
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtended-ensemble docking to probe dynamic variation of ligand binding sites during large-scale structural changes of proteins†
Karan
Kapoor
 ,
Sundar
Thangapandian
and
Emad
Tajkhorshid
*
,
Sundar
Thangapandian
and
Emad
Tajkhorshid
*
Theoretical and Computational Biophysics Group, NIH Center for Macromolecular Modeling and Bioinformatics, Beckman Institute for Advanced Science and Technology, Department of Biochemistry, Center for Biophysics and Quantitative Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: emad@illinois.edu
First published on 16th March 2022
Abstract
Proteins can sample a broad landscape as they undergo conformational transition between different functional states. At the same time, as key players in almost all cellular processes, proteins are important drug targets. Considering the different conformational states of a protein is therefore central for a successful drug-design strategy. Here we introduce a novel docking protocol, termed extended-ensemble docking, pertaining to proteins that undergo large-scale (global) conformational changes during their function. In its application to multidrug ABC-transporter P-glycoprotein (Pgp), extensive non-equilibrium molecular dynamics simulations employing system-specific collective variables are first used to describe the transition cycle of the transporter. An extended set of conformations (extended ensemble) representing the full transition cycle between the inward- and the outward-facing states is then used to seed high-throughput docking calculations of known substrates, non-substrates, and modulators of the transporter. Large differences are predicted in the binding affinities to different conformations, with compounds showing stronger binding affinities to intermediate conformations compared to the starting crystal structure. Hierarchical clustering of the binding modes shows all ligands preferably bind to the large central cavity of the protein, formed at the apex of the transmembrane domain (TMD), whereas only small binding populations are observed in the previously described R and H sites present within the individual TMD leaflets. Based on the results, the central cavity is further divided into two major subsites, first preferably binding smaller substrates and high-affinity inhibitors, whereas the second one shows preference for larger substrates and low-affinity modulators. These central subsites along with the low-affinity interaction sites present within the individual TMD leaflets may respectively correspond to the proposed high- and low-affinity binding sites in Pgp. We propose further an optimization strategy for developing more potent inhibitors of Pgp, based on increasing its specificity to the extended ensemble of the protein, instead of using a single protein structure, as well as its selectivity for the high-affinity binding site. In contrast to earlier in silico studies using single static structures of Pgp, our results show better agreement with experimental studies, pointing to the importance of incorporating the global conformational flexibility of proteins in future drug-discovery endeavors.
1 Introduction
Modern drug discovery is centered around the identification of suitable protein targets that play important roles in a specific disease, and the development of small molecules designed to either attenuate or promote the function of those targets.1 Structural determination techniques such as X-ray crystallography and cryo-EM have led to a large number of structurally known proteins that can help guide the drug-discovery process.2,3 Experimentally resolved structures, however, generally represent starting or end states in the functional cycle of a protein, as the conformational intermediates (usually shorter-lived) arising during the function are often inaccessible under the experimental conditions.4 This reduces the possible structural diversity of the target that can be utilized in designing more efficient drug-discovery strategies. Generating and targeting these conformational intermediates still remains a major challenge in successful drug-discovery campaigns.Computational methods are increasingly used to complement experimental drug-discovery strategies.5,6 Structure-based drug-discovery methods like docking utilize the available three-dimensional structure(s) of the target protein to make predictions for the best binding ligand candidates.7,8 The approach can significantly reduce the number of chemotypes to experimentally synthesize and test, compared to a high-throughput screening approach utilized alone without prior knowledge of the potential interactions between the target and small molecules.9
Traditionally, and given the structural determination challenges described above, only a single experimental (e.g., crystal or cryo-EM) structure or a single computational model (e.g., a homology model) of the target protein has been used for docking purposes, in an approach we refer to as single-point docking10 (Fig. 1). As stated above, these structures generally represent only a static snapshot of one conformational state of the target protein. More recently, the single-point docking approach has been improved to take into account thermal fluctuations of the target protein in an approach referred to as ensemble docking, which has been shown to enhance both the quality and the efficiency of lead prediction11 and successfully applied to a number of protein targets12–15 (Fig. 1). Ensemble docking makes use of molecular dynamics (MD) simulations for sampling the conformational basin of the protein target in the vicinity of a starting structure.16–19 In order to improve the sampling of the local conformational basin, some studies have combined equilibrium MD simulations with enhanced sampling techniques, e.g., metadynamics, to broaden the range of protein conformations that can be then used in the following docking step.20
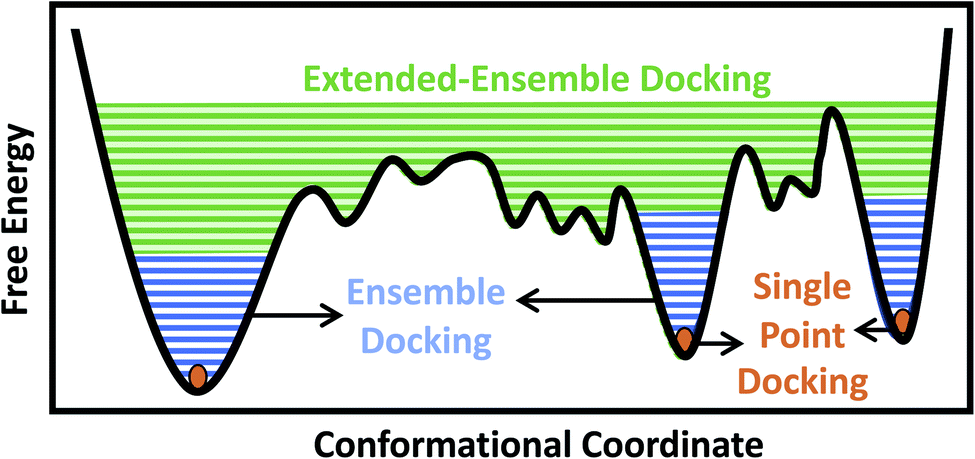 | ||
| Fig. 1 Conformational domains targeted by different docking approaches. Single-point docking utilizes a single structure of the protein target, restricting the sampling to a single point (orange dots) in the conformational landscape. Ensemble docking utilizes an ensemble of protein structures, often generated using MD simulations, taking into account thermal fluctuations within a local conformational basin in the vicinity of the starting experimental structure (blue lines). Extended-ensemble docking, the method introduced here, aims at taking into account the full functional cycle of the protein, generated, e.g., through the application of biasing techniques to transition between the major functional states of the protein (green lines). | ||
In the case of larger and more flexible protein targets though, which often display a complex conformational landscape with multiple local minima,4,21,22 the limited timescales of equilibrium MD simulations, even when combined with enhanced sampling techniques, may not cover the large conformational heterogeneity of the target protein.23 Large-scale motions and state transitions are of high relevance to the function of many macromolecular systems, e.g., the transmembrane transport of substrates by membrane transporters, activation of receptors, and gating of channels.4,24,25 Non-equilibrium methods allow us to expand our sampling of the phase space and to visit intermediate conformations formed during the transition between major functional states of a protein.26,27 This forms the basis of what we term here as the extended-ensemble docking, which uses enhanced sampling offered by non-equilibrium MD for generating an extended ensemble of the protein (in comparison to the more ‘limited’ ensemble of the protein usually generated in ensemble docking), sampling the full conformational transition pathway between functional end states (Fig. 1). The extended ensemble of the protein can then be used to seed high-throughput docking calculations for specific drug-discovery applications.
Here we report the first application of this approach to the investigation of ligand binding in the ABC exporter, P-glycoprotein (Pgp),28 a transporter that is over-expressed in the plasma membrane of cancer cells and is a major cause for the development of multi-drug resistance.29,30 Pgp accomplishes its function as a cellular ‘vacuum cleaner’ by binding of different classes of molecules31,32 to its large central drug binding pocket (DBP), formed between two opposing, pseudosymmetric transmembrane domains (TMDs) (Fig. S1†). As an active transporter, Pgp follows the general ‘alternating-access’ model,33,34 thus transitioning between inward-facing (IF) and outward-facing (OF) states, alternatively exposing the DBP (and any bound substrate) to the cytoplasmic and extracellular sides of the membrane. The large-scale structural transitions of the transporter are fueled by and coupled to ATP-driven dimerization and opening/closing motions of its two nucleotide binding domains (NBDs) connected to the TMDs35–37 (Fig. S1†).
A number of computational studies employing molecular docking,38–40 pharmacophore mapping,41 machine learning,42 and MD-based approaches43 have characterized the promiscuity of the ligand-binding sites in Pgp. However, these studies have generated vastly conflicting results to each other and to earlier mutational studies;44–46 it still remains debatable as to whether diverse molecules bind to distinct or overlapping binding sites in Pgp. A major limitation of in silico approaches to ligand binding is that they often neglect structural heterogeneity inherent to a flexible, multidomain protein such as Pgp by targeting only a single crystal structure/homology model, providing only a snapshot and ignoring the myriad of conformations arising during the function of a protein.
In the present study, we use non-equilibrium, driven MD simulations employing system-specific reaction coordinates describing the alternate-access transitions of ABC transporters, in order to sample the full transition cycle of Pgp between the IF and OF states. Docking of diverse small molecules, including substrates, non-substrates, and modulators, to the generated extended ensemble of the protein reveals that different classes of compounds may bind distinctly to different conformational states/intermediates of Pgp. The captured differential ligand-binding properties of different “subsites” in Pgp, whose accessibility and size are modulated by the protein conformational changes as it undergoes transition between the IF and OF states may be a determining factor in the broad substrate specificity of the transporter. Our results show a close relationship with previous experimental studies and additionally allow us to make suggestions for developing more potent inhibitors of this multidrug resistance transporter.
2 Computational approach
The main distinction of extended-ensemble docking from other docking approaches is to first generate structural models for the intermediate conformational states of the protein that arise during the transition between its structurally known states, e.g., through the application of biased simulation techniques. Then, instead of targeting a single experimental/modeled structure, the entire extended ensemble of the protein's conformations is targeted by high-throughput docking calculations (extended-ensemble docking). Following are the details of the different steps involved in the protocol used to generate the target conformational ensemble and its specific application to Pgp (Fig. 2). We also present a suitable method for clustering the predicted binding modes of compounds to the ensemble of structures, in order to obtain relevant results that are related to the mechanism of the protein.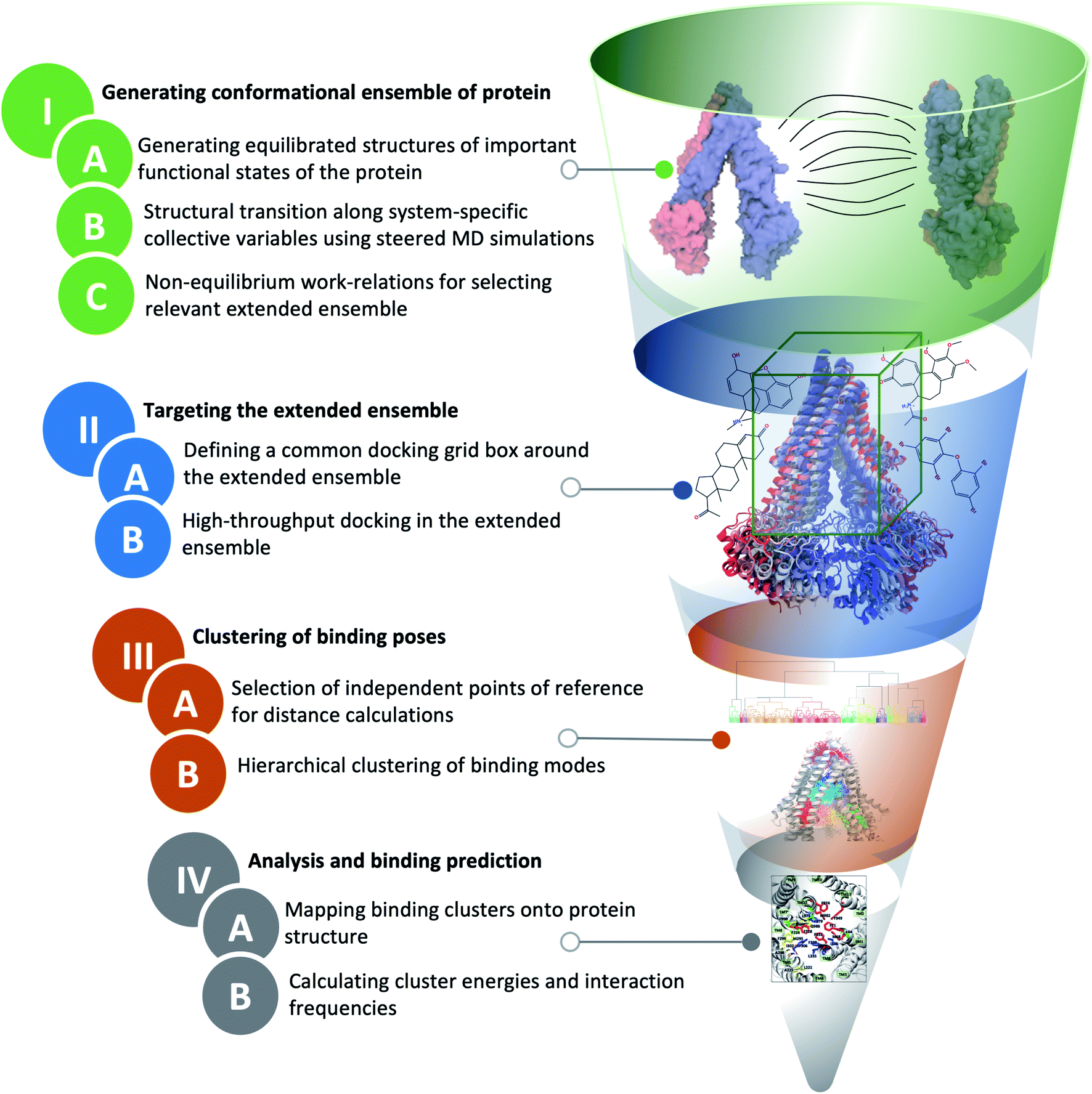 | ||
| Fig. 2 Extended-ensemble docking protocol. A flow diagram showing different steps involved in the extended-ensemble docking approach. The approach involves the targeting of an extended ensemble of the protein conformations, generated along its functional cycle, by docking small molecules, followed by clustering of the predicted binding poses for each representative conformation. See Methods for details of each step. | ||
2.1 General strategy to generate an extended conformational ensemble for the target protein
We start with the available (known/modeled) structures of major functional states (end states) of the protein. In order to obtain relaxed structures, which are required for optimal sampling of the transition pathway between them, one would need to first equilibrate them in a native environment, e.g., solution for globular proteins or a lipid bilayer for membrane proteins.Subsequently, in order to describe conformational transition between major states, we define a set of system-specific collective variables (CVs) that capture the essential conformational changes in the protein during the transition. These CVs need to be closely related to important/relevant structural features of the different states; by applying time-dependent biases along these CVs, for example by using biased methods such as steered MD (SMD),47 we can generate transition pathways connecting the end states.22,48 The amount of non-equilibrium work needed to induce the conformational change during a driven transition along a particular pathway (obtained from multiple simulation replicates) provides an approximate initial metric for the likelihood and relevance of the sampled transition pathway.49,50 At the same time, the RMSD of the resulting structure from the target structure provides a measure for the effectiveness of the prescribed protocol to complete the transition. Low RMSD to the target and low non-equilibrium work values for a pathway/mechanism point towards the efficiency and quality of the applied transition protocol. An extended conformational ensemble of the protein can then be generated by selecting structures spanning the distance (along the CV space) between the two end states, for example, by uniformly selecting representative conformations along the optimal transition pathway.
In the following sections we describe each of these steps for the specific example of Pgp.
As for the OF state, recently a cryo-EM structure of Pgp in this state was published.57 Due to a Q/E mutation at the catalytic site of the structure, it has been suggested to be a ‘dead mutant’, a non-catalytic form of Pgp incapable of substrate transport.58 Other structural studies have instead suggested this structure to represent a post-transport, collapsed/occluded state of the transporter.59 Additionally, studies using DEER spectroscopy have shown that Pgp samples a wider opening (not seen in the cryo-EM structure) on the extracellular side of the TMDs in its OF state during the catalytic cycle.60 Due to these reasons, we have generated our own equilibrated model of the Pgp OF state using careful sequence alignment, homology modeling, and SMD simulations, and tested its stability in multiple MD simulations.60 The distances between different regions of the transporter in this state were found to match well with the DEER spectroscopy results.60
SMD simulations were performed starting from 20 equilibrated ATP/Mg2+-bound IF structures of Pgp that were already embedded in POPC/cholesterol lipid bilayers, solvated and neutralized with Na+ and Cl− ions,55 and targeting the equilibrated model of the OF state,60 using the 4 transition protocols (P1, P2, P3 and P4). This amounted to a total of 80 (20 starting structures × 4 transition protocols) independent SMD runs. All simulations were performed using NAMD62,63 with the CHARMM36 force field representing protein, lipid, and nucleic acids64–66 and the TIP3P model for water.67 The simulations were carried out in an NPT ensemble, with the temperature maintained at 310 K using Langevin dynamics68 with a damping coefficient of γ = 0.5 ps−1, and the pressure maintained at 1 bar using the Nosé–Hoover Langevin piston method.69,70 Periodic boundary conditions were used, and long-range electrostatic forces were calculated using the particle mesh Ewald (PME) method.71 The non-bonded interactions were calculated with switching and cutoff distances of 10 Å and 12 Å, respectively. A 2 fs integration timestep was used for calculating the forces. Eighty independent SMD runs were performed each for 30 ns to bring the starting structure close to the target orientations and distances (Table S1†). Harmonic force constants of 105 kcal mol−1 rad−2 and 2 kcal mol−1 Å−2 were used for the quaternion-based and distance-based CVs, respectively. Subsequently, non-equilibrium work profiles and Cα RMSD with respect to the target OF structure were calculated using in-house TCL scripts in VMD.72
2.2 Targeting the extended ensemble by molecular docking
In extended-ensemble docking, we utilize the conformations and intermediate states generated along the global transition pathway of a protein for docking. The selection of a suitable docking methodology generally depends on the specific goal of the study. In the following section we describe the steps involved in the docking calculations carried out in the extended ensemble of Pgp.In order to compare the results obtained from our extended-ensemble approach with other in silico studies, we used a previous docking study39 conducted on the crystal structure of the IF state of Pgp (PDB: 4M1M31) as a point of reference. From this study, we obtained a set of 14 compounds from different ligand classes for Pgp (substrates, non-substrates and high- and low-affinity modulators). These molecules have been shown to bind to overlapping sites within the TMD by mutational studies.44,45 Docking of compounds, while allowing flexibility around all rotatable bonds, was carried out using Autodock Vina74 in the extended ensemble of pgp along with the starting crystal structure of the IF state (Fig. 3). All structures were superimposed on the starting crystal structure, centered at (0, 0, 0), to discount for the translation and rotation of the protein during the simulation. A common docking grid box was then defined around the extended ensemble covering the TMD apex (putative binding sites) of the protein. This box centered at (0, 0, 40) had dimensions of 80, 54, and 80 Å in the x, y and z directions, respectively (Fig. 3). Docking of compounds was carried out in this grid box in an agnostic manner, i.e., the compounds were allowed to sample the entire binding space within the box without any constraints. A total of nine binding poses were generated for each docked compound in each protein conformation.
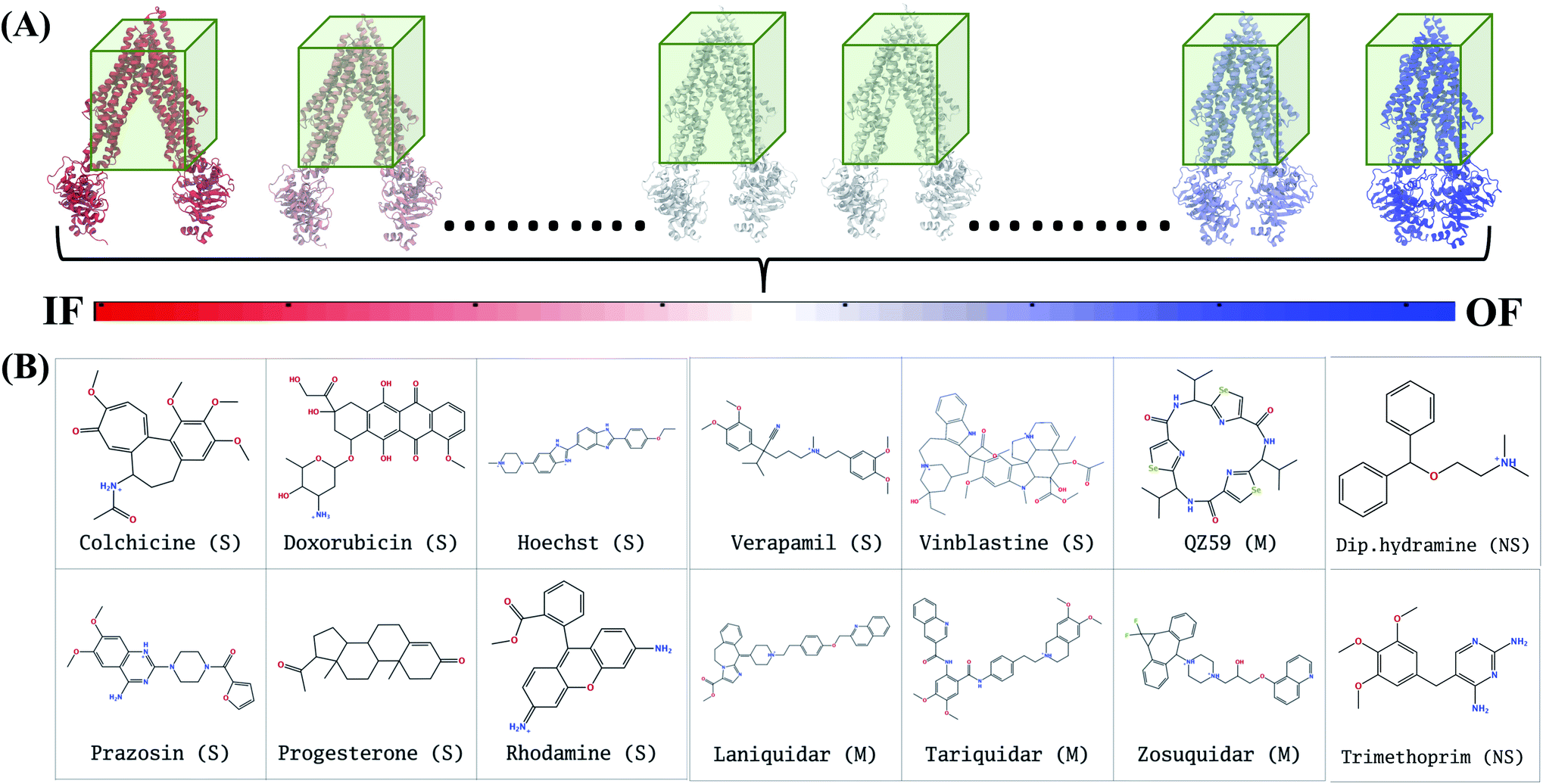 | ||
| Fig. 3 Docking in the extended ensemble. (A) The extended ensemble of Pgp (with color changing from red to blue between states depending on the position in the trajectory), generated by taking 50 snapshots nearly equally distributed along the CV phase-space defining the IF to OF transition. Molecular docking was carried out in the same docking grid box (shown in green) defined around the TMD of the protein in all conformations. (B) The chemical structures of the 14 compounds selected for docking in the extended ensemble of Pgp are shown. These compounds include known substrates (S), modulators (M) and non-substrates (NS) of Pgp. | ||
In Autodock Vina, the docking calculation consists of a number of independent runs each consisting of several sequential steps (the number of steps in a run is determined heuristically, based on the size and flexibility of the ligand). Each independent run is started with a ligand in random conformation, orientation and torsions inside the docking grid box.75 Due to the larger size of the grid box used for the TMD of the protein, we have set the exhaustiveness parameter to a large value of 200. This translates to a total of 200 independent docking runs, each starting from a different random ligand configuration in the grid box, thus providing 25 times more sampling compared to the default sampling rate (default exhaustiveness value of 8), thereby decreasing the possibility of missing relevant poses. In order to check the reproducibility of the results at the selected sampling level, we have also repeated the docking runs 4 times for 4 representative compounds with the employed exhaustiveness value of 200; the results show close similarity indicating that at this exhaustiveness, one can expect a high level of reproducibility.
2.3 Clustering of binding poses
Given the large structural diversity of conformations obtained from the extended ensemble, geometrical clustering of the predicted binding poses can be challenging. To overcome this problem, we first defined a hybrid system of reference, which combines independent points in space with protein-dependent points to assign positions/distances to the bound ligand. The resulting distances were then used in clustering. This clustering strategy can be generalized to other similarly flexible protein targets.For each protein conformation in the extended ensemble, 6 reference points were defined (Fig. S3†): four fixed points at the centers of the four faces of the grid box along the y and z axes (−y, +y, −z, and +z; these points are the same for all protein conformations), and two protein-dependent points used to track the main structural change of the protein along the transition pathway, namely opening and closing of the cytoplasmic mouth (−x, +x, corresponding to the cytoplasmic ends of two of the TMD helices). For the fixed points, we used the docking grid box defined around the TMD region (centered at (0, 0, 40) with dimensions of 80, 54, and 80 Å in the x, y and z directions, respectively); the four fixed points lied at (0, 27, 40), (0, −27, 40), (0, 0, 0) and (0, 0, 80), respectively. The two variable points in ± x were the position of the Cα atoms of the first residues of TM1 (L45) and TM7 (P705) helices, respectively. The six-dimensional distance vector between the center of mass of the docked compound and these reference points were used for the hierarchical clustering method in MATLAB.76
In the first step of clustering, a pairwise dissimilarity matrix was composed (based on the distances defined above) for all the docked poses. This symmetric dissimilarity matrix consisted of 63002 elements (6300 = 50 protein conformations × 14 compounds × 9 binding poses/compound). In the second step, a hierarchical tree was constructed by linking proximal poses (based on the dissimilarity matrix) using the average linkage method.77 In the third step, a distance cutoff was used for pruning the branches of the hierarchical tree, allowing partitioning of all points lying below a specific branch into individual clusters. A cutoff of 15 Å resulted in a suitable partitioning of the hierarchical tree with different pruned branches representing the clusters, representing binding to different sites/regions of the protein.
2.4 Analysis and binding predictions
The binding of different compounds to the extended ensemble of the Pgp was characterized in terms of the predicted binding affinities for the different binding modes of each compound in each cluster, as well as the relative population distribution of these compounds in each cluster. The predicted binding affinities for each binding mode were obtained from the Autodock Vina output. Mapping of these clusters onto the TMD of Pgp allowed identification of the corresponding binding sites in the protein along with the residues with the highest overall contact frequencies. The protein residues interacting with the predicted binding modes were identified using in-house scripts in VMD;72 any protein heavy atom within 4 Å of a ligand heavy atom was considered to form a contact and to contribute to the binding site.3 Results
Here, we describe the results of the extended-ensemble docking approach to the multi-drug transporter Pgp. The results pertinent to the generation of the extended ensemble using SMD simulations are provided in the ESI (Fig. S4–S8†). The reproducibility of the docking results was checked by repeating the docking runs and also provided as the ESI (Fig. S9†). Here, we first describe the results evaluating possible binding sites formed in Pgp as it transitions from the IF to OF state. The results highlight differential binding modes and affinities to the different conformations of Pgp. Furthermore, the calculations allowed us to differentiate between the different classes of compounds based on their binding preference for specific sites in Pgp.3.1 Dynamic variation of binding pockets in Pgp during IF to OF transition
Analysis of the characterized binding pockets allowed us to evaluate their response to the global changes of the protein. Three binding pockets are formed in the membrane encompassing region of the TMDs (Fig. 4). Out of these, the pocket at the TMD apex (where the two TMD leaflets meet; shown in blue in Fig. 4) displays a high ligand binding propensity in all conformations except in snapshot 50 representing the OF state of Pgp (Table S2†). The disappearance of this binding pocket in the OF state can be attributed to its significant shape change in this state, due to the rearrangement of TMD helices (Fig. 4G). The largest size for this pocket (along with the highest ligand-binding propensity) is observed in snapshot 30 (Fig. 4E) which is an IF-like state similar (RMSD of 3.2 Å) to the starting crystal structure (Fig. 4A). The other two binding pockets in the DBP lie within the individual TMD leaflets (shown in red and green in Fig. 4), and show relatively smaller sizes and lower ligand-binding propensities in different protein conformations compared to the apex binding pocket. Additional small binding pockets exhibiting low ligand-binding propensities are also observed to form in the late conformations in the Pgp extended ensemble, either on the cytoplasmic side, where the two TMD leaflets come together and close the cytoplasmic entry to the protein lumen (shown in pink in Fig. 4F) or on the extracellular side, where the two TMD leaflets separate and lead to an extracellular opening (shown in yellow in Fig. 4F and G).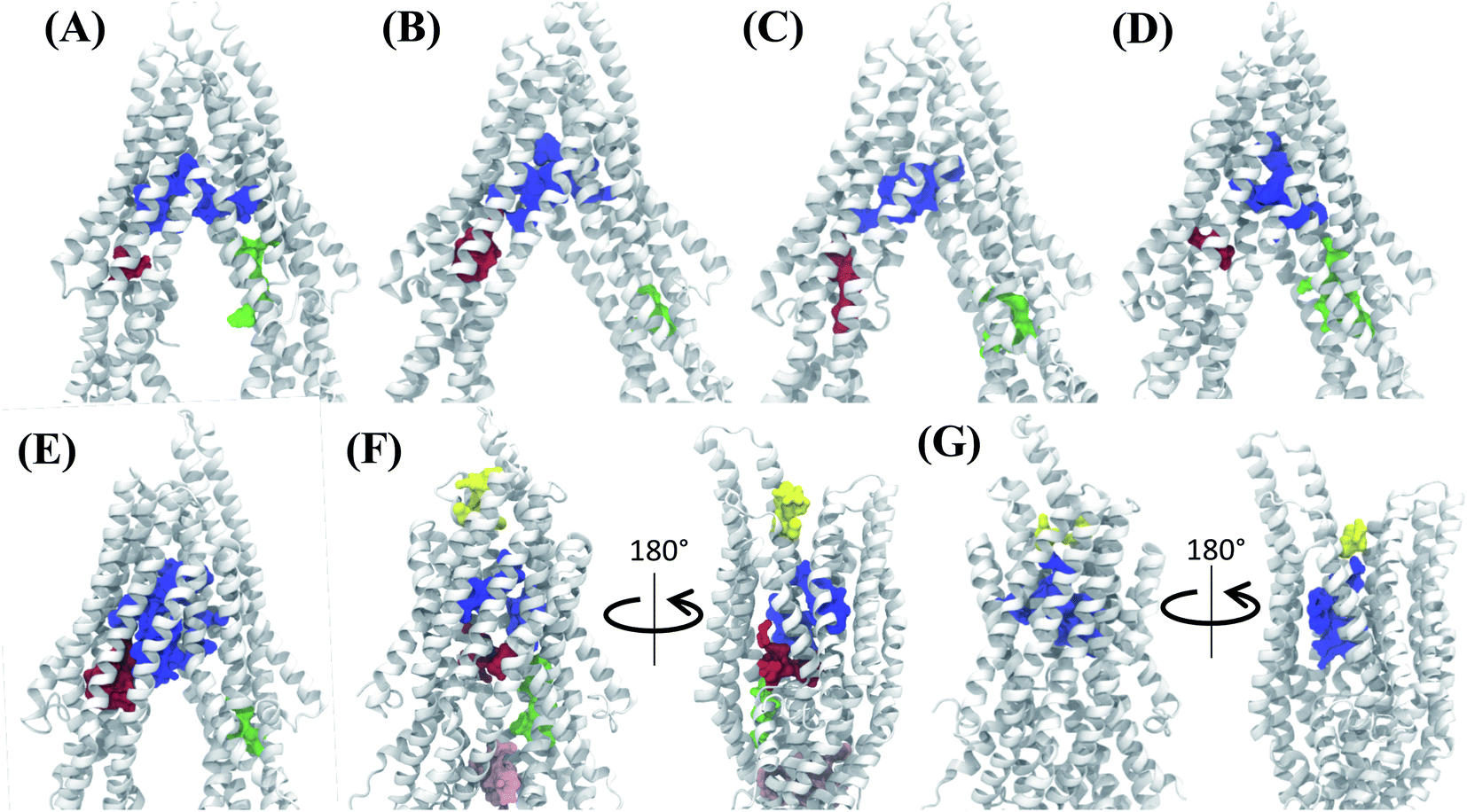 | ||
| Fig. 4 Binding pocket predictions during Pgp IF to OF transition. (A) The active binding pockets predicted for the starting IF crystal structure are shown. The binding pocket residues are shown by space filling representations in different colors. (B–G) Binding pockets predicted for snapshots 1, 10, 20, 30, 40 and 50 of the extended ensemble of Pgp, respectively. The large central binding region (blue) in the apex of the DBP shows the highest ligand binding propensity and is present in all protein conformations. | ||
3.2 Differential binding affinities to different protein conformations
The tested compounds show different binding affinities to different protein conformations (Fig. 5 and S10†). Interestingly, all the compounds show stronger binding affinities (more negative docking scores) in conformations other than the starting IF crystal structure, in some cases reaching binding affinities as much as −4.1 kcal mol−1 better than the crystal structure (laniquidar shown in Fig. S10†). In general, high-affinity Pgp modulators (laniquidar, tariquidar and zosuquidar) show stronger binding affinities than low-affinity modulators (QZ59), large substrates (doxorubicin and vinblastine), and small substrates (hoechst, verapamil, progesterone, prazosin, rhodamine, and colchicine). Consistent with their known properties, non-substrate ligands (diphenhydramine, trimethoprim) show the weakest binding affinities among all the compounds.Overall for the majority of the compounds, we observe an increasing trend in predicted binding affinities (becoming stronger binders) for protein snapshots 30–40 (Fig. 5 and S10†). Pgp transitions from open IF to closed IF-like conformations in these snapshots. Correspondingly, the DBP changes from a ‘flatter’ shape, in which part of the ligand may not form favorable interactions with the protein and instead remain solvent-exposed (on the cytoplasmic side), to a more compact shape, that may allow favorable interaction with TM helices in the DBP. Furthermore, the binding affinities become weaker for most of the compounds in protein conformations 40–50 (Fig. 5 and S10†). Pgp transforms from the IF into an OF-like conformation during this phase of the transition, exhibiting large-scale rearrangements in the DBP, which becomes solvent exposed from the extracellular side, and as a result shows decreased binding affinities.
3.3 Preferential binding to two major sites
The hierarchical clustering of binding modes obtained for the tested compounds allowed us to characterize their preference for different binding regions of Pgp. Using a cutoff distance of 15 Å the resulting hierarchical tree was partitioned into 12 separate clusters (Fig. S11†). The relative populations of the clusters showed that clusters 4 and 5 together constituted majority (∼78%) of all predicted binding modes. Mapping of the clusters onto the protein TMDs showed that clusters 1, 2 and 3 (named E1, E3 and E2 sites, respectively) are within the extracellular side of the TMDs, clusters 4, 5 and 11 (named M1, M2, and M3 sites, respectively) are within the apex of the TMDs, clusters 6 and 7 (named R2 and R1, respectively) represent binding to TMD2, Cluster 8 (named S1) represents binding to membrane-facing surface of TMD2, Cluster 9 (named S2) describes binding to the intracellular end of the TMDs, and clusters 10 and 12 (named H1 and H2, respectively) are related to binding to TMD1. The naming convention for binding sites assigned is further described in Discussion.High-affinity modulators (laniquidar, tariquidar and zosuquidar) and small substrates (hoechst, verapamil, progesterone, prazosin, rhodamine, and colchicine) show high populations at the M1 site at the apex of the DBP, whereas the low-affinity modulator (QZ59) and large substrates (doxorubicin, vinblastine) show preferential binding to M2, which is also located at the apex of DBP but closer to TMD2 (Fig. 6, S12† and Table 1). Overall, M1 and M2 binding sites show relatively stronger binding affinities for all tested compounds. The M3 site, below M1 and towards TMD1, is generally only sparsely populated, except for large substrates and low-affinity modulators, which show moderate populations at this site (Fig. 6, S12† and Table 1). This binding site (M3) only forms during the late transition to the OF state (Fig. 7 and S13†). R1 and R2 sites are below the M2 site in TMD2, whereas H1 and H2 sites are present below the M3 site in TMD1 (Fig. 6 and S12†). Low-affinity modulators and large substrates display moderate binding populations to R1/R2 sites, while small substrates like prazosin show moderate binding to H1 (Table 1). The H2 site, on the other hand, shows marginal binding populations for all compounds. In general, R1 and H1 are seen to form in protein conformations 1–30, while the protein is still in IF-like conformations, whereas R2 and H2 sites are sampled more during the later phase of the transition (conformations 30–50), with the protein in OF-like conformations (Fig. 7 and S13†). Extracellular sites E1 and E2 are found in OF-like conformations (conformations 30–50), where the DBP opens up towards the extracellular side (Fig. 7 and S13†). Most compounds bind with substantial populations to the E2 site, whereas E1 shows only marginal binding (Fig. 6, S12† and Table 1).
| Compounds | M1 | M2 | M3 | H1 | H2 | R1 | R2 | E1 | E2 | E3 | S1 | S2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrates | Colchicine | 42.4 | 22.0 | 1.8 | 0.9 | 0.7 | 2.0 | 3.1 | 0.0 | 15.7 | 0.9 | 8.3 | 2.2 |
| Doxorubicin | 22.3 | 40.0 | 5.1 | 5.3 | 0.9 | 8.3 | 3.6 | 0.0 | 9.8 | 1.8 | 1.8 | 1.1 | |
| Hoechst | 58.5 | 27.1 | 1.1 | 2.2 | 0.0 | 3.8 | 0.9 | 2.4 | 4.0 | 0.0 | 0.0 | 0.0 | |
| Prazosin | 46.0 | 28.5 | 0.4 | 7.4 | 0.2 | 5.3 | 0.5 | 0.5 | 7.6 | 2.5 | 0.9 | 0.2 | |
| Progesterone | 71.3 | 24.0 | 0.0 | 0.2 | 0.2 | 0.2 | 0.0 | 0.2 | 3.4 | 0.0 | 0.5 | 0.0 | |
| Rhodamine | 60.0 | 15.4 | 2.7 | 2.4 | 1.3 | 1.3 | 0.9 | 0.7 | 11.4 | 0.7 | 1.6 | 1.6 | |
| Verapamil | 75.3 | 16.4 | 0.0 | 1.1 | 0.0 | 1.6 | 0.5 | 0.9 | 2.0 | 0.2 | 1.8 | 0.2 | |
| Vinblastine | 10.2 | 51.1 | 8.5 | 1.3 | 1.8 | 5.3 | 14.7 | 0.0 | 4.4 | 0.9 | 1.3 | 0.5 | |
| Modulators | QZ59 | 16.1 | 54.3 | 7.4 | 2.5 | 0.0 | 4.5 | 4.7 | 0.0 | 1.8 | 2.2 | 4.5 | 2.0 |
| Laniquidar | 68.2 | 19.6 | 0.7 | 0.7 | 0.0 | 0.2 | 2.0 | 1.3 | 3.8 | 0.0 | 3.1 | 0.4 | |
| Tariquidar | 56.2 | 27.2 | 0.5 | 1.3 | 2.9 | 4.0 | 1.8 | 1.8 | 3.6 | 0.0 | 0.5 | 0.2 | |
| Zosuquidar | 70.0 | 17.9 | 2.2 | 1.3 | 1.1 | 0.0 | 1.4 | 0.5 | 3.8 | 0.0 | 0.5 | 1.3 | |
| Non-substrates | Diphenhydramine | 80.2 | 15.4 | 0.0 | 0.4 | 0.0 | 0.7 | 0.0 | 3.3 | 0.0 | 0.0 | 0.0 | 0.0 |
| Trimethoprim | 28.5 | 31.0 | 1.1 | 10.2 | 0.0 | 9.1 | 0.0 | 1.1 | 14.6 | 1.4 | 3.0 | 0.0 |
Some of the compounds also show binding to an additional extracellular site, E3, on the extracellular surface of the TMDs. This site is found only during the early phase of the protein transition (snapshots 1–30), i.e., in IF-like states, and hence it may not represent a physiologically important site for the substrates as the DBP in these conformations is inaccessible from the extracellular side.
Other than the above ten binding sites, some compounds show binding to two additional subsidiary sites S1 and S2, with S1 located on the membrane-facing surface of TMD2 between the elbow helix and the broken (unstructured) region of TM12 (Fig. 6, S12† and Table 1), and S2 at the bottom of the TMDs between TM4 and TM6. S2 is found only in the late protein conformations (snapshots 40–50), i.e., in OF-like states, and, similar to E3, may not represent a physiologically important site as the DBP may become inaccessible in these conformation due to the closing of the TMDs on the intracellular side during the IF-OF transition.
3.4 Important binding residues in major sites
The contacts formed by the protein residues with each compound were further analyzed and quantified as interaction frequencies. Comparatively, high-affinity modulators (laniquidar, tariquidar and zosuquidar) and some substrates (progesterone and verapamil) show higher interaction frequencies with the same set of residues as the low-affinity modulator (QZ59) and both large (doxorubicin and vinblastine) and small substrates (colchicine, hoechst, prazosin, and rhodamine) (Fig. 8 and S14†). The non-substrate trimethoprim shows the lowest interaction frequency among all compounds, relating to its low selectivity for Pgp. Overall, most compounds form highly favorable interactions with residues in TM1, TM5, TM6, TM7, TM11, and TM12, which come together to form the DBP, with the exception of large substrates and the low-affinity modulator that instead display higher interactions with TM4 residues.Based on the most frequently (top 10) interacting residues, all compounds display binding preference for a set of core aromatic/hydrophobic residues present at the apex of the DBP: Y306, L335, I336, F339 and F979 (Fig. 9 and Table 2). High-affinity modulators and small substrates additionally show binding preference for a set of majorly aromatic residues, also lying close to the apex of the DBP: M68, F71, F332, F728, Y949, F974 and M982. The low-affinity modulator and large substrates, on the other hand, show preference for a different set of mostly hydrophobic residues of the DBP present in the TMD2: I221, I225, I295, I298, I299, M986 and M990. As high-affinity modulators/smaller substrates and low-affinity modulator/large substrates do not share any binding residues other than the core ones, these compounds likely bind to distinct but overlapping sites in Pgp. The predicted binding residues for the docked compounds were further compared with the available experimental studies for these compounds in Pgp, including binding and transport mutagenesis studies, cysteine-scanning mutagenesis, photoaffinity labeling and X-ray crystallography and cryoEM structures. The data have been presented in Table 2.
| Compound | Top 10 binding residues | Top 10–20 binding residues |
|---|---|---|
| a No data available. b Mutagenesis. c Cysteine-scanning mutagenesis. d Phootoaffinity labeling. e X-ray crystal structure. f CryoEM structure. | ||
| Colchicine | L64,99b M68, F71, F332, L335,95c I336, F728, F974, F979, M982 (ref. 115)b | F299, I302, Y306, F339, F724,100b M945, Y949, L971,95,101b,c A983,115b Q986 |
| Doxorubicin | L221, F299, I302, Y306, L335, I336, F339, F979,106d Q986,106d F990 (ref. 106)d | M68, M295, F332, A338,105b G342,105b Q721, F724, M982,106d A983,106d V987 (ref. 106)d |
| Hoechst | F71, I302, Y306, F332, L335, F339,100b F728, Y949, F974, F979 | M68, S218, L221, I336, A338, G342, F724,100b F953, L971, M982 |
| Prazosin | M68, Y306,116d F332,101,116bd L335,116d I336,116d F339,101,116bd F724, Y949, F974, F979 | L64, F71, F299,116d I302,116d Y303,116d Q721, F728, M982,101b A983, V987 |
| Progesterone | M68, F71, Y306, F332, L335, I336, F339, F728, F974, F979 | F299, I302, Y303, Q721, F724, Y949, L971, M982, A983, V987 (ref. 98)c |
| Rhodamine | M68, F71, F332,115b I336,117b F728, Y949,118b L971,117b F974, F979,115b M982 (ref. 115)b | L64,117b M67, Y306, L335,107b F339,107b F724,100b M945, S975, V978,117b Q986 |
| Verapamil | M68, F71, Y306, F332,101b L335,96b I336, F728, F974, F979, M982 (ref. 101)b | L64,99b M67, I302,97b F339,101b Q721, F724,100b M945, Y949, L971,95,101bc A983 |
| Vinblastine | L221, A225, M295, A298, F299, I302,97,107b Y306, F339,101b Q986, F990 | Y303, L335,95c G342, Q343, P346, Q721, F979, M982,101b A983, V987 |
| QZ59 | L221, M295,31e A298, F299,31e I302,31e Y306,31e L335,31e F339,31e F979,31e Q986 (ref. 31)e | G222, A225, I336,31e A338, G342, Q343, M982,31e A983,31e V987,31e F990 |
| Laniquidara | M68, F71, Y306, F332, L335, I336, F728, Y949, F974, F979 | L64, M67, F299, I302, F339, F724, M945, M982, A983, Q986 |
| Tariquidar | M68,90f Y306,90f F332,90f L335,90f I336,90f F339,90f F728,90f F974,90f F979,90f M982 (ref. 90)f | L64,90f F71,90f F299,90f I302,90f F724,90f M945,90f Y949,90f L971,90f A983, Q986 (ref. 90)f |
| Zosuquidar | M68, F71, F332,90f L335,90f I336,90f F728, Y949,90f F974, F979,90f M982 (ref. 90)f | L64,90f I302,90f Y306,90f F339,90f F724, M945,90f L971, S975, V978, Q986 (ref. 90)f |
| Diphenhydramine | M68, F71, F332, F724, F728, Y949, F953, L971, F974, F979 | M67, Y303, Y306, L335, I336, F339, A952, V970, S975, M982 |
| Trimethoprim | F299, I302, Y303, Y306, F339, Q721, Q834, F979, A983, V987 | M68, F332, L335, I336, N717, G718, L720, F724, F766, Q986 |
3.5 Differential binding of substrates, modulators and non-substrates
Ranking of the compounds based on their predicted binding affinities to the highly populated M1 site shows that high-affinity modulators bind with the strongest affinities to the extended ensemble, whereas non-substrates display the weakest binding affinities (Fig. 10A). The substrates and the low-affinity modulator lie between these two extremes. High-affinity modulators and most substrates also display high binding populations in the M1 site, ranging between 40 and 80% for the different compounds (Fig. 10B). In contrast, the low-affinity modulator and large substrates show low binding populations in the M1 site, and instead display higher binding populations in the second major binding site of the protein, the M2 site (Table 1).4 Discussion
We have put forth a protocol (Fig. 2), termed extended-ensemble docking, that can be used to characterize the binding of small molecules to proteins undergoing large-scale conformational changes during their function. Employing the technique to the case of the multidrug resistance protein, Pgp, here we make use of a specifically designed SMD protocol utilizing system-specific CVs to describe the structural transitions of the protein and to generate an extended ensemble to be used in docking. Multiple runs with different CV protocols allowed us to identify a low-work transition pathway, likely corresponding to the most mechanistically relevant transition pathway connecting the two major functional states of Pgp (IF and OF). An extended ensemble of the protein was then generated along this transition pathway followed by a series of docking and clustering calculations, aiming at further characterization of the different binding sites in Pgp as they evolve during the transport cycle of the transporter.The global conformational changes during the transport cycle of Pgp modulate the size and shape as well as the ligand-binding propensities of the binding pockets (Fig. 4). Radioligand binding assays characterizing the binding of Pgp substrates as well as a study showing the conformational sensitivity of drug-binding affinity and reactivity to an antibody have shown that binding sites in Pgp can display differential affinities for substrates depending on the conformation of the transporter that might arise during its catalytic cycle.78,79 In the present study, these differences are reflected in the large variations observed in the predicted ligand binding affinities to the protein's extended ensemble (Fig. 5). All compounds display stronger binding affinities to conformations other than its crystal structure, suggesting that experimentally determined protein structures may not necessarily represent the best interaction between the transporter and its substrates.
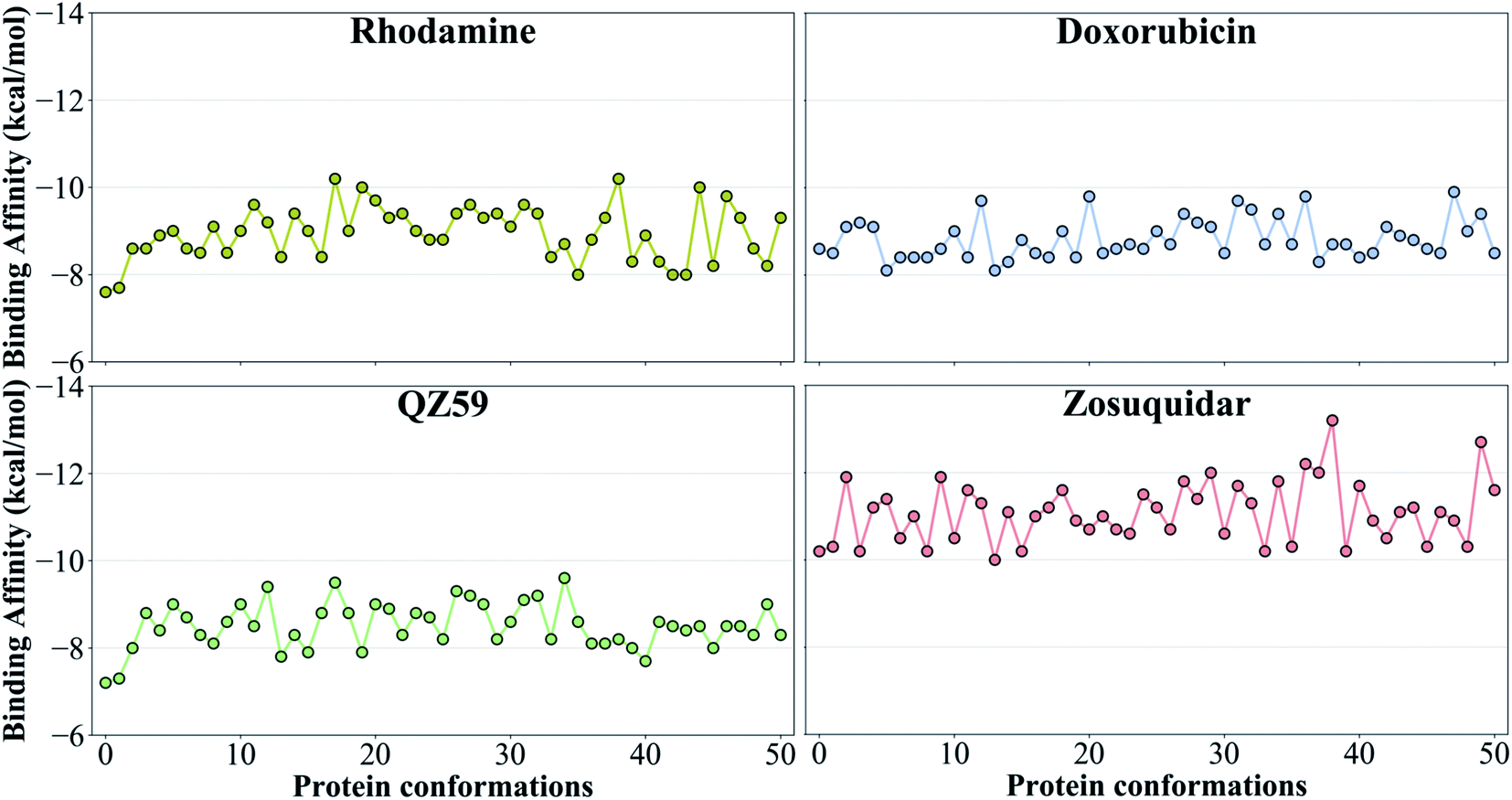 | ||
| Fig. 5 Binding affinities to the extended ensemble. The highest predicted binding affinities for 4 representative compounds (small substrate: rhodamine; large substrate: doxorubicin; low-affinity modulator: QZ59; high-affinity modulator: zosuquidar) to each conformation in the extended ensemble of Pgp are shown. Data for the other compounds are presented in Fig. S10.† Conformation 0 is the starting IF crystal structure, and the selected conformations along the IF to OF transition pathway are numbered 1 to 50. The predicted binding affinities fluctuate between different protein conformations, with the high-affinity modulator showing the highest binding affinities among all the compounds. | ||
Previous drug–drug interaction studies utilizing fluorescence spectroscopy,80,81 and kinetic measurements of substrate binding and transport82,83 as well as competitive binding assays84,85 have postulated at least 3 binding sites within the TMDs of Pgp. These sites have been termed as H and R sites80,81 showing preference for substrates hoechst and rhodamine, respectively, and the modulatory M site showing preference for vinblastine45,84 and verapamil,82,83 two compounds that have been described to function as both modulators and substrates of Pgp.86–89 A previous single-point docking study targeting the crystal structure of Pgp in the IF state assigned the above sites to different regions in the TMDs but showed that the docked compounds do not display binding preference for any specific site (i.e., the compounds showed similar binding affinities to all 3 sites).39 Here, in contrast, we show that all compounds show binding preference for only one major substrate/modulator binding site, analogous to the M site, encompassing the apex of the DBP, whereas the sites in the lower half of the DBP and in individual TMD leaflets, analogous to the H and R sites described above, show low binding populations and comparatively weaker binding affinities for all tested compounds. The H and R sites may thus only function as poly-specific interaction sites in Pgp. We also report an interaction site on the membrane-facing surface of the TMD2, named the S site, as well as additional sites formed on the extracellular side of TMDs which form as the transporter undergoes transition to the OF state (E sites) and may assist in the extrusion of molecules out of the transporter.
Based on mapping of the clustering results onto the protein structure, the M site is found to consist of 3 subsites, which we term M1, M2 and M3. The M1 subsite is positioned in a central location at the apex of the TMDs, allowing interactions with residues from multiple helices from both TMD leaflets. The M1 subsite may represent the central binding site for substrate molecules (Fig. 6). High-affinity modulators are also predicted to bind to M1 but with relatively stronger binding affinities and higher interaction frequencies than the substrates, pointing to a more selective binding mode for these compounds (Fig. 7 and 8). The top binding residues predicted for these compounds show excellent correlation with the recent cryo-EM structures of Pgp in the complex with these compounds90 (Table 2). These modulators, showing IC50 values in the nanomolar range,91–93 can also function as strong third-generation inhibitors of Pgp and may directly compete with binding of low-affinity substrates. Alternatively, as suggested by a recent MD study, the stable binding of a third-generation inhibitor (tariquidar in this case) inside the DBP may promote increased lipid diffusion inside the lumen, locking Pgp in a catalytically inactive open IF-like state.94 This may further enhance the inhibitory function of these molecules.
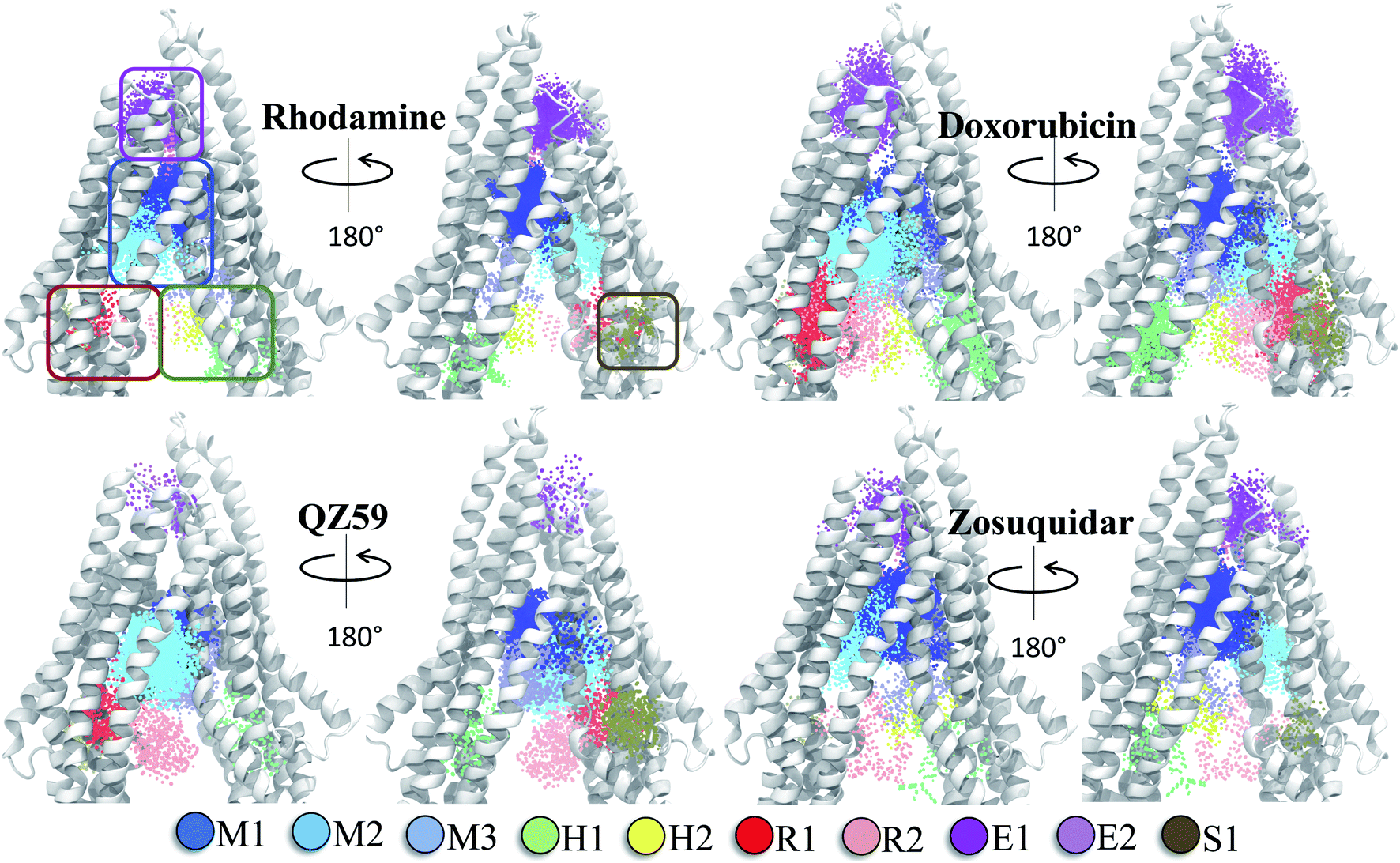 | ||
| Fig. 6 Clustering of binding modes generated by docking. Clustering of the binding modes for 4 representative compounds to the extended ensemble of Pgp is shown (the results for other compounds are shown in Fig. S12†). Only one representative, IF-like conformation of the protein is shown here for clarity. The main binding sites in the TMDs are marked by colored rectangles (different shades of blue: modulator or M site; green and yellow: hoechst-binding or H site; red and salmon: rhodamine-binding or R site; purple: extracellular or E site; brown: subsidiary or S site) shown for the first representative compound (rhodamine). Binding clusters (or binding subsites) within the main binding regions are highlighted with colored points (as indicated in the legend at the bottom), representing the heavy atoms of the clustered binding modes (E3 and S2 sites are not shown as they may not represent sites important for substrate binding/transport). The density of points in each cluster represents the cluster population. M1 and M2 subsites at the apex of the TMDs show the highest cluster populations in all compounds. | ||
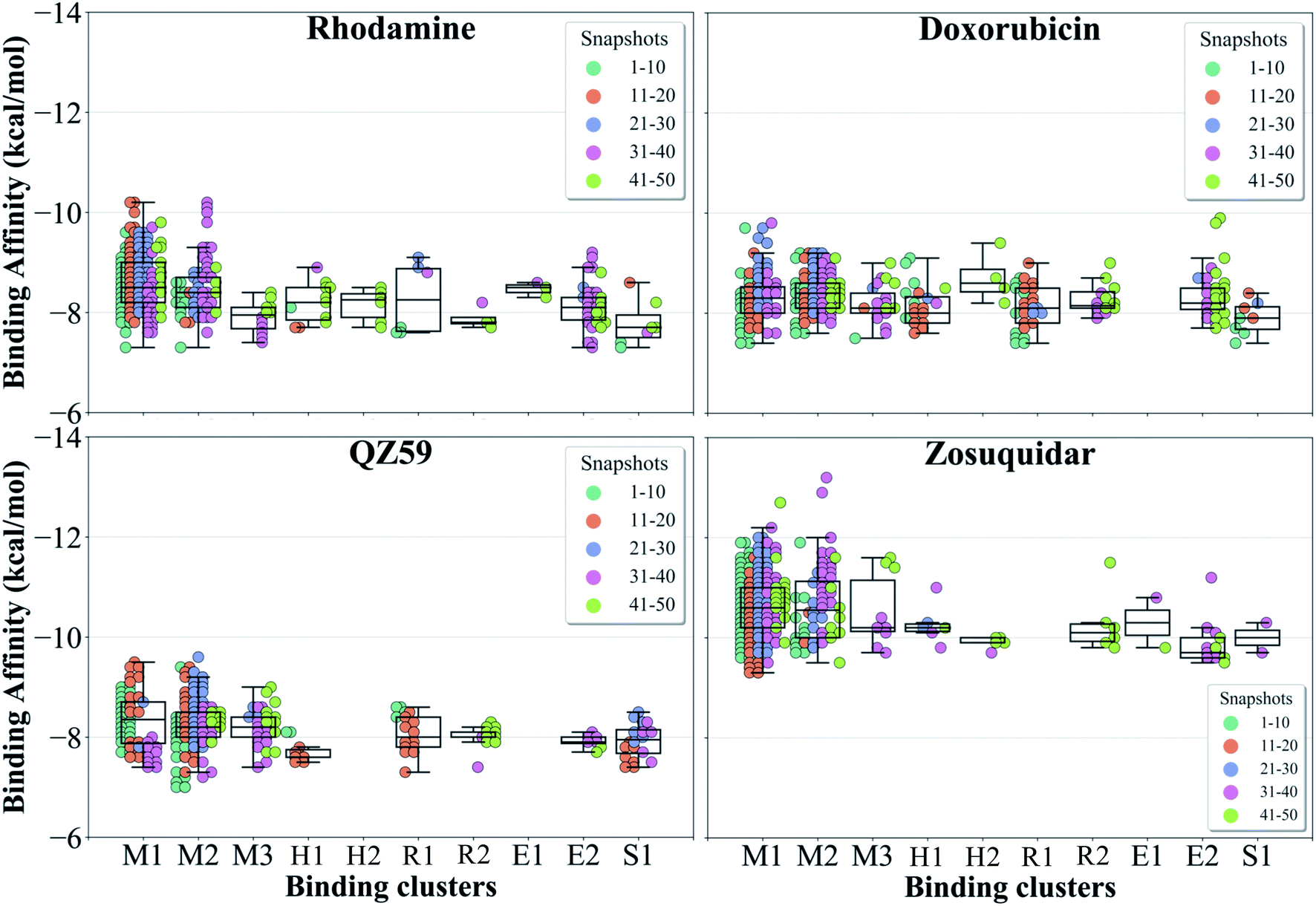 | ||
| Fig. 7 Binding cluster energies. The predicted binding affinities in each cluster are shown as a swarm plot for 4 representative compounds (data for the other compounds are shown in Fig. S13†). 1–50 represent protein conformations arising during the IF-OF transition (shown with different colors defined in the legend). Additionally, a boxplot providing the median cluster values, Q1 and Q3 quartiles, as well as minimum (Q1 − 1.5× interquartile range) and maximum (Q3 + 1.5× interquartile range) binding affinity values, is overlaid on top of the swarm plot for each cluster. M1 and M2 binding clusters show the highest populations and display the strongest binding affinities for all compounds. | ||
 | ||
| Fig. 8 Frequency of Pgp residues interacting with ligands. The normalized interaction frequencies of Pgp's binding residues for all binding modes predicted of 4 representative compounds are shown (data for the other compounds are shown in Fig. S14†). The residues are considered to interact with the ligand if their heavy atoms are within 4 Å. The orange arrows point to the regions of the protein showing differences in their interaction patterns for different classes of compounds. High-affinity modulators like zosuquidar display the highest interaction frequencies with the binding residues pointing to a more specific mode of their binding. | ||
Substrates verapamil and progesterone bind to the M1 subsite with high interaction frequencies comparable to high-affinity modulators, but with relatively weaker binding affinities. Binding residues predicted in our study, especially for verapamil, have been shown to be important for binding in several mutagenesis studies95–101 (Table 2). These molecules show IC50 values in the micromolar range and were previously characterized as first-generation inhibitors of Pgp.102 They require high concentrations to fully inhibit the transporter, to the degree that they start to display toxicity and thus not suitable for administration.103 A recently published cryo-EM structure shows binding of a substrate molecule taxol to the M1 subsite,104 whereas another structure shows two zosuquidar molecules bound to the M site of Pgp, with the first molecule occupying the M1 subsite and the second one occupying the M2/M3 subsites.59 These structural studies provide further evidence that the M1 subsite described in our study may represent the central substrate/modulator binding site in Pgp.
Larger low-affinity modulator QZ59 shows preference for the M2 subsite, with some binding in the M1 and M3 subsites. The crystal structure of Pgp with this modulator shows two copies of the ligand bound, with the first copy located in a site similar to the M2 subsite described in our study (partially within the TMD2 leaflet of Pgp), and the second copy located in the M1 subsite.31 As expected, the top binding residues predicted in our study show good correlation with the above crystal structure binding site (Table 2). In multiple crystal structures of Pgp in the complex with structural analogs of QZ59, simultaneous binding of two ligands has also been observed to the M2/M1 or M3/M1 subsites.53
Larger substrates like doxorubicin and vinblastine also show a similar binding behavior to the low-affinity modulator studied here, with a preferential binding for the M2 subsite, though some binding was also observed for the M1 and M3 subsites. The binding residues predicted for these molecules are further supported by several mutagenesis and photoaffinity experiments95,97,101,105–107 (Table 2). Similar to the QZ59 analogs, it is possible that two (or more) substrates more stably bind together to the multiple M subsites.
Mapping of the clustering results on the protein structure further showed that the H and R sites identified in the separate TMD leaflets (showing much lower binding populations compared to the M site), each consisted of two subsites, H1 and H2, and R1 and R2, respectively (Fig. 6). The H1 and R1 subsites are found in open IF-like conformations and may be involved in initial interactions with the molecules as they partition from the membrane through the two proposed drug-entry portals located on either sides of Pgp.51 An increase in the local concentration of molecules at these interaction sites may facilitate their binding to the central binding site (M1). The subsidiary S site (consisting of the S1 subsite) on the membrane-facing side of TMD2 may similarly facilitate formation of encounter complexes of substrates with the transporter, and it may function as an entry site for these molecules into Pgp. A similar interaction site has also been reported in the crystal structure of Pgp in complex with QZ-Valine,53 a derivative of QZ59, in MD simulations of Pgp in cholesterol-rich lipid bilayers,108 as well as in site-specific spin labeling studies of MsbA (a bacterial homolog of Pgp).109
The extracellular E site, consisting of E1 and E2 subsites, represents novel binding sites identified in our study that partially overlap with the M1 subsite in the DBP and are formed as the TMDs open to the extracellular side during the formation of the OF state. These sites may play a role in assisting the diffusion of the substrate out of the transporter, as the DBP becomes exposed to the extracellular environment in the OF state.
Experiments using photoaffinity labeling of Pgp substrates as well as crystal structures have suggested the presence of high- and low-affinity binding sites in Pgp.53,110,111 Potential of mean force calculations using umbrella sampling also identified multiple overlapping binding locations for different substrates in Pgp.112,113 The results of our extended-ensemble docking provide further support for this proposal, where the M1 and M2 subsites serve as the main binding sites for the high-affinity modulators/small substrates and large, low-affinity modulators/substrates, respectively (Fig. 11A). These subsites form overlapping sites within the DBP of Pgp, with all modulators and substrates showing preference for the same set of core residues (Fig. 9). The low-affinity binding sites proposed in the above studies may consist of the H, R and S sites located below the M site in the individual TMDs of Pgp. Correspondingly, the predicted binding affinities display an increasing trend between these low- and high-affinity binding sites (Fig. 11B), likely promoting the diffusion of molecules towards the central binding site (M1) and their eventual extrusion through the extracellular sites.
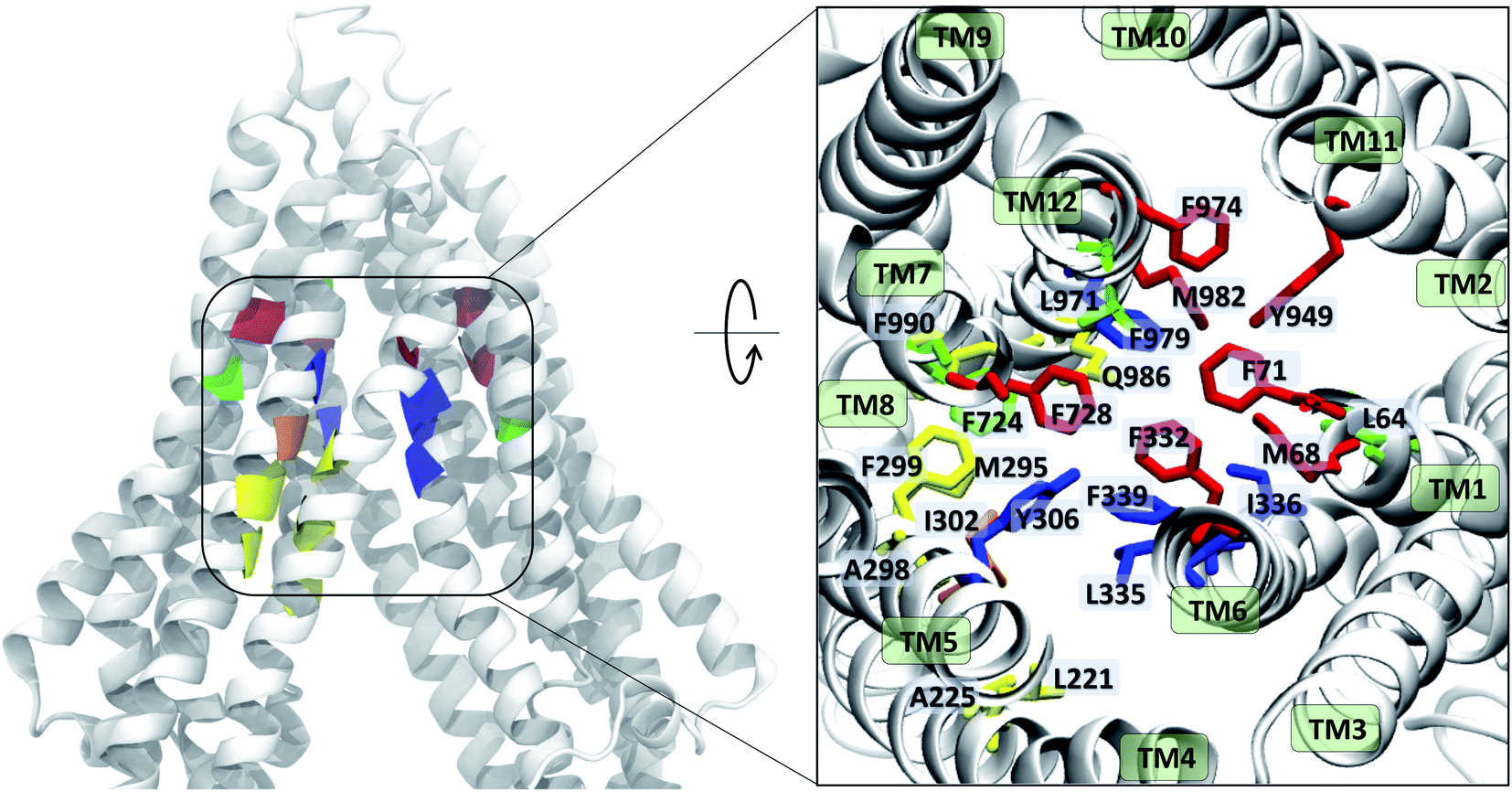 | ||
| Fig. 9 Ligand binding residues identified in Pgp. Binding residues in Pgp showing the highest (top 10) interaction frequencies (from Fig. 8 and S14†) with the tested compounds are shown in color within a cartoon representation of the protein backbone (left) and in stick representation (inset, right). The TM helices are individually labeled in the inset. The binding residues common to all compounds are shown in blue, residues common to binding of small substrates/high-affinity modulators in red, residues common to low-affinity modulators/large substrates in yellow, those common to small substrates/low-affinity modulators in orange, and residues showing preference for only small substrates are shown in green. High-affinity modulators share all binding residues with small substrates and show binding preference for the M1 subsite, whereas low-affinity modulators and large substrates show binding preference for residues forming the M2 subsite, lying below the M1 subsite and partially overlapping with it. | ||
The differential binding behavior of the compounds to the above subsites in the extended ensemble of the protein further allowed us to differentiate between different classes of compounds (Fig. 10). The availability of different binding sites, modulated by the conformational changes in the transporter, as well as modulation of their binding propensities may form the basis of poly-specificity of Pgp. Furthermore, information on molecules showing stronger binding affinity (specificity) to the protein extended ensemble instead of a single structure may provide a more complete framework for the development of specific, fourth-generation inhibitors of Pgp.
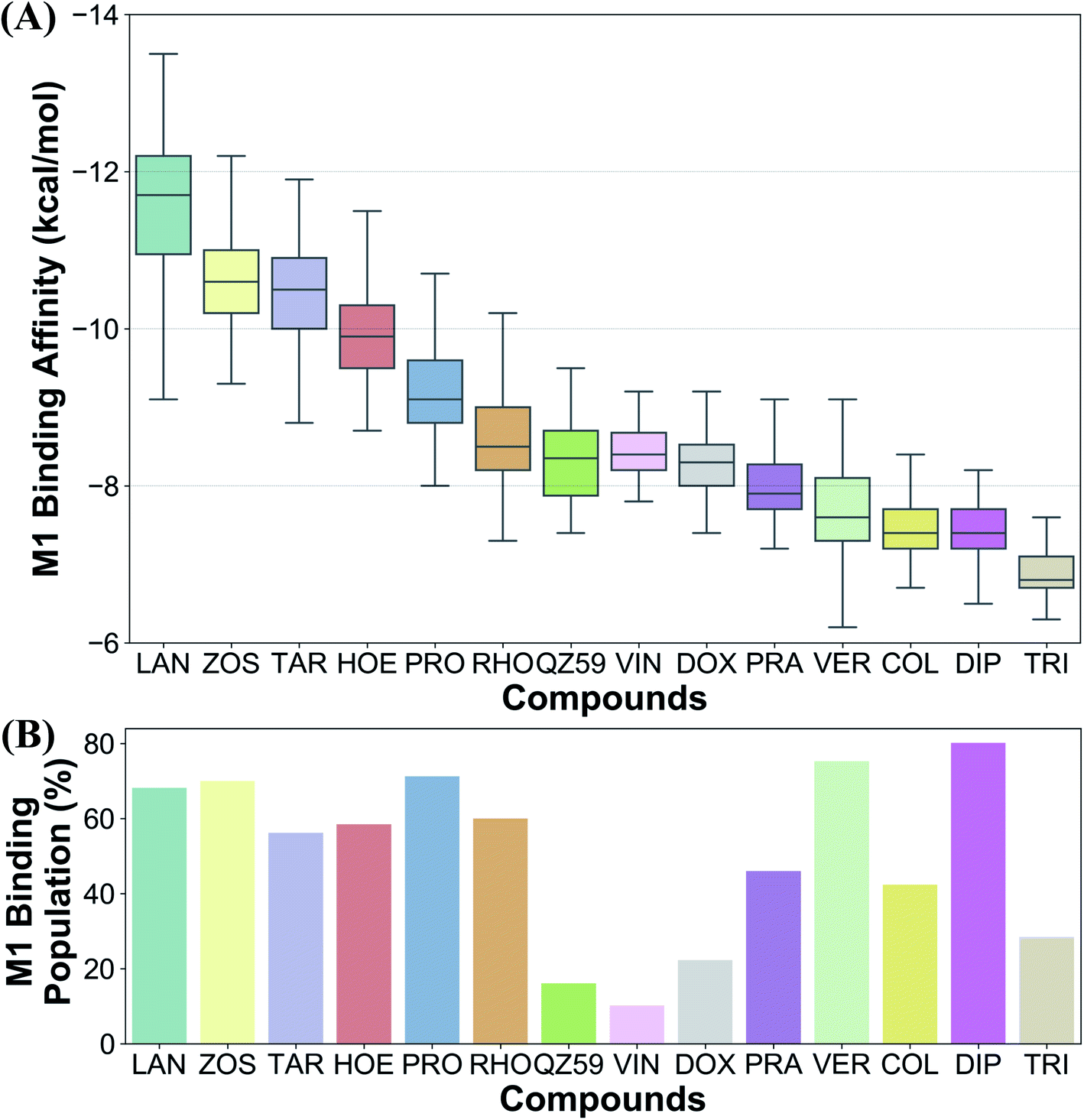 | ||
| Fig. 10 Differentiating between different classes of ligands. The binding site preference of different compounds was evaluated in terms of (A) the predicted binding affinities calculated for the extended ensemble in the M1 subsite, and (B) the respective populations in the M1 site. Ranking the compounds based on their binding affinities places the high-affinity modulators and the non-substrates at the two extremes, with low-affinity modulators and substrates lying between them. Comparison of the binding site population in the M1 subsite further distinguished the high-affinity modulators and small substrates (showing high populations in the M1 subsite) from low-affinity modulators and large substrates (showing higher populations in the M2 subsite instead). | ||
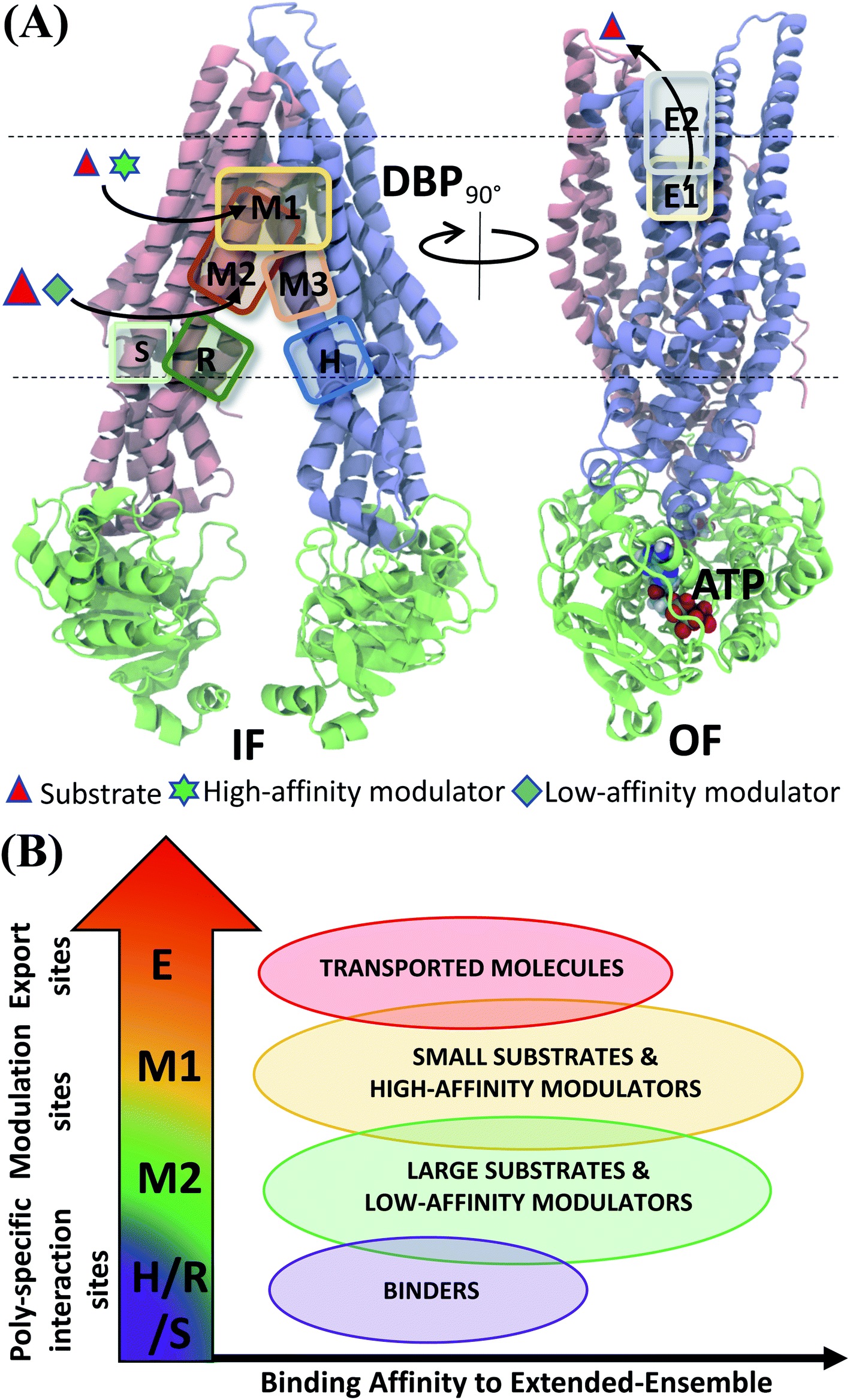 | ||
| Fig. 11 Classification of ligand binding sites in Pgp identified by extended ensemble docking. (A) Different binding sites are shown in representative IF (left) and OF (right) conformations. The two TMD leaflets are shown in blue (TMD1) and pink (TMD2), respectively, and the NBDs are shown in green. The major substrate-modulator binding site (M1), as well as the low-affinity modulator/large substrate binding site (M2) are observed throughout the conformational transition of Pgp, whereas extracellular sites (E1 and E2) are only observed in the OF-like states. (B) Combining the predicted binding affinities of all compounds in the different binding sites obtained in the extended ensemble (Fig. 7 and S13†), we observe differences in the relative binding affinities of the poly-specific interaction sites (H/R/S), modulation sites (M1/M2) and extracellular sites (E). These differences may facilitate the transport of the molecules from the inside to the outside of the cell. | ||
5 Conclusion
Large flexible proteins like Pgp have proven to be challenging drug targets, as a number of recent ventures in developing new drug molecules targeting it have failed. Further optimization strategies using crystal or cryo-EM structures can be technically difficult, as these proteins fluctuate between different conformational states, concurrently leading to changes in the available/characterized binding sites. This challenge also represents a major roadblock for developing accurate structural–activity relationship models for conformationally heterogeneous proteins. Future endeavours in structure-based drug discovery targeting these proteins mandates new in silico strategies, taking into account the inherent flexibility and crosstalk between the different domains of the protein as it undergoes transitions between major functional states. We have described here a new docking protocol pertaining to these systems, allowing high-throughput virtual screening of an extended ensemble derived along the protein's transition pathway, instead of targeting a single crystal/cryo-EM structure. This has allowed us to provide novel insights into the differential binding behaviors of different classes of compounds in Pgp. Future applications to other flexible protein systems may open a new frontier in in silico drug design targeting these important drug targets.In addition to the use of SMD simulations applied in the present study, use of other enhanced sampling methods, for example, accelerated MD, has been suggested for improving the sampling of the protein phase space that can then be used for structure-based drug discovery.114 Furthermore, free energy calculations incorporating Boltzmann reweighting of the conformations obtained from the biasing technique will provide additional enhancements to the approach outlined in this study. Post-processing the docking results based on the free energies of the different conformations constituting the extended-ensemble of the protein should take this approach even closer to the measured ligand binding properties of the protein.
Data availability
The full data set used in docking, including the extended-ensemble of Pgp and the ligands used, is made available in Open Science Framework (OSF), https://osf.io/v7y8e/?view_only=0f00e9fe832b4a1ea35e40e39fb9e28c.Author contributions
KK and ET designed the research. KK and ST carried out all simulations. KK analyzed the data and wrote the manuscript draft. KK and ET edited and revised the manuscript.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
The authors acknowledge support by the National Institutes of Health under award numbers P41-GM104601 and R01-GM123455 (to E. T.). We also acknowledge computing resources provided by Blue Waters at National Center for Supercomputing Applications (NCSA).References
- M. Schenone, V. Dančík, B. K. Wagner and P. A. Clemons, Target identification and mechanism of action in chemical biology and drug discovery, Nat. Chem. Biol., 2013, 9, 232 CrossRef CAS PubMed.
- J. R. Helliwell, New developments in crystallography: exploring its technology, methods and scope in the molecular biosciences, Biosci. Rep., 2017, 37(4) DOI:10.1042/BSR20170204.
- E. Nogales, The development of cryo-EM into a mainstream structural biology technique, Nat. Methods, 2015, 13, 24 CrossRef PubMed.
- G. Wei, W. Xi, R. Nussinov and B. Ma, Protein ensembles: how does nature harness thermodynamic fluctuations for life? The diverse functional roles of conformational ensembles in the cell, Chem. Rev., 2016, 116, 6516–6551 CrossRef CAS PubMed.
- S.-S. Ou-Yang, J.-Y. Lu, X.-Q. Kong, Z.-J. Liang, C. Luo and H. Jiang, Computational drug discovery, Acta Pharmacol. Sin., 2012, 33, 1131–1140 CrossRef CAS PubMed.
- R. R. Ramsay, M. R. Popovic-Nikolic, K. Nikolic, E. Uliassi and M. L. Bolognesi, A perspective on multi-target drug discovery and design for complex diseases, Clin. Transl. Med., 2018, 7, 3 Search PubMed.
- A. C. Anderson, The process of structure-based drug design, Chem. Biol., 2003, 10, 787–797 CrossRef CAS PubMed.
- M. Batool, B. Ahmad and S. Choi, A structure-based drug discovery paradigm, Int. J. Mol. Sci., 2019, 20, 2783 CrossRef CAS PubMed.
- J. Antel, Integration of combinatorial chemistry and structure-based drug design, Curr. Opin. Drug Discov. Dev, 1999, 2, 224–233 CAS.
- R. L. Van Montfort and P. Workman, Structure-based drug design: aiming for a perfect fit, Essays Biochem., 2017, 61, 431–437 CrossRef PubMed.
- S. R. Ellingson, Y. Miao, J. Baudry and J. C. Smith, Multi-Conformer Ensemble Docking to Difficult Protein Targets, J. Phys. Chem. B, 2015, 119, 1026–1034 CrossRef CAS PubMed , PMID: 25198248..
- L. S. Cheng, R. E. Amaro, D. Xu, W. W. Li, P. W. Arzberger and J. A. McCammon, Ensemble-Based Virtual Screening Reveals Potential Novel Antiviral Compounds for Avian Influenza Neuraminidase, J. Med. Chem., 2008, 51, 3878–3894 CrossRef CAS PubMed.
- R. E. Amaro, A. Schnaufer, H. Interthal, W. Hol, K. D. Stuart and J. A. McCammon, Discovery of drug-like inhibitors of an essential RNA-editing ligase in Trypanosoma brucei, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 17278–17283 CrossRef CAS PubMed.
- K. Kapoor, N. McGill, C. B. Peterson, H. V. Meyers, M. N. Blackburn and J. Baudry, Discovery of novel nonactive site inhibitors of the prothrombinase enzyme complex, J. Chem. Inf. Model., 2016, 56, 535–547 CrossRef CAS PubMed.
- M. Pi, K. Kapoor, R. Ye, D.-J. Hwang, D. D. Miller, J. C. Smith, J. Baudry and L. D. Quarles, Computationally identified novel agonists for GPRC6A, PLoS One, 2018, 13, e0195980 CrossRef PubMed.
- R. E. Amaro, R. Baron and J. A. McCammon, An improved relaxed complex scheme for receptor flexibility in computer-aided drug design, J. Comput.-Aided Mol. Des., 2008, 22, 693–705 CrossRef CAS PubMed.
- W. Evangelista, R. L. Weir, S. R. Ellingson, J. B. Harris, K. Kapoor, J. C. Smith and J. Baudry, Ensemble-based docking: From hit discovery to metabolism and toxicity predictions, Bioorg. Med. Chem., 2016, 24, 4928–4935 CrossRef PubMed.
- R. E. Amaro, J. Baudry, J. Chodera, O. Demir, J. A. McCammon, Y. Miao and J. C. Smith, Ensemble Docking in Drug Discovery, Biophys. J., 2018, 114, 2271–2278 CrossRef CAS PubMed.
- A. Acharya, et al., Supercomputer-based ensemble docking drug discovery pipeline with application to COVID-19, J. Chem. Inf. Model., 2020, 60, 548–557 CrossRef PubMed.
- A. Basciu, G. Malloci, F. Pietrucci, A. M. Bonvin and A. V. Vargiu, Holo-like and druggable protein conformations from enhanced sampling of binding pocket volume and shape, J. Chem. Inf. Model., 2019, 59, 1515–1528 CrossRef CAS PubMed.
- K. Henzler-Wildman and D. Kern, Dynamic personalities of proteins, Nature, 2007, 450, 964–972 CrossRef CAS PubMed.
- M. Moradi and E. Tajkhorshid, Mechanistic picture for conformational transition of a membrane transporter at atomic resolution, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 18916–18921 CrossRef CAS PubMed.
- D. M. Zuckerman, Equilibrium sampling in biomolecular simulations, Annu. Rev. Biophys., 2011, 40, 41–62 CrossRef CAS PubMed.
- L. Orellana, Large-scale conformational changes and protein function: breaking the in silico barrier, Front. mol. biosci., 2019, 6, 117 CrossRef CAS PubMed.
- I. Chen, S. Pant, Q. Wu, R. Cater, M. Sobti, R. Vandenberg, A. G. Stewart, E. Tajkhorshid, J. Font and R. Ryan, Glutamate transporters have a chloride channel with two hydrophobic gates, Nature, 2021, 591, 327–331 CrossRef CAS PubMed.
- R. C. Bernardi, M. C. R. Melo and K. Schulten, Enhanced Sampling Techniques in Molecular Dynamics Simulations of Biological Systems, Biochim. Biophys. Acta, 2015, 1850, 872–877 CrossRef CAS PubMed.
- M. Moradi and E. Tajkhorshid, Computational recipe for efficient description of large-scale conformational changes in biomolecular systems, J. Chem. Theory Comput., 2014, 10, 2866–2880 CrossRef CAS PubMed.
- I. B. Holland and M. A. Blight, ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans, J. Mol. Biol., 1999, 293, 381–399 CrossRef CAS PubMed.
- O. Fardel, V. Lecureur and A. Guillouzo, The P-glycoprotein multidrug transporter, Gen. Pharmacol., 1996, 27, 1283–1291 CrossRef CAS PubMed.
- F. J. Sharom, The P-glycoprotein multidrug transporter, Essays Biochem., 2011, 50, 161–178 CrossRef CAS PubMed.
- J. Li, K. F. Jaimes and S. G. Aller, Refined structures of mouse P-glycoprotein, Protein Sci., 2014, 23, 34–36 CrossRef CAS PubMed.
- F. J. Sharom, Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function, Front. Oncol., 2014, 4, 1–19 Search PubMed.
- P. Mitchell, A general theory of membrane transport from studies of bacteria, Nature, 1957, 180, 134–136 CrossRef CAS PubMed.
- O. Jardetzky, Simple allosteric model for membrane pumps, Nature, 1966, 211, 969–970 CrossRef CAS PubMed.
- M. L. Oldham, A. L. Davidson and J. Chen, Structural insights into ABC transporter mechanism, Curr. Opin. Struct. Biol., 2008, 18, 726–733 CrossRef CAS PubMed.
- K. P. Locher, Structure and mechanism of ATP-binding cassette transporters, Philos. Trans. R. Soc. Lond. B Biol. Sci., 2009, 364, 239–245 CrossRef CAS PubMed.
- S. Dehghani-Ghahnaviyeh, K. Kapoor and E. Tajkhorshid, Conformational changes in the nucleotide-binding domains of P-glycoprotein induced by ATP hydrolysis, FEBS Lett., 2021, 595, 735–749 CrossRef CAS PubMed.
- Á. Tarcsay and G. M. Keserű, Homology modeling and binding site assessment of the human P-glycoprotein, Future Med. Chem., 2011, 3, 297–307 CrossRef PubMed.
- R. J. Ferreira, M.-J. U. Ferreira and D. J. dos Santos, Molecular docking characterizes substrate-binding sites and efflux modulation mechanisms within P-glycoprotein, J. Chem. Inf. Model., 2013, 53, 1747–1760 CrossRef CAS PubMed.
- E. E. Chufan, K. Kapoor, H.-M. Sim, S. Singh, T. T. Talele, S. R. Durell and S. V. Ambudkar, Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1), PLoS One, 2013, 8, e82463 CrossRef PubMed.
- S. Silvente-Poirot and M. Poirot, Cholesterol and cancer, in the balance, Science, 2014, 373, 1445–1446 CrossRef PubMed.
- F. Klepsch, P. Vasanthanathan and G. F. Ecker, Ligand and structure-based classification models for prediction of P-glycoprotein inhibitors, J. Chem. Inf. Model., 2014, 54, 218–229 CrossRef CAS PubMed.
- J. Zhang, T. Sun, L. Liang, T. Wu and Q. Wang, Drug promiscuity of P-glycoprotein and its mechanism of interaction with paclitaxel and doxorubicin, Soft Matter, 2014, 10, 438–445 RSC.
- S. V. Ambudkar, S. Dey, C. A. Hrycyna, M. Ramachandra, I. Pastan and M. M. Gottesman, Biochemical, cellular, and pharmacological aspects of the multidrug transporter, Annu. Rev. Pharmacol. Toxicol., 1999, 39, 361–398 CrossRef CAS PubMed.
- A. R. Safa, Identification and Characterization of the Binding Sites of P-Glycoprotein for Multidrug Resistance-Related Drugs and Modulators, Curr. Med. Chem. Anti Cancer Agents, 2004, 4, 1–17 CrossRef CAS PubMed.
- T. W. Loo, M. C. Bartlett and D. M. Clarke, Processing Mutations Disrupt Interactions between the Nucleotide Binding and Transmembrane Domains of P-glycoprotein and the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), J. Biol. Chem., 2008, 283, 28190–28197 CrossRef CAS PubMed.
- B. Isralewitz, J. Baudry, J. Gullingsrud, D. Kosztin and K. Schulten, Steered Molecular Dynamics Investigations of Protein Function, J. Mol. Graph. Model., 2001, 19, 13–25 CrossRef CAS PubMed.
- M. Moradi, G. Enkavi and E. Tajkhorshid, Atomic-level characterization of transport cycle thermodynamics in the glycerol-3-phosphate:phosphate transporter, Nat. Commun., 2015, 6, 8393 CrossRef CAS PubMed.
- M. Moradi, C. Sagui and C. Roland, Calculating relative transition rates with driven nonequilibrium simulations, Chem. Phys. Lett., 2011, 518, 109–113 CrossRef CAS.
- M. Moradi and E. Tajkhorshid, Mechanistic picture for conformational transition of a membrane transporter at atomic resolution, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 18916–18921 CrossRef CAS PubMed.
- S. G. Aller, J. Yu, A. Ward, Y. Weng, S. Chittaboina, R. Zhuo, P. M. Harrell, Y. T. Trinh, Q. Zhang, I. L. Urbatsch and G. Chang, Structure of P-Glycoprotein reveals a molecular basis for poly-specific drug binding, Science, 2009, 323, 1718–1722 CrossRef CAS PubMed.
- A. B. Ward, et al., Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain, Proc. Natl. Acad. Sci. U.S.A., 2013, 110, 13386–13391 CrossRef CAS PubMed.
- P. Szewczyk, H. Tao, A. P. McGrath, M. Villaluz, S. D. Rees, S. C. Lee, R. Doshi, I. L. Urbatsch, Q. Zhang and G. Chang, Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein, Acta Crystallogr. D, 2015, 71, 732–741 CrossRef CAS PubMed.
- L. Esser, F. Zhou, K. M. Pluchino, J. Shiloach, J. Ma, W.-k. Tang, C. Gutierrez, A. Zhang, S. Shukla, J. P. Madigan, T. Zhou, P. D. Kwong, S. V. Ambudkar, M. M. Gottesman and D. Xia, Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity, J. Biol. Chem., 2017, 292, 446–461 CrossRef CAS PubMed.
- S. Thangapandian, K. Kapoor and E. Tajkhorshid, Probing cholesterol binding and translocation in P-glycoprotein, Biochim. Biophys. Acta Biomembr., 2020, 1862, 183090 CrossRef CAS PubMed.
- P.-C. Wen, B. Verhalen, S. Wilkens, H. S. Mchaourab and E. Tajkhorshid, On the Origin of Large Flexibility of P-glycoprotein in the Inward-facing State, J. Biol. Chem., 2013, 288, 19211–19220 CrossRef CAS PubMed.
- Y. Kim and J. Chen, Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation, Science, 2018, 359, 915–919 CrossRef CAS PubMed.
- R. Dastvan, S. Mishra, Y. B. Peskova, R. K. Nakamoto and H. S. Mchaourab, Mechanism of allosteric modulation of P-glycoprotein by transport substrates and inhibitors, Science, 2019, 364, 689–692 CrossRef CAS PubMed.
- A. Alam, J. Kowal, E. Broude, I. Roninson and K. P. Locher, Structural insight into substrate and inhibitor discrimination by human P-glycoprotein, Science, 2019, 363, 753–756 CrossRef CAS PubMed.
- B. Verhalen, R. Dastvan, S. Thangapandian, Y. Peskova, H. A. Koteiche, R. K. Nakamoto, E. Tajkhorshid and H. S. Mchaourab, Energy Transduction and Alternating Access of the Mammalian ABC Transporter P-glycoprotein, Nature, 2017, 543, 738–741 CrossRef CAS PubMed.
- J. Zaitseva, S. Jenewein, T. Jumpertz, I. B. Holland and L. Schmitt, H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB, EMBO J., 2005, 24, 1901–1910 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- J. C. Phillips, et al., Scalable molecular dynamics on CPU and GPU architectures with NAMD, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and A. D. MacKerell, Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell Jr and R. W. Pastor, Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- N. Foloppe and A. D. MacKerell Jr, All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data, J. Comput. Chem., 2000, 21, 86–104 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-Alkanes, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- G. J. Martyna, D. J. Tobias and M. L. Klein, Constant pressure molecular dynamics algorithms, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- S. E. Feller, Y. Zhang and R. W. Pastor, Constant pressure molecular dynamics simulation: The Langevin piston method, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, VMD – Visual Molecular Dynamics, J. Mol. Graph., 1996, 14, 33–38 CrossRef CAS.
- Molecular Operating Environment, 2019.01, Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2021 Search PubMed.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- Matlab Version 6.5 Release 13, The MathWorks, Inc., 3 Apple Hill Dr., Natick, MA 01760-2098, 508/647-7000, Fax 508/647-7001, 2002 Search PubMed.
- O. Maimon and L. RokachData mining and knowledge discovery handbook, Springer, 2005 Search PubMed.
- V. Martel, C. Racaud-Sultan, S. Dupe, C. Marie, F. Paulhe, A. Galmiche, M. R. Block and C. Albiges-Rizo, Conformation, Localization, and Integrin Binding of Talin Depend on Its Interaction with Phosphoinositides, J. Biol. Chem., 2001, 276, 21217–21227 CrossRef CAS PubMed.
- O. Bársony, G. Szalóki, D. Türk, S. Tarapcsák, Z. Gutay-Tóth, Z. Bacsó, I. J. Holb, L. Székvölgyi, G. Szabó, L. Csanády, G. Szakács and K. Goda, A single active catalytic site is sufficient to promote transport in P-glycoprotein, Sci. Rep., 2016, 6, 24810 CrossRef PubMed.
- A. B. Shapiro and V. Ling, Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities, Eur. J. Biochem., 1997, 250, 130–137 CrossRef CAS PubMed.
- A. B. Shapiro and V. Ling, Transport of LDS-751 from the cytoplasmic leaflet of the plasma membrane by the rhodamine-123-selective site of P-glycoprotein, Eur. J. Biochem., 1998, 254, 181–188 CrossRef CAS PubMed.
- T. W. Loo and D. M. Clarke, Determining the dimensions of the drug-binding domain of human P-glycoprotein using thiol cross-linking compounds as molecular rulers, J. Biol. Chem., 2001, 276, 36877–36880 CrossRef CAS PubMed.
- T. W. Loo, M. C. Bartlett and D. M. Clarke, Simultaneous Binding of Two Different Drugs in the Binding Pocket of the Human Multidrug Resistance P-glycoprotein, J. Biol. Chem., 2003, 278, 39706–39710 CrossRef CAS PubMed.
- C. Pascaud, M. Garrigos and S. Orlowski, Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents, Biochem. J., 1998, 333, 351–358 CrossRef CAS PubMed.
- R. Callaghan and J. R. Riordan, Synthetic and natural opiates interact with P-glycoprotein in multidrug-resistant cells, J. Biol. Chem., 1993, 268, 16059–16064 CrossRef CAS.
- T. Tsuruo, H. Iida, S. Tsukagoshi and Y. Sakurai, Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil, Cancer Res., 1981, 41, 1967–1972 CAS.
- S.-Y. Wang, K. Bonner, C. Russell and G. K. Wang, Tryptophan scanning of D1S6 and D4S6 C-termini in voltage-gated sodium channels, Biophys. J., 2003, 85, 911–920 CrossRef CAS PubMed.
- X. Q. Wang, L. N. Li, W. R. Chang, J. P. Zhang, L. L. Gui, B. J. Guo and D. C. Liang, Structure of C-phycocyanin from Spirulina platensis at 2.2 Å resolution: a novel monoclinic crystal form for phycobiliproteins in phycobilisomes, Acta Crystallogr. D, 2001, 57, 784–792 CrossRef CAS PubMed.
- J. Rautio, J. E. Humphreys, L. O. Webster, A. Balakrishnan, J. P. Keogh, J. R. Kunta, C. J. Serabjit-Singh and J. W. Polli, In vitro P-glycoprotein inhibition assays for assessment of clinical drug interaction potential of new drug candidates: a recommendation for probe substrates, Drug Metabol. Dispos., 2006, 34, 786–792 CrossRef CAS PubMed.
- K. Nosol, K. Romane, R. N. Irobalieva, A. Alam, J. Kowal, N. Fujita and K. P. Locher, Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 26245–26253 CrossRef CAS PubMed.
- P. Mistry, A. J. Stewart, W. Dangerfield, S. Okiji, C. Liddle, D. Bootle, J. A. Plumb, D. Templeton and P. Charlton, In vitro and in vivo reversal of P-glycoprotein-mediated multidrug resistance by a novel potent modulator, XR9576, Cancer Res., 2001, 61, 749–758 CAS.
- A. H. Dantzig, R. L. Shepard, J. Cao, K. L. Law, W. J. Ehlhardt, T. M. Baughman, T. F. Bumol and J. J. Starling, Reversal of P-glycoprotein-mediated multidrug resistance by a potent cyclopropyldibenzosuberane modulator, LY335979, Cancer Res., 1996, 56, 4171–4179 CAS.
- L. van Zuylen, A. Sparreboom, A. van der Gaast, K. Nooter, F. Eskens, E. Brouwer, C. Bol, R. De Vries, P. Palmer and J. Verweij, Disposition of docetaxel in the presence of P-glycoprotein inhibition by intravenous administration of R101933, Eur. J. Cancer, 2002, 38, 1090–1099 CrossRef CAS PubMed.
- K. Kapoor, S. Pant and E. Tajkhorshid, Active participation of membrane lipids in inhibition of multidrug transporter P-glycoprotein, Chem. Sci., 2021, 12, 6293–6306 RSC.
- T. W. Loo and D. M. Clarke, Identification of residues in the drug-binding site of human P-glycoprotein using a thiol-reactive substrate, J. Biol. Chem., 1997, 272, 31945–31948 CrossRef CAS PubMed.
- T. W. Loo and D. M. Clarke, Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil, J. Biol. Chem., 2001, 276, 14972–14979 CrossRef CAS PubMed.
- T. W. Loo, M. C. Bartlett and D. M. Clarke, Permanent activation of the human P-glycoprotein by covalent modification of a residue in the drug-binding site, J. Biol. Chem., 2003, 278, 20449–20452 CrossRef CAS PubMed.
- T. W. Loo, M. C. Bartlett and D. M. Clarke, Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein: direct evidence for the substrate-induced fit mechanism for drug binding, J. Biol. Chem., 2003, 278, 13603–13606 CrossRef CAS PubMed.
- T. W. Loo, M. C. Bartlett and D. M. Clarke, Transmembrane segment 1 of human P-glycoprotein contributes to the drug-binding pocket, Biochem. J., 2006, 396, 537–545 CrossRef CAS PubMed.
- T. W. Loo, M. C. Bartlett and D. M. Clarke, Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket, Biochem. J., 2006, 399, 351–359 CrossRef CAS PubMed.
- A. Sajid, S. Lusvarghi, M. Murakami, E. E. Chufan, B. Abel, M. M. Gottesman, S. R. Durell and S. V. Ambudkar, Reversing the direction of drug transport mediated by the human multidrug transporter P-glycoprotein, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 29609–29617 CrossRef CAS PubMed.
- D. Balayssac, N. Authier, A. Cayre and F. Coudore, Does inhibition of P-glycoprotein lead to drug–drug interactions?, Toxicol. Lett., 2005, 156, 319–329 CrossRef CAS PubMed.
- M. Kurosawa, M. Okabe, N. Hara, K. Kawamura, S. Suzuki, K. Sakurada and M. Asaka, Reversal effect of itraconazole on adriamycin and etoposide resistance in human leukemia cells, Ann. Hematol., 1996, 72, 17–21 CrossRef CAS PubMed.
- A. Alam, R. Kung, J. Kowal, R. A. McLeod, N. Tremp, E. V. Broude, I. B. Roninson, H. Stahlberg and K. P. Locher, Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 1973–1982 CrossRef PubMed.
- T. W. Loo and D. M. Clarke, Mutations to amino acids located in predicted transmembrane segment 6 (TM6) modulate the activity and substrate specificity of human P-glycoprotein, Biochemistry, 1994, 33, 14049–14057 CrossRef CAS PubMed.
- X. Zhang, K. I. Collins and L. M. Greenberger, Functional Evidence That Transmembrane 12 and the Loop between Transmembrane 11 and 12 Form Part of the Drug-binding Domain in P-glycoprotein Encoded by MDR1, J. Biol. Chem., 1995, 270, 5441–5448 CrossRef CAS PubMed.
- T. W. Loo, M. C. Bartlett and D. M. Clarke, Suppressor mutations in the transmembrane segments of P-glycoprotein promote maturation of processing mutants and disrupt a subset of drug-binding sites, J. Biol. Chem., 2007, 282, 32043–32052 CrossRef CAS PubMed.
- S. Thangapandian, K. Kapoor and E. Tajkhorshid, Probing Cholesterol Binding and Translocation in P-glycoprotein, Biochim. Biophys. Acta Biomembr., 2020, 1862, 183090 CrossRef CAS PubMed.
- P. Z. Smriti and H. S. Mchaourab, Mapping daunorubicin-binding sites in the ATP-binding cassette transporter MsbA using site-specific quenching by spin labels, J. Biol. Chem., 2009, 284, 13904 CrossRef CAS PubMed.
- S. Dey, M. Ramachandra, I. Pastan, M. M. Gottesman and S. V. Ambudkar, Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein, Proc. Natl. Acad. Sci. U.S.A., 1997, 94, 10594–10599 CrossRef CAS PubMed.
- M. K. Al-Shawi and H. Omote, The remarkable transport mechanism of P-glycoprotein: a multidrug transporter, J. Bioenerg. Biomembr., 2005, 37, 489–496 CrossRef CAS PubMed.
- N. Subramanian, K. Condic-Jurkic, A. E. Mark and M. L. O'Mara, Identification of possible binding sites for morphine and nicardipine on the multidrug transporter P-glycoprotein using umbrella sampling techniques, J. Chem. Inf. Model., 2015, 55, 1202–1217 CrossRef CAS PubMed.
- N. Subramanian, A. Schumann-Gillett, A. E. Mark and M. L. O'Mara, Probing the pharmacological binding sites of P-glycoprotein using umbrella sampling simulations, J. Chem. Inf. Model., 2018, 59, 2287–2298 CrossRef PubMed.
- I. G. Rodriguez-Bussey, U. Doshi and D. Hamelberg, Enhanced molecular dynamics sampling of drug target conformations, Biopolymers, 2016, 105, 35–42 CrossRef CAS PubMed.
- T. W. Loo, M. C. Bartlett and D. M. Clarke, Identification of residues in the drug translocation pathway of the human multidrug resistance P-glycoprotein by arginine mutagenesis, J. Biol. Chem., 2009, 284, 24074–24087 CrossRef CAS PubMed.
- D. I. Morris, L. M. Greenberger, E. P. Bruggemann, C. Cardarelli, M. M. Gottesman, I. Pastan and K. B. Seamon, Localization of the forskolin labeling sites to both halves of P-glycoprotein: similarity of the sites labeled by forskolin and prazosin, Mol. Pharmacol., 1994, 46, 329–337 CAS.
- T. W. Loo and D. M. Clarke, Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein, J. Biol. Chem., 2002, 277, 44332–44338 CrossRef CAS PubMed.
- Y. D. Cakil, N. Khunweeraphong, Z. Parveen, D. Schmid, M. Artaker, G. F. Ecker, H. H. Sitte, O. Pusch, T. Stockner and P. Chiba, Pore-exposed tyrosine residues of P-glycoprotein are important hydrogen-bonding partners for drugs, Mol. Pharmacol., 2014, 85, 420–428 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc00841f |
| This journal is © The Royal Society of Chemistry 2022 |