
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBridging D–A type photosensitizers with the azo group to boost intersystem crossing for efficient photodynamic therapy†
Boyi
Hao
,
Jiaxin
Wang
 ,
Chao
Wang
,
Ke
Xue
,
Minghui
Xiao
,
Shuyi
Lv
and
Chunlei
Zhu
*
,
Chao
Wang
,
Ke
Xue
,
Minghui
Xiao
,
Shuyi
Lv
and
Chunlei
Zhu
*
Key Laboratory of Functional Polymer Materials of Ministry of Education, State Key Laboratory of Medicinal Chemical Biology, Institute of Polymer Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: chunlei.zhu@nankai.edu.cn
First published on 14th March 2022
Abstract
Photodynamic therapy (PDT) has attracted much attention in disease treatments. However, the exploration of a novel method for the construction of outstanding photosensitizers (PSs) with stimuli-responsiveness remains challenging. In this study, we, for the first time, report a novel and effective strategy to boost reactive oxygen species (ROS) generation by bridging donor–acceptor (D–A) type PSs with the azo group. In contrast to the counterpart without azo-bridging, the azo-bridged PSs exhibit remarkably enhanced ROS generation via both type-I and type-II photochemical reactions. Theoretical calculations suggest that azo-bridging leads to a prominent reduction in ΔEST, thereby enabling enhanced ROS generation via efficient intersystem crossing (ISC). The resulting azo-bridged PS (denoted as Azo-TPA-Th(+)) exhibits a particularly strong bactericidal effect against clinically relevant drug-resistant bacteria, with the killing efficiency up to 99.999999% upon white light irradiation. Since azo-bridging generates an azobenzene structure, Azo-TPA-Th(+) can undergo trans-to-cis isomerization upon UV irradiation to form emissive aggregates by shutting down the ISC channel. By virtue of the fluorescence turn-on property of unbound Azo-TPA-Th(+), we propose a straightforward method to directly discern the effective photodynamic bactericidal dose without performing the tedious plate-counting assay. This study opens a brand-new avenue for the design of advanced PSs with both strong ROS generation and stimuli-responsiveness, holding great potential in high-quality PDT with rapid prediction of the therapeutic outcome.
Introduction
Photodynamic therapy (PDT) has attracted much attention in disease treatments due to its prominent advantages, such as non-invasiveness, high spatiotemporal controllability, and low systemic toxicity.1,2 In general, PDT primarily relies on three key components, that is, photosensitizers (PSs), a light source, and oxygen. In a typical process, PSs are excited to the excited singlet state (Sn, n ≥ 1) by absorbing light energy, followed by intersystem crossing (ISC) to the excited triplet state (Tn, n ≥ 1). The excitation energy is then consumed via an electron transfer process (type-I reaction3) and/or an energy transfer process (type-II reaction4) to the surrounding molecular oxygen (3O2) to generate reactive oxygen species (ROS) (e.g., superoxide radical anion (O2˙−), hydroxyl radical (·OH), and singlet oxygen (1O2)), giving rise to the irreversible oxidation of bioactive molecules and thus bacterial/cell death.5In view of the importance of PSs in PDT, a variety of PSs have been developed, such as porphyrins,6 boron-dipyrromethene derivatives,7,8 phthalocyanines,9 and bacteriochlorins.10 However, these traditional PSs are prone to aggregation when used in aqueous solutions due to the hydrophobic and rigid planar structures, leading to pronounced fluorescence quenching (known as aggregation-caused quenching (ACQ)) and reduced ROS generation.11 In recent years, aggregation-induced emission (AIE) luminogens have emerged as an alternative class of PSs to overcome the ACQ problem.12–15 Unlike traditional ACQ PSs, AIE PSs exhibit both strong fluorescence and efficient ROS generation in the aggregated state, which results from the remarkably suppressed nonradiative decay and thus favored ISC due to restricted intramolecular motions.16,17 According to perturbation theory, the rate constant of ISC (kISC) is described by the following equation:18,19
| kISC ∝ ξST2/exp(ΔEST2) |
Herein, we, for the first time, reported a novel and effective strategy to remarkably enhance photosensitization by covalently coupling D–A type PSs with the azo group for efficient PDT. First of all, we synthesized a D–A type AIE PS (denoted as TPA-Th(+)) as well as its azo-bridging counterpart (denoted as Azo-TPA-Th(+)). Surprisingly, the azo-bridged Azo-TPA-Th(+) exhibited remarkably enhanced ROS generation when compared to TPA-Th(+) (Scheme 1). To authenticate the applicability of this approach, we also prepared another pair of D–A type AIE PSs (denoted as TPA(+) and Azo-TPA(+)) with shortened conjugation length. Even for the particularly weak PS (i.e., TPA(+)), azo-bridging remained an effective strategy to enhance ROS generation. Theoretical calculations suggested that azo-bridging markedly decreased the ΔEST of the resulting PSs, thereby facilitating ISC to boost the subsequent photosensitization process. Considering the prominent ROS generation capability of Azo-TPA-Th(+), we further evaluated its PDT effect against clinically relevant methicillin-resistant Staphylococcus aureus (MRSA). Under optimal conditions, the bacterial killing efficiency was determined to be 99.999999%. Due to the introduction of an azo group, an azobenzene structure31–33 was formed in the resulting Azo-TPA-Th(+). Interestingly, UV irradiation initiated the photoisomerization of Azo-TPA-Th(+) from trans to cis configurations, leading to the formation of emissive aggregates due to the exposure of accessible hydrophobic domains. By virtue of the responsiveness of Azo-TPA-Th(+) to UV light, it was convenient to discern whether there existed excess, unbound Azo-TPA-Th(+) molecules via simple fluorescence tests, which actually reflected the binding threshold of Azo-TPA-Th(+) toward bacteria. In this case, the effective PDT bactericidal dose could be forecasted within 1 h but without performing the tedious plate-counting assay that typically requires 24 h.
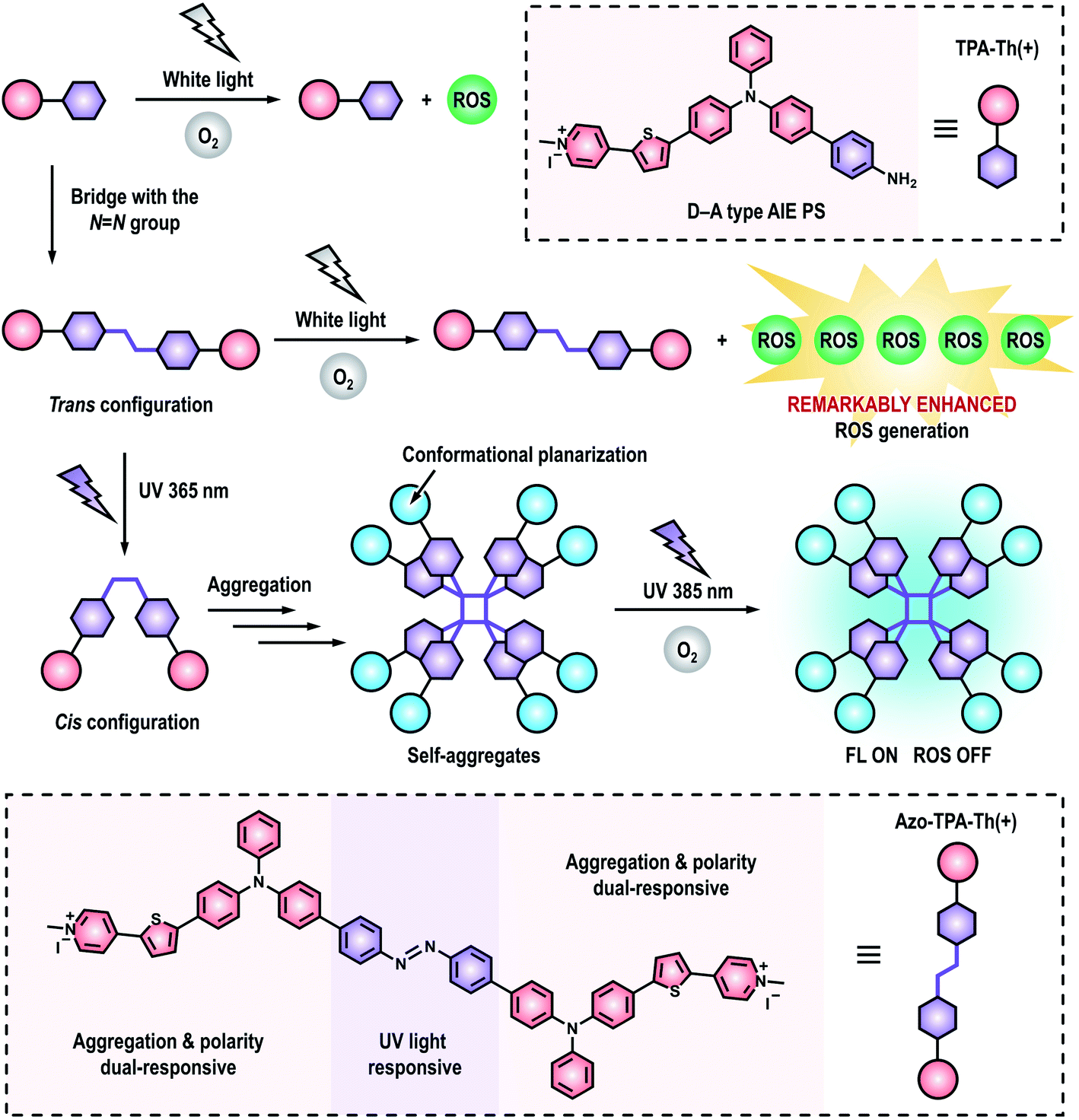 | ||
| Scheme 1 Schematic illustration showing the enhanced ROS generation by bridging D–A type PSs with the azo group as well as the responsiveness of the azo-bridged PSs to UV light. | ||
Results and discussion
The synthetic routes to TPA-Th(+) and Azo-TPA-Th(+) are shown in Scheme 2. To construct D–A type AIE PSs, electron-donating and electron-withdrawing units as well as molecular rotators should be introduced into the molecular backbone. In terms of the synthesis of TPA-Th(+), compound 1 and compound 2 were reacted under Suzuki coupling conditions to give compound 3. Next, aniline was attached to compound 3 through Suzuki coupling to yield compound 4, which was followed by quaternization with iodomethane to obtain TPA-Th(+). For the synthesis of Azo-TPA-Th(+), two molecules of compound 3 were bridged by compound 5 through Suzuki coupling to give compound 6, which was followed by quaternization with iodomethane to yield Azo-TPA-Th(+). The chemical structures of all compounds were verified by nuclear magnetic resonance spectrometry and high-resolution mass spectrometry (Fig. S1–S15†).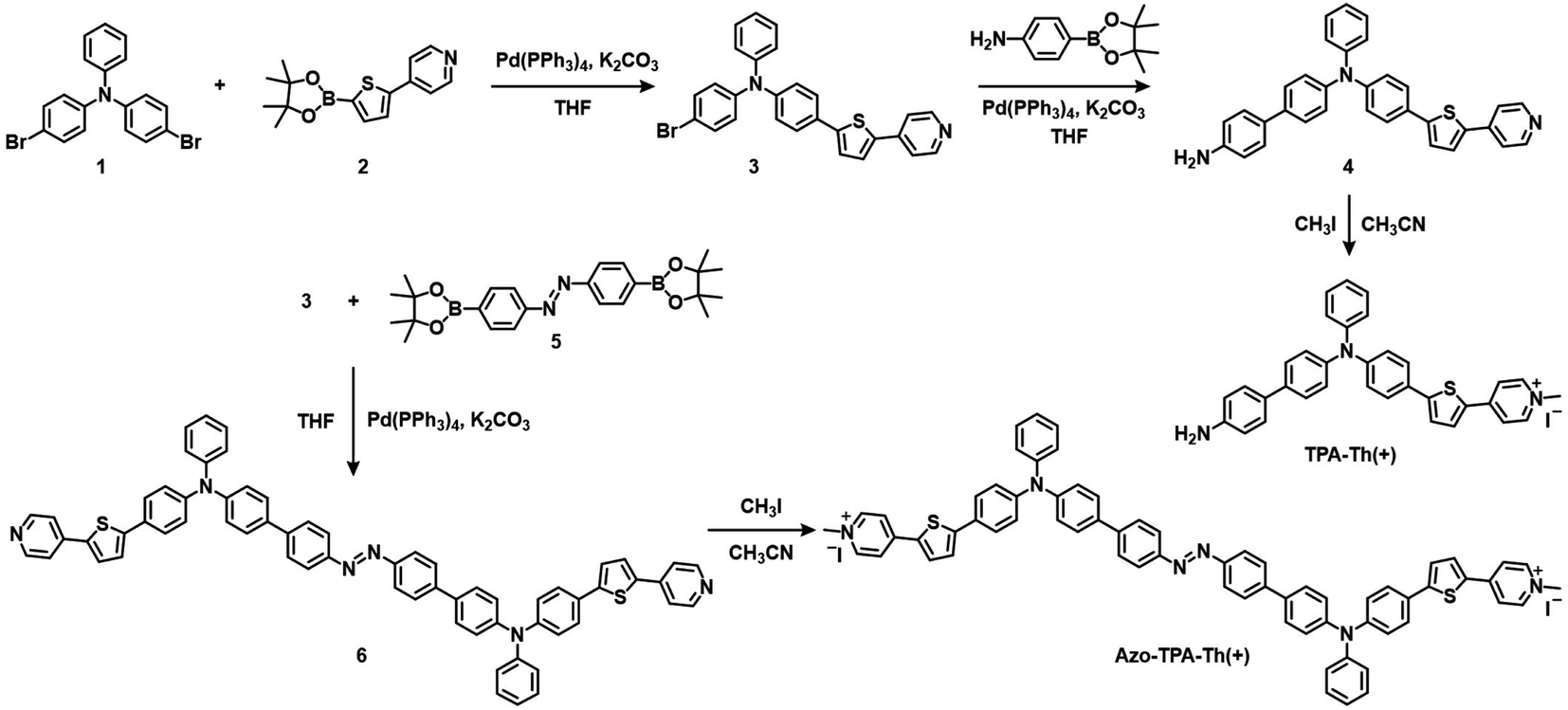 | ||
| Scheme 2 Synthetic routes to TPA-Th(+) and Azo-TPA-Th(+). | ||
We first characterized the basic photophysical properties of TPA-Th(+) and Azo-TPA-Th(+). As shown in Fig. 1A, Azo-TPA-Th(+) and TPA-Th(+) exhibited a broad absorption in the range of 300–550 nm, with the absorption peaks located at ca. 465 nm. To study the AIE properties, a series of dimethyl sulfoxide (DMSO) solutions with different toluene fractions (ft) were prepared. As ft increased, TPA-Th(+) and Azo-TPA-Th(+) started to form aggregates, resulting in enhanced emission in the range of 550–800 nm with the emission maxima located at ca. 600 and 630 nm, respectively (Fig. S31A–D†). Furthermore, the dynamic light scattering (DLS) data showed that the hydrodynamic diameters of TPA-Th(+) and Azo-TPA-Th(+) at ft = 99% were predominantly located at ca. 220 and 530 nm, respectively (Fig. S31E and F†). Taking the emission intensity at 600 and 630 nm as the variable parameters, respectively, the emission intensity (I) of TPA-Th(+) and Azo-TPA-Th(+) in solutions with different ft was compared with that in pure DMSO (I0) to plot the AIE curves. As shown in Fig. 1B, when ft was increased to 99%, the enhancement ratios for TPA-Th(+) and Azo-TPA-Th(+) were ca. 13- and 8-fold, respectively, suggesting the typical AIE feature. Notably, the smaller I/I0 for Azo-TPA-Th(+) was presumably attributed to the fast trans-to-cis isomerization of azobenzene that typically causes fluorescence quenching.34 To confirm the photoisomerization of the azobenzene in Azo-TPA-Th(+), we performed a photoirradiation experiment to check the changes of its absorption spectra (Fig. S32†). However, both the π–π* and n–π* absorptions of azobenzene largely overlapped with the strong absorption of the TPA-Th(+) unit, making the spectral changes post π–π* and n–π* excitations hardly identifiable (Fig. S32A†). In this case, we enlarged the spectral regions that correspond to the π–π* and n–π* transitions of azobenzene. As shown in Fig. S32B and C,† after irradiation of Azo-TPA-Th(+) with 365 nm light, the intensities of the absorption peaks located between 310–350 nm and 450–490 nm exhibited opposite variation tendencies. Further irradiation with 450 nm light caused the absorption spectrum to basically return to its original state. Such a phenomenon was consistent with the trans-to-cis isomerization of azobenzene, suggesting the occurrence of photoisomerization.
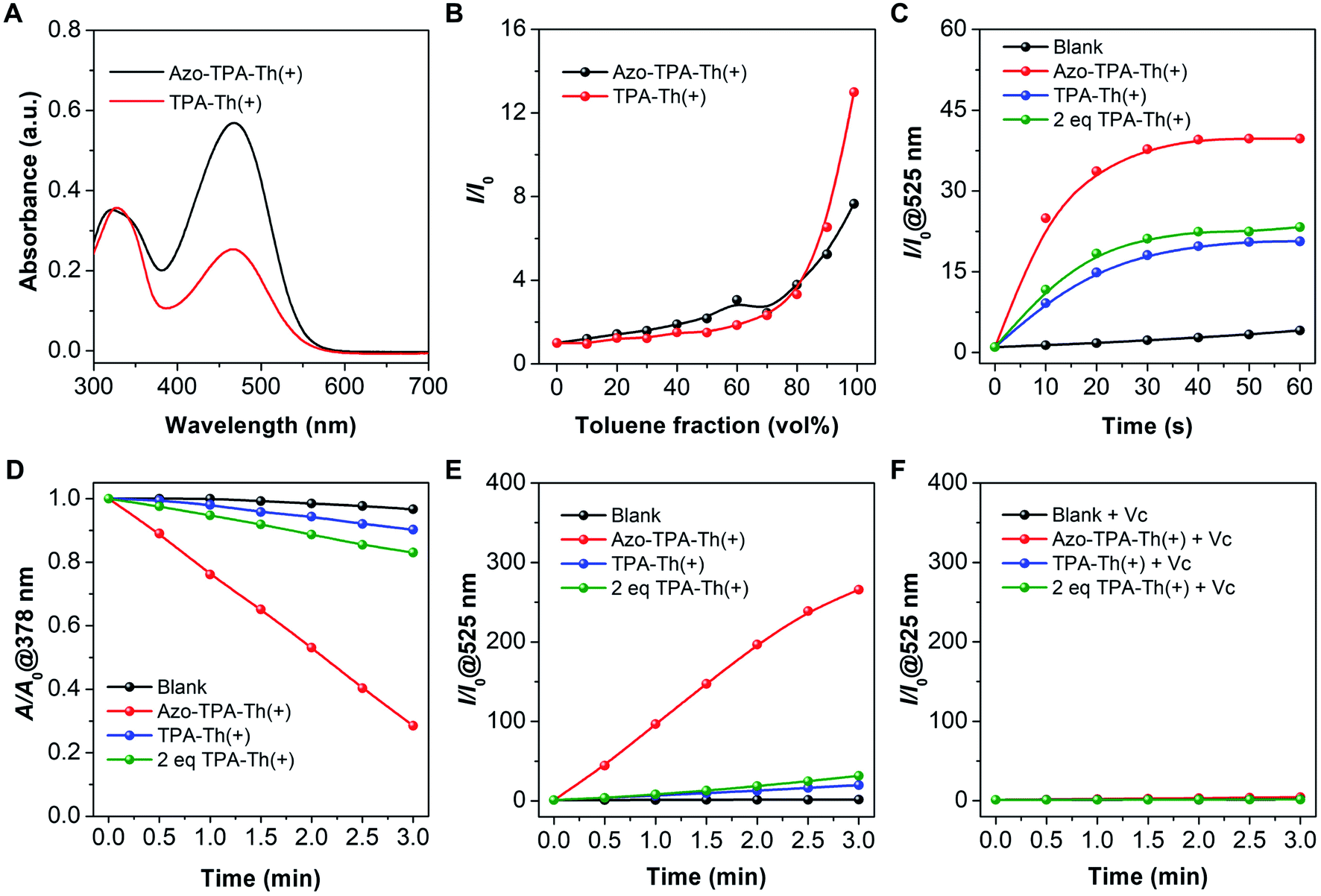 | ||
| Fig. 1 Photophysical and photochemical characterizations of TPA-Th(+) and Azo-TPA-Th(+). (A) UV-vis spectra of Azo-TPA-Th(+) and TPA-Th(+) in DMSO (20 μM). (B) Plots of the relative emission intensity (I/I0) of Azo-TPA-Th(+) at 630 nm and TPA-Th(+) at 600 nm as a function of toluene fractions (ft) in DMSO/toluene mixtures (20 μM, Ex = 488 nm). (C) Plots of the relative emission intensity (I/I0) at 525 nm of DCFH in the presence of Azo-TPA-Th(+) and TPA-Th(+) upon white light irradiation. (D) Decomposition rate of ABDA in the presence of Azo-TPA-Th(+) and TPA-Th(+) upon white light irradiation. A0 and A represent the absorbance at 378 nm before and after white light irradiation, respectively. (E) Plots of the relative emission intensity (I/I0) at 525 nm of DHR-123 in the presence of Azo-TPA-Th(+) and TPA-Th(+) as a function of irradiation time. (F) Plots of the relative emission intensity (I/I0) at 525 nm of DHR-123 in the presence of Azo-TPA-Th(+) and TPA-Th(+) supplemented with Vc as a function of irradiation time. | ||
We next assessed the ROS generation of TPA-Th(+) and Azo-TPA-Th(+) using three commercial indicators, 2′,7′-dichlorofluorescin (DCFH),35 9,10-anthracenediyl-bis(methylene)-dimalonic acid (ABDA),36 and dihydrorhodamine 123 (DHR-123)37 (Fig. S33–S36†). DCFH is a non-fluorescent probe that responds to all kinds of ROS by converting to highly fluorescent 2′,7′-dichlorofluorescein (DCF). Taking the emission intensity at 525 nm as the variable parameter, we recorded the relative emission intensity (I/I0) of DCFH in the presence of TPA-Th(+) and Azo-TPA-Th(+) upon white light irradiation. As shown in Fig. 1C, as the irradiation time prolonged, the emission signals for both groups gradually increased and reached a plateau after 40 s. It should be pointed out that the enhancement ratio for the Azo-TPA-Th(+) group was ca. 40-fold, which was much higher than that for the TPA-Th(+) group (ca. 20-fold) as well as that for the group with two equivalents of TPA-Th(+) (2 eq. TPA-Th(+), ca. 23-fold), suggesting the remarkably enhanced photosensitization post azo-bridging. To further specify the type of generated ROS, ABDA and DHR-123 were utilized to reflect the production of type-II and type-I ROS, respectively. In the presence of 1O2, ABDA is transformed into its endoperoxide form, leading to a decrease in absorbance. In this case, the proportion of the absorbance at 378 nm after and before irradiation (A/A0) is inversely correlated with the amount of 1O2. As shown in Fig. 1D, the Azo-TPA-Th(+) group exhibited the lowest A/A0 value (0.28), which was much lower than that of the TPA-Th(+) group (0.9) as well as that of the 2 eq. TPA-Th(+) group (0.8). In the presence of type-I ROS, DHR-123 is oxidized to the fluorescent rhodamine 123. Taking the emission intensity at 525 nm as the variable parameter, we recorded the relative emission intensity (I/I0) of DHR-123 in the presence of Azo-TPA-Th(+) and TPA-Th(+) upon white light irradiation. Surprisingly, the enhancement ratio of the Azo-TPA-Th(+) group was up to 265-fold, which was astonishingly higher than those of the TPA-Th(+) (20-fold) and 2 eq. TPA-Th(+) (31-fold) groups (Fig. 1E). Besides, the addition of vitamin C (Vc) completely inhibited the conversion of DHR-123 into its fluorescent form due to the efficient consumption of type-I ROS via a redox reaction (Fig. 1F). To further confirm the generation of O2˙− by the sensitization of Azo-TPA-Th(+), we also performed electron spin resonance (ESR) experiments with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the spin trapping agent. As shown in Fig. S37,† in the presence of DMPO and Azo-TPA-Th(+), there was a characteristic ESR signal for the DMPO/O2˙− adduct under white light irradiation, suggesting the generation of O2˙−. These results altogether demonstrated that bridging D–A type PSs with the azo group was an effective strategy to boost the generation of both type-II and type-I ROS.
To confirm the applicability of azo-bridging enhanced photosensitization, another pair of D–A type PSs with shortened conjugation length (denoted as TPA(+) and Azo-TPA(+)) were prepared using the synthetic routes similar to TPA-Th(+) and Azo-TPA-Th(+) (Scheme S1 and Fig. S16–S30†). As anticipated, the absorption maxima of TPA-Th(+) and Azo-TPA-Th(+) were blue-shifted to ca. 430 nm as a result of the decreased π conjugation (Fig. S38A†). Similarly, TPA-Th(+) exhibited a typical AIE phenomenon, with the enhancement ratio of ca. 16 (Fig. S38B and S39A and C†). However, only 1-fold enhancement was observed for Azo-TPA(+) (Fig. S38B and S39B and D†), which likewise resulted from the quenching effect of azobenzene. The DLS data also suggested the formation of large aggregates at ft = 99% for TPA(+) and Azo-TPA(+), respectively (Fig. S39E and F†). In addition, ROS measurements demonstrated that Azo-TPA(+) showed the strongest capability in the generation of total, type-II, and type-I ROS (particularly type-I ROS) when compared to TPA(+) and two equivalents of TPA(+) (2 eq. TPA(+)) (Fig. S38C–F and S40–S42†). Due to the weakened D–A effect, TPA(+) and Azo-TPA(+) showed decreased ROS generation relative to TPA-Th(+) and Azo-TPA-Th(+), respectively. However, azo-bridging transformed a remarkably weak PS (i.e., TPA(+)) into a moderately strong PS (i.e., Azo-TPA(+)), which once again evidenced the effectiveness of this strategy in boosting ROS generation. It should be noted that azo-bridging did not contribute to the extension of effective conjugation, which could be clearly indicated when comparing the absorption spectra of the azo-bridged and non-bridged PSs. As shown in Fig. 1A and S38A,† the absorption maxima in the visible region of all azo-bridged PSs were basically identical to those of the corresponding non-bridged PSs. However, when comparing the absorption maxima of Azo-TPA(+)/TPA(+) and Azo-TPA-Th(+)/TPA-Th(+), there was a 30 nm redshift after the introduction of thiophene. Such changes indicated that the insertion of a π-conjugated unit rather than azo-bridging led to the extension of effective conjugation for D–A type PSs. By referring to the calculated frontier molecular orbitals (FMOs) shown in Fig. S43,† we inferred that the strong D–A interactions in the symmetric side of Azo-TPA-Th(+) played a decisive role in affecting electron delocalization, thereby making the absorption profiles of the azo-bridged PSs resemble those of the corresponding non-bridged ones.
To gain more insight into the mechanism of enhanced photosensitization, we performed theoretical calculations on the two pairs of D–A type PSs, in which both the trans and cis forms of Azo-TPA-Th(+) and Azo-TPA(+) were calculated. The oscillator strength (f), energies of Sn and Tn (ESn/ETn), ΔEST, and ξST are summarized in Tables S1–S6.† According to the calculated f values, the S1 of trans-Azo-TPA(+) and cis-Azo-TPA(+) and the S2 of cis-Azo-TPA-Th(+) were specified as the dark state (f = 0, 0.05, and 0, respectively). In terms of trans-Azo-TPA-Th(+), both S1 and S2 were the bright state (f = 1.94 and 1.12, respectively). However, the ξST between S1 and Tn (n = 1, 2, 3) indicated that the ISC from S1 was less likely to occur (ξS1–T1 = 0.02 cm−1, ξS1–T2 = 0.02 cm−1, and ξS1–T3 = 0 cm−1, respectively). In addition, the minimal ΔEST between S1 and T3 (0.26 eV) was much larger than that between S2 and T4 (0.01 eV) (Fig. 2A). Given the small minimal ΔEST and large ξST (ξS2–T4 = 0.28 cm−1), we inferred that the ISC from S2 to T4 was the primary pathway for trans-Azo-TPA-Th(+). In contrast, the minimal ΔEST of TPA-Th(+) was calculated to be 0.79 eV between S1 and T1, accompanied by ξS1–T1 of 0.32 cm−1 (Fig. 2B). Such a comparison suggested that azo-bridging caused a prominent reduction in the minimal ΔEST, which enabled the increase of kISC and thus efficient ISC. In this case, the excited Azo-TPA-Th(+) in the singlet state easily reached the triplet state via ISC, which was followed by type-II and type-I photochemical reactions to generate considerable ROS. Besides, the minimal ΔEST of cis-Azo-TPA-Th(+) was calculated to be 0.58 eV between S1 and T3, with ξS1–T3 of 0.08 cm−1. In this case, the ISC process for the cis configuration was less likely to occur, which instead favored fluorescence turn-on in the aggregated state (Fig. 2C). Similarly, the minimal ΔEST of trans-Azo-TPA(+), TPA(+), and cis-Azo-TPA(+) was determined to be 0.51 eV between S2 and T4, 0.74 eV between S1 and T1, and 0.88 eV between S2 and T3, respectively, together with ξS2–T4 of 0.17 cm−1, ξS1–T1 of 0.14 cm−1, and ξS2–T3 of 0.04 cm−1 (Fig. S44†). These values once again suggested the decreased ΔEST in azo-bridged trans-compounds and increased ΔEST in azo-bridged cis-compounds. To computationally demonstrate the feasibility of type-I photosensitization by all PSs, we calculated their vertical ionization potentials (VIPs) and vertical electron affinities (VEAs) (Table S7†). As depicted in the equation of PS(T1) + 3O2 → PS(T1)˙+ + O2˙−, T1 state PSs can sensitize the generation of O2˙− by donating an electron to 3O2. The prerequisite for this reaction is that the summation of the VIP for the T1 state (VIPT1) of PSs and the adiabatic electron affinity (AEA) of 3O2 (AEAO2; −3.91 eV in water) should be negative.38,39 As shown in Table S7,† the summation of VIPT1 and AEAO2 for all PSs was negative, indicating that O2˙− could be generated by the photosensitization of all PSs. In terms of type-II photochemical reactions, the ET1 of all PSs were higher than 0.98 eV which corresponded to the lowest energy required to promote 3O2 to 1O2, indicating that all PSs could sensitize the generation of 1O2via a direct energy transfer pathway.40 Taken together, in contrast to unbridged D–A type PSs, azo-bridged trans-compounds exhibited decreased ΔEST, which favored enhanced generation of both type-I and type-II ROS via efficient ISC. For azo-bridged cis-compounds, the ΔEST was increased and the corresponding ξST was decreased. Both changes led to suppressed ISC, which could be employed for the construction of light-responsive fluorescence turn-on systems.
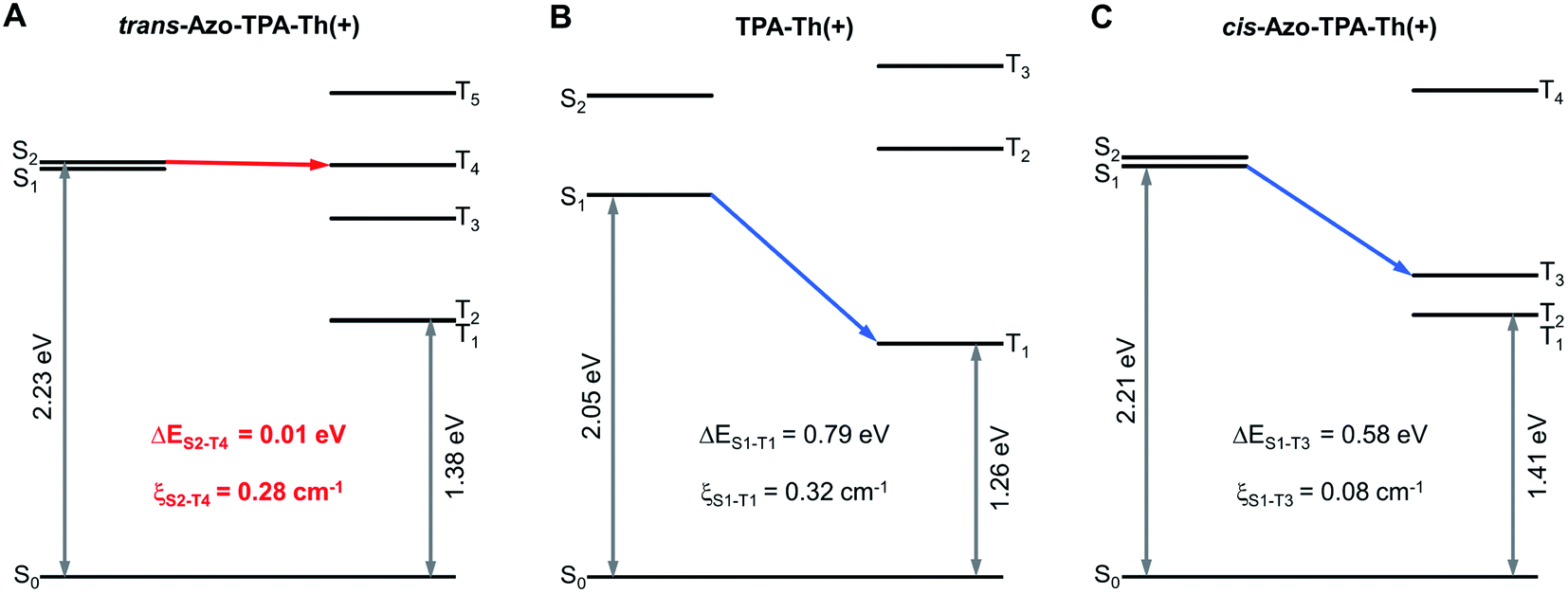 | ||
| Fig. 2 Energy level diagrams and calculated ξST between different singlet–triplet channels for (A) trans-Azo-TPA-Th(+), (B) TPA-Th(+), and (C) cis-Azo-TPA-Th(+). | ||
The strong ROS generation capability of Azo-TPA-Th(+) encouraged us to further explore its potential in PDT. As a proof-of-concept demonstration, we studied the application of Azo-TPA-Th(+) in photodynamic killing of drug-resistant bacteria, in which Gram-positive MRSA was selected in view of its responsibility for the largest outbreak of hospital-acquired infection.41,42 Due to the inherent positive charges, Azo-TPA-Th(+) tended to associate with negatively charged bacteria through electrostatic and/or hydrophobic interactions.43,44 The quantitative bacterial killing by Azo-TPA-Th(+) was evaluated by plate-counting assay in the absence and presence of white light irradiation. As shown in Fig. 3A, in the absence of white light, there was a moderate concentration-dependent inhibition on bacterial viability, which was attributed to the membrane-damaging effect of cationic species.45 Upon white light irradiation, the bacterial viability significantly decreased due to the additional destructive effect of the generated ROS. In particular, when the concentration of Azo-TPA-Th(+) reached 10 μM, the bacterial killing efficiency was as high as 99.999999%, indicating its outstanding PDT effect. In addition, the corresponding photographs of bacterial colonies formed on agar plates clearly suggested the irradiation-caused enhancement in the inactivation of MRSA (Fig. 3B and S45†). To visualize the antibacterial effect of Azo-TPA-Th(+), we also performed a live/dead fluorescent staining assay. As shown in Fig. 3C, in contrast to the blank group with green fluorescence, the Azo-TPA-Th(+) groups without and with light irradiation showed a pronounced shift in the emission color, in which light irradiation transformed all green-emissive bacteria into red-emissive bacteria, indicating the destructive effect of Azo-TPA-Th(+) on MRSA. Furthermore, we examined the morphology of MRSA without and with light irradiation by scanning electron microscopy (SEM). As shown in Fig. 3D, the smooth bacterial surface became rough after incubation with Azo-TPA-Th(+) in the absence of light irradiation. In contrast, light irradiation gave rise to obvious bacterial damage with serious membrane splitting and deformation due to the strong photodynamic effect of Azo-TPA-Th(+). These results altogether demonstrated that Azo-TPA-Th(+) was able to photodynamically inactivate drug-resistant bacteria with high killing efficiency.
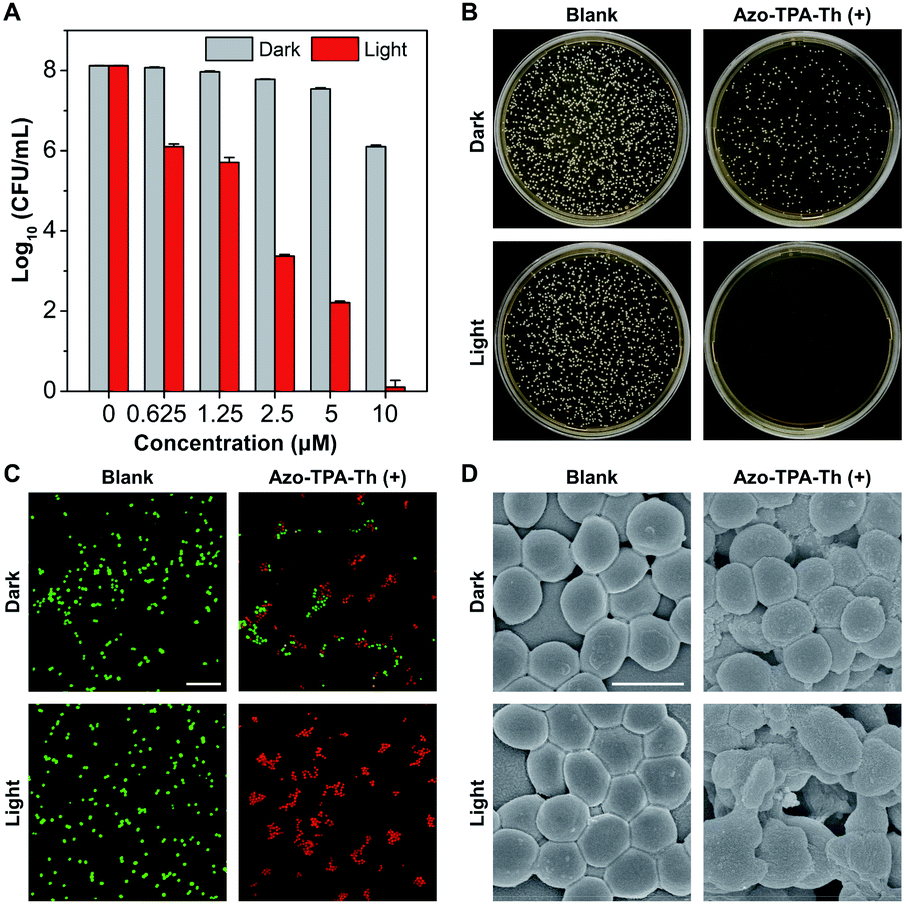 | ||
| Fig. 3 In vitro antibacterial behaviors of Azo-TPA-Th(+) against MRSA. (A) Bacterial viability by the logarithmic number as a function of the concentration of Azo-TPA-Th(+) without and with white light irradiation. (B) Photographs of bacterial colonies formed on agar plates after incubation with Azo-TPA-Th(+) (5 μM) without and with white light irradiation. (C) Live/dead staining of bacteria with different treatments, where live and dead bacteria are shown in green and red, respectively. Scale bar: 10 μm. (D) SEM images of bacteria with different treatments. Scale bar: 1 μm. | ||
Since azo-bridging of two molecules of TPA-Th(+) generated an azobenzene structure, the resulting Azo-TPA-Th(+) could respond to UV irradiation by experiencing trans-to-cis isomerization.46–48 According to the theoretical calculations, cis-Azo-TPA-Th(+) should be potentially efficient in fluorescence emission rather than ROS generation. To verify our hypothesis, we studied the photophysical properties of Azo-TPA-Th(+) in phosphate buffered saline (PBS) before and after UV irradiation. As shown in Fig. 4A, as the irradiation time prolonged, the absorption maximum gradually blue-shifted from 450 to 400 nm, which was accompanied by decreased absorbance. Meanwhile, the emission peak at ca. 460 nm started to appear (Fig. 4B). It should be mentioned that the excitation wavelength for Azo-TPA-Th(+) post UV irradiation was fixed at 385 nm as it was the excitation maximum as shown in Fig. S46.† After irradiation for 30 min, the dispersion of Azo-TPA-Th(+) turned from light yellow to near colorless; meanwhile, a pronounced blue emission was observed (Fig. 4C and D). We inferred that the blue-shifted and decreased absorption as well as the fluorescence turn-on phenomenon was attributed to the trans-to-cis isomerization of Azo-TPA-Th(+),49 which gave rise to the formation of larger aggregates to activate the AIE phenomenon. Since the absorption peak of TPA-Th(+) largely overlapped with the characteristic absorption of azobenzene, it was difficult to confirm the configurational transition of Azo-TPA-Th(+) from the UV-vis spectra. However, theoretical calculations indicated that the ROS generation by cis-Azo-TPA-Th(+) should be remarkably decreased when compared to the trans-counterpart due to the larger ΔEST and smaller ξST. To exclude the potential interference of cis-Azo-TPA-Th(+) on fluorescent ROS indicators, we performed 1O2 measurements before and after UV irradiation with ABDA to indirectly reflect the UV-triggered trans-to-cis isomerization. As shown in Fig. 4E, upon UV irradiation, ROS generation was remarkably inhibited, with the value of A/A0 varying from 0.28 to 0.93. In addition, in contrast to the relatively planar trans-Azo-TPA-Th(+), the distorted cis-configuration was more prone to aggregation in an aqueous environment due to the reduced electrostatic repulsion between pyridinium cations to favor hydrophobic interactions (Fig. S43†). As anticipated, UV irradiation caused the hydrodynamic diameter of Azo-TPA-Th(+) to change from ca. 140 nm to ca. 400 nm (Fig. 4F). Transmission electron microscopy (TEM) characterization further indicated that the small aggregates of Azo-TPA-Th(+) merged into larger aggregates post UV irradiation (Fig. 4G and H). To verify that the aforementioned changes did not result from UV-caused molecular decomposition, we also conducted exactly the same experiment using DMSO as the solvent. As shown in Fig. S47,† both the absorption and emission intensities basically remained unchanged, suggesting that the chemical structure of Azo-TPA-Th(+) was not destroyed under experimental conditions. Besides, due to the typical D–A structure, Azo-TPA-Th(+) intrinsically possessed twisted intramolecular charge transfer (TICT) property.50–52 As shown in Fig. 4I, Azo-TPA-Th(+) exhibited a pronounced solvatochromic phenomenon, with the emission maximum blue-shifting from 533 nm in DMSO to 453 nm in toluene, indicating that Azo-TPA-Th(+) could sense the polarity of its residing environment via TICT. The calculated FMOs also suggested the spatial separation of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) electron clouds, which enabled intramolecular charge transfer from azobenzene and triphenylaniline to pyridinium (Fig. S43†). Due to the decreased polarity within the larger aggregates, cis-Azo-TPA-Th(+) displayed blue-shifted emission, which accounted for the blue fluorescence of Azo-TPA-Th(+) post UV irradiation.
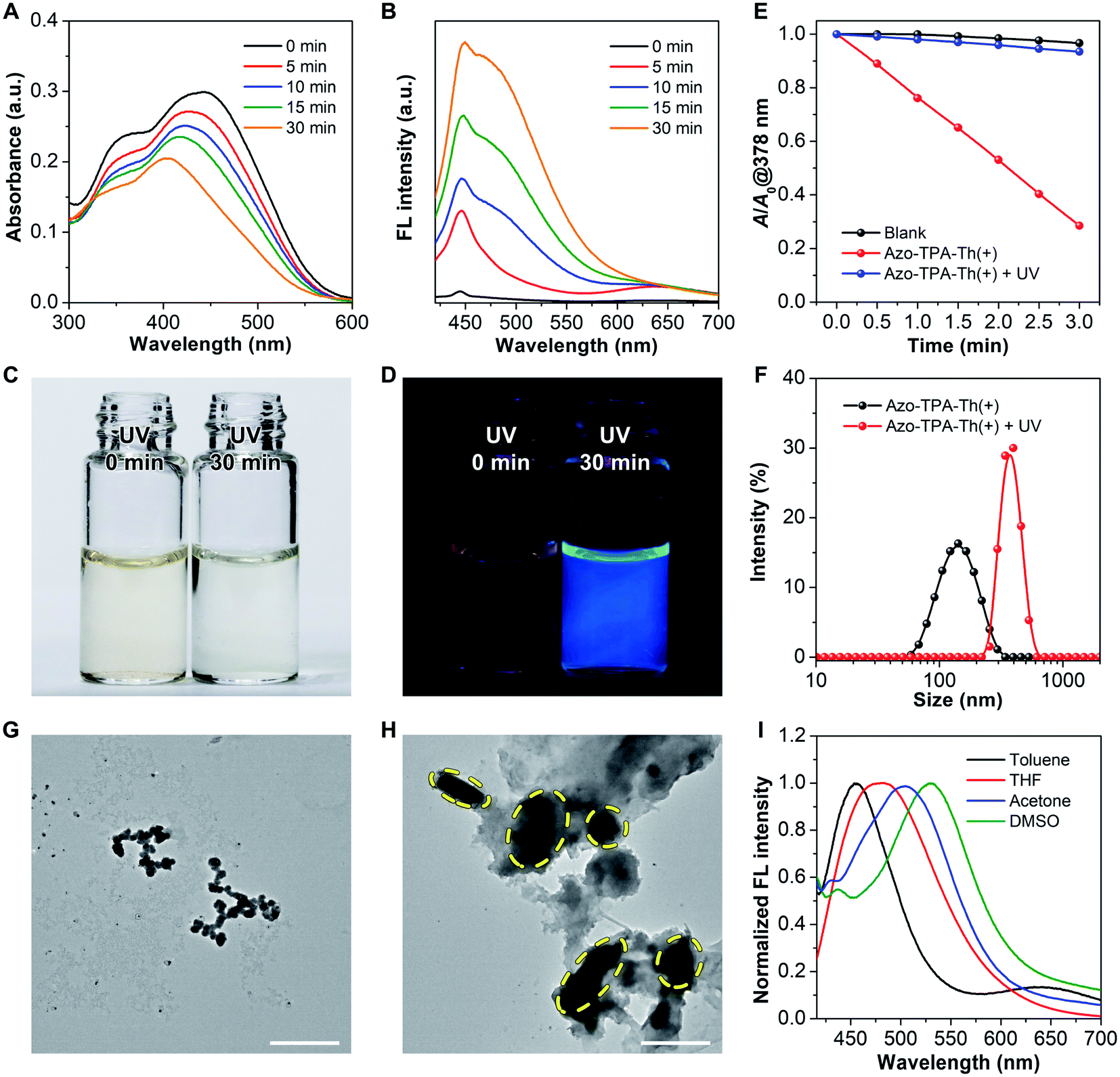 | ||
| Fig. 4 Responsiveness of Azo-TPA-Th(+) upon 365 nm UV irradiation. (A and B) (A) Absorption and (B) emission spectra of Azo-TPA-Th(+) irradiated for different periods of time. Ex = 385 nm. (C and D) Photographs of Azo-TPA-Th(+) before and after UV irradiation taken under (C) a filament lamp and (D) a 365 nm UV lamp. (E) 1O2 measurements before and after UV irradiation. (F) Particle size distribution of Azo-TPA-Th(+) before and after UV irradiation. (G and H) TEM images of Azo-TPA-Th(+) (G) before and (H) after UV irradiation. Scale bars: 500 nm. (I) Normalized emission spectra of Azo-TPA-Th(+) in different organic solvents. | ||
Motivated by the responsiveness of Azo-TPA-Th(+) to UV light, we postulated that if Azo-TPA-Th(+) is added to bacterial suspensions at a concentration higher than a binding threshold, the excess molecules should undergo photoisomerization upon UV irradiation and thus emit blue fluorescence (Scheme 3). In contrast, if the concentration is lower than a binding threshold, Azo-TPA-Th(+) will associate with the bacteria through electrostatic and/or hydrophobic interactions and locate in the cell membrane. In this case, no evident fluorescence signals should be detected even after UV irradiation due to restricted configurational transformation. As such, we can directly discern whether the concentration of Azo-TPA-Th(+) is sufficiently high for effective bacterial killing based on the appearance of fluorescence post UV irradiation. We first measured the emission spectra of Azo-TPA-Th(+) inoculated by bacteria at different concentrations. The relative concentration, that is, the amount of Azo-TPA-Th(+) per optical density at 600 nm (OD), was employed to indicate different groups. As shown in Fig. 5A–C, when the relative concentrations were set at 50, 25, and 20 μM per OD, respectively, we observed a time-dependent fluorescence turn-on phenomenon post UV irradiation, with the groups at high concentrations exhibiting remarkable emission enhancement, suggesting the presence of excess Azo-TPA-Th(+). In contrast, when the relative concentrations were fixed at 10, 6.7, and 5 μM per OD, no evident fluorescence turn-on phenomenon was observed, indicating the absence of unbound Azo-TPA-Th(+) for photoisomerization (Fig. 5D–F). Conveniently, such changes could be directly visualized under a 365 nm UV lamp. As shown in Fig. 5G and H, when the relative concentration was ≥20 μM per OD, the blue fluorescence signals became visible to the naked eye, which was in accordance with the aforementioned result. Furthermore, we plotted a curve to portray the quantitative relationship between emission enhancement and the relative concentration of Azo-TPA-Th(+) (Fig. 5I). According to the results shown in Fig. 5A–H, the binding threshold of Azo-TPA-Th(+) toward MRSA was set at 20 μM per OD, at which the enhancement ratio was calculated to be 2.2-fold. When the relative concentration reached or exceeded the binding threshold, there should exist unbound Azo-TPA-Th(+) to isomerize upon UV irradiation, resulting in the turn-on fluorescence. However, when the relative concentration was less than the binding threshold, Azo-TPA-Th(+) was primarily associated with MRSA with restricted configurational transformation, and, thus, no fluorescence appeared post UV irradiation. Due to the prominent bactericidal capability of Azo-TPA-Th(+), the appearance of fluorescence also implied highly efficient bacterial killing (≥99.999999%), as reflected in Fig. 5J. In this case, by conducting a simple fluorescence measurement or simply observing under a 365 nm UV lamp, we could directly discern the effective PDT bactericidal dose within 1 h without performing the time-consuming plate-counting assay, indicating its practicality in real-world applications.
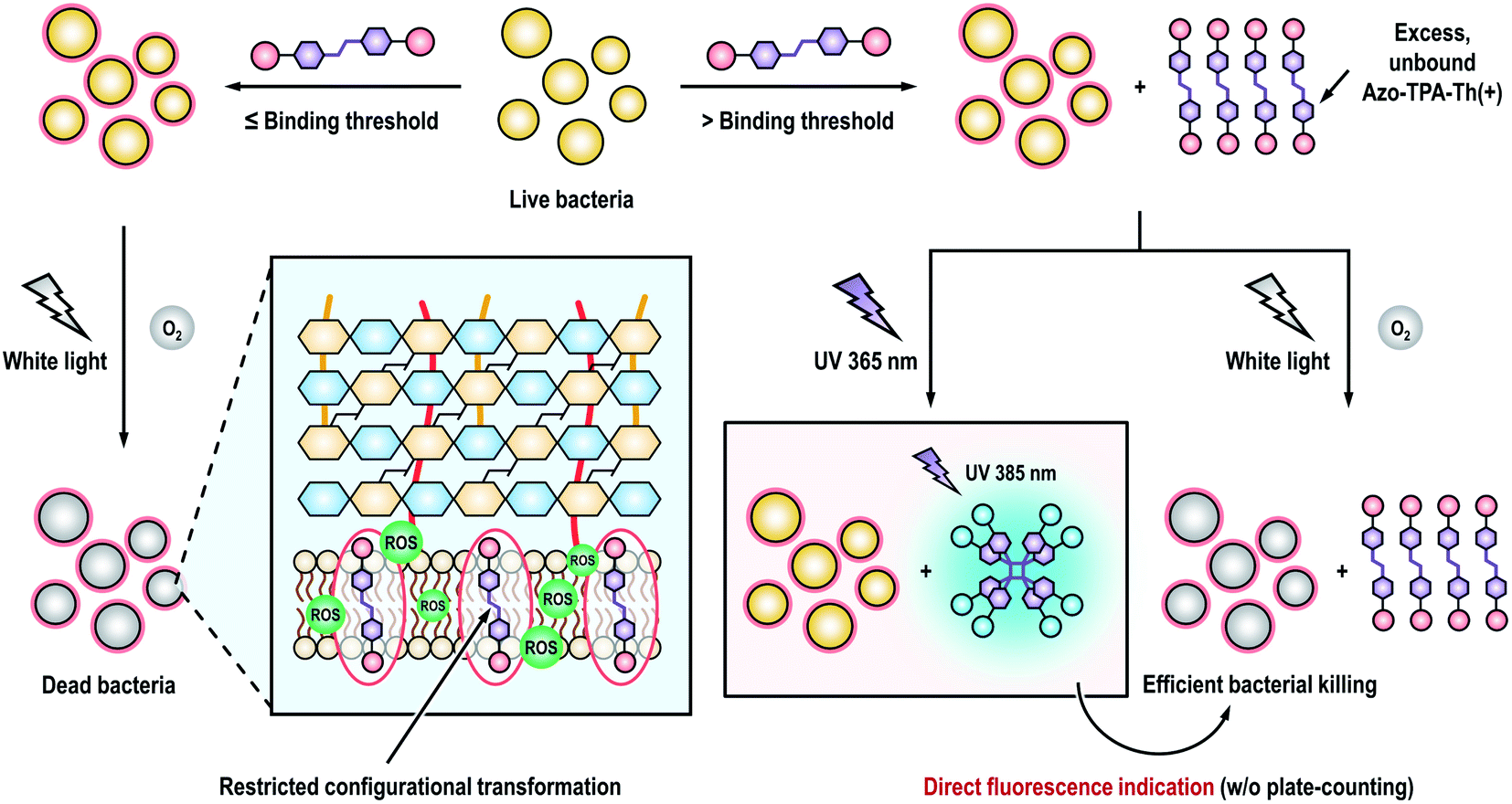 | ||
| Scheme 3 Schematic illustration showing the procedure of direct determination of the effective photodynamic bactericidal dose through the appearance of fluorescence. | ||
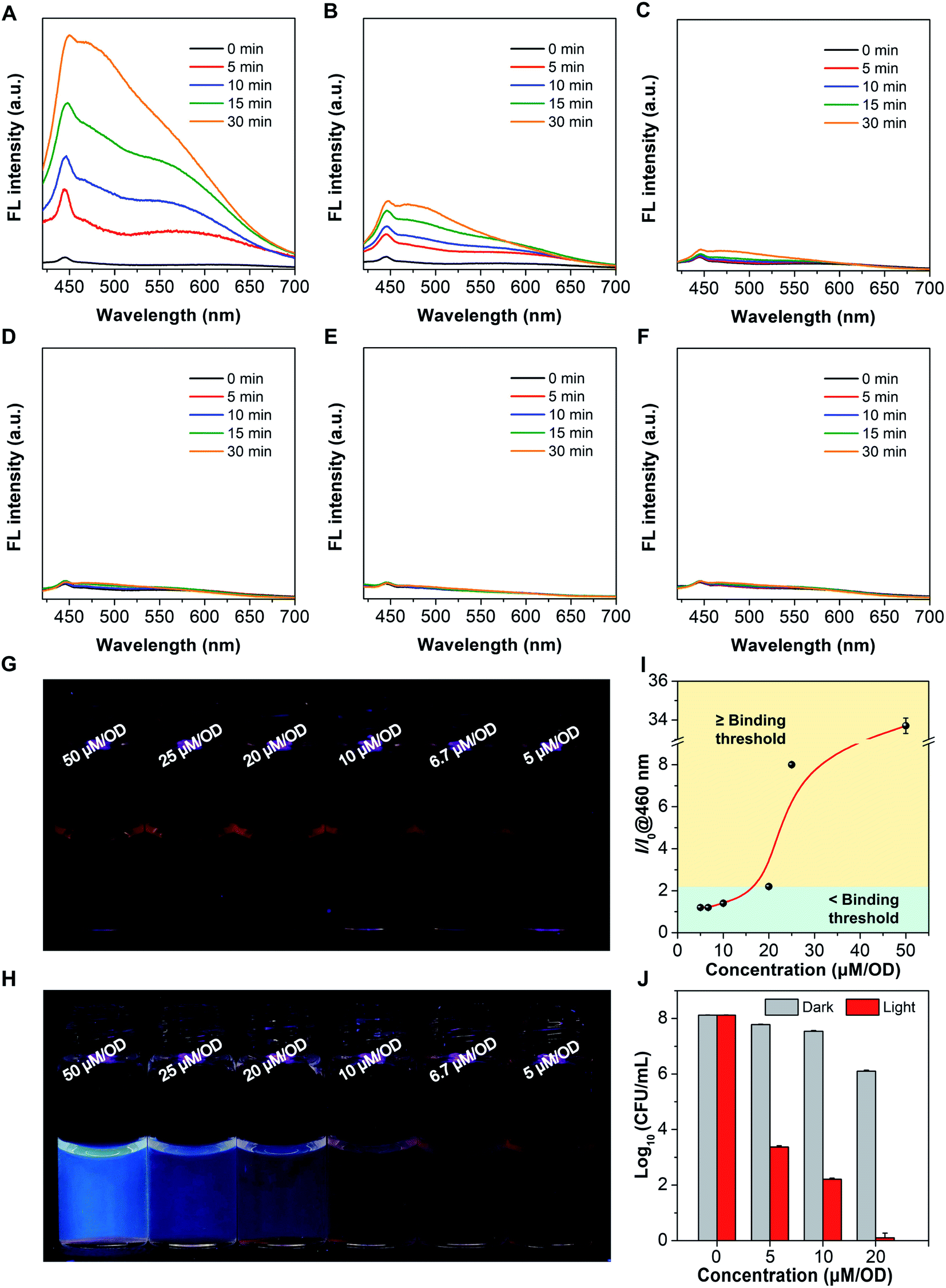 | ||
| Fig. 5 Fluorescence determination of the threshold for efficient photodynamic bacterial killing. (A–F) Emission spectra of bacterial suspensions in the presence of Azo-TPA-Th(+) at the relative concentrations of (A) 50 μM per OD, (B) 25 μM per OD, (C) 20 μM per OD, (D) 10 μM per OD, (E) 6.7 μM per OD, and (F) 5 μM per OD irradiated with 365 nm UV light for different periods of time. Ex = 385 nm. (G and H) Photographs of bacterial suspensions in the presence of Azo-TPA-Th(+) at different relative concentrations (G) before and (H) after irradiation for 30 min taken under a 365 nm UV lamp. (I) Relative emission intensity (I/I0) of bacterial suspensions in the presence of Azo-TPA-Th(+) at different relative concentrations, in which I0 and I represented the emission intensity at 460 nm upon irradiation for 0 min and 30 min, respectively. (J) Bacterial viability by the logarithmic number as a function of the concentration of Azo-TPA-Th(+) without and with white light irradiation. | ||
Conclusions
In summary, we have, for the first time, developed a novel and effective strategy to boost ROS generation by bridging D–A type PSs with the azo group. Theoretical calculations suggested that azo-bridging led to the significant decrease in ΔEST and thus efficient ISC. Such a strategy not only transformed moderately strong PSs into particularly strong PSs, but also converted particularly weak PSs into moderately strong PSs. In particular, the azo-bridged Azo-TPA-Th(+) exhibited a particularly strong bactericidal effect against clinically relevant MRSA, with the killing efficiency up to 99.999999% upon white light irradiation. Due to the presence of an azobenzene structure, Azo-TPA-Th(+) could undergo trans-to-cis isomerization upon UV irradiation to form emissive aggregates. By virtue of the fluorescence turn-on property, it was possible to determine the efficient PDT bactericidal dose within 1 h. It should be mentioned that the proof-of-concept azo-bridged PSs are less suited for in vivo applications due to their short-wavelength absorption. However, this issue can be addressed by using the well-established approaches to bathochromically shift the absorption spectra of D–A type PSs into the therapeutic window, such as extending the conjugation length and/or enhancing intramolecular D–A interactions.53–55 This study opens a brand-new avenue for the design of advanced PSs with strong ROS generation and stimuli-responsiveness, holding great potential in high-quality PDT with rapid prediction of the therapeutic outcome.Data availability
The data supporting the findings of this study are available within the article and in the ESI.†Author contributions
C. Z. and B. H. conceived the project. B. H. carried out the experiments. J. W. performed the theoretical calculations. C. W. participated in the synthesis of chemical compounds. K. X. participated in the live/dead staining experiment. M. X. and S. L. participated in the plate-counting experiment. C. Z. and B. H. wrote the manuscript. All authors discussed the results and contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52003123 and 92163126) and the Fundamental Research Funds for the Central Universities (Nankai University, 63213121 and 63213058).Notes and references
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- Z. Shen, Q. Ma, X. Zhou, G. Zhang, G. Hao, Y. Sun and J. Cao, NPG Asia Mater., 2021, 13, 39 CrossRef CAS.
- D. Wang, L. Niu, Z.-Y. Qiao, D.-B. Cheng, J. Wang, Y. Zhong, F. Bai, H. Wang and H. Fan, ACS Nano, 2018, 12, 3796–3803 CrossRef CAS PubMed.
- A. Turksoy, D. Yildiz and E. U. Akkaya, Coord. Chem. Rev., 2019, 379, 47–64 CrossRef CAS.
- B. Yuan, H. Wang, J.-F. Xu and X. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 26982–26990 CrossRef CAS PubMed.
- I. Roy, S. Bobbala, R. M. Young, Y. Beldjoudi, M. T. Nguyen, M. M. Cetin, J. A. Cooper, S. Allen, O. Anamimoghadam, E. A. Scott, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 12296–12304 CrossRef CAS PubMed.
- T. Luo, K. Ni, A. Culbert, G. Lan, Z. Li, X. Jiang, M. Kaufmann and W. Lin, J. Am. Chem. Soc., 2020, 142, 7334–7339 CrossRef CAS PubMed.
- Z. He, S. Tian, Y. Gao, F. Meng and L. Luo, Front. Chem., 2021, 9, 672917 CrossRef CAS PubMed.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- C. Zhu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, ACS Appl. Bio Mater., 2018, 1, 1768–1786 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- X. Liu, C. Zhu and B. Z. Tang, Acc. Chem. Res., 2022, 55, 197–208 CrossRef CAS PubMed.
- S. Xu, Y. Duan and B. Liu, Adv. Mater., 2020, 32, 1903530 CrossRef CAS PubMed.
- S. Liu, G. Feng, B. Z. Tang and B. Liu, Chem. Sci., 2021, 12, 6488–6506 RSC.
- W. Zhao, Z. He, Q. Peng, J. W. Y. Lam, H. Ma, Z. Qiu, Y. Chen, Z. Zhao, Z. Shuai, Y. Dong and B. Z. Tang, Nat. Commun., 2018, 9, 3044 CrossRef PubMed.
- Y. Chong, X. Zhang, B. Chen, R. Liu, Z. Wu, G. Zhang, J. Jiang, S. Mukamel and G. Zhang, J. Phys. Chem. A, 2021, 125, 3088–3094 CrossRef CAS PubMed.
- L. A. Ortiz-Rodríguez, S. J. Hoehn, A. Loredo, L. Wang, H. Xiao and C. E. Crespo-Hernández, J. Am. Chem. Soc., 2021, 143, 2676–2681 CrossRef PubMed.
- H. Zou, J. Zhang, C. Wu, B. He, Y. Hu, H. H. Y. Sung, R. T. K. Kwok, J. W. Y. Lam, L. Zheng and B. Z. Tang, ACS Nano, 2021, 15, 9176–9185 CrossRef CAS PubMed.
- K. M. Farrell, M. M. Brister, M. Pittelkow, T. I. Sølling and C. E. Crespo-Hernández, J. Am. Chem. Soc., 2018, 140, 11214–11218 CrossRef CAS PubMed.
- W. Piao, K. Hanaoka, T. Fujisawa, S. Takeuchi, T. Komatsu, T. Ueno, T. Terai, T. Tahara, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2017, 139, 13713–13719 CrossRef CAS PubMed.
- S. Liu, H. Zhang, Y. Li, J. Liu, L. Du, M. Chen, R. T. K. Kwok, J. W. Y. Lam, D. L. Phillips and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 15189–15193 CrossRef CAS PubMed.
- S. Xu, Y. Yuan, X. Cai, C.-J. Zhang, F. Hu, J. Liang, G. Zhang, D. Zhang and B. Liu, Chem. Sci., 2015, 6, 5824–5830 RSC.
- S. Xu, W. Wu, X. Cai, C.-J. Zhang, Y. Yuan, J. Liang, G. Feng, P. Manghnani and B. Liu, Chem. Commun., 2017, 53, 8727–8730 RSC.
- F. Hu, S. Xu and B. Liu, Adv. Mater., 2018, 30, 1801350 CrossRef PubMed.
- W. Wu, D. Mao, S. Xu, Kenry, F. Hu, X. Li, D. Kong and B. Liu, Chem, 2018, 4, 1937–1951 CAS.
- T. Zhou, R. Hu, L. Wang, Y. Qiu, G. Zhang, Q. Deng, H. Zhang, P. Yin, B. Situ, C. Zhan, A. Qin and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9952–9956 CrossRef CAS PubMed.
- S. Wang, W. Wu, P. Manghnani, S. Xu, Y. Wang, C. C. Goh, L. G. Ng and B. Liu, ACS Nano, 2019, 13, 3095–3105 CrossRef CAS PubMed.
- L. Schweighauser, M. A. Strauss, S. Bellotto and H. A. Wegner, Angew. Chem., Int. Ed., 2015, 54, 13436–13439 CrossRef CAS PubMed.
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Nat. Rev. Chem., 2022, 6, 51–69 CrossRef.
- L. Dong, Y. Feng, L. Wang and W. Feng, Chem. Soc. Rev., 2018, 47, 7339–7368 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- L. Bourré, S. Thibaut, A. Briffaud, N. Rousset, S. Eléouet, Y. Lajat and T. Patrice, J. Photochem. Photobiol., B, 2002, 67, 23–31 CrossRef.
- Z. Liu, H. Zou, Z. Zhao, P. Zhang, G.-G. Shan, R. T. K. Kwok, J. W. Y. Lam, L. Zheng and B. Z. Tang, ACS Nano, 2019, 13, 11283–11293 CrossRef CAS PubMed.
- M. Li, T. Xiong, J. Du, R. Tian, M. Xiao, L. Guo, S. Long, J. Fan, W. Sun, K. Shao, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2019, 141, 2695–2702 CrossRef CAS PubMed.
- L. Shen, H.-F. Ji and H.-Y. Zhang, J. Mol. Struct.: THEOCHEM, 2008, 851, 220–224 CrossRef CAS.
- J. Llano, J. Raber and L. A. Eriksson, J. Photochem. Photobiol., A, 2003, 154, 235–243 CrossRef CAS.
- K. Wen, H. Tan, Q. Peng, H. Chen, H. Ma, L. Wang, A. Peng, Q. Shi, X. Cai and H. Huang, Adv. Mater., 2022, 34, 2108146 CrossRef CAS PubMed.
- J. Xie, M. Zhou, Y. Qian, Z. Cong, S. Chen, W. Zhang, W. Jiang, C. Dai, N. Shao, Z. Ji, J. Zou, X. Xiao, L. Liu, M. Chen, J. Li and R. Liu, Nat. Commun., 2021, 12, 5898 CrossRef CAS PubMed.
- K. Xue, C. Yang, C. Wang, Y. Liu, J. Liu, L. Shi and C. Zhu, CCS Chem., 2022, 4, 272 CrossRef.
- X. Liu, M. Xiao, K. Xue, M. Li, D. Liu, Y. Wang, X. Yang, Y. Hu, R. T. K. Kwok, A. Qin, C. Zhu, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 19222–19231 CrossRef CAS PubMed.
- M. Kang, C. Zhou, S. Wu, B. Yu, Z. Zhang, N. Song, M. M. S. Lee, W. Xu, F.-J. Xu, D. Wang, L. Wang and B. Z. Tang, J. Am. Chem. Soc., 2019, 141, 16781–16789 CrossRef CAS PubMed.
- Y. Li, F. Liu, J. Zhang, X. Liu, P. Xiao, H. Bai, S. Chen, D. Wang, S. H. P. Sung, R. T. K. Kwok, J. Shen, K. Zhu and B. Z. Tang, Adv. Sci., 2021, 8, 2001750 CrossRef CAS PubMed.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234–3243 CrossRef CAS PubMed.
- S. Mehrparvar, Z. N. Scheller, C. Wölper and G. Haberhauer, J. Am. Chem. Soc., 2021, 143, 19856–19864 CrossRef CAS PubMed.
- H.-B. Cheng, S. Zhang, J. Qi, X.-J. Liang and J. Yoon, Adv. Mater., 2021, 33, 2007290 CrossRef CAS PubMed.
- M. R. Han, Y. Hirayama and M. Hara, Chem. Mater., 2006, 18, 2784–2786 CrossRef CAS.
- C. Wang, X. Zhao, H. Jiang, J. Wang, W. Zhong, K. Xue and C. Zhu, Nanoscale, 2021, 13, 1195–1205 RSC.
- K. Xue, C. Wang, J. Wang, S. Lv, B. Hao, C. Zhu and B. Z. Tang, J. Am. Chem. Soc., 2021, 143, 14147–14157 CrossRef CAS PubMed.
- C. Wang, W. Chi, Q. Qiao, D. Tan, Z. Xu and X. Liu, Chem. Soc. Rev., 2021, 50, 12656–12678 RSC.
- Z. Zhang, W. Xu, M. Kang, H. Wen, H. Guo, P. Zhang, L. Xi, K. Li, L. Wang, D. Wang and B. Z. Tang, Adv. Mater., 2020, 32, 2003210 CrossRef CAS PubMed.
- W. Wu, D. Mao, S. Xu, M. Panahandeh-Fard, Y. Duan, F. Hu, D. Kong and B. Liu, Adv. Funct. Mater., 2019, 29, 1901791 CrossRef CAS.
- W. Zhu, M. Kang, Q. Wu, Z. Zhang, Y. Wu, C. Li, K. Li, L. Wang, D. Wang and B. Z. Tang, Adv. Funct. Mater., 2021, 31, 2007026 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00381c |
| This journal is © The Royal Society of Chemistry 2022 |