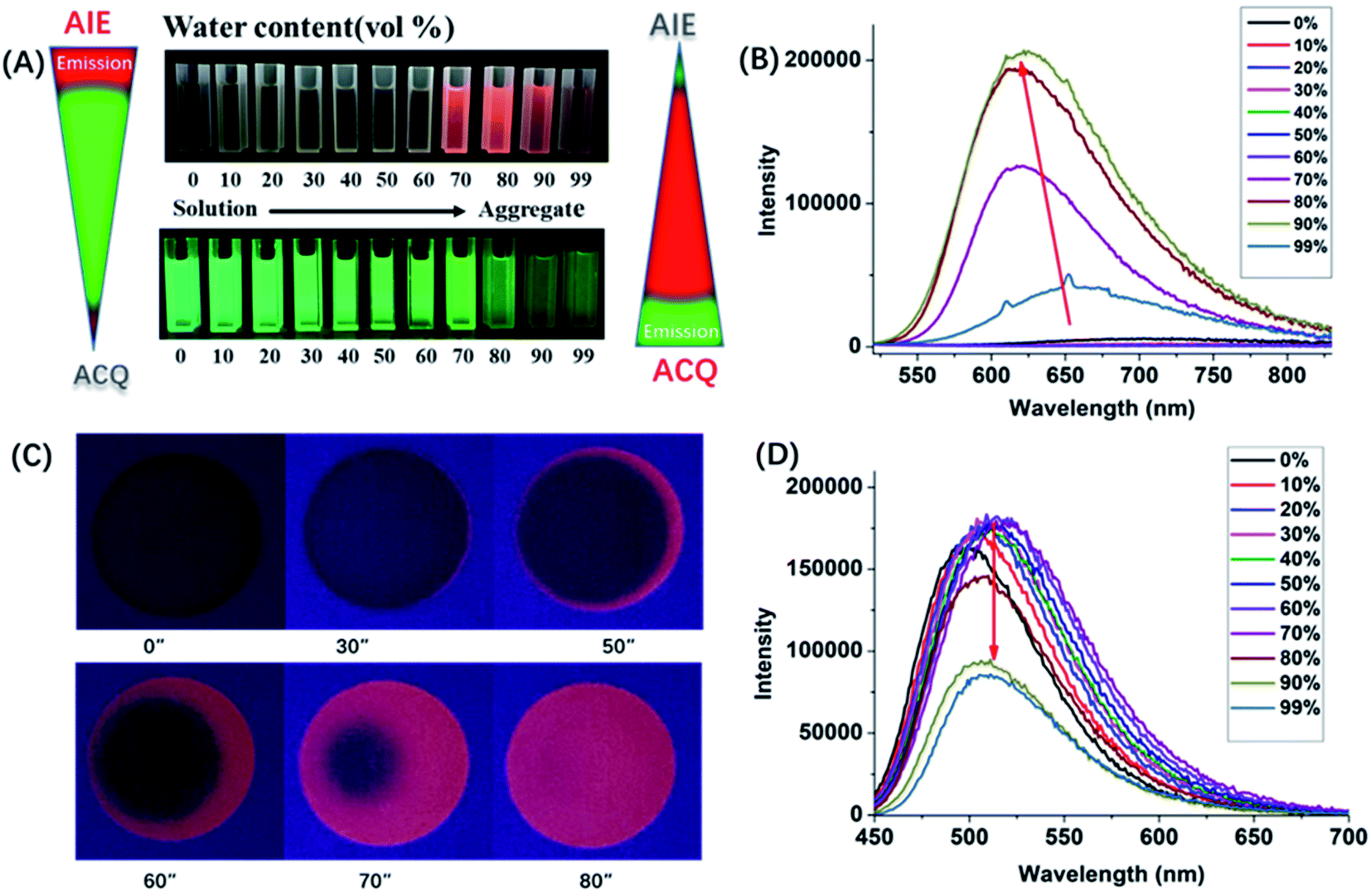 | ||
| Fig. 4 (A) Fluorescence photographs of solutions or suspensions of DPA-BN-BFT (down) and DMA-DPA-BN-BFT (up) in THF/water mixtures with different water contents; (B) PL spectra of DMA-DPA-BN-BFT in THF/water mixtures with different water fractions (fw). Concentration: 30 μM; excitation wavelength: 420 nm; (C) digital photos of one drop of DMA-DPA-BN-BFT solution (in CH2Cl2) on thin layer chromatography plate with different evaporation timescales at room temperature (under irradiation with UV lamp @ 365 nm); (D) PL spectra of DPA-BN-BFT in THF/water mixtures with different water fractions (fw). Concentration: 30 μM; excitation wavelength: 420 nm. | ||
Determination of the underlying mechanisms for the AIE phenomenon is of great importance to a fundamental understanding of photophysics, which will help deepen our understanding of luminescence processes and guide our endeavors to design novel AIE systems. It is worth noting that the crystal configurations play an important role in bridging the natural properties of single molecules and the macroscopic optoelectronic performance of organic materials.17 Therefore, to gain insight into the cause of the different behaviors in the aggregate state, we selected DPA-BN-BFT and DMA-DPA-BN-BFT as the representative examples, and their crystal structures were investigated in detail.
Single crystals of DMA-DPA-BN-BFT were obtained by recrystallization in methanol and chloroform solutions. Single crystal analyses reveal that molecule DMA-DPA-BN-BFT is constructed based on a monoclinic crystal system, crystallizing in the space group P21/n with four molecules in one cell unit, while molecule DPA-BN-BFT is P21/c. The most notable differences between the two structures are the torsion angles and noncovalent interactions (Fig. 5A). Regarding DPA-BN-BFT, crystal data show that the torsion angles between the BN embedded benzo[f]tetraphene core and phenyl rings at the N position were 30.6° (θ1) and 34.1° (θ2), respectively. In terms of DMA-DPA-BN-BFT, the corresponding torsion angles were 36.8° (θ3) and 60.5° (θ4). This difference in torsion angles results in totally different interactions in the crystals. First, such a large twist angle of DMA-DPA-BN-BFT could efficiently impede the π–π stacking between the five BN fused cores to avoid emission quenching. Indeed, its dimer structure showed that the two intermolecular BN-benzo[f]tetraphene rings were facing each other, while the centroid distance was 9.378 Å (Fig. 5B), which was long enough to impede π–π stacking. Thus, the ACQ effect was largely diminished, mitigating emission quenching. In addition, benefitting from the relatively large centroid distance, the N,N′-dimethyl diphenylamine groups can be effectively embedded in them, resulting in abundant intermolecular interactions in the crystal of DMA-DPA-BN-BFT. As revealed in Fig. 5C, C–H (N–CH3)…π, C–H (CH)…π interactions have distances from 2.698 Å to 3.446 Å. These intense intermolecular interactions help rigidize the molecular conformation and block the nonradiative pathway by restricting molecular motions. However, for compound DPA-BN-BFT, as shown in Fig. 5D, the intermolecular interactions are obviously weakened, and the quantity of the intermolecular interactions is significant reduced, indicating a relatively loose molecular packing mode. Based on these data, we concluded that the increasingly strong noncovalent interactions in crystal DMA-DPA-BN-BFT make it pack in a more rigid molecular conformation than DPA-BN-BFT and impede intramolecular rotations, which largely reduces the energy loss by suppressing the rotations of their N,N′-dimethyl diphenylamine groups in the solid state. In dilute solutions, DMA-DPA-BN-BFT exists in a more relaxed environment, and all the substituents are linked with binding atoms by single bonds. When excited by irradiation, the rotors of isolated molecules have great freedom to rotate against the axes of the single bonds and transform the photonic energy to thermal energy, leading to nonradiative relaxation and resulting in negligible fluorescence in the dilute solution state.
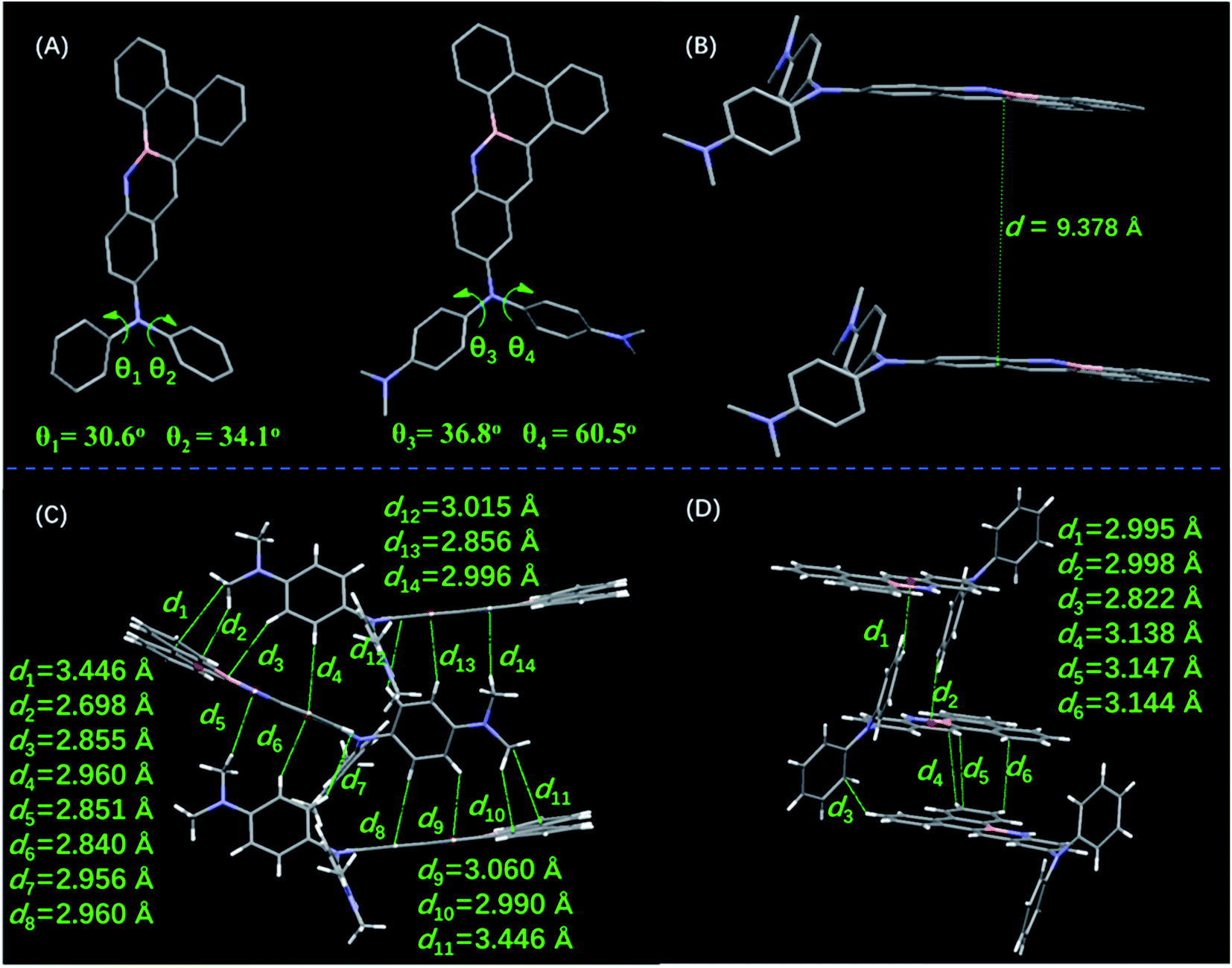 | ||
| Fig. 5 (A) Molecular structures and torsion angles of DPA-BN-BFT (left), and DMA-DPA-BN-BFT (right); (B) π–π distances of DMA-DPA-BN-BFT; intermolecular interactions for DMA-DPA-BN-BFT (C) and DPA-BN-BFT (D). | ||
This result raises another intriguing question, namely, why did the very similarly structured DPA-BN-BFT and DMA-DPA-BN-BFT show completely different stacking patterns? We predicted that the molecular charge distribution and dipole moment may have an important impact on crystal packing. Therefore, to determine the reason for the change in crystal packing of the two derivatives, the molecular electrostatic potential surface maps (ESPs) and dipole moment of the two molecules were calculated to visualize and understand the molecular packing modes.18 As shown in Fig. 6A, for molecule DPA-BN-BFT, significant differences between positive and negative potentials were not observed. However, compared with DPA-BN-BFT, the charge separation between the heads and tails of molecule DMA-DPA-BN-BFT was very remarkable. Benefitting from the polarization between the heads and tails of the molecule, the adjacent molecules tend to associate with each other in an edge-to-face herringbone arrangement mode with severe slippage (Fig. S9†). Such a edge-to-face herringbone packing mode can significantly maximize electrostatic attraction and minimize electrostatic repulsion as well as avoid steric hindrance between N,N′-dimethyl groups. Moreover, a much stronger electron donor substituent can increase the electron density, leading to a stronger intermolecular electrostatic repulsion and thus eliminating π–π interactions, which is highly consistent with the crystal data. The single crystal stacking mode of DPA-BN-BFT is very different from that of DMA-DPA-BN-BFT. Instead, it shows a slipped parallel stacks mode along a axis (Fig. S10†). This is mainly due to the relatively weak negative and positive potential on the conjugated backbone, which is unable to provide enough electrostatic attraction interaction. Furthermore, the dipole moment was introduced to discuss the molecular polarity. As shown in Fig. 6B, the dipole moment of DMA-DPA-BN-BFT (9.9843 Debye) is much larger than that of DPA-BN-BFT (4.0002 Debye), which is consistent with the calculated ESP results for DFT optimized molecules.
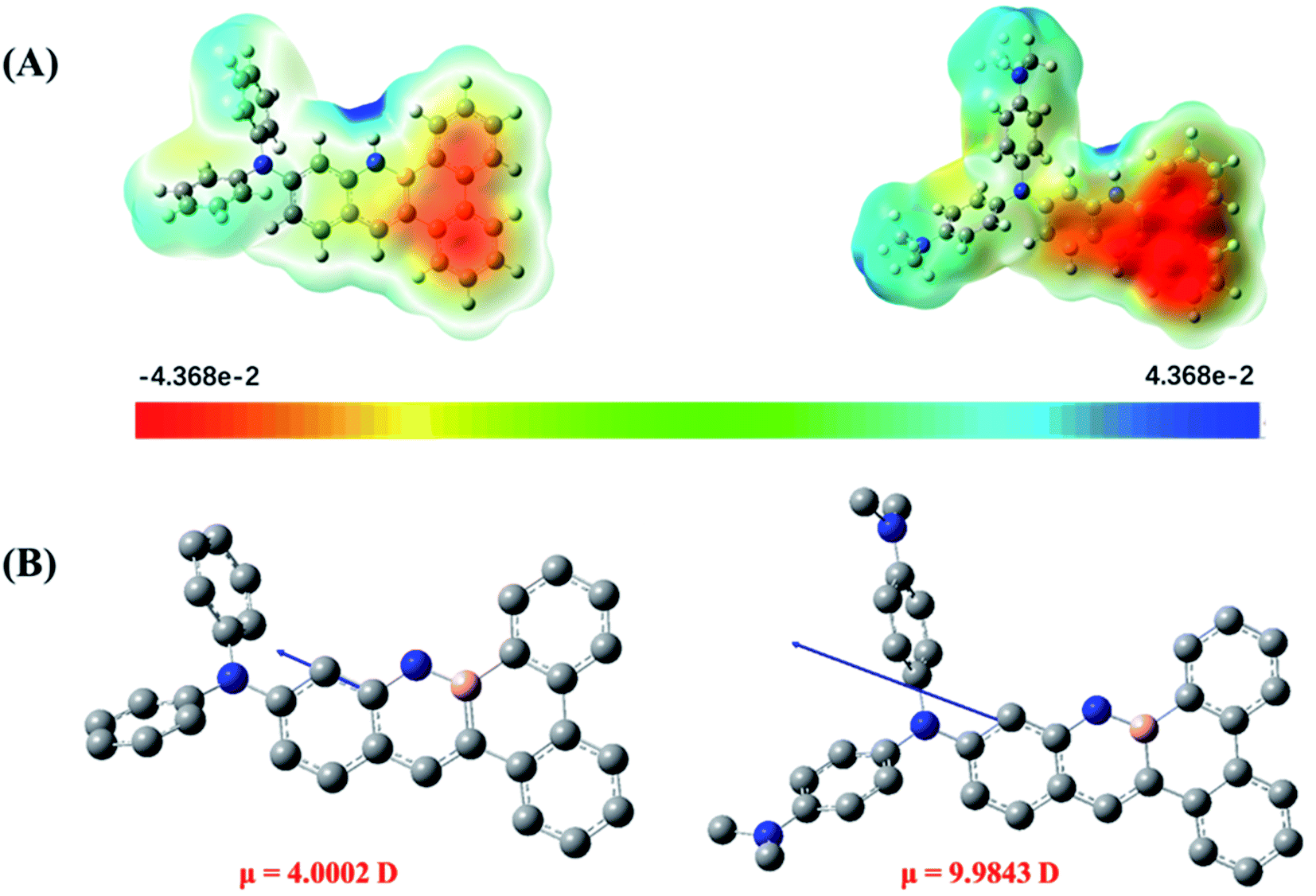 | ||
| Fig. 6 (A) ESP maps of DPA-BN-BFT (left), DMA-DPA-BN-BFT (right), (B) the dipole moment of DPA-BN-BFT (left) and DMA-DPA-BN-BFT (right). | ||
The above results indicate that BN modified polycyclic aromatics indeed present a promising potential in creating new materials and generating unique properties. The BN substitution has shown to be effective in modulating the photophysical properties as well as intermolecular interactions of conjugated molecules. In this work, the distinct photophysical properties should be ascribed to their different stacking patterns and the distinguishing charge levels in molecules. DMA-DPA-BN-BFT possesses a well segregated surface electrostatic potential and large dipole moment characteristics, which contribute to a strong intermolecular interaction and make it a good AIEgen, while infinitesimal surface electrostatic potentials combined with a suitable dipole moment endow DPA-BN-BFT with a relatively loose molecular packing mode. Such studies not only expand the variety of AIEgens but also provide a deep understanding of the structure-packing-performance relationship, guiding the design of organic molecules with AIE properties.
LDs are subcellular organelles containing neutral lipids such as triacylglycerols or cholesteryl esters surrounded by a phospholipid monolayer.19 Previously, LDs were regarded as an inert aggregate of neutral lipids for simple storage purpose.20 Recent studies have demonstrated that LDs not only are a simple warehouse for lipid storage but also play a very important role in the regulation of various physiological processes, including membrane formation and maintenance, protein binding and degradation, and signal transduction lipid.21 Their regulatory functions and levels have a close association with metabolic diseases, such as obesity, fatty liver disease, and type II diabetes.22 Thus, the development of highly sensitive and easy-to-handle imaging technologies to visualize the accumulation of LDs in biological systems is highly desirable. Fluorescence techniques are an effective means to study the cell imaging in living cells and tissues due to their advantages, such as strong labeling capability, excellent biocompatibility, large absorptivity and high brightness in the design of bioprobes. Enlightened by the D–A structural features of the developed LDs dyes, such as Nile red and Seoul Fluor-based fluorogens,23 we proposed that the D–A effect and AIE activity of DMA-DPA-BN-BFT may make it as a new agent for LDs specific imaging. Therefore, their practical applications in fluorescent imaging in organelles were investigated. We explored the potential applications of DMA-DPA-BN-BFT in cancer cell imaging.
The MCF-7 cells were incubated with DMA-DPA-BN-BFT (10 μM) and monitored by confocal fluorescence microscopy (Fig. 6). After 30 min, the aggregates of DMA-DPA-BN-BFT specifically entered the cells and light up the LDs region, rendering the reticulum structures of LDs clearly visible with bright orange fluorescence. The specificity was further verified by comparison with the image obtained from the commercial LDs probe BODIPY 493/503. We acquired fluorescent images of MCF-7 cells costained with DMA-DPA-BN-BFT and BODIPY 493/503. Merging both photos generates a chartreuse image, from which the shape, position, and amount of LDs stained by DMA-DPA-BN-BFT are found to be the same as those stained by commercial LDs probes, suggesting that the observed fluorescence from DMA-DPA-BN-BFT is localized to the LDs of living MCF-7 cells. In addition, the imaging effect of DMA-DPA-BN-BFT was also studied (Fig. 7). After the living MCF-7 cells were treated with DPA-BN-BFT for the same experimental conditions, the LDs of living MCF-7 cells were lit up with green color. Such studies not only expand the variety of organelle visualizers but also provide more insight into their molecular design. We conclude that the selectivity may be ascribed to the inherent lipophilic nature of LDs, indicating that molecular lipophilicity is one of the predominant factors for specific LDs targeting.
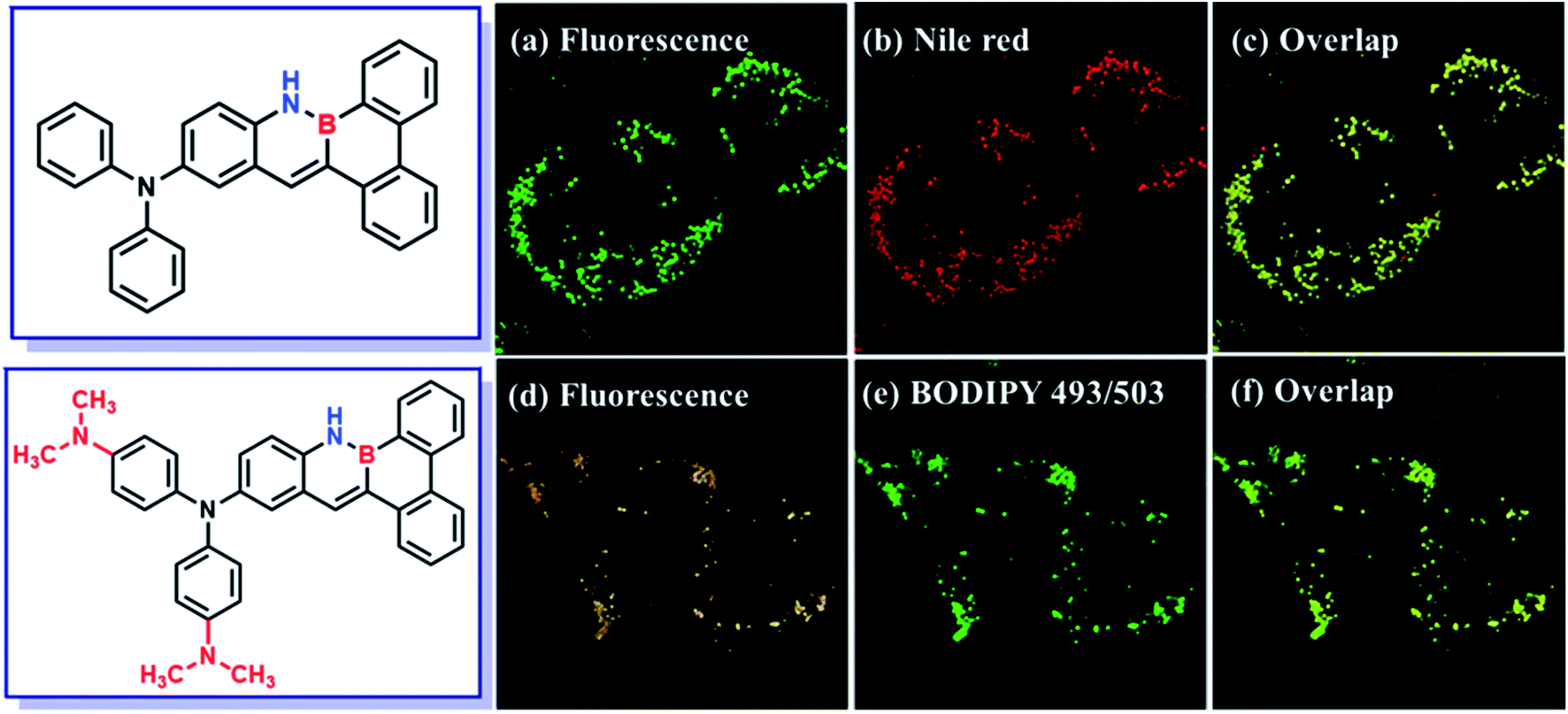 | ||
| Fig. 7 Co-localization experiments of MCF-7 cells stained with BPA-BN-BFT (a–c) and DMA-DPA-BN-BFT (d–f), and then co-stained with commercial LDs probes BODIPY 493/503 and Nile red. | ||
Conclusion
We successfully designed and synthesized three novel BN-functionalized molecules by regioselective functional-ization engineering and systematically investigated their luminescence behavior and potential applications. Impressively, molecular DPA-BN-BFT and MeO-DPA-BN-BFT exhibited ACQ behaviors. However, with subtle structural modifications, the introduction of N,N′-dimethylaniline units, which function as conformation regulators to effectively manipulate the molecular arrangements and packing mode, that results in the formation of the expectant AIE-active DMA-DPA-BN-BFT. The very similar structures of DPA-BN-BFT, MeO-DPA-BN-BFT and DMA-DPA-BN-BFT with distinctly different photometric characteristics in this work can help us to obtain a deep understanding of the structure-packing-performance relationship, guiding the design of organic molecules with AIE characteristics. Due to their unusual photophysical characteristics and lipophilic nature, DPA-BN-BFT and DMA-DPA-BN-BFT can act as fluorescent visualizers for specific staining of LDs in living cells with excellent sensitivity and selectivity. In summary, these results indicated that BN-embedded PAHs were robust enough for and tolerant to a variety of further chemical transformations. This work not only provides a new class of AIE molecules with tunable properties but also encourages the strategy of regioselective functionalization of BN-containing PAHs for rapid diversification and fine-tuning of their molecular properties. It is also possible to achieve tailor made materials for various scientific and biological applications in the future.Data availability
All the data have been included in the ESI.†Author contributions
Huanan Huang and Jianguo Wang conceptualized the project methodology, wrote the original draft and supervised the investigation. Huanan Huang and Han Xu analysed the data and built data pipeline. Lingxiu Liu, Ying Zhou, Huanan Hu, Xinglin Ye, Guochang Liu, Zhixiong Xu, Wen Yang, Yawei Wang, You Peng, Pinghu Yang, Jianqi Sun, Ping Yan collected experimental data. Ben Zhong Tang supervised the investigation and co-wrote the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21861022, 21864015), the Key Project of Natural Science Foundation of Jiangxi Province (20202ACBL213003), Youth Science Fund Project of Jiangxi Province (2020BABL213009), and the Education Department of Jiangxi Province Foundation of China (No. GJJ201829, GJJ211836).Notes and references
- (a) M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891–4946 CrossRef CAS PubMed; (b) C. Li, M. Liu, N. G. Pschirer, M. Baumgarten and K. Müllen, Chem. Rev., 2010, 110, 6817–6855 CrossRef CAS PubMed; (c) G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS; (d) Y. Wang, Z. Yan, H. Guo, M. A. Uddin, S. Ling, X. Zhou, H. Su, J. Dai, H. Y. Woo and X. Guo, Angew. Chem., Int. Ed., 2017, 56, 15304–15308 CrossRef CAS PubMed; (e) M. Chu, J.-X. Fan, S. Yang, D. Liu, C. F. Ng, H. Dong, A.-M. Ren and Q. Miao, Adv. Mater., 2018, 30, 1803467 CrossRef PubMed; (f) Z. Jin, Z.-F. Yao, K. P. Barker, J. Pei and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 2034–2039 CrossRef CAS PubMed; (g) H. Fu, Z. Wang and Y. Sun, Angew. Chem., Int. Ed., 2019, 58, 4442–4453 CrossRef CAS PubMed; (h) Z. Yao, X. Liao, K. Gao, F. Lin, X. Xu, X. Shi, L. Zuo, F. Liu, Y. Chen and A. K.-Y. Jen, J. Am. Chem. Soc., 2018, 140, 2054–2057 CrossRef CAS PubMed.
- (a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed; (b) Q. Miao, Adv. Mater., 2014, 26, 5541–5549 CrossRef CAS PubMed; (c) X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed; (d) K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed; (e) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed.
- For leading reviews on BN aromatics, see: (a) Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed; (b) M. M. Morgan and W. E. Piers, Dalton Trans., 2016, 45, 5920–5924 RSC; (c) M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef; (d) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed; (e) X.-Y. Wang, J.-Y. Wang and J. Pei, Chem.–Eur. J., 2015, 21, 3528–3539 CrossRef CAS PubMed; (f) J.-Y. Wang and J. Pei, Chin. Chem. Lett., 2016, 27, 1139–1146 CrossRef CAS.
- (a) S. N. Wang, D.-T. Yang, J. S. Lu, H. Shimogawa, S. L Gong, X. Wang, S. K. Mellerup, A. Wakamiya, Y.-L. Chang, C. L. Yang and Z. H. Lu, Angew. Chem., Int. Ed., 2015, 54, 15074–15078 CrossRef CAS PubMed; (b) H. P. Wei, Y. L. Liu, T. Y. Gopalakrishna, H. Phan, X. B. Huang, L. P. Bao, J. Guo, J. Zhou, S. L. Luo, J. S. Wu and Z. B. Zeng, J. Am. Chem. Soc., 2017, 139, 15760–15767 CrossRef CAS PubMed; (c) X. G. Liu, Y. Z. Zhang, B. Li, L. N. Zakharov, M. Vasiliu, D. A. Dixon and S.-Y. Liu, Angew. Chem., Int. Ed., 2016, 55, 8333–8337 CrossRef CAS PubMed; (d) S. M. Xu, T. C. Mikulas, L. N. Zakharov, D. A. Dixon and S.-Y. Liu, Angew. Chem., Int. Ed., 2013, 52, 7527–7531 CrossRef CAS PubMed; (e) Y. G. Shi, J. W. Wang, H. J. Li, G. F. Hu, X. Li, S. K. Mellerup, N. Wang, T. Peng and S. N. Wang, Chem. Sci., 2018, 9, 1902–1911 RSC; (f) S. N. Wang, K. Yuan, M. F. Hu, X. Wang, T. Peng, N. Wang and Q. S. Li, Angew. Chem., Int. Ed., 2018, 57, 1073–1077 CrossRef CAS PubMed; (g) S. K. Mellerup, C. Li, T. Peng and S. N. Wang, Angew. Chem., Int. Ed., 2017, 56, 6093–6097 CrossRef CAS PubMed.
- (a) T. Hatakeyama, S. Hashimoto, S. Seki and M. Nakamura, J. Am. Chem. Soc., 2011, 133, 18614–18617 CrossRef CAS PubMed; (b) X.-Y. Wang, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 7668–7671 CrossRef CAS PubMed; (c) X.-Y. Wang, F.-D. Zhuang, R.-B. Wang, X.-C. Wang, X.-Y. Cao, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2014, 136, 3764–3767 CrossRef CAS PubMed; (d) H. N. Huang, Y. Zhou, M. Wang, J. Y. Zhang, X. H. Cao, S. T. Wang, D. P. Cao and C. M. Cui, Angew. Chem., Int. Ed., 2019, 58, 10132–10137 CrossRef CAS PubMed; (e) H. N. Huang, Y. Zhou, Y. W. Wang, X. H. Cao, C. Han, G. C. Liu, Z. X. Xu, C. C. Zhan, H. H. Hu, Y. Peng, P. Yan and D. P. Cao, J. Mater. Chem. A, 2020, 8, 22023–22031 RSC; (f) W.-M. Wan, D. Tian, Y.-N. Jing, X.-Y. Zhang, W. Wu, H. Ren and H.-L. Bao, Angew. Chem., Int. Ed., 2018, 57, 15510–15516 CrossRef CAS PubMed.
- (a) Y. J. Chen, W. N. Chen, Y. J. Qiao, X. F. Lu and G. Zhou, Angew. Chem., Int. Ed., 2020, 59, 7122–7130 CrossRef CAS PubMed; (b) C. Zhang, L. Zhang, C. Sun, W. F. Sun and X. G. Liu, Org. Lett., 2019, 21, 3476–3480 CrossRef CAS PubMed; (c) Y. L. Zhang, C. Zhang, Y. K. Guo, J. C. Ye, B. Zhen, Y. Chen and X. G. Liu, J. Org. Chem., 2021, 86, 6322–6330 CrossRef CAS PubMed; (d) L. J. Zi, J. Y. Zhang, C. L. Li, Y. Qu, B. Zhen, X. G. Liu and L. Zhang, Org. Lett., 2020, 22, 1499–1503 CrossRef CAS PubMed; (e) C. L. Li, Y. M. Liu, Z. Sun, J. Y. Zhang, M. Y. Liu, C. Zhang, Q. Zhang, H. J. Wang and X. G. Liu, Org. Lett., 2018, 20, 2806–2810 CrossRef CAS PubMed; (f) Q. Zhang, Z. Sun, L. Zhang, M. Y. Li, L. J. Zi, Z. Y. Liu, B. Zhen, W. F. Sun and X. G. Liu, J. Org. Chem., 2020, 85, 7877–7883 CrossRef CAS PubMed.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- (a) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (b) Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (c) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC.
- (a) J. G. Wang, X. G. Gu, P. F. Zhang, X. B. Huang, X. Y. Zheng, M. Chen, H. T. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974–16979 CrossRef CAS PubMed; (b) D. Y. Yan, Q. Wu, D. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 15724–15742 CrossRef CAS PubMed; (c) Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC.
- H. Lu, T. Nakamuro, K. Yamashita, H. Yanagisawa, O. Nureki, M. Kikkawa, H. Gao, J. W. Tian, R. Shang and E. Nakamura, J. Am. Chem. Soc., 2020, 142, 18990–18996 CrossRef CAS PubMed.
- A. W. Baggett, F. Guo, B. Li, S.-Y. Liu and J. k. Frieder, Angew. Chem., Int. Ed., 2015, 54, 11191–11195 CrossRef CAS PubMed.
- (a) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900 RSC; (b) R. Xavi, C–H and C–X Bond Functionalization, Catalysis Series, The Royal Society of Chemistry, 2013, pp. 1–471 Search PubMed; (c) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC; (d) J. J. Topczewski and M. S. Sanford, Chem. Sci., 2015, 6, 70–76 RSC; (e) M. M. Lorion, K. Maindan, A. R. Kapdi and L. Ackermann, Chem. Soc. Rev., 2017, 46, 7399–7420 RSC; (f) J. A. Leitch and C. G. Frost, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC; (g) T. Gensch, M. J. James, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 2296–2306 CrossRef CAS PubMed; (h) R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS PubMed.
- K. K. Laali, T. Okazaki and R. G. Harvey, J. Org. Chem., 2001, 66, 3977–3983 CrossRef CAS PubMed.
- R. Pino-Rios, D. Inostroza, G. Cárdenas-Jirón and W. Tiznado, J. Phys. Chem. A, 2019, 123, 10556–10562 CrossRef CAS PubMed.
- (a) G. Y. Zhang, J. B. Zhao, P. C. Y. Chow, K. Jiang, J. Q. Zhang, Z. L. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447–3507 CrossRef CAS PubMed; (b) P. Data, P. Pander, M. Okazaki, Y. Takeda, S. Minakata and A. P. Monkman, Angew. Chem., Int. Ed., 2016, 55, 5739–5744 CrossRef CAS PubMed; (c) S. Izumi, H. F. Higginbotham, A. Nyga, P. Stachelek, N. Tohnai, P. Silva, P. Data, Y. Takeda and S. Minakata, J. Am. Chem. Soc., 2020, 142, 1482–1491 CrossRef CAS PubMed; (d) A. Belyaev, Y.-H. Cheng, Z.-Y. Liu, A. J. Karttunen, P.-T. Chou and I. O. Koshevoy, Angew. Chem., Int. Ed., 2019, 58, 13456–13465 CrossRef CAS PubMed.
- (a) Z. L. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Eichhorn, A. Steffen, Y. Wang and T. B. Marder, Chem.–Eur. J., 2015, 21, 177–190 CrossRef CAS PubMed; (b) N. Ando, T. Kushida and S. Yamaguchi, Chem. Commun., 2018, 54, 5213–5216 RSC.
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- (a) C. J. Pace and J. Gao, Acc. Chem. Res., 2013, 46, 907–915 CrossRef CAS PubMed; (b) S. E. Wheeler, J. Am. Chem. Soc., 2011, 133, 10262–10274 CrossRef CAS PubMed; (c) L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS PubMed; (d) J.-H. Dou, Y.-Q. Zheng, Z.-F. Yao, Z.-A. Yu, T. Lei, X. Shen, X.-Y. Luo, J. Sun, S.-D. Zhang, Y.-F. Ding, G. Han, Y. Yi, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137, 15947–15956 CrossRef CAS PubMed.
- D. J. Murphy, Prog. Lipid Res., 2001, 40, 325–438 CrossRef CAS PubMed.
- R. V. Farese Jr and T. C. Walther, Cell, 2009, 139, 855–860 CrossRef PubMed.
- H. Zhang, Y. Wang, J. Li, J. Yu, J. Pu, L. Li, H. Zhang, S. Zhang, G. Peng, F. Yang and P. Liu, J. Proteome Res., 2011, 10, 4757–4768 CrossRef CAS PubMed.
- K. G. M. M. Alberti, P. Zimmet and J. Shaw, Lancet, 2005, 366, 1059–1062 CrossRef.
- (a) Y. Lee, S. Na, S. Lee, N. L. Jeon and S. B. Park, Mol. BioSyst., 2013, 9, 952–956 RSC; (b) S. Lee, E. Kim and S. B. Park, Chem. Sci., 2013, 4, 3282–3287 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2099085 and 2099086. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00380e |
| This journal is © The Royal Society of Chemistry 2022 |