
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptical resolution of 1,16-dihydroxytetraphenylene by chiral gold(III) complexation and its applications as chiral ligands in asymmetric catalysis†
Jia
Guo
a,
Wen-Bin
Xiong
a,
Hao-Ran
Ma
ab,
Luoyi
Fan
a,
You-Yun
Zhou
a,
Henry N. C.
Wong
 abc and
Jian-Fang
Cui
*a
abc and
Jian-Fang
Cui
*a
aDepartment of Chemistry, Southern University of Science and Technology, 1088 Xueyuan Blvd, Shenzhen 518055, China. E-mail: cuijf@sustech.edu.cn
bSchool of Science and Engineering, The Chinese University of Hong Kong (Shenzhen), 2001 Longxiang Blvd, Shenzhen 518172, China
cDepartment of Chemistry, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong SAR, China
First published on 24th March 2022
Abstract
We report herein a novel approach involving optical resolution of (±)-1,16-dihydroxytetraphenylene (DHTP) by chiral gold(III) complexation. This method features several key advantages, i.e., recyclability of chiral resolution reagents, feasibility of scaling up to gram quantities, and operational simplicity. On the basis of this method, which led to optically pure DHTP, a library of 2,15-diaryl (S)-DHTPs and several (S)-DHTP-derived phosphoramidite ligands were synthesized. Finally, the superior performance of a (S)-DHTP phosphoramidite ligand was demonstrated by efficient iridium-catalyzed asymmetric allylic alkynylation reactions.
Introduction
Tetraphenylene (1)1 is a nonplanar saddle-shaped molecule, featuring four benzene rings that are arranged alternatively above and below its eight-membered ring (Scheme 1A).2 As a molecule with a D2d molecular symmetry, 1 is achiral in nature. However, chiral tetraphenylenes can be obtained by introducing appropriate substituents into the benzene rings due to the loss of D2d symmetry, and also due to their extraordinarily high molecular inversion barrier.3 Owing to their unique geometry and rigid molecular frameworks, chiral tetraphenylenes are endowed with great potential to function as excellent chiral ligands/catalysts and building blocks.3b,4 It is therefore of great interest to design and prepare optically pure tetraphenylenes as novel ligands/catalysts for asymmetric reactions.1c In 2005, our group reported for the first time the use of tetraphenylene-derived phosphoramidite as chiral ligands in a Rh-catalyzed asymmetric hydrogenation.4c As a result, α-acylaminoacrylates were hydrogenated, leading to quantitative yields with excellent enantioselectivities (94–99% ee). In 2010, we also designed and synthesized tetraphenylene-derived organocatalysts for the Diels–Alder cycloaddition reaction between anthrones and maleimides.5 However, applications of chiral tetraphenylenes in asymmetric catalysis have still not been attractive, presumably due to the difficulty in obtaining enantiopure tetraphenylenes through asymmetric synthesis3b,4a,6 or by chiral resolution.4c,d,7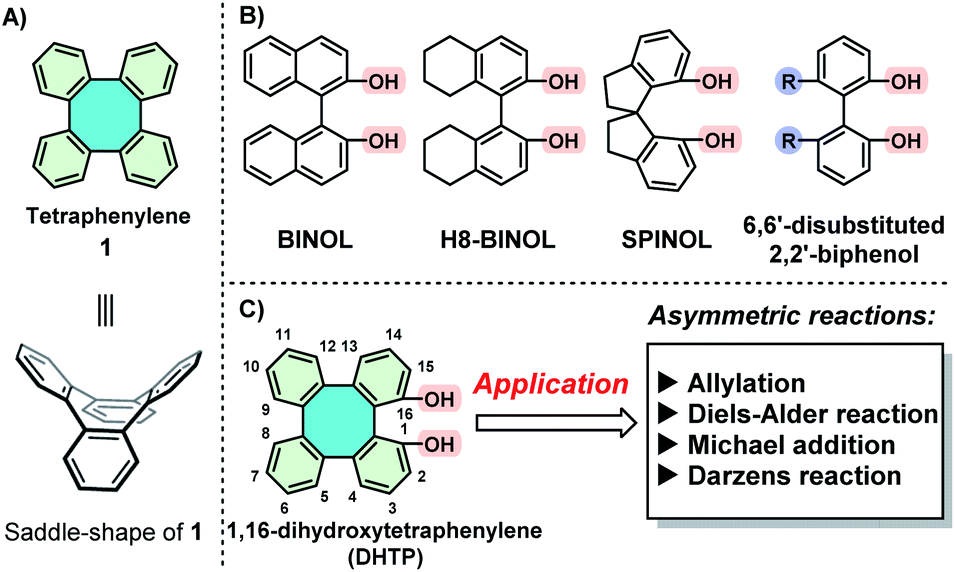 | ||
| Scheme 1 (A) Structure of tetraphenylene; (B) structure of BINOL, H8-BINOL, SPINOL and 6,6′-disubstituted 2,2′-biphenol; (C) structure of 1,16-dihydroxytetraphenylene (DHTP). | ||
Over the past few decades, atropisomeric biphenols, such as BINOL (1,1′-bi-2-naphthol), H8-BINOL, SPINOL (1,1′-spirobiindane-7,7′-diol) and 6,6′-disubstituted-2,2′-biphenol (Scheme 1B), have been well-developed into privileged scaffolds for the design and synthesis of a variety of chiral ligands/catalysts.8 Inspired by the architectural similarity of these structurally floppy biphenols, we opine that it is thought-provoking to apply structurally rigid 1,16-dihydroxytetraphenylene (DHTP)4b,9 as a platform for comparison, in order to explore their application in asymmetric catalysis (Scheme 1C). Consequently, the development of enantiopure DHTPs as chiral ligands/catalysts in various asymmetric reactions, including asymmetric allylation, the Diels–Alder reaction, Michael addition and the Darzens reaction, has recently witnessed significant progress.10 Nonetheless, the utilization of chiral DHTPs in asymmetric catalysis is limited and still in its infancy because an efficient approach to enantiomeric (S)-DHTP and (R)-DHTP in large quantities is still challenging.1d,e,g
To the best of our knowledge, so far there are only two available pathways to access enantiopure (S)-DHTP and (R)-DHTP. The first pathway is the diastereomeric separation of a pair of bis-(S)-camphorsulfonates of (±)-DHTP by a conventional column chromatographic separation on silica gel (Scheme 2A).4b This method suffers from the use of an expensive and non-recyclable chiral resolution reagent, as well as from the tedious and time-consuming chromatographic purification of the resulting diastereomeric camphorsulfonates. The second pathway is the enantiomeric separation of (±)-DHTP on a chiral stationary phase by preparative high performance liquid chromatography (Scheme 2B).10a Although this method is straightforward, avoiding multistep reactions, its high-cost due to the use of large amounts of expensive HPLC grade solvents is definitely discouraging, not to mention the lengthy chromatographic process. Furthermore, the primary limitation of this approach is that these preparative HPLC instruments and chiral columns, are usually available only in commercial laboratories. Therefore, attempts to look for practical and reliable methods to access enantiopure (S)-DHTP and (R)-DHTP in large quantities are urgent and essential. Herein we report a newly developed approach for the optical resolution of (±)-DHTP by chiral gold(III) complexes (Scheme 2C). This method not only circumvents expensive and time-consuming chromatographic steps, but most importantly features noteworthy advantages including (a) the recyclability of chiral resolution reagents, (b) the feasibility of gram scale manipulation, and (c) the simplicity of the operation.
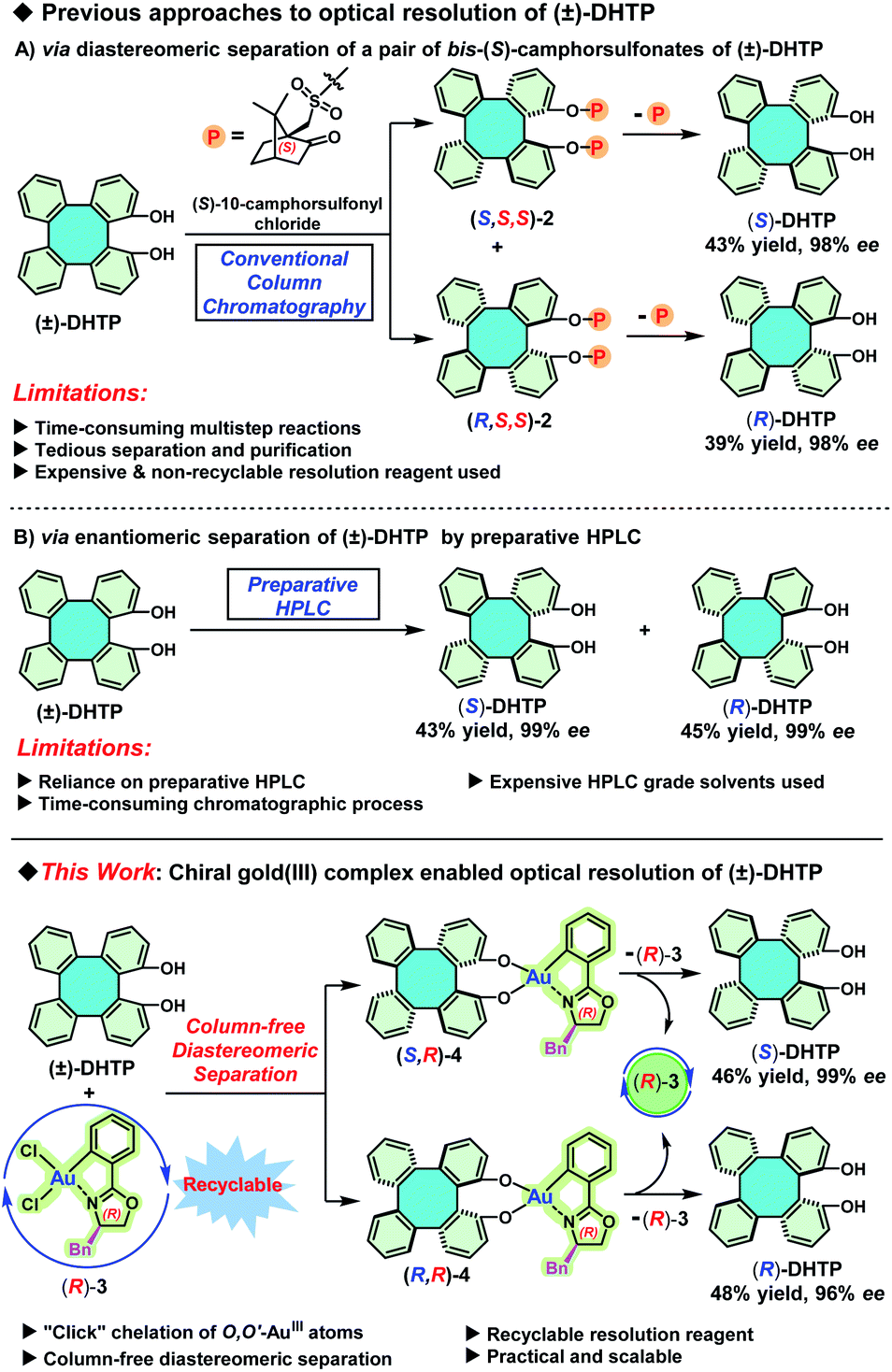 | ||
| Scheme 2 Previous approaches to optical resolution of (±)-DHTP and our newly developed strategy. Bn = benzyl. | ||
Results and discussion
We have previously demonstrated that BINOLs and 2,2′-biphenols can be well-chelated with chiral oxazoline-based C^N-cyclometalated gold(III) dichloride [(C^N)AuCl2, C^N = 2-aryl oxazolinyl] to form C,O- and O,O′-AuIII chelated stable chiral gold(III) complexes, respectively.11 Hence, we reasoned that (±)-DHTP might react with an enantiopure oxazoline-based C^N-cyclometalated gold(III) dichloride, resulting probably in two separable diastereomers as access to optically pure (S)-DHTP and (R)-DHTP, respectively. Our investigation commenced with the reaction between (±)-DHTP and enantiopure oxazoline-based cyclometalated gold(III) dichloride (R)-3.11a Thus, reaction of (±)-DHTP (0.5 mmol) with (R)-3 (0.5 mmol) in the presence of Cs2CO3 in CH2Cl2 at room temperature (25 °C) gave rapidly a yellow precipitate (Scheme 3A). Interestingly, it was found that the solubility of (S,R)-4 and (R,R)-4 in CH2Cl2 was significantly different: (S,R)-4 is almost insoluble in CH2Cl2, leading to the precipitate, while (R,R)-4 showed exceptional solubility in CH2Cl2. The precipitate was collected by filtration and washed with H2O and CH2Cl2 to give (S,R)-4. Subsequently, (S,R)-4 was treated with 6 N HCl in CH2Cl2, followed by column chromatography to give (S)-DHTP in 44% yield and the recovered (R)-3 in 43% yield.11a The optical purity of the obtained (S)-DHTP was determined to be 99% ee by HPLC analysis (Scheme 3B).10a Then, treatment of the filtrate with 6 N HCl afforded (R)-DHTP in 49% yield with 90% ee, and (R)-3 was recovered in 51% yield. Notably, this optical resolution specifically depended on the use of a benzyl (Bn) substituted chiral oxazoline-based gold(III) complex, because phenyl-, isopropyl- or tertiary butyl-substituted analogs did not lead to the formation of a precipitate, therefore optical resolution could not be realized.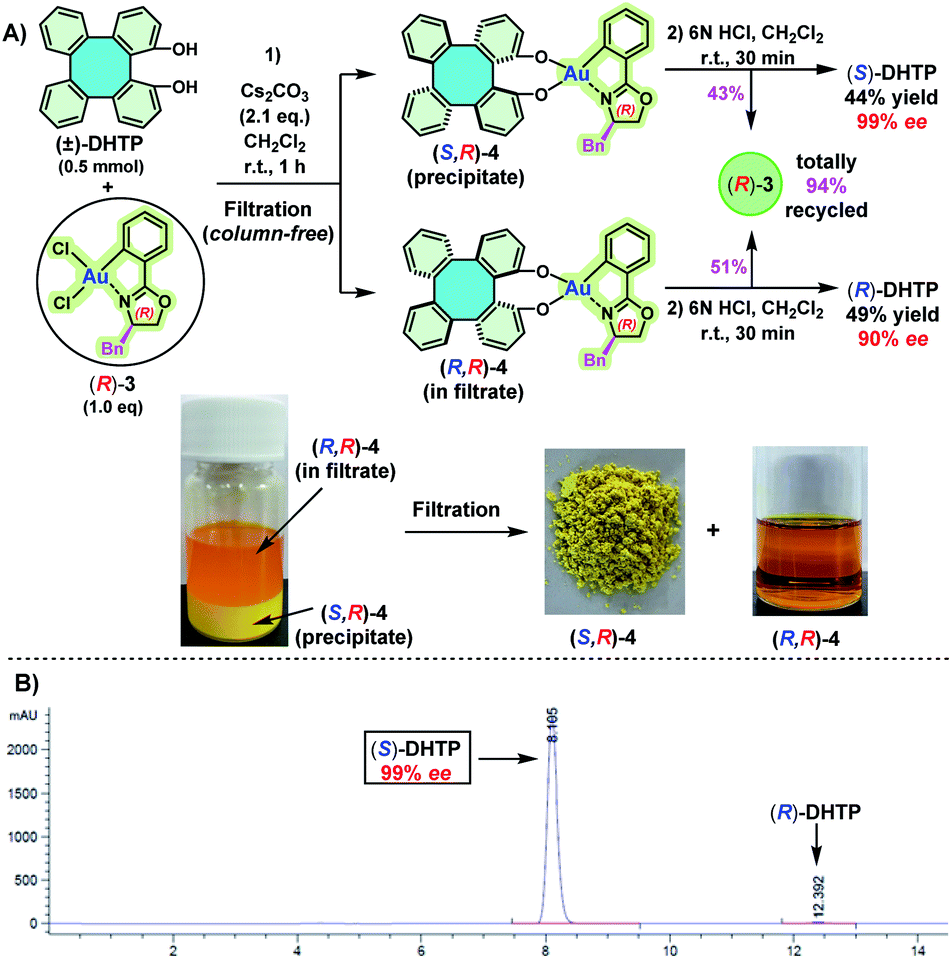 | ||
| Scheme 3 (A) Optical resolution of (±)-DHTP by using enantiopure (R)-3. Reaction conditions: step (1): (±)-DHTP (0.5 mmol), (R)-3 (0.5 mmol), Cs2CO3 (2.1 eq.), CH2Cl2 (10 mL), room temperature (25 °C), reaction time: 1 h; step (2): CH2Cl2 (10 mL), HCl (6 N, aq.), room temperature, reaction time: 30 min. Yield of the isolated product. (B) HPLC spectrum of the obtained (S)-DHTP. Bn = benzyl. | ||
To demonstrate the scalability and practicability of this process, a 30 mmol reaction was first carried out (Table 1, entry 1). Treatment of (±)-DHTP (30 mmol, 10.1 g) with (R)-3 (30 mmol, 15.1 g) by the standard procedure provided (S)-DHTP in 45% yield with 99% ee, (R)-DHTP in 49% yield with 88% ee, and (R)-3 in 98% yield (14.8 g).
|
|
||||
|---|---|---|---|---|
| Entry | Scale of (±)-DHTP (mmol) | Yield and ee of (S)-DHTP | Yield and ee of (R)-DHTP | Recovery yield of (R)-3 (%) |
| Yield [ee] (%) | Yield [ee] (%) | |||
| a Reaction conditions: step (1): (±)-DHTP (1.0 eq.), (R)-3 (1.0 eq.), Cs2CO3 (2.1 eq.), CH2Cl2 ([substrate] = 0.1 M), room temperature (25 °C), reaction time: 1 h; step (2): CH2Cl2 ([substrate] = 0.1 M), HCl (6 N, aq.), room temperature, reaction time: 30 min. b Yield of the isolated product. c ee% was determined by chiral HPLC analysis. d (R)-3 was obtained from the last reaction cycle. | ||||
| 1 | 30 | 45 [99] | 49 [88] | 98 (14.8 g) |
| 2d | 25 | 47 [99] | 50 [95] | 99 |
| 3d | 20 | 45 [99] | 50 [89] | 98 |
| 4d | 15 | 46 [99] | 49 [93] | 98 |
| 5 | 10 | 46 [99] | 48 [96] | 99 |
| 6d | 5 | 48 [99] | 50 [87] | 98 |
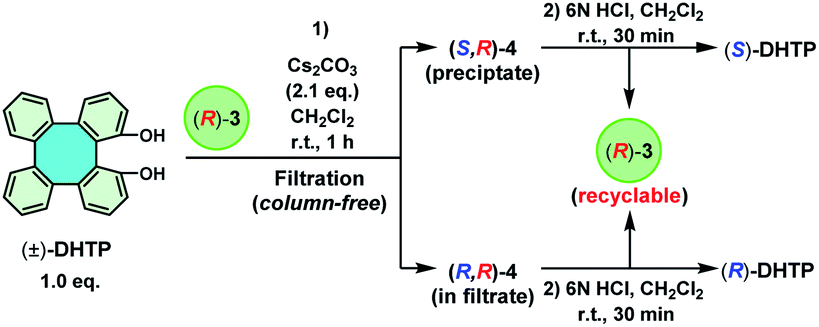
An important advantage of employing (R)-3 instead of (S)-10-camphorsulfonyl chloride is its recyclability. Based on recovered (R)-3 (14.8 g) from the 30 mmol reaction, the recyclability of (R)-3 was examined. A series of resolution steps for (±)-DHTP in 25, 20, 15, 10 and 5 mmol quantities were carried out by using (R)-3 recovered from the last step, respectively (Table 1, entries 2–6). The results showed that (R)-3 could be repeatedly used for 6 cycles without any adverse effect on resolution performance to give (S)-DHTP in excellent yield (45–48%) and optical purity (99% ee), as well as in excellent recovery yield (98–99%) of (R)-3 in each cycle. It is noteworthy that the 10 mmol scale resolution process, providing (S)-DHTP in 46% yield with 99% ee and (R)-DHTP in 48% yield with 96% ee, was the optimal manipulation result (Table 1, entry 5). To our delight, (R)-3 was still recovered in 98% yield at the end of the 6th cycle. These results indicated that (R)-3 is extremely stable during the optical resolution process and could even be promisingly reused for many more cycles.
The use of (R)-3 only offered (R)-DHTP with a meager 96% ee. In order to obtain (R)-DHTP with a higher optical purity, enantiopure (S)-3 was employed to perform the optical resolution of (±)-DHTP (Scheme 4). In the reaction of (±)-DHTP (10 mmol) with (S)-3 (10 mmol) in the presence of Cs2CO3 in CH2Cl2 within 1 h, a yellow precipitate was similarly formed. (R,S)-4 as the precipitate was collected by filtration, which was subsequently treated with 6 N HCl to give (R)-DHTP in excellent yield (46%) and in high optical purity (>99% ee). Complex (S)-3 was recovered in 45% yield. Likewise, treatment of the filtrate containing (S,S)-4 with 6 N HCl furnished (S)-DHTP in 52% yield with 90% ee, and (S)-3 in 52% yield.
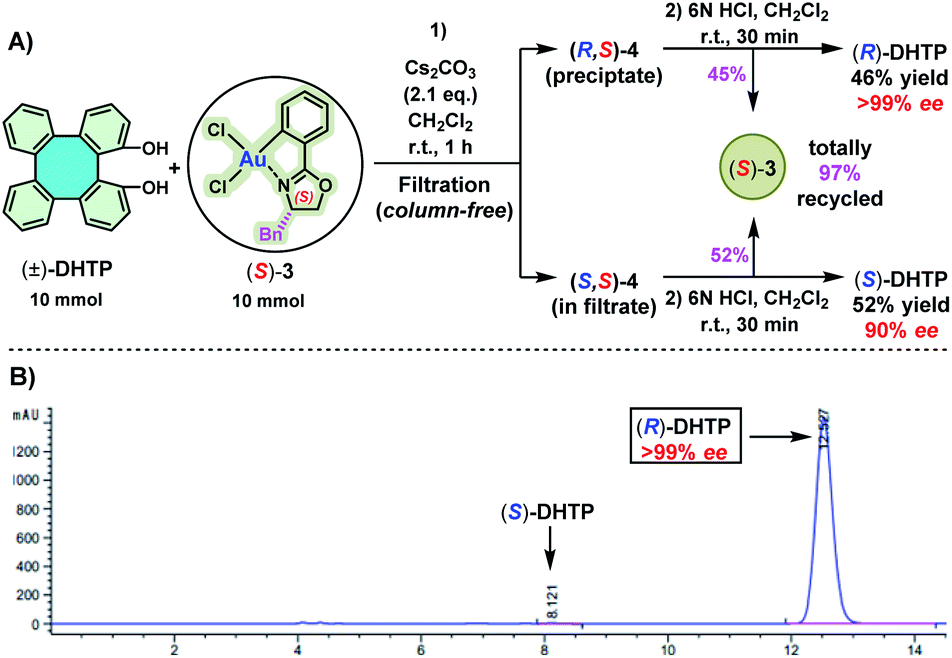 | ||
| Scheme 4 (A) Optical resolution of (±)-DHTP by using enantiopure (S)-3. Reaction conditions: step (1): (±)-DHTP (10 mmol), (S)-3 (10 mmol), Cs2CO3 (2.1 eq.), CH2Cl2 (100 mL), room temperature (25 °C), reaction time: 1 h; step (2): CH2Cl2 (50 mL), HCl (6 N, aq.), room temperature, reaction time: 30 min. Yield of the isolated product. (B) HPLC spectrum of the obtained (R)-DHTP. Bn = benzyl. | ||
To obtain an insight into the chelated mode of (S)-DHTP and (R)-DHTP with (S)-3 and (R)-3, respectively, the preparation of four DHTP/oxazoline Au(III) complexes (S,R)-4, (R,S)-4, (S,S)-4 and (R,R)-4 by using enantiopure DHTP and enantiopure 3 was studied (Scheme 5A). Thus, the treatment of (S)-DHTP and (R)-DHTP with (S)-3 and (R)-3 in the presence of Cs2CO3 in methanol at room temperature (25 °C) successfully afforded four diastereomers (S,R)-4, (R,S)-4, (S,S)-4 and (R,R)-4, respectively, in 86–92% yields. The four resulting chiral Au(III) complexes were light, moisture, and heat insensitive. They remained intact even upon exposure to air for months. These chiral Au(III) complexes were characterized by 1H and 13C NMR spectroscopic studies, and high-resolution ESI-MS. Furthermore, the configurations of (S,R)-4, (R,S)-4, (S,S)-4 and (R,R)-4, all adopting an O,O′-chelating mode, were confirmed by X-ray crystallographic analyses (Scheme 5B).12 Circular dichroism (CD) spectroscopy was carried out to give additional chiroptical evidence of these four diastereomers. The symmetry of the CD spectra clearly demonstrated the enantiomeric relationship of (S,R)-4 and (R,S)-4, as well as (S,S)-4 and (R,R)-4 (Scheme 5C).
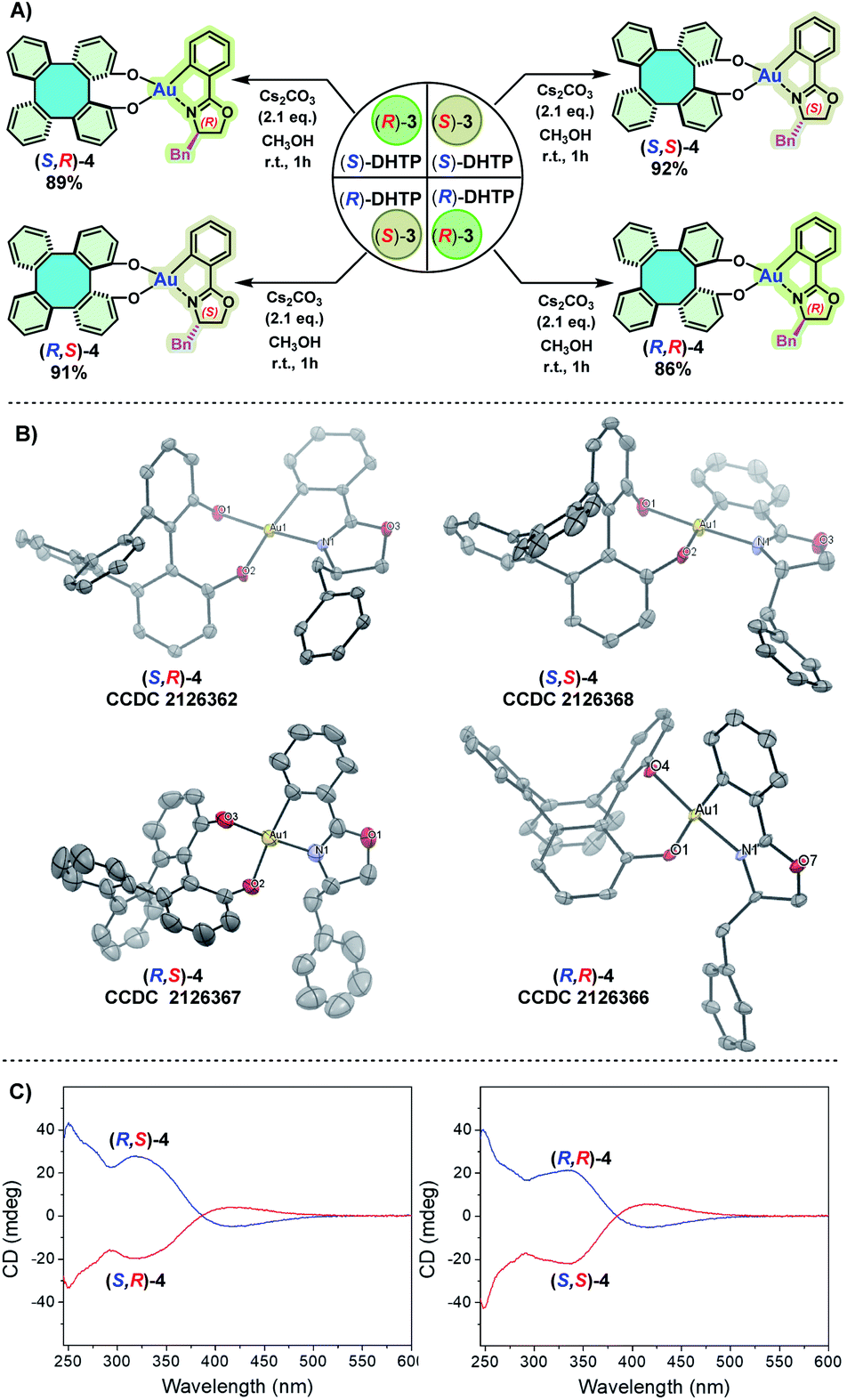 | ||
| Scheme 5 (A) Synthesis of enantiopure DHTP/oxazoline Au(III) complexes. Reaction conditions: DHTP (0.1 mmol), 3 (0.11 mmol), Cs2CO3 (2.1 eq.), CH3OH (2 mL), room temperature (25 °C), reaction time: 1 h. Yield of the isolated product. (B) X-ray crystal structures of (S,R)-4, (R,S)-4, (S,S)-4 and (R,R)-4. Displacement ellipsoids are drawn at the 50% probability level. Solvent molecules and H atoms are omitted for clarity. (C) Circular dichroism (CD) spectra of (S,R)-4 and (R,S)-4, as well as (S,S)-4 and (R,R)-4 in CHCl3 at a concentration of 2.0 × 10−5 M. Bn = benzyl. | ||
A lot of studies have demonstrated that aryl substituents at C-3 and C-3′ of the BINOL skeleton display significant influences towards a variety of asymmetric reactions.13–18 For this reason, 3,3′-diaryl BINOLs are notably privileged skeletons for the construction of valuable chiral ligands/catalysts,13 such as phosphoramidites,14 phosphoric acids,15 phosphoramides,16 and phase-transfer catalysts.17 However, due to the synthetic intricacy in realizing enantiopure DHTP in significant quantities, an efficient preparative method towards enantiopure 2,15-diaryl DHTPs has remained an almost unexplored territory. Consequently, only a very few examples have been reported.10c Inspired by the availability of a larger amount of enantiopure DHTP, we next turned our attention to constructing a library of 2,15-diaryl DHTPs as chiral ligands/catalysts. In accordance with the well-established strategy for preparing 3,3′-diaryl BINOLs, a key intermediate, namely, 2,15-bis-borylated (S)-MOM-DHTP [(S)-6] was synthesized (Scheme 6).18 Thus, first the methoxymethylation of (S)-DHTP (>99% ee) by methoxymethyl chloride in the presence of NaH in THF afforded (S)-MOM-DHTP [(S)-5] in 95% yield. Subsequently, a direct ortho-lithiation of (S)-5 by n-BuLi in dry THF, followed by borylation with iPrOBPin (2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane) provided the key intermediate (S)-6 as a white solid in 90% yield with 99% ee. Notably, the borylation process was carried out on a 20 mmol scale without difficulties.
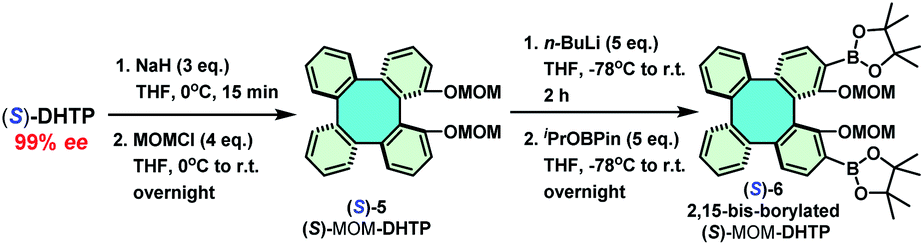 | ||
| Scheme 6 Synthetic routes for methoxymethylation and borylation of (S)-DHTP. MOM = methoxymethyl, Pin = pinacol, and iPrOBPin = 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane. | ||
With an ample quantity of (S)-6 in hand, we started to construct the library of 2,15-diaryl DHTPs by Suzuki coupling reactions between (S)-6 and a series of aryl bromides (Table 2).18 The Suzuki coupling reaction of (S)-6 and phenyl bromide was first performed with Pd(PPh3)4 in 1,4-dioxane/H2O at 80 °C using K2CO3 as the base, whose coupling product was subsequently treated with concentrated hydrochloric acid to provide 2,15-diphenyl (S)-DHTP [(S)-8a] in 90% yield with 99% ee. Then, various 4-substituted phenyl bromides bearing either electron-donating (methyl, t-butyl, phenyl and methoxyl) or electron-withdrawing (Cl, F, CF3, and NO2) groups were employed to expand the scope of 2,15-diaryl DHTPs, providing (S)-8b–i with good to excellent yields (70–90%). In addition, 2-naphthyl, 9-anthryl, as well as several di- and tri-substituted aryl bromides were also found to be compatible with this coupling process, affording (S)-8j–p in 75–90% yield. It was uncovered that sterically hindered aryl bromides, such as 2,6-(Me)2C6H3Br and 2,4,6-(Me)3C6H2Br, only led to (S)-8q and (S)-8r with relatively low yields (56% and 42%, respectively), and extremely sterically hindered aryl bromides, such as 2,4,6-(iPr)3C6H2Br and 2,4,6-(Cy)3C6H2Br even failed to furnish the corresponding products (S)-8s and (S)-8t under standard conditions. Nonetheless, (S)-8s and (S)-8t were obtained in good yields (75% and 70%, respectively) under a modified condition in which a catalytic system Pd(PPh3)4/Pd(dba)2/(±)-BIDIME was employed and K3PO4 was used as the base in toluene at 100 °C.18c,19,20 It has been known that the concomitant limitation in the synthesis of enantiomerically pure 3,3′-diaryl BINOLs under either basic or acidic conditions is their potential racemization.13a,21 However, due to the rigidity of the DHTPs, their racemization has never been observed. The configurations and optical purities (98–99% ee) of all DHTPs remained consistent throughout the synthetic process. For example, (S)-8 showed no racemization even at 60 °C or 80 °C for 6 h or 24 h during the demethoxymethylation step with conc. HCl.
a Reaction conditions: step (1): (S)-6 (1 mmol), 7 (4.0 eq.), Pd(PPh3)4 (10 mol%) K2CO3 (6.0 eq.), 1,4-dioxane (16 mL), H2O (4 mL), 80 °C, reaction time: 6 h; step (2): THF (10 mL), conc. HCl (2 mL), 80 °C, reaction time: 6 h.
b Yield of the isolated product.
c ee% was determined by chiral HPLC analysis.
d Reaction time of step 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24 h.
e Modified conditions: step (1): (S)-6 (1 mmol), 7 (4.0 eq.), Pd(PPh3)4 (5 mol%), Pd(dba)2 (10 mol%), (±)-BIDIME (20 mol%), K3PO4 (6.0 eq.), toluene (20 mL), 100 °C, reaction time: 24 h; step (2): 1,4-dioxane (10 mL), conc. HCl (2 mL), 80 °C, reaction time: 24 h. BIDIME = 3-(tert-butyl)-4-(2,6-dimethoxy-phenyl)-2,3-dihydrobenzo[d][1,3]oxaphosphole. :24 h.
e Modified conditions: step (1): (S)-6 (1 mmol), 7 (4.0 eq.), Pd(PPh3)4 (5 mol%), Pd(dba)2 (10 mol%), (±)-BIDIME (20 mol%), K3PO4 (6.0 eq.), toluene (20 mL), 100 °C, reaction time: 24 h; step (2): 1,4-dioxane (10 mL), conc. HCl (2 mL), 80 °C, reaction time: 24 h. BIDIME = 3-(tert-butyl)-4-(2,6-dimethoxy-phenyl)-2,3-dihydrobenzo[d][1,3]oxaphosphole.
|
|---|
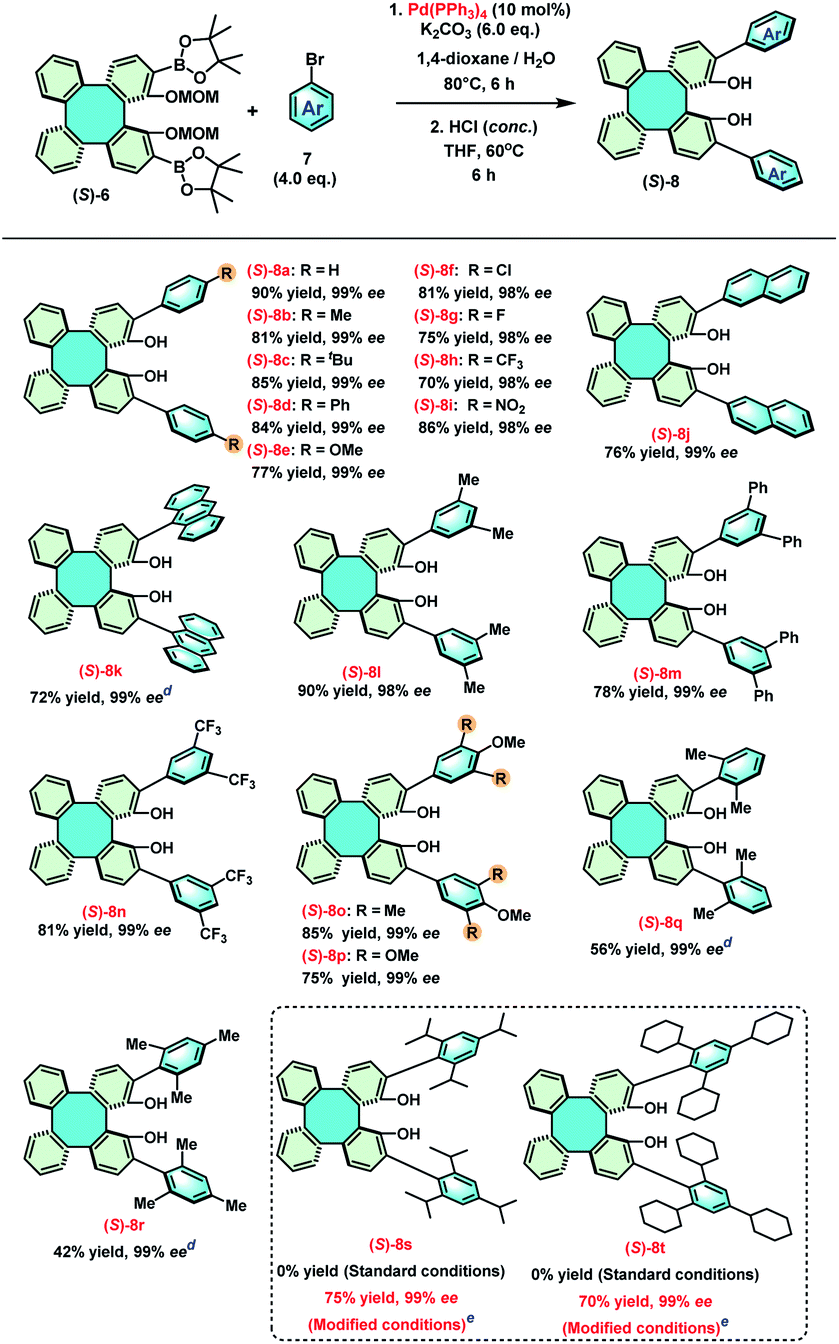
|
Chiral phosphoramidites have been demonstrated as highly versatile and privileged ligands in metal-catalyzed asymmetric reactions.14 Thus, the synthesis of (S)-DHTP-derived phosphoramidite ligands was performed to further demonstrate the potential application of chiral DHTP in asymmetric catalysis. Treatment of enantiopure (S)-DHTP (>99% ee) with PCl3 in the presence of Et3N with the subsequent addition of the corresponding amine, phosphoramidite ligands (S)-L1, (S)-L2, (S,S,S)-L3, (S)-L4 and (S)-L5 were obtained in good yields (72–85%), respectively (Scheme 7).22 In addition, the framework and absolute configuration of (P, olefin)-ligand (S)-L1 were well-defined by X-ray crystallographic analysis (CCDC 2124171†).12
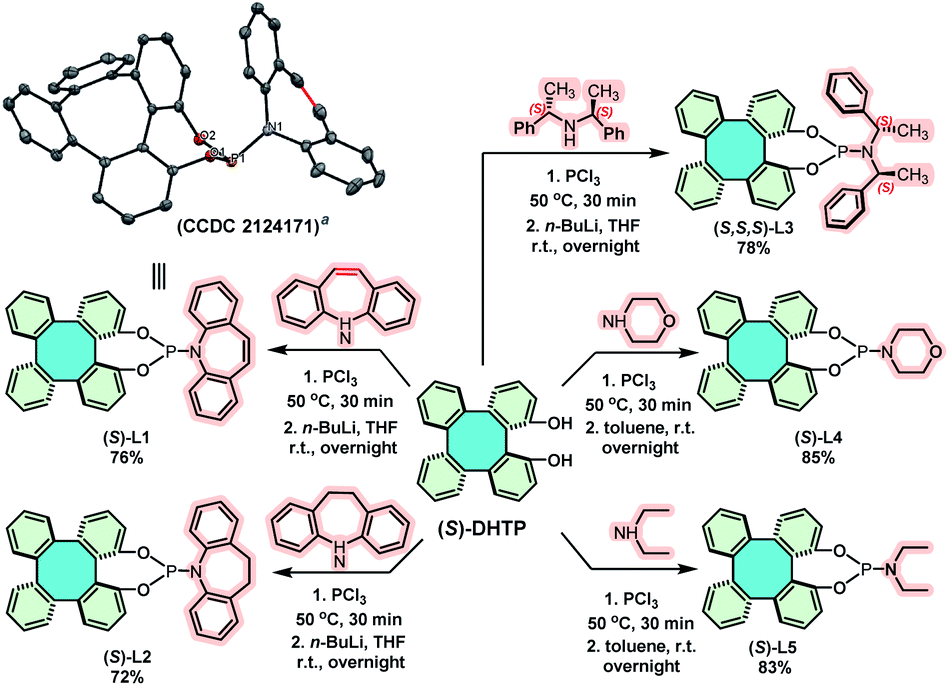 | ||
| Scheme 7 Synthesis of (S)-DHTP derived phosphoramidite ligands. a X-ray crystal structures of (S)-L1, displacement ellipsoids are drawn at the 50% probability level and H atoms are omitted for clarity. | ||
Next, we selected an iridium-catalyzed allylic alkynylation between racemic allylic alcohols and potassium alkynyl-trifluoroborates, previously reported by Carreira using BINOL-derived (S)-L7 as the chiral ligand, to test the efficiency of (S)-DHTP-derived phosphoramidite ligands (Scheme 8).14c,23 Employing (S)-L1 as the chiral ligand, the iridium-catalyzed allylic alkynylation of 9a and 10a afforded 11a in 86% yield with 99% ee. However, 11a was not obtained when (S)-L2 was used as the ligand under the same conditions. (S,S,S)-L3 afforded 11a only in 14% yield with 3% ee. These preliminary results indicated that the olefin moiety of the amine might be essential. When a SPINOL-based phosphoramidite (S)-L6 was used as the chiral ligand, slightly lower yield (79%) and enantioselectivity (97% ee) of 11a were achieved, as compared with those resulting from (S)-L1. For comparison, Carreira reported that the BINOL-derived phosphoramidite (S)-L7 gave 11a in 88% yield with 99% ee.23
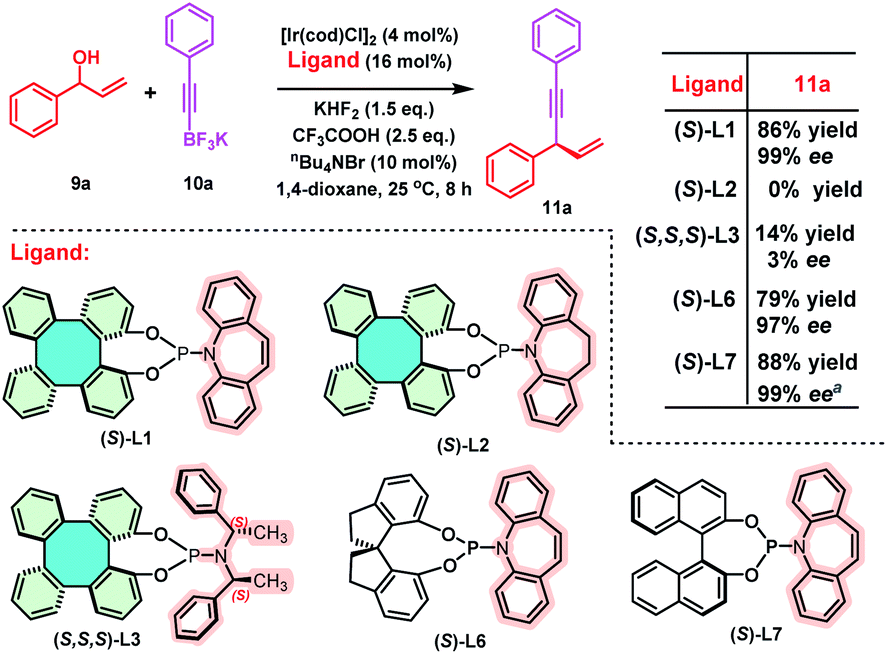 | ||
| Scheme 8 Iridium-catalyzed enantioselective allylic alkynylation. Reaction conditions: 9a (0.2 mmol, 1.0 eq.), 10a (0.3 mmol, 1.5 eq.), [Ir(cod)Cl]2 (4 mol%), ligand (16 mol%), 1,4-dioxane (0.4 mL), KHF2 (1.5 eq.), CF3COOH (2.5 eq.), nBu4NBr (10 mol%). Yield of the isolated product. ee% was determined by chiral HPLC analysis. a Reaction time: 6 h (see ref. 23). | ||
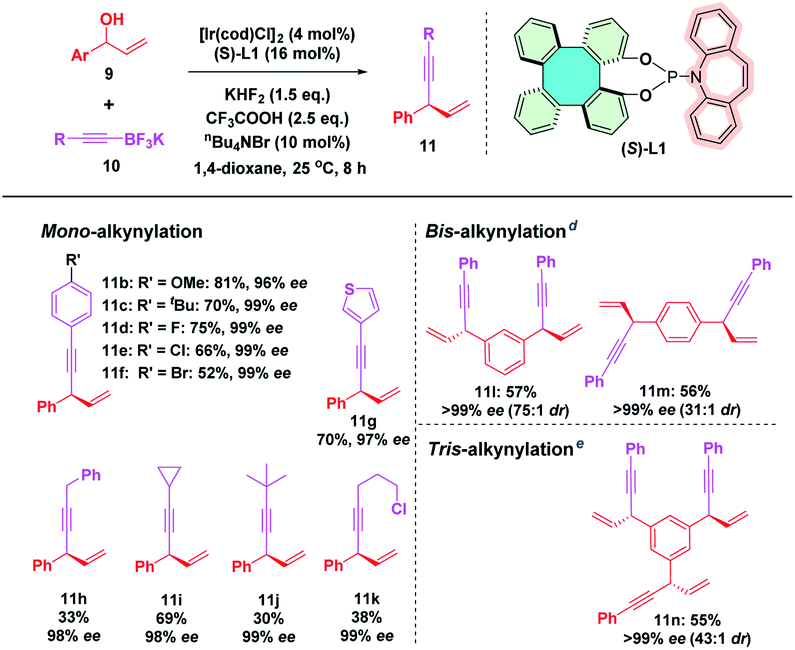
The results shown by (S)-L1 and BINOL-derived (S)-L7 encouraged us to expand the substrate scope to further evaluate the efficiency of (S)-L1 in this iridium-catalyzed enantioselective allylic alkynylation (Table 3). To our delight, a range of phenyl alkynyltrifluoroborates bearing either electron-donating (methoxyl and t-butyl) or electron-withdrawing (F, Cl, and Br) groups reacted smoothly under the Ir/(S)-L1 catalytic system, affording the corresponding products 11b–f in good yields and excellent enantioselectivities. In addition, heteroaromatic (3-thienyl) and aliphatic alkynyltrifluoroborates also gave products 11g–k in high enantioselectivities although the yields were relatively low. It is noteworthy that substrates containing two allylic alcohol moieties proceeded smoothly, leading to the formation of the corresponding products 11l and 11m in good yields (57% and 56%) and in 99% ee. Moreover, a triallylic alcohol substrate also afforded the tris-alkynylation product 11n containing three chiral centers in good yield (55%) and excellent stereoselectivity (>99% ee with 43:1 dr). Even though relatively low yields of some cases were obtained, the high level of enantiocontrol indicates that (S)-L1 indeed exhibits excellent efficiency as BINOL-derived phosphoramidite (S)-L7 towards the enantioselective iridium-catalyzed allylic alkynylation reaction. The comparison of X-ray crystallographic structural data of (S)-L1 (CCDC 2124171†) with those of SPINOL-derived (S)-L6 (CCDC 1439599) and BINOL-derived (S)-L7 (CCDC 694272) indicates that (S)-L1 has a larger dihedral angle (54.6°) between the two phenol rings than the corresponding angles of (S)-L6 (53.2°) and (S)-L7 (53.1°).24 The larger dihedral angle and the rigid skeleton of tetraphenylene might render (S)-L1 rather sensitive towards steric repulsion from the substrate, thereby leading to excellent enantiocontrol. This outcome points to the prospect that (S)-L1 might be a fine alternative to BINOL-derived (S)-L7 in asymmetric catalysis.
| a Reaction conditions: 9 (0.4 mmol, 1.0 eq.), 10 (0.6 mmol, 1.5 eq.), 1,4-dioxane (0.8 mL). b Yield of the isolated product. c ee% was determined by chiral HPLC analysis. d 9 (0.4 mmol, 1.0 eq.), 10 (1.2 mmol, 3.0 eq.), [Ir(cod)Cl]2 (8 mol%), (S)-L1 (32 mol%), 1,4-dioxane (1.6 mL), KHF2 (3.0 eq.), CF3COOH (5.0 eq.), nBu4NBr (20 mol%). e 9 (0.4 mmol, 1.0 eq.), 10 (1.8 mmol, 4.5 eq.), [Ir(cod)Cl]2 (12 mol%), (S)-L1 (48 mol%), 1,4-dioxane (2.4 mL), KHF2 (4.5 eq.), CF3COOH (7.5 eq.), nBu4NBr (30 mol%). |
|---|
|
Conclusions
In summary, we have established a novel approach to optical resolution of (±)-1,16-dihydroxytetraphenylene (DHTP) using chiral gold(III) complexes. This efficient and reliable method can provide (S)-DHTP and (R)-DHTP in large quantities. Accordingly, a library of 2,15-diaryl (S)-DHTPs and several (S)-DHTP-derived phosphoramidite ligands were synthesized for the first time. The outstanding performance of (S)-DHTP-derived phosphoramidite ligands was demonstrated by an iridium-catalyzed asymmetric allylic alkynylation reaction. It is envisioned that this practical and scalable approach to optically pure DHTP and well-established synthesis of 2,15-diaryl (S)-DHTPs will open a new synthetic avenue, leading to further development in the application of chiral DHTP-derived ligands/catalysts. In this connection, preparations of more (S)-DHTP-derived mono- and bis-phosphine ligands, as well as chiral phosphoric acid catalysts are currently underway in our laboratory.Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.† Crystallographic data for compounds (S)-L1, (S,R)-4, (R,R)-4, (R,S)-4 and (S,S)-4 have been deposited in the Cambridge Crystallographic Data Centre under accession numbers CCDC 2124171, 2126362, 2126366–2126368.†Author contributions
J.-F. C. and H. N. C. W. conceived the project. J.-F. C. and J. G. performed the experimental work. H.-R. M., W.-B. X. and L. F. synthesized some starting materials, and collected and analysed the spectroscopic data. J.-F. C., H. N. C. W. and Y.-Y. Z. wrote the manuscript. All of the authors discussed the results and contributed to the preparation of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support of the Research Start-up Fund from Southern University of Science and Technology, the Innovation and Technology Commission (Hong Kong) in the form of a subsidy to the State Key Laboratory of Synthetic Chemistry, the University Development Fund Grants from The Chinese University of Hong Kong (Shenzhen). We thank Dr Xiaoyong Chang for X-ray crystallographic analysis. H. N. C. W. acknowledges with gratitude the appointment as a Long-term Distinguished Visiting Professor by Southern University of Science and Technology.Notes and references
- (a) X.-L. Hou, H. Huang and H. N. C. Wong, Synlett, 2005, 1073–1089 CAS; (b) A. Rajca, S. Rajca, M. Pink and M. Miyasaka, Synlett, 2007, 1799–1822 CrossRef CAS; (c) H. Huang, C.-K. Hau, C. C. M. Law and H. N. C. Wong, Org. Biomol. Chem., 2009, 7, 1249–1257 RSC; (d) A. Rajca and S. Rajca, Angew. Chem., Int. Ed., 2010, 49, 672–674 CrossRef CAS PubMed; (e) J.-W. Han, H. Wong, J.-X. Chen, X. Li and X.-S. Peng, Synlett, 2013, 2188–2198 CrossRef CAS; (f) J.-W. Han, X. Li and H. N. C. Wong, Chem. Rec., 2015, 15, 107–131 CrossRef CAS PubMed; (g) J.-W. Han, X.-S. Peng and H. N. C. Wong, Natl. Sci. Rev., 2017, 4, 892–916 CrossRef CAS.
- (a) I. L. Karle and L. O. Brockway, J. Am. Chem. Soc., 1944, 66, 1974–1979 CrossRef CAS; (b) H. Irngartinger and W. R. K. Reibel, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 1724–1728 CrossRef.
- (a) P. Rashidi-Ranjbar, Y. M. Man, J. Sandstroem and H. N. C. Wong, J. Org. Chem., 1989, 54, 4888–4892 CrossRef CAS; (b) A. Rajca, A. Safronov, S. Rajca and J. Wongsriratanakul, J. Am. Chem. Soc., 2000, 122, 3351–3357 CrossRef CAS; (c) S. M. Bachrach, J. Org. Chem., 2009, 74, 3609–3611 CrossRef CAS PubMed; (d) H. Huang, T. Stewart, M. Gutmann, T. Ohhara, N. Niimura, Y.-X. Li, J.-F. Wen, R. Bau and H. N. C. Wong, J. Org. Chem., 2009, 74, 359–369 CrossRef CAS PubMed.
- (a) A. Rajca, H. Wang, P. Bolshov and S. Rajca, Tetrahedron, 2001, 57, 3725–3735 CrossRef CAS; (b) J.-F. Wen, W. Hong, K. Yuan, T. C. W. Mak and H. N. C. Wong, J. Org. Chem., 2003, 68, 8918–8931 CrossRef CAS PubMed; (c) H.-Y. Peng, C.-K. Lam, T. C. W. Mak, Z. Cai, W.-T. Ma, Y.-X. Li and H. N. C. Wong, J. Am. Chem. Soc., 2005, 127, 9603–9611 CrossRef CAS PubMed; (d) F. Lin, H.-Y. Peng, J.-X. Chen, D. T. W. Chik, Z. Cai, K. M. C. Wong, V. W. W. Yam and H. N. C. Wong, J. Am. Chem. Soc., 2010, 132, 16383–16392 CrossRef CAS PubMed; (e) C. Cheng, Z. Cai, X.-S. Peng and H. N. C. Wong, J. Org. Chem., 2013, 78, 8562–8573 CrossRef CAS PubMed; (f) C.-L. Deng, X.-S. Peng and H. N. C. Wong, Tetrahedron, 2017, 73, 3606–3611 CrossRef CAS.
- C.-K. Hau, H. He, A. W. M. Lee, D. T. W. Chik, Z. Cai and H. N. C. Wong, Tetrahedron, 2010, 66, 9860–9874 CrossRef CAS.
- T. Shibata, T. Chiba, H. Hirashima, Y. Ueno and K. Endo, Angew. Chem., Int. Ed., 2009, 48, 8066–8069 CrossRef CAS PubMed.
- H. Kaku, A. Mitarai, N. Okamoto, K. Tanaka, S. Ichikawa, T. Yamamoto, M. Inai, T. Nishii, M. Horikawa and T. Tsunoda, Eur. J. Org. Chem., 2018, 2018, 6991–6999 CrossRef CAS.
- (a) M. Shibasaki and S. Matsunaga, in Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, Wiley-VCH, Weinheim, 2011, ch. 8, pp. 295−332 Search PubMed; (b) S.-F. Zhu and Q.-L. Zhou, in Privileged Chiral Ligands and Catalysts, ed. Q.-L Zhou, Wiley-VCH, Weinheim, 2011, ch. 4, pp. 137−170 CrossRef; (c) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed.
- J.-F. Cui, H. Huang and H. N. C. Wong, Synlett, 2011, 1018–1022 CAS.
- (a) G.-L. Chai, J.-W. Han and H. N. C. Wong, Synthesis, 2017, 49, 181–187 CAS; (b) G.-L. Chai, A. Q. Sun, D. Zhai, J. Wang, W.-Q. Deng, H. N. C. Wong and J. Chang, Org. Lett., 2019, 21, 5040–5045 CrossRef CAS PubMed; (c) G.-L. Chai, B. Zhu and J. Chang, J. Org. Chem., 2019, 84, 120–127 CrossRef CAS PubMed; (d) G.-L. Chai, Y. Qiao, P. Zhang, R. Guo, J. Wang and J. Chang, Org. Lett., 2020, 22, 8023–8027 CrossRef CAS PubMed.
- (a) J.-F. Cui, H.-M. Ko, K.-P. Shing, J.-R. Deng, N. C.-H. Lai and M.-K. Wong, Angew. Chem., Int. Ed., 2017, 56, 3074–3079 CrossRef CAS PubMed; (b) J. Rodriguez and D. Bourissou, Angew. Chem., Int. Ed., 2018, 57, 386–388 CrossRef CAS PubMed; (c) J.-F. Cui, B. Yang, Q. Yu, N. C.-H. Lai, H. Chen and M.-K. Wong, ChemistrySelect, 2019, 4, 1476–1482 CrossRef CAS; (d) J.-J. Jiang, J.-F. Cui, B. Yang, Y. Ning, N. C.-H. Lai and M.-K. Wong, Org. Lett., 2019, 21, 6289–6294 CrossRef CAS PubMed; (e) R. Jouhannet, S. Dagorne, A. Blanc and P. Frémont, Chem.–Eur. J., 2021, 27, 9218–9240 CrossRef CAS PubMed; (f) G. Zuccarello, I. Escofet, U. Caniparoli and A. M. Echavarren, ChemPlusChem, 2021, 86, 1283–1296 CrossRef CAS PubMed.
- CCDC 2124171 [(S)-L1], 2126362 [(S,R)-4], 2126366 [(R,R)-4], 2126367 [(R,S)-4] and 2126368 [(S,S)-4] contain the supplementary crystallographic data for this paper†.
- (a) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3212 CrossRef CAS PubMed; (b) J. M. Brunel, Chem. Rev., 2005, 105, 857–898 CrossRef CAS PubMed.
- (a) J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486–2528 CrossRef CAS PubMed; (b) Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed; (c) S. L. Rössler, D. A. Petrone and E. M. Carreira, Acc. Chem. Res., 2019, 52, 2657–2672 CrossRef PubMed.
- (a) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (b) M. Rueping, A. Kuenkel and I. Atodiresei, Chem. Soc. Rev., 2011, 40, 4539–4549 RSC; (c) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- D. Nakashima and H. Yamamoto, J. Am. Chem. Soc., 2006, 128, 9626–9627 CrossRef CAS PubMed.
- (a) T. Hashimoto and K. Maruoka, Chem. Rev., 2007, 107, 5656–5682 CrossRef CAS PubMed; (b) S. Shirakawa and K. Maruoka, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed.
- (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566–1568 CrossRef CAS PubMed; (b) R. Z. Jin, Z. Bian, C. Q. Kang, H. Q. Guo and L. X. Gao, Synth. Commun., 2005, 35, 1897–1902 CrossRef CAS; (c) F. Romanov-Michailidis, L. Guénée and A. Alexakis, Angew. Chem., Int. Ed., 2013, 52, 9266–9270 CrossRef CAS PubMed; (d) Y. Xu, S. Yu, Q. Chen, X. Chen, Y. Li, X. Yu and L. Pu, Chem.–Eur. J., 2016, 22, 12061–12067 CrossRef CAS PubMed.
- W. Tang, A. G. Capacci, X. Wei, W. Li, A. White, N. D. Patel, J. Savoie, J. J. Gao, S. Rodriguez, B. Qu, N. Haddad, B. Z. Lu, D. Krishnamurthy, N. K. Yee and C. H. Senanayake, Angew. Chem., Int. Ed., 2010, 49, 5879–5883 CrossRef CAS PubMed.
- Under the Pd(dba)2/(±)-BIDIME catalytic system under the same conditions without Pd(PPh3)4, the reactions provided (S)-8s in 46% yield, but only gave a trace amount of (S)-8t (∼5% yield). We speculated that additive Pd(PPh3)4 bearing a “small” phosphine ligand (PPh3) might have facilitated the oxidative addition step between Pd(0) and the extremely sterically hindered aryl bromides in the Suzuki-Miyaura coupling process.
- (a) L. Meca, D. Řeha and Z. Havlas, J. Org. Chem., 2003, 68, 5677–5680 CrossRef CAS PubMed; (b) J.-F. Yang, R.-H. Wang, Y.-X. Wang, W.-W. Yao, Q.-S. Liu and M. Ye, Angew. Chem., Int. Ed., 2016, 55, 14116–14120 CrossRef CAS PubMed.
- (a) A. Duursma, J.-G. Boiteau, L. Lefort, J. A. F. Boogers, A. H. M. De Vries, J. G. De Vries, A. J. Minnaard and B. L. Feringa, J. Org. Chem., 2004, 69, 8045–8052 CrossRef CAS PubMed; (b) M. Lafrance, M. Roggen and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3470–3473 CrossRef CAS PubMed.
- J. Y. Hamilton, D. Sarlah and E. M. Carreira, Angew. Chem., Int. Ed., 2013, 52, 7532–7535 CrossRef CAS PubMed.
- (a) R. Mariz, A. Briceño, R. Dorta and R. Dorta, Organometallics, 2008, 27, 6605–6613 CrossRef CAS; (b) A. Herrera, A. Linden, F. W. Heinemann, R.-C. Brachvogel, M. von Delius and R. Dorta, Synthesis, 2016, 48, 1117–1121 CrossRef CAS; (c) R. Zhang, S. Ge and J. Sun, J. Am. Chem. Soc., 2021, 143, 12445–12449 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2124171, 2126362, 2126366–2126368. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00388k |
| This journal is © The Royal Society of Chemistry 2022 |