
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe oxygen-resistant [FeFe]-hydrogenase CbA5H harbors an unknown radical signal†
Melanie
Heghmanns‡
 a,
Andreas
Rutz‡
b,
Yury
Kutin
a,
Vera
Engelbrecht
b,
Martin
Winkler
c,
Thomas
Happe
*b and
Müge
Kasanmascheff
*a
a,
Andreas
Rutz‡
b,
Yury
Kutin
a,
Vera
Engelbrecht
b,
Martin
Winkler
c,
Thomas
Happe
*b and
Müge
Kasanmascheff
*a
aTU Dortmund University, Department of Chemistry and Chemical Biology, Otto-Hahn-Straße 6, 44227 Dortmund, Germany. E-mail: muege.kasanmascheff@tu-dortmund.de
bRuhr University Bochum, Faculty of Biology and Biotechnology, Photobiotechnology, Universitätsstr. 150, 44801 Bochum, Germany. E-mail: thomas.happe@ruhr-uni-bochum.de
cTechnical University of Munich Campus Straubing for Biotechnology and Sustainability, Professorship for Electrobiotechnology, Uferstrasse 53, 94315 Straubing, Germany
First published on 7th June 2022
Abstract
[FeFe]-hydrogenases catalyze the reversible conversion of molecular hydrogen into protons and electrons with remarkable efficiency. However, their industrial applications are limited by their oxygen sensitivity. Recently, it was shown that the [FeFe]-hydrogenase from Clostridium beijerinckii (CbA5H) is oxygen-resistant and can be reactivated after oxygen exposure. In this work, we used multifrequency continuous wave and pulsed electron paramagnetic resonance (EPR) spectroscopy to characterize the active center of CbA5H, the H-cluster. Under oxidizing conditions, the spectra were dominated by an additional and unprecedented radical species. The generation of this radical signal depends on the presence of an intact H-cluster and a complete proton transfer pathway including the bridging azadithiolate ligand. Selective 57Fe enrichment combined with isotope-sensitive electron-nuclear double resonance (ENDOR) spectroscopy revealed a spin density distribution that resembles an H-cluster state. Overall, we uncovered a radical species in CbA5H that is potentially involved in the redox sensing of CbA5H.
Introduction
Molecular hydrogen is a promising alternative to fossil fuels in meeting the world's increasing energy demand.1,2 Unlike platinum-based systems, metalloenzymes such as [FeFe]-hydrogenases use only earth-abundant metals to produce and oxidize H2 under mild conditions and with high turnover rates (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecules per second).3,4 The catalytic properties of these enzymes inspired the development of cheap and efficient H2 catalysts for carbon-neutral hydrogen production.5 Yet, their industrial application is hindered by their intrinsic O2-sensitivity.6,7 Despite the extensive research, engineering [FeFe]-hydrogenases with improved oxygen stability has not been entirely successful.8
000 molecules per second).3,4 The catalytic properties of these enzymes inspired the development of cheap and efficient H2 catalysts for carbon-neutral hydrogen production.5 Yet, their industrial application is hindered by their intrinsic O2-sensitivity.6,7 Despite the extensive research, engineering [FeFe]-hydrogenases with improved oxygen stability has not been entirely successful.8
The active site of [FeFe]-hydrogenases harbors the so-called H-cluster: a cubane [4Fe4S] cluster ([4Fe]H) linked by a cysteine to a unique [2Fe2S]-subsite ([2Fe]H).3 The distal (Fed) and proximal (Fep) iron atoms of [2Fe]H are coordinated by two CN− and three CO ligands and are bridged by an azadithiolate (adt) ligand.9,10 The binding of O2 to the open coordination site at Fed initiates a degradative process causing irreversible damage to the H-cluster.7,11–13 Partial reduction and protonation of dioxygen, leading to H-cluster destruction, was assumed to be inherent to all [FeFe]-hydrogenases until the discovery of the hydrogenase from Clostridium beijerinckii, termed CbA5H (Fig. 1).14 The H-cluster of CbA5H can reversibly switch from the oxygen-sensitive and active Hox state to the oxygen-stable but inactive Hinact state.14,15 Recently, the air-exposed crystal structure revealed that this outstanding ability is reached by the binding of a conserved cysteine residue (C367) to Fed shielding the cofactor from O2.16 Structural elucidation of the Hinact state combined with spectroscopic and electrochemical investigations14,15 of CbA5H strongly indicate a novel oxygen resistance mechanism that does not involve direct O2 binding to the H-cluster. Understanding the unusual oxygen resistance mechanism of CbA5H might present an important step toward facilitating the use of [FeFe]-hydrogenases as carbon-neutral energy carriers.
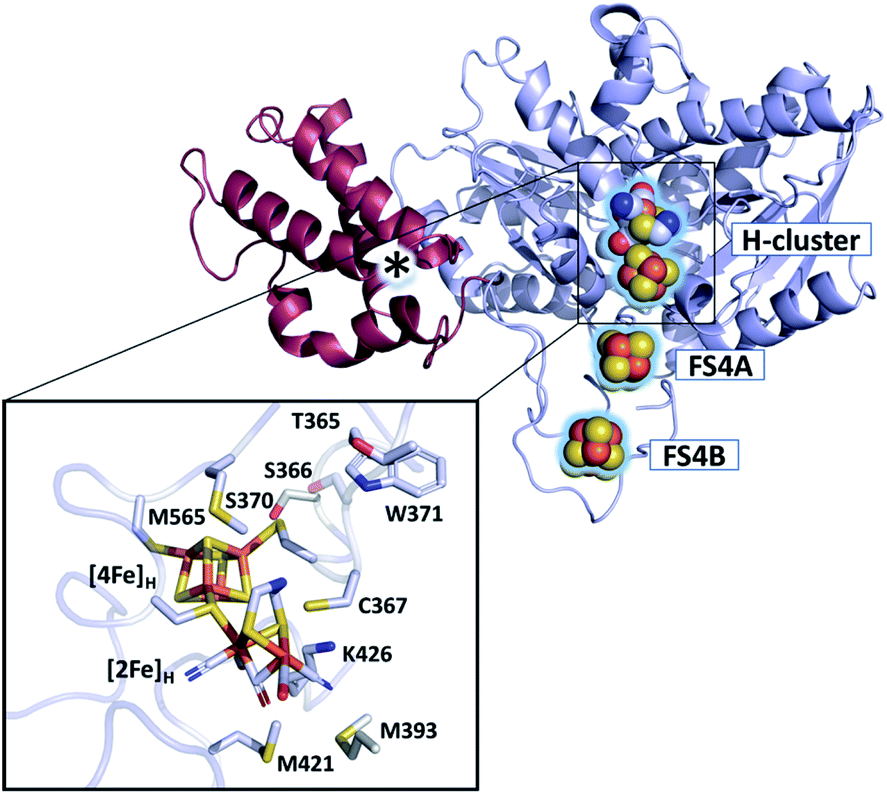 | ||
| Fig. 1 Structure of CbA5H (PDB ID 6TTL). X-ray structure of CbA5H displayed as a monomer with SLBB – (soluble ligand-binding beta-grasp) domain (dark-red), and H- and F-cluster containing domain (light-blue). Positions of the FeS clusters are highlighted and depicted as spheres. The asterisk indicates electron density whose nature could not be unambiguously identified yet. The lower panel shows the active site of CbA5H representing [4Fe]H and [2Fe]H embedded in the protein environment. | ||
The work presented here expands our understanding of CbA5H and its exceptional oxygen resistance by characterizing its paramagnetic centers under oxidative and reductive conditions using isotope-sensitive electron paramagnetic resonance (EPR) spectroscopy. Along with the well-known H-cluster states in the active CbA5H, we detected an unusual radical species dominating the oxygen-treated spectra, which has not been reported in other [FeFe]-hydrogenases under similar conditions. Our investigation suggests that this radical is unique to CbA5H and potentially plays a role in the redox-sensing of the enzyme.
Results and discussion
Analysis of reduced states of apo- and holo-CbA5H
First, to investigate the paramagnetic H-cluster states, the enzyme was reduced either with H2 or with varying concentrations of sodium dithionite (CbA5HNaDT). Additionally, we performed NaDT-free control measurements (see ESI†). Analysis of the respective EPR spectra is facilitated by using elevated temperatures at which signals from fast-relaxing, accessory FeS clusters, the so-called F-clusters, are undetectable (20 K vs. 10 K spectra in Fig. 2, S1 and S2†).17 The EPR spectrum of the active CbA5H recorded at 20 K exhibits the characteristic, well-known H-cluster states Hox,18–27 Hox–CO,12,23,24,28 and Hhyd29–31 (Fig. 2, S1, S2 and Tables S2 and S3†). Their presence is also confirmed via Fourier-transform infrared (FTIR) spectroscopy (Fig. S3 and ref. 15†). Interestingly, an additional signal at g ≈ 2.01, which can be observed even up to 180 K, is also detected (Fig. S2, S4 and S5†). The origin of this unidentified species, termed R˙ox, is discussed below.
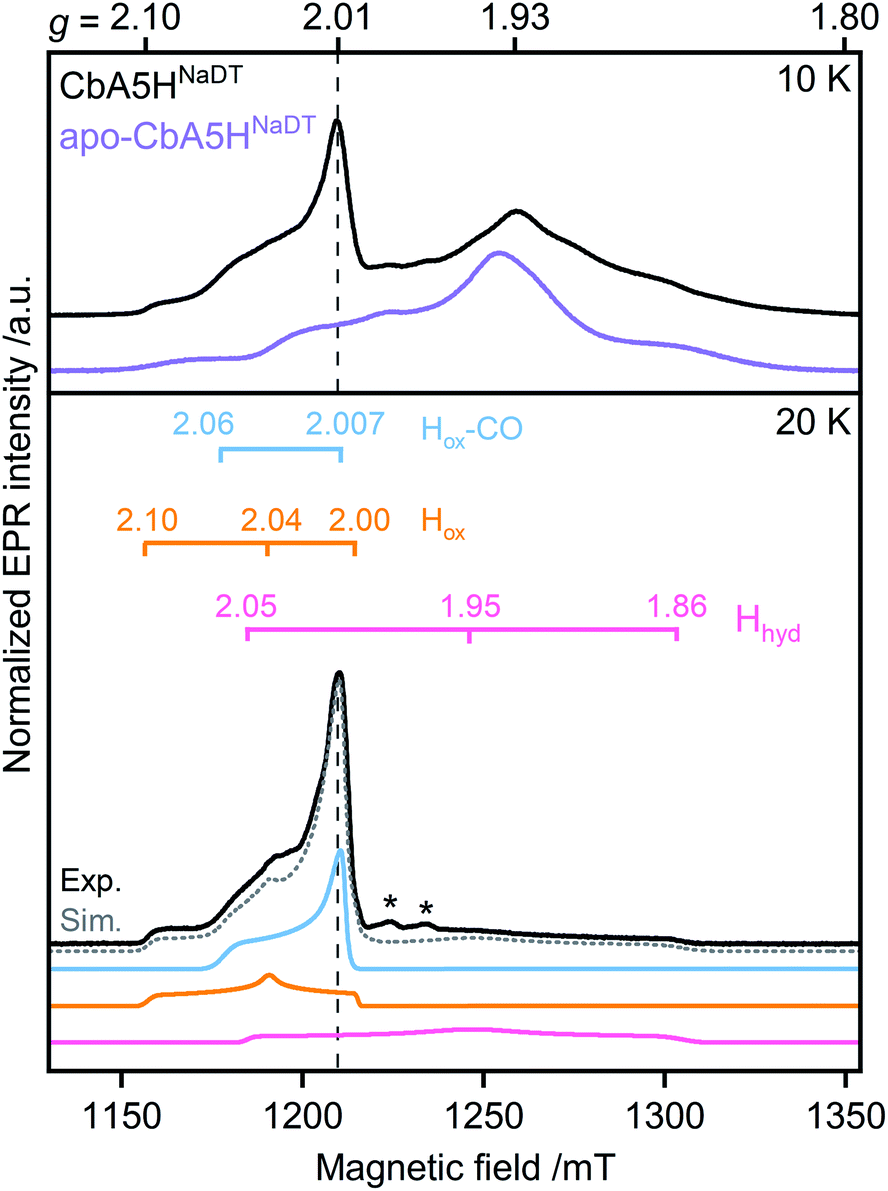 | ||
| Fig. 2 Pulsed EPR spectra (34 GHz) of CbA5H (black) and apo-CbA5H (purple). The samples were reduced with 10 mM NaDT and measured at 10, and 20 K. Suppression of fast-relaxing FeS clusters at 20 K enabled the complete simulation of holo-CbA5H (dotted grey trace). Hox, Hox–CO, Hhyd and R˙ox were included in the simulation (Fig. S1†). Details of the experiments and analysis are given in ESI.† Dashed vertical lines mark the additional signal at g ≈ 2.01. A Mn2+ impurity signal is marked with asterisks. | ||
Next, to characterize the accessory FeS clusters, we employed multi-frequency EPR on the inactive apoenzyme that lacks the [2Fe]H subsite but harbors the F-clusters and [4Fe]H (apo-CbA5H) (Fig. 2, S2 and S6†). The EPR spectrum at 10 K (Fig. 2, purple trace) is dominated by a broad signal centered around g = 1.93. This signal is broadened beyond detection at 20 K, confirming the presence of fast-relaxing [4Fe4S]1+ clusters (Fig. S2†).32,33 Furthermore, its spectral features are frequency-dependent (Fig. S6†). In conjunction with the signal's significant width, the frequency dependence clearly indicates spin–spin interaction between the F-clusters.35,36 This observation is not surprising as the clusters are adjacent (Fig. 1).16 Spectral simulation using parameters for two FeS clusters similar to those reported in the literature resulted in a good fit for the experimental apo-CbA5HNaDT spectrum (Fig. S2†). Note that the R˙ox signal found in the holoenzyme of CbA5H is absent in the apoprotein. This relates R˙ox to the presence of an intact H-cluster.
The H2–O2 cycle
Unlike other [FeFe]-hydrogenases (from Desulfovibrio desulfuricans (DdHydAB) and Desulfovibrio vulgaris Hildenborough), the inactive Hinact state of CbA5H can undergo several cycles of oxidative inactivation and reductive reactivation.14,15 We investigated this reversible transformation by monitoring the spectral changes of the anaerobically isolated enzyme repeatedly treated with H2 and O2 (termed CbA5HH2 and CbA5HO2, respectively) (Fig. 3 and S4†). At cryogenic temperatures, EPR spectra of CbA5HH2 reveal a complex line shape (Fig. 3) arising from paramagnetic H-cluster states and F-clusters (see also Fig. 2). The overall signal intensity of CbA5HH2 is reduced by approximately 40% after the first cycle (see ESI† on the H2–O2 cycle). This observation agrees with activity assays and FTIR spectra (Fig. S3, S7 and ref. 15†), confirming the partial reactivation of CbA5H after O2-treatment.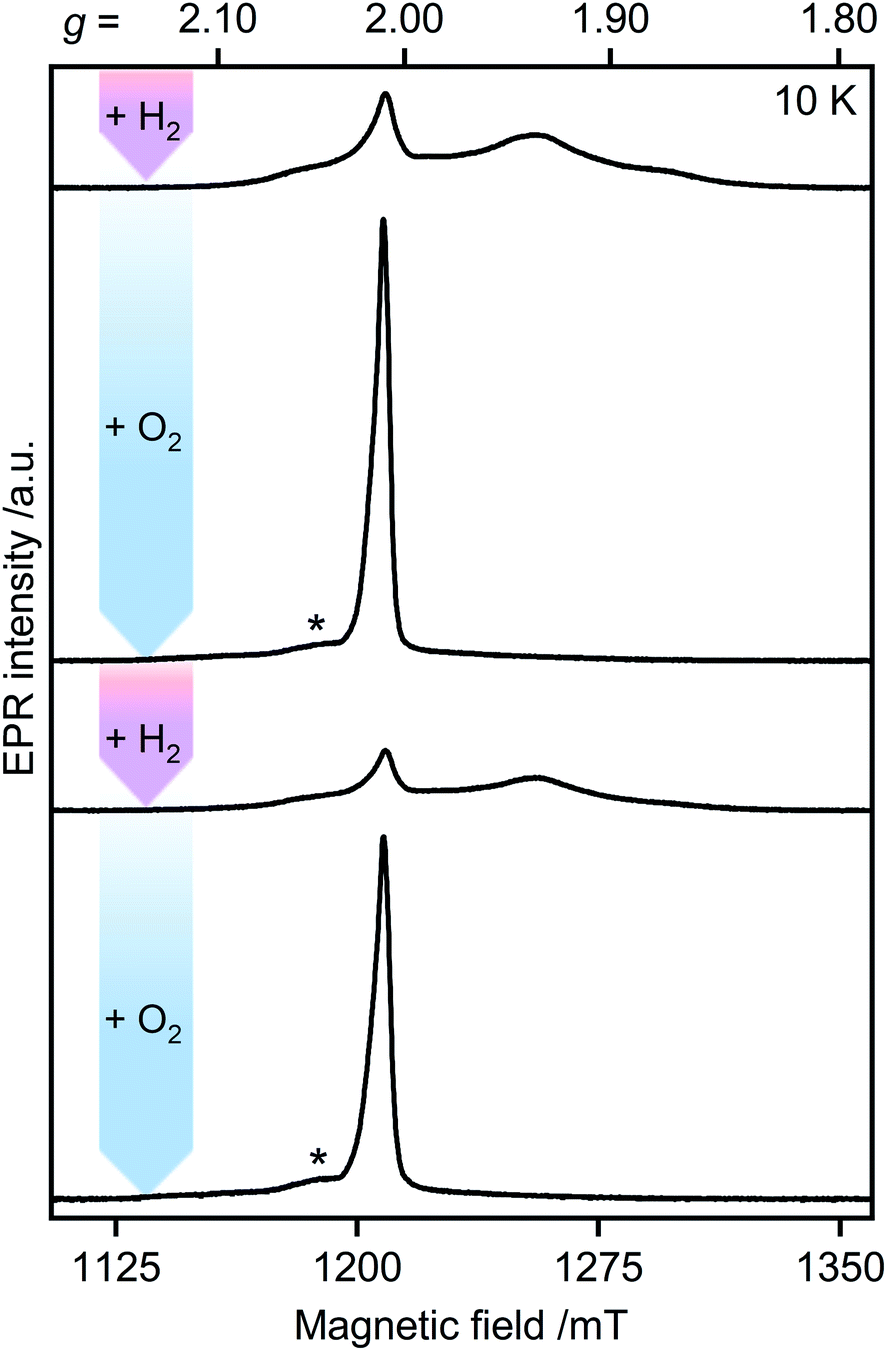 | ||
| Fig. 3 Pulsed EPR spectra of CbA5H (34 GHz). As-isolated CbA5H was treated alternately with H2 (pink arrows) and O2 (blue arrows). Asterisks indicate residual Hox–CO signals (see also ESI† on the H2–O2-cycle). | ||
When CbA5HH2 is exposed to O2, all identified signals from the F-clusters and H-cluster disappear. This was expected as most FeS clusters are EPR-inactive in the oxidized state, and Hinact, the only H-cluster state present in CbA5HO2 (see Fig. S3†), is suggested to be EPR-silent.15 The EPR spectra of CbA5HO2 (Fig. 3), however, are dominated by the nearly isotropic signal at g = 2.01 (Fig. 4). Its EPR signature is distinct from typical signals of Hox–CO and degraded FeS clusters (Fig. S4 and S7†). Its three principal g-values were determined as g = 2.019, 2.010, 2.006 via a global fit at the X- and Q-band frequencies (see Table S2† for details). Surprisingly, its intensity decreases only by 5–13% after two H2–O2 cycles and cryo-annealing, while the amount of active protein drops significantly (ESI on the H2–O2 cycle, Fig. 3 and S4–S7†). Even though R˙ox is present in CbA5HH2, its EPR-intensity is considerably higher in the oxidized enzyme (Fig. 3 and S4†). The high stability of R˙ox contradicts the observed degradation of Hinact during the cycle, as shown via FTIR (Fig. S3†), indicating that the Hinact state itself is not the source of R˙ox. Moreover, the generation of R˙ox by treatment with the mild oxidant hexamine ruthenium(III) chloride (HAR) showed that O2 is not the only catalyst triggering its formation (Fig. S9†). Similar results were reported for the generation of Hinact,15 emphasizing the connection between Hinact formation and the R˙ox signal. This is reminiscent of the EPR signals observed for the proximal [4Fe3S] cluster of O2-tolerant [NiFe]-hydrogenases under oxidative conditions, even in the absence of O2,37 which turn out to be a strong indicator for the underlying O2 tolerance mechanism. These intriguing properties of the R˙ox species prompted us to investigate its identity further.
 | ||
| Fig. 4 Continuous-wave (cw) EPR spectra of oxygen-treated samples (9.7 GHz, T = 100 K). CbA5H, C367D, CpI, apo-CbA5H, and CbA5H(pdt) were treated with O2. Blue lines represent simulations of R˙ox detected in CbA5H and variant C367D, and of Hox–CO detected in CpI. The asterisk indicates a change in the R˙ox line shape for variant C367D (see Fig. S15†). | ||
Investigation of R˙ox formation under various conditions
First, we analyzed the EPR spectra of O2-exposed CbA5H compared to CpI (Fig. 4 and S10†). CpI, a ‘standard’ [FeFe]-hydrogenase, was purified and oxidized using the same procedure for CbA5H. In agreement with reported CpI data,34 R˙ox was not detected in EPR spectra of CpIO2, demonstrating that the signal is unique to CbA5H. These data also exclude the possibility of R˙ox being an artifact related to the protein preparation procedures or a radical species generated due to external factors such as the buffers used. This conclusion is further supported by (i) the almost identical EPR signature of R˙ox in aerobically and anaerobically isolated CbA5H (Fig. S11†) and (ii) the absence of the R˙ox signal in EPR spectra of apo-CbA5H (reduced or oxidized). The only signal detected in the oxidized apoenzyme originates from a [3Fe4S]+ cluster in substoichiometric amounts, possibly from mild oxidative damage to the FeS clusters (Fig. S6, S8 and S12†).We, therefore, explored whether an active enzyme is necessary to generate R˙ox. Apo-CbA5H was maturated with a chemically altered cofactor ([Fe2(pdt)(CO4)(CN2)]2−, CbA5H(pdt)) yielding a catalytically inactive enzyme due to the non-protonatable bridgehead.35 This artificial ligand does not interfere with the native structure of the hydrogenases but disrupts the proton-transfer pathway to and from Fed.36,38,39 The complex EPR spectrum of H2-reduced CbA5H(pdt) (Fig. S13†) resembles the corresponding spectrum reported for CpI(pdt).17 The spectrum of O2-treated CbA5H(pdt) shows substoichiometric amounts of a [3Fe4S]+ cluster (Fig. S13†), similar to the one detected in apo-CbA5HO2. Strikingly, none of the EPR spectra recorded for CbA5H(pdt) exhibit the R˙ox signal (Fig. 4 and S13†), although the formation of the Hinact state in CbA5H(pdt) was verified by FTIR measurements (Fig. S14†). This again excludes Hinact as the source of R˙ox.
To this point, our results on EPR characteristics (g-values and temperature-dependency) of R˙ox hint at a protein-based radical species whose generation is dependent on the presence of an active H-cluster and the native adt ligand, ensuring an intact proton transfer pathway. The cysteine residue C367 is one of the closest amino acids to the H-cluster and is involved in the Hinact formation.16 Therefore, we investigated whether the oxidation of C367 results in the R˙ox signal.
We recorded EPR spectra of the O2-treated CbA5H in which C367 is replaced with aspartate (C367D) (Fig. 4 and S15†). This variant prevented the formation of the Hinact state while retaining 20% of the H2-production activity compared to the wild-type enzyme.16 Temperature-dependent spectra exhibit the R˙ox signal but with a narrower line shape and decreased intensity. Although these results exclude C367 as the source of R˙ox, they show that the identity of residue 367 affects its electronic structure.
Furthermore, the characteristics of our EPR and UV-vis data conflict with those of typical (i) amino acid radicals, (ii) sulfur-based radicals, (iii) peroxyl radicals, and (iv) semiquinone radicals (Fig. S16 and ESI Discussion† on the identity of R˙ox). Yet, an unusual organic radical near, on, or bound to the H-cluster with anomalous spectroscopic properties, as observed with the tryptophane cation radical found in cytochrome c peroxidase40,41 cannot be ruled out. Candidates that might be considered are residues M393, S370 or M565, M421, and W371 (Fig. 1), of which the last three represent highly conserved positions among other [FeFe]-hydrogenases.
57Fe-labeling and isotope-sensitive studies of R˙ox
EPR studies combined with 57Fe labeling provided invaluable information on iron-containing metalloproteins, including elucidation of the electronic structure of H-cluster states from different organisms.9,27,42,43 The presence of a 57Fe nucleus with a nuclear spin I = ½ results in EPR line broadening due to hyperfine interaction (hfi). To investigate whether R˙ox is associated with the H-cluster, we selectively labeled the apoprotein ([4Fe]H and F-clusters) with 57Fe and subsequently maturated it with 56Fe–[2Fe]H. The g-value of R˙ox did not change upon labeling. However, 57Fe enrichment resulted in EPR line broadening of the R˙ox spectra due to strong hfis between 57Fe nuclei and R˙ox (Fig. S17†).Next, we recorded orientation-selective 57Fe electron-nuclear double resonance (ENDOR) spectra of R˙ox (Fig. 5). The ENDOR line shape shows three broad features symmetrically centered around |A|/2 (A is the hyperfine coupling constant) and split by twice the Larmor frequency ν57Fe due to strong hfis with several 57Fe nuclei. At least three 57Fe nuclei needed to be introduced to simulate the experimental ENDOR line shape (Fig. S18†). At this point, however, a complete and unique analysis of the orientation-selective 57Fe ENDOR pattern is not possible, as only a few features of the overlapping signals are resolved at Q-band. Nonetheless, the hf couplings of all observed 57Fe nuclei are in the range of 25–35 MHz, very similar to those observed for the [4Fe]H subcluster in the Hox–CO state from other hydrogenases.9,43 These data suggest that R˙ox is either a unique H-cluster state or located close to the intact H-cluster and thus coupled to [4Fe]H. Note that the F-clusters in their native conformation can be excluded as the source of observed hfis because (i) R˙ox is not generated in the absence of an intact H-cluster, i.e., in apo-CbA5H, (ii) slow relaxation behavior of R˙ox is inconsistent with a typical [4Fe4S]1+ cluster, (iii) substituting the C367 residue residing close to [2Fe]H perturbs the EPR line shape of R˙ox, and (iv) the isotropic hfis determined for R˙ox are significantly different from the ones observed for the standard [4Fe4S]1+ clusters displaying |Aiso| of around 15 MHz for their ferrous Fe2+–Fe2+ pair.44
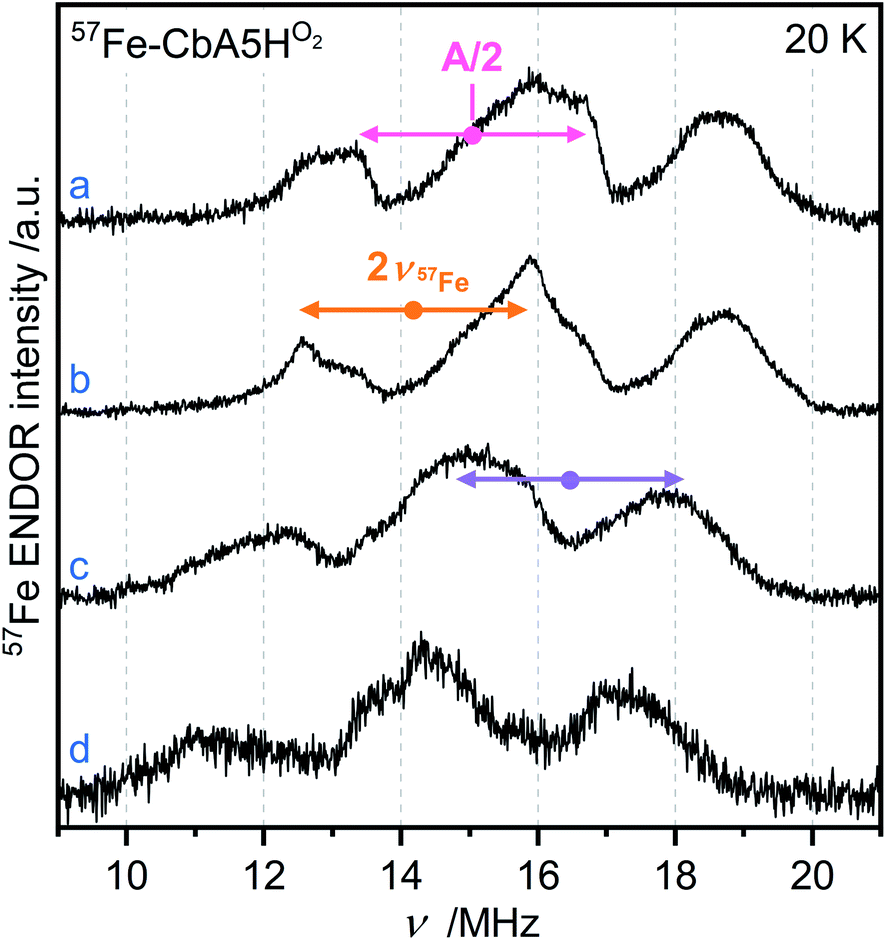 | ||
| Fig. 5 Orientation-selective Davies ENDOR spectra of 57Fe-labeled CbA5HO2 recorded at 20 K, Q-band and (a) g = 2.008 (b) g = 2.012 (c) g = 2.018 (d) g = 2.022. At least three different 57Fe hyperfine couplings are observable, centered at A/2 (dots), and split by twice the Larmor frequency (arrows). All experimental details are given in ESI.† | ||
Next, we performed orientation-selective proton (1H) ENDOR experiments on R˙ox (Fig. S19†). The spectra revealed overlapping signals due to contributions from several protons having hfis not larger than 9 MHz. As our 57Fe EPR data showed effective spin density on the H-cluster, we compared 1H ENDOR spectra of R˙ox with those of Hox, Hox–CO, and Hhyd from different [FeFe]-hydrogenases.45–47 As with the 57Fe data, the strength of the hfis and the spectral shape of R˙ox 1H ENDOR resemble those detected with Hox–CO assigned to β-protons of the cysteine ligands of [4Fe]H.45–47
To demonstrate that the detected 57Fe and 1H hfis do not arise from the underlying Hox–CO species, we recorded FTIR spectra for oxygen-treated 57Fe–CbA5H (Fig. S3†) and 1H ENDOR spectra with CbA5Hair whose EPR spectrum is composed of R˙ox and Hox–CO (Fig. S19†). The absence of an Hox–CO FTIR signature in 57Fe–CbA5HO2 and the presence of additional features in 1H ENDOR spectra of CbA5Hair showed that the observed hfis belong to R˙ox. The similarities detected in hfis strongly imply a similar spin density distribution for R˙ox and Hox–CO, which features a paramagnetic [2Fe]H (S = 1/2) exchange coupled to the diamagnetic [4Fe]H2+.
Interestingly, the F-cluster-truncated form of the [FeFe]-hydrogenase from Megasphaera elsdenii, harboring the H-cluster, displayed an EPR signal similar to R˙ox upon CO-treatment.6 This signal was attributed to the Hox–CO state. However, the EPR signature differs substantially from the Hox–CO state observed for the as-isolated enzyme and other known [FeFe]-hydrogenases. As the FTIR spectrum of this redox state was similar to those of other hydrogenases, the unusual change in the EPR spectrum could not be explained. Here, we can exclude the well-known Hox–CO state as the origin of the R˙ox signal because we can distinguish the features of Hox–CO and R˙ox in our temperature-dependent EPR and ENDOR data (Fig. S8 and S19†).
Lastly, we investigated the presence of exchangeable protons by recording 1H ENDOR spectra of R˙ox in the D2O buffer. Indeed, we detected differences in the proton ENDOR spectra of R˙ox in H2O or D2O buffers. In addition, the 2H Mims ENDOR spectrum revealed at least two proton hfis arising from the exchangeable protons (see Fig. S19–S20†). These associate R˙ox with coordinating water or solvent-derived protonated species, e.g., an amino acid residue with an exchangeable proton. One might hypothesize that the flexible loop around the H-cluster of CbA5H facilitates the movement of conserved water/s that is part of the proton-transfer pathway (see Fig. S6 in ref. 16†).
Overall, our EPR and ENDOR data show a spin density distribution at the H-cluster similar to the Hox–CO state and exclude C367 and Hinact as the source of R˙ox. Therefore, it would be intriguing to interpret R˙ox as an H-cluster state distinct from Hox, Hox–CO, Hinact, and Hhyd. However, the absence of the corresponding FTIR data that could be associated with R˙ox prevents an unambiguous assignment at this moment.
Conclusions
In this study, we provided direct spectroscopic evidence for the presence of an unprecedented radical species in CbA5H, named R˙ox. We showed that the formation of R˙ox under oxidizing conditions is dependent on the presence of an active H-cluster harboring the native adt ligand that ensures an intact proton transfer pathway. While advanced spectroscopic and biochemical studies are underway aiming to reveal the identity of R˙ox, the combined results of our temperature-dependent and isotope-sensitive spectroscopic investigations already narrow down its location within the protein to either the H-cluster or its immediate vicinity. In line with the early onset of anaerobic oxidative inactivation and the formation of Hinact, the R˙ox signal appears either in the presence of O2 or upon applying oxidative conditions in the absence of it. Similar to the intact proton transfer pathway to Fed, R˙ox formation seems to be part of the redox sensing process that determines the reversible formation of Hinact in CbA5H.Data availability
All experimental data associated with the paper can be found in the article or in the ESI.†Author contributions
MH (first shared author): data curation, formal analysis, investigation, writing – original draft. AR (first shared author): data curation, formal analysis, investigation. YK: supervision, data curation, validation, writing – review & editing. VE: writing – review & editing. MW: conceptualization, writing – review & editing. TH (corresponding author): supervision, validation, resources, funding acquisition, conceptualization, writing – review & editing. MK (corresponding author): methodology, validation, supervision, resources, funding acquisition, conceptualization, writing – original draft, review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Shanika Yadav and Ulf-Peter Apfel for the adt and pdt ligands. We thank Silke Leimkühler for the ICP measurement. T. H. thanks the Deutsche Forschungsgemeinschaft for funding (RTG 2341 and HA 2555/10-1). We also thank the Studienstiftung des deutschen Volkes for supporting V. E. and A. R. This work is funded by the Deutsche Forschungsgemeinschaft (German Research Foundation) under Germany's Excellence Strategy (EXC 2033-390677874-RESOLV).References
- J. Tollefson, Nature, 2010, 464, 1262–1264 CrossRef CAS PubMed.
- M. K. Singla, P. Nijhawan and A. S. Oberoi, Environ. Sci. Pollut. Res., 2021, 28, 15607–15626 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- B. R. Glick, W. G. Martin and S. M. Martin, Can. J. Microbiol., 1980, 26, 1214–1223 CrossRef CAS PubMed.
- S. Gao, W. Fan, Y. Liu, D. Jiang and Q. Duan, Int. J. Hydrogen Energy, 2020, 45, 4305–4327 CrossRef CAS.
- G. Caserta, C. Papini, A. Adamska-Venkatesh, L. Pecqueur, C. Sommer, E. Reijerse, W. Lubitz, C. Gauquelin, I. Meynial-Salles, D. Pramanik, V. Artero, M. Atta, M. del Barrio, B. Faivre, V. Fourmond, C. Léger and M. Fontecave, J. Am. Chem. Soc., 2018, 140, 5516–5526 CrossRef CAS PubMed.
- J. Esselborn, L. Kertess, U. P. Apfel, E. Hofmann and T. Happe, J. Am. Chem. Soc., 2019, 141, 17721–17728 CrossRef CAS PubMed.
- A. Dubini and D. Gonzalez-Ballester, in Algae Biotechnology, 2016, pp. 165–193 Search PubMed.
- A. Silakov, E. J. Reijerse, S. P. J. Albracht, E. C. Hatchikian and W. Lubitz, J. Am. Chem. Soc., 2007, 129, 11447–11458 CrossRef CAS PubMed.
- Z.-P. P. Liu and P. Hu, J. Am. Chem. Soc., 2002, 124, 5175–5182 CrossRef CAS PubMed.
- A. Kubas, C. Orain, D. De Sancho, L. Saujet, M. Sensi, C. Gauquelin, I. Meynial-Salles, P. Soucaille, H. Bottin, C. Baffert, V. Fourmond, R. B. Best, J. Blumberger and C. Léger, Nat. Chem., 2017, 9, 88–95 CrossRef CAS PubMed.
- K. D. Swanson, M. W. Ratzloff, D. W. Mulder, J. H. Artz, S. Ghose, A. Hoffman, S. White, O. A. Zadvornyy, J. B. Broderick, B. Bothner, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2015, 137, 1809–1816 CrossRef CAS PubMed.
- S. Mebs, R. Kositzki, J. Duan, L. Kertess, M. Senger, F. Wittkamp, U. P. Apfel, T. Happe, S. T. Stripp, M. Winkler and M. Haumann, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 28–41 CrossRef CAS PubMed.
- S. Morra, M. Arizzi, F. Valetti and G. Gilardi, Biochemistry, 2016, 55, 5897–5900 CrossRef CAS PubMed.
- P. S. Corrigan, J. L. Tirsch and A. Silakov, J. Am. Chem. Soc., 2020, 142, 12409–12419 CrossRef CAS PubMed.
- M. Winkler, J. Duan, A. Rutz, C. Felbek, L. Scholtysek, O. Lampret, J. Jaenecke, U.-P. Apfel, G. Gilardi, F. Valetti, V. Fourmond, E. Hofmann, C. Léger and T. Happe, Nat. Commun., 2021, 12, 756 CrossRef CAS PubMed.
- J. H. Artz, D. W. Mulder, M. W. Ratzloff, C. E. Lubner, O. A. Zadvornyy, A. X. Levan, S. G. Williams, M. W. W. Adams, A. K. Jones, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2017, 139, 9544–9550 CrossRef CAS PubMed.
- H. J. Grande, W. R. Dunham, B. Averill, C. van Dijk and R. H. Sands, Eur. J. Biochem., 1983, 136, 201–207 CrossRef CAS PubMed.
- W. R. Hagen, A. van Berkel-Arts, K. M. Krüse-Wolters, W. R. Dunham and C. Veeger, FEBS Lett., 1986, 201, 158–162 CrossRef CAS PubMed.
- M. W. Adams, J. Biol. Chem., 1987, 262, 15054–15061 CrossRef CAS PubMed.
- A. J. Pierik, W. R. Hagen, J. S. Redeker, R. B. G. Wolbert, M. Boersma, M. F. J. M. Verhagen and H. J. Grande, Eur. J. Biochem., 1992, 209, 63–72 CrossRef CAS PubMed.
- B. Bennett, B. J. Lemon and J. W. Peters, Biochemistry, 2000, 39, 7455–7460 CrossRef CAS PubMed.
- S. P. J. Albracht, W. Roseboom and E. C. Hatchikian, J. Biol. Inorg Chem., 2006, 11, 88–101 CrossRef CAS PubMed.
- C. Kamp, A. Silakov, M. Winkler, E. J. Reijerse, W. Lubitz and T. Happe, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 410–416 CrossRef CAS PubMed.
- A. Adamska, A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2012, 51, 11458–11462 CrossRef CAS PubMed.
- G. Caserta, L. Pecqueur, A. Adamska-Venkatesh, C. Papini, S. Roy, V. Artero, M. Atta, E. Reijerse, W. Lubitz and M. Fontecave, Nat. Chem. Biol., 2017, 13, 779–784 CrossRef CAS PubMed.
- G. Rao and R. D. Britt, Inorg. Chem., 2018, 57, 10935–10944 CrossRef CAS PubMed.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, J. Biol. Inorg Chem., 2006, 11, 102–118 CrossRef CAS PubMed.
- D. W. Mulder, M. W. Ratzloff, M. Bruschi, C. Greco, E. Koonce, J. W. Peters and P. W. King, J. Am. Chem. Soc., 2014, 136, 15394–15402 CrossRef CAS PubMed.
- D. W. Mulder, Y. Guo, M. W. Ratzloff and P. W. King, J. Am. Chem. Soc., 2017, 139, 83–86 CrossRef CAS PubMed.
- M. Winkler, M. Senger, J. Duan, J. Esselborn, F. Wittkamp, E. Hofmann, U. P. Apfel, S. T. Stripp and T. Happe, Nat. Commun., 2017, 8(16115) Search PubMed.
- R. Cammack, Adv. Inorg. Chem., 1992, 38, 281–322 CrossRef CAS.
- H. Rupp, K. K. Rao, D. O. Hall and R. Cammack, Nat. Commun., 1978, 537, 255–269 CAS.
- M. W. W. Adams, Biochim. Biophys. Acta, Bioenerg., 1990, 1020, 115–145 CrossRef CAS.
- G. Berggren, A. Adamska, C. Lambertz, T. R. Simmons, J. Esselborn, M. Atta, S. Gambarelli, J. M. Mouesca, E. Reijerse, W. Lubitz, T. Happe, V. Artero and M. Fontecave, Nature, 2013, 499, 66–69 CrossRef CAS PubMed.
- M. M. Roessler, R. M. Evans, R. A. Davies, J. Harmer and F. A. Armstrong, J. Am. Chem. Soc., 2012, 134, 15581–15594 CrossRef CAS PubMed.
- J. Esselborn, C. Lambertz, A. Adamska-Venkatesh, T. Simmons, G. Berggren, J. Noth, J. Siebel, A. Hemschemeier, V. Artero, E. Reijerse, M. Fontecave, W. Lubitz and T. Happe, Nat. Chem. Biol., 2013, 9, 607–609 CrossRef CAS PubMed.
- W. Lubitz, E. Reijerse and M. van Gastel, Chem. Rev., 2007, 107, 4331–4365 CrossRef CAS PubMed.
- J. Esselborn, N. Muraki, K. Klein, V. Engelbrecht, N. Metzler-Nolte, U. P. Apfel, E. Hofmann, G. Kurisu and T. Happe, Chem. Sci., 2016, 7, 959–968 RSC.
- M. Sivaraja, D. B. Goodin, M. Smith and B. M. Hoffman, Science, 1989, 245, 738–740 CrossRef CAS PubMed.
- A. L. P. Houseman, P. E. Doan, D. B. Goodin and B. M. Hoffman, Biochemistry, 1993, 32, 4430–4443 CrossRef CAS.
- C. V. Popescu and E. Münck, J. Am. Chem. Soc., 1999, 121, 7877–7884 CrossRef CAS.
- R. Gilbert-Wilson, J. F. Siebel, A. Adamska-Venkatesh, C. C. Pham, E. Reijerse, H. Wang, S. P. Cramer, W. Lubitz and T. B. Rauchfuss, J. Am. Chem. Soc., 2015, 137, 8998–9005 CrossRef CAS.
- J. M. Mouesca, L. Noodleman, D. A. Case and B. Lamotte, Inorg. Chem., 1995, 34, 4347–4359 CrossRef CAS.
- J. Telser, M. J. Benecky, M. W. Adams, L. E. Mortenson and B. M. Hoffman, J. Biol. Chem., 1987, 262, 6589–6594 CrossRef CAS.
- A. Silakov, E. J. Reijerse and W. Lubitz, Eur. J. Inorg. Chem., 2011, 1056–1066 CrossRef CAS.
- A. Adamska-Venkatesh, T. R. Simmons, J. F. Siebel, V. Artero, M. Fontecave, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2015, 17, 5421–5430 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Material and methods, sample descriptions, experimental details and additional experimental data. See https://doi.org/10.1039/d2sc00385f |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |