
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical synthesis of human selenoprotein F and elucidation of its thiol-disulfide oxidoreductase activity†
Peisi
Liao
a,
Hongmei
Liu
 *bc and
Chunmao
He
*a
*bc and
Chunmao
He
*a
aSchool of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, China. E-mail: hecm@scut.edu.cn
bHubei Key Laboratory of Bioinorganic Chemistry and Materia Medica, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China. E-mail: hongmeiliuhust@hust.edu.cn
cShenzhen Huazhong University of Science and Technology Research Institute, Shenzhen 518057, China
First published on 6th May 2022
Abstract
Selenoprotein F (SelF) is an endoplasmic reticulum-residing eukaryotic protein that contains a selenocysteine (Sec) residue. It has been suggested to be involved in a number of physiological processes by acting as a thiol-disulfide oxidoreductase, but the exact role has remained unclear due to the lack of a reliable production method. We document herein a robust synthesis of the human SelF through a three-segment two-ligation semisynthesis strategy. Highlighted in this synthetic route are the use of a mild desulfurization process to protect the side-chain of the Sec residue from being affected and the simultaneous removal of acetamidomethyl and p-methoxybenzyl protection groups by PdCl2, thus facilitating the synthesis of multi-milligrams of homogenous SelF. The reduction potential of SelF was determined and the thiol-disulfide oxidoreductase activity was further supported by its ability to catalyze the reduction and isomerization of disulfide bonds.
Introduction
Selenoprotein F (SelF), or the 15 kDa selenoprotein (Sep15), is an endoplasmic reticulum (ER)-residing eukaryotic protein containing the 21st essential (proteinogenic) amino acid selenocysteine (Sec, U).1 An increasing number of studies have linked SelF gene polymorphisms and SelF dysregulation to various diseases, including several types of cancer, AIDS, and neurodegeneration, which reveals the importance of SelF's physiological functions.2 As shown in Fig. 1A, the mature human SelF consists of 134 amino acid (aa) residues, including an N-terminal Cys-rich domain and a C-terminal thioredoxin (Trx)-like domain. Although with no typical ER retention peptide sequence, SelF is able to bind the UDP-glucose: glycoprotein glucosyltransferase (UGGT)—a large chaperone protein in the ER, via its N-terminal Cys-rich domain, thus also called the UGGT binding domain;3 the C-terminal Trx-like domain contains a unique CGU redox motif in a dynamic loop, a key structure rendering SelF a competent thiol-disulfide oxidoreductase in the ER.4 As such, SelF has been suggested to play a role in the quality control of the ER, by either rearranging (isomerase function) or reducing incorrectly formed disulfide bonds (reductase function) in misfolded glycoproteins bound to UGGT,5,6 but its exact biological function is yet to be elucidated. Along this line, the in vitro characterization of SelF is generally missing due to the lack of reliable recombinant expression techniques, and most studies are carried out with its Sec-to-Cys homologue.4 In this context, the disulfide pairing mode of SelF(U65C) has been elucidated in our previous work through site-directed mutagenesis and enzymatic digestion (see Fig. 1B),7 the clear evidence of a selenenylsulfide bond in the CGU motif is, however, still lacking. Notably, a few examples have been reported using the genetic code expansion technology to incorporate Sec site-specifically.8–11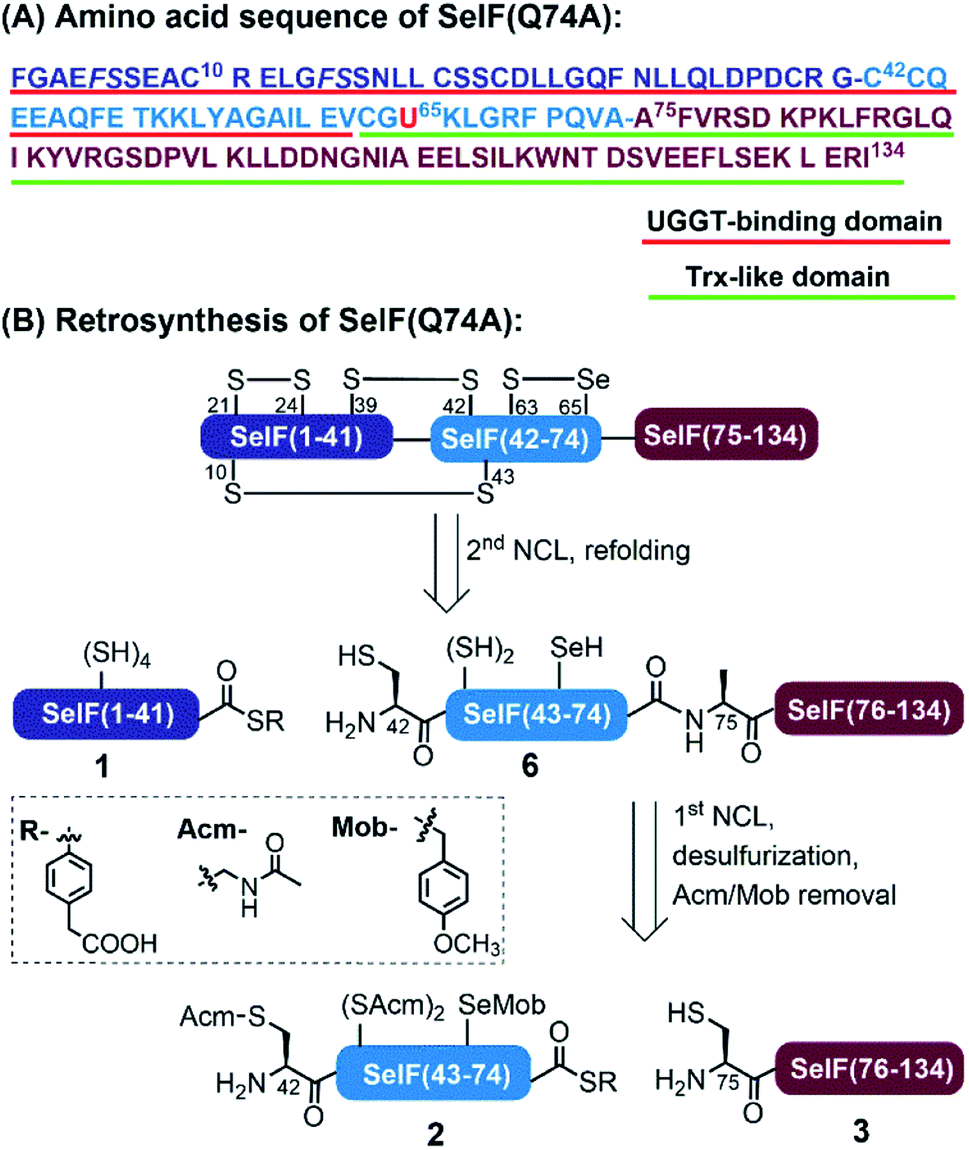 | ||
| Fig. 1 (A) Sequence of the human SelF(Q74A). The red and green underlined areas are UGGT-binding domain and Trx-like domain, respectively. The pseudoproline dipeptides used were in italic. (B) Retrosynthesis strategy. | ||
Chemical protein synthesis (CPS), enabled by chemo-selective peptide ligation reactions like the native chemical ligation (NCL),12,13 has contributed a large number of synthetic proteins which easily incorporate non-natural amino acids or protein modifications.14,15 The development of expressed protein ligation (EPL) technologies has further powered the synthesis of (especially) large proteins.16,17 A number of native selenoproteins and Sec-containing analogue proteins have thus been generated by CPS, as listed in Table S1.†18–30 Despite these significant achievements, the currently used synthetic routes usually involve either no or limited post-ligation treatment, as a result most of the synthetic Sec-proteins either are small in size (usually <100 aa and with few Cys/Sec residues) or contain a Sec residue at the C-terminal region, which can be obtained by EPL through ligation with a synthetic peptide bearing an N-terminus Sec (Table S1,† entries 4, 8 and 11). As such, there is a clear need to develop a more straight-forward strategy for the synthesis of complex selenoproteins having multiple Cys and Sec residues, like in the case of SelF (7 Cys and 1 Sec).
We disclose herein the synthesis of SelF, where the Sec65 residue resides in the middle of its sequence, making it inconvenient to apply EPL. To maximize the overall synthetic yield, we adopted a three-segment two-ligation strategy, where Ala75 was chosen as one of the ligation sites as there is no Cys available in the C-terminal region, and a desulfurization step will be required (Fig. 1B). While selective deselenization in the presence of Cys has been routinely carried out using tris(2-carboxyethyl)phosphine (TCEP),31,32 the selective desulfurization in the presence of Sec has not been reported to the best of our knowledge, which is one of the key challenges in our synthetic endeavour. With the current strategy, multi-milligram of homogeneous synthetic SelF was obtained, which allowed the elucidation of its thiol-disulfide oxidoreductase activity, thus providing evidences for its involvement in the quality control of the ER.
Results and discussion
Synthetic strategy
In analogy to the synthesis of the SelF(U65C) analogue,7 the full-length protein was disconnected into three segments at Gly41–Cys42 and Gln74–Ala75. Noted that the Gln74 was mutated to an Ala residue in order to obtain a stable peptide hydrazide segment, and according to previous experience7 this mutation should not affect the protein folding and function (vide infra). Unlike most of the reported synthetic Sec-containing proteins where the Sec residue is placed in the N-terminus of the peptide, i.e., the ligation site, we opted to place it as an internal residue in segment 2 and the side-chain was protected with a p-methoxybenzyl (Mob) group. Moreover, the Cys75 residue of segment 3 will have to be desulfurized to give the native Ala residue, and for this purpose, all the Cys residues in segment 2 were protected with an acetamidomethyl (Acm) group, which also prevents the lactamization of the resulting thioester. As such, segments 1 and 2 were obtained with standard Fmoc solid-phase peptide synthesis (SPPS), and segment 3 through N-terminal His-SUMO fusion protein expression and Ulp1 cleavage,33,34 as our initial attempt to synthesise this fragment containing 60 aa directly by Fmoc-SPPS failed, partially due to aspartimide formation at the DDN sequence (see the ESI†).35Chemical synthesis of SelF and its Trx-like domain
With all peptide segments in hands, the first NCL reaction was carried out between segments 2 and 3, which completed within 2 h (Fig. 2). At this stage, the initial plan for a one-pot desulfurization at Cys75 failed due to the increased amount of 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) required in this case (vide infra). Instead, peptide 4 was purified and subjected to standard desulfurization conditions (i.e., 30–40 eq. VA-044, 200 mM TCEP and 5% t-BuSH) (t-BuSH = tert-butylthiol).36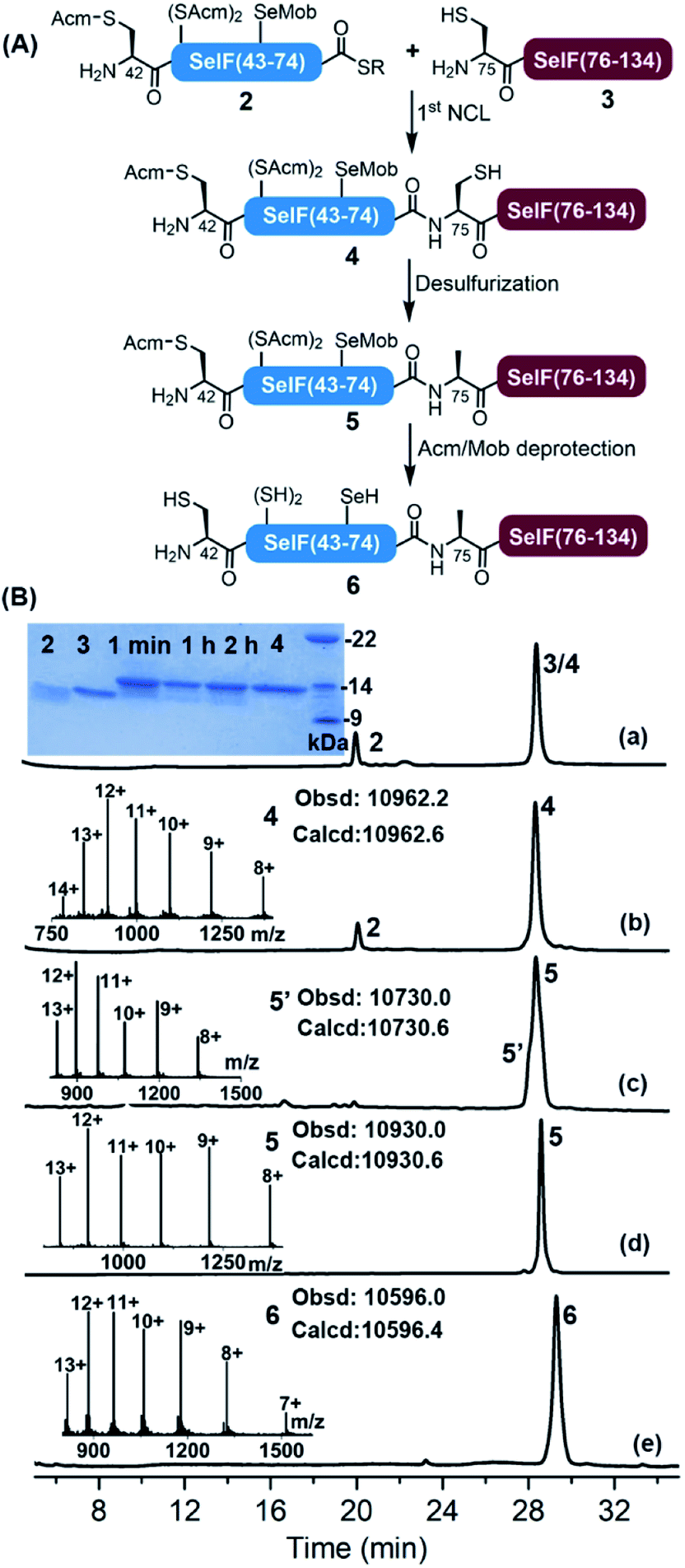 | ||
| Fig. 2 (A) Scheme for the synthesis of the peptide 6. Reaction conditions: 1st NCL, 6.0 M Gdn·HCl, 0.2 M Na2HPO4, 10 mM TCEP, 20 mM MPAA, pH 6.5, RT, 2 h; desulfurization, 6.0 M Gdn·HCl, 0.2 M Na2HPO4, 0.2 M TCEP, 5% t-BuSH, 2.5 eq. VA-044, pH 6.5, 37 °C, 30 min; Acm/Mob deprotection, 6.0 M Gdn·HCl, 0.2 M Na2HPO4, 150 eq. PdCl2, pH 6.9, 37 °C, 2 h, and followed by quenching with DTT and further reducing by TCEP and sodium ascorbate for 30 min. (B) (a and b) Analytical HPLC traces of NCL at 1 min and 2 h, respectively. Inset a: SDS-PAGE analysis of the ligation reaction. Inset b: ESI-MS of peptide 4. (c) Analytical HPLC trace of desulfurization at 30 min and ESI-MS of the by-product 5′. (d) Analytical HPLC trace and ESI-MS of purified 5. (e) HPLC trace of PdCl2-mediated Acm/Mob removal in 2 h and ESI-MS of the product 6. | ||
Surprisingly, the unwanted peptide 5′ with desulfurization at Cys75 and deselenization at Sec65—despite being Mob-protected, was the major product (Fig. S14 and S15†). The peptide S2 (the hydrazide precursor of segment 2) was then used as a model peptide and its stability was tested against each desulfurization component, and no significant change was observed when incubating with TCEP or t-BuSH (Fig. S16 and S17†), which led us to conclude that VA-044 could most probably be the reason. To our delight, when reducing the amounts of VA-044 (to 2.5 eq.), the desired desulfurized peptide 5 was obtained as the major product at 30 min, with an isolated yield of 39.5% (Fig. S23†).37 It seems that the presence of the Mob-protected Sec residue in the sequence speeded up the desulfurization, as the same process would usually take longer (4–24 h) and need more VA-044 (e.g., 50 eq.) in the Cys-containing homologue peptide.7
Next, the global Acm removal of peptide 5 was attempted, and the Mob group of Sec65 was supposed to be retained at this stage to prevent possible deselenizaiton in the following NCL. While the Acm-removal with AgOAc went well during the synthesis of SelF(U65C),7 it gave a complex mixture in this case, with all Acm and Mob groups being removed. And even worse, severe peptide truncation between Val73–Ala74 was observed, due to a currently unknown cause (Fig. S25 and S26†). We then switched to the PdCl2–dithiothreitol (DTT) method,38,39 and by accident we noticed that an increased amount of Pd2+ could lead to a simultaneous Acm and Mob removal of a model peptide S5 (Fig. S27 and S28†). It is worth noting that there is literature presence using Pd0 for the deprotection of an allyl group from Sec in aqueous solution.40 Gratifyingly, when using 150 eq. of PdCl2 quantitative global Acm/Mob removal of peptide 5 was accomplished (Fig. S30†), and subsequently, the excess Pd reagent was removed by DTT and extensive washing. Noted that at this stage, ascorbate and TCEP were added to the mixture to reduce any possible peptide diselenide dimer to afford peptide 6 in free selenol/thiol form. The discovery that the Mob group can be facilely removed by aqueous PdCl2 solution is remarkable considering the generally harsh conditions required in the literature procedures,41,42 like the use of dimethyl sulfoxide (DMSO)43 or 2,2′-dithiobis(5-nitropyridine) (DTNP)44,45 in TFA. We envision that the chemistries relating to the selective desulfurization in the presence of Sec and simultaneous Acm/Mob removal disclosed herein may find further applications in the CPS field, where the use of Sec either for selenoprotein synthesis or as a Cys analogue, has become an increasingly popular strategy.28,32,46
The second ligation between peptide 1 and 6 was carried out in the presence of 0.1 M ascorbate and a reduced concentration of TCEP (5 mM) to prevent the now-free selenol side-chain from being removed,47–49 furnishing the full-length protein 7. After 12 h, the ligation mixture was directly subjected to folding trials. The initial attempt at folding involved sequential dialysis; however, it led to no obvious folding product as judged by HPLC-MS (see the ESI†). We therefore switched to a rapid dilution strategy where the ligation mixture was added directly to a redox buffer (0.4 M Arg·HCl, 0.2 M Tris, 0.1 M (NH4)2SO4, 2 mM EDTA, 0.2 mM GSSG, and 1 mM GSH, pH 8.2), and the refolding was allowed for 12 h at 4 °C. Following this procedure, the refolded protein 8 was obtained after HPLC purification with an isolated yield of 13.7% over two steps (∼1.5 mg). Noted that the presence of thiolactone derivative of peptide 1 (indicated with * in Fig. 3) was probably one of the reasons for the slow ligation rate and the slightly lowered recovery yield. In principle installation of Acm protection group at the Cys sidechain during the synthesis of peptide 1 could avoid the formation of thiolactone, we have, however, observed significant aspartimide formation at the Asp–Cys(Acm) sequence (data not shown), as also reported in the literature.35
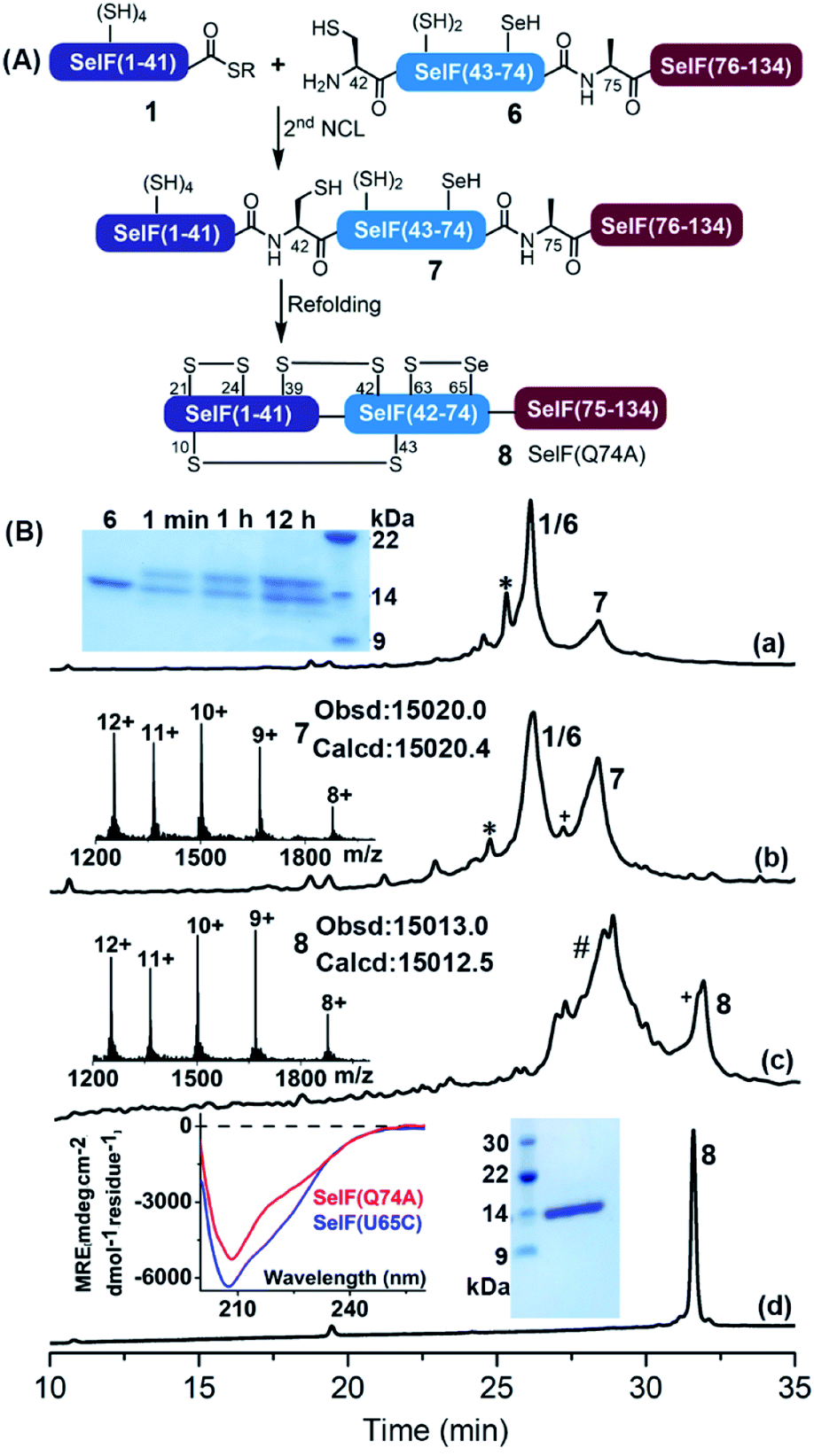 | ||
| Fig. 3 (A) Scheme for the second ligation and folding to afford the SelF(Q74A) (8). Reaction conditions: 2nd NCL, 6.0 M Gdn·HCl, 0.2 M Na2HPO4, 5 mM TCEP, 0.1 M sodium ascorbate, 50 mM MPAA, pH 6.5, RT, 12 h; refolding reaction, 0.4 M Arg·HCl, 0.2 M Tris, 0.1 M (NH4)2SO4, 2 mM EDTA, 0.2 mM GSSG, 1 mM GSH, pH 8.2, 4 °C, 12 h. (B) (a and b) Analytical HPLC traces of ligation at 1 min and 12 h, respectively. * denotes the thiolactone derivative of peptide 1. Inset a: SDS-PAGE analysis of the ligation reaction. Inset b: ESI-MS of 7. (c) Analytical HPLC trace of the refolding at 12 h and ESI-MS of 8. # denotes a mixture of hydrolyzed 1, unreacted peptide 6 and the misfolded by-products, etc. + denotes a small amount of deselenized by-product. (d) Analytical HPLC trace of the purified 8. Inset: CD spectra of the synthetic SelF(Q74A) and the expressed SelF(U65C) (left), SDS-PAGE (right) of 8. | ||
Nevertheless, with the current route enough amounts of protein was obtained for further studies, and the residual Pd-content in 8 determined using ICP-MS was negligible (<0.061%). The proper folding of 8 was confirmed by ESI-MS (∼−8 Da vs.7) and the CD spectrum (vs. the expressed Cys analogue7). And importantly, the presence of a mixed selenenylsulfide bond between Cys63–Sec65 was established via a consecutive trypsin/chymotrypsin digestion (Fig. S52†), which agrees well the reported NMR structure of the fruit fly Sep15 protein as well as the SelF(U65C) homologue (both with a disulfide bond instead).4,7
Meanwhile, following a similar synthetic strategy, e.g., NCL between a short Sec(Mob)-containing peptide thioester segment S9 and segment 3, selective desulfurization, simultaneous Acm/Mob removal and refolding, the Trx-like domain SelF(63–134)(Q74A) 9 was obtained conveniently (Fig. S39–S49†), which set the stage for a comparative functional study with the full-length protein 8 (vide infra).
Probing of the thiol-disulfide reductase activity of SelF
With the synthetic protein in hand, it is now possible to determine its redox potential, a key parameter gauging its ability to act as a native thiol-disulfide oxidoreductase.50,51 Using a glutathione (GSH)–glutathione disulfide (GSSG) reference buffer, the fractions of the reduced SelF at selected GSH/GSSG ratios were measured by HPLC and plotted against the redox potential poised by the corresponding redox buffers (Fig. 4A and S55†). The resulting data was fitted by Nernst equation and the potential of the SelF was established as −222 mV, whereas the Cys homologue has a redox potential of −205 mV. It agrees well with the reported value of the fruit fly Sep15 (−225 mV), which has a disulfide rather than a selenenylsulfide in the active-site redox motif.4 Encouraged by this result, we probed the thiol-disulfide oxidoreductase activities of the synthetic SelF protein. Firstly, the disulfide reductase activity was determined using insulin as a model substrate,52 where the cleavage of the disulfide bond connecting chain A and chain B will lead to protein aggregation and the resulting turbidity can be followed by absorption spectroscopy for assessment. As shown in Fig. 4B, S58 and 59†, SelF leads to a quick increase of the turbidity (onset, 16 min) with a kobs value of 0.034 min−1, whereas the Cys homologue SelF(U65C) shows a much lower reaction rate (onset, 32 min and kobs = 0.008 min−1). It is worth noting that the double mutant SelF(U65C/Q74A) shows a similar, albeit low, reactivity compared to SelF(U65C), suggesting that the effect of mutation at this site (Gln74) is minimum. Interestingly, the Trx-like domain of SelF 9, with the key CGU motif, is also active in reducing the disulfides of insulin. Altogether these data indicate that SelF is a viable disulfide reductase and the presence of Sec in the redox motif is clearly the key for this activity. Moreover, we also tested the protein disulfide isomerase activity of the synthetic SelF by assessing its ability to catalyze the refolding of the scrambled RNase A. As shown in Fig. S62,† only in the case of SelF a small but appreciable amount of folded RNase A can be observed after 2 h incubation. While RNase A may not be the native substrate as presented in the ER,4 the current data suggests that SelF could indeed contribute in the isomerization of disulfide bonds of the (misfolded) glycoprotein substrates of UGGT.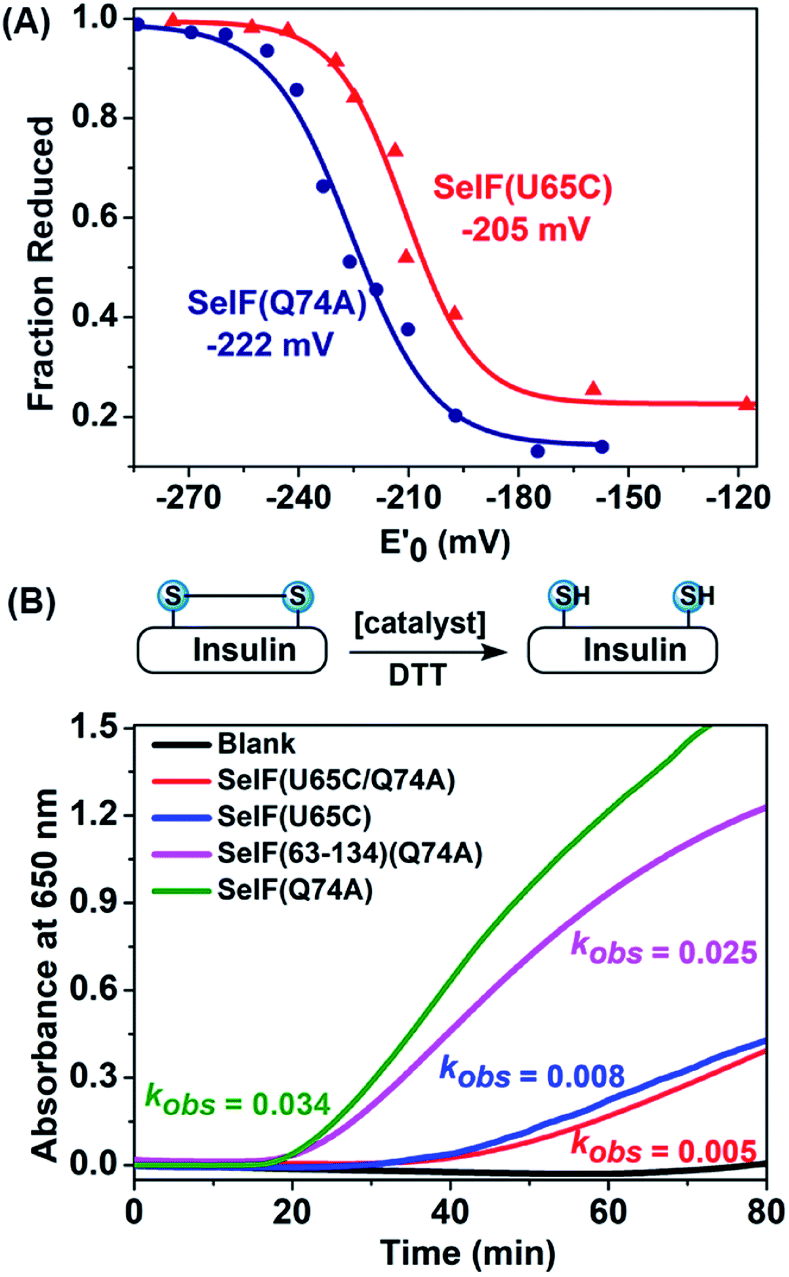 | ||
| Fig. 4 (A) Determination of the redox potentials of SelF(U65C) and SelF(Q74A). (B) Reductase activity assays via recording the turbidity caused by insulin's aggregation. Protein conc.: 2 μM. | ||
Conclusion
In summary, we have developed a robust synthetic strategy affording the full-length human SelF protein that contains an internal Sec residue and seven other Cys residues. Notable challenges addressed in the synthetic route are (1) the use of a reduced amount of VA-044 during desulfurization to protect the side-chain of the Sec residue from being affected and (2) the simultaneous removal of Acm and Mob protection groups by PdCl2, thus facilitating the synthesis of multi-milligrams of homogenous SelF for biological studies. The critical selenenylsulfide bond Cys63–Sec65 in the CGU motif of SelF was unambiguously established, representing the first experimental evidence for such connectivity in SelF. The redox potential of the synthetic protein was determined to be −222 mV, typical for those of the thiol-disulfide oxidoreductase.4,53,54 We demonstrate that SelF is capable of catalyzing the disulfide reduction and to a less extent, isomerisation, in vitro, and the Sec residue is the key for these functions. These data suggest that SelF, together with UGGT, can indeed play a crucial role in the quality control of ER. Moreover, the synthetic strategy developed herein may find broad applications in the synthesis of other complex selenoproteins with multiple Cys/Sec residues, and thus helping in the elucidation of their physiological functions.Data availability
Detailed experimental procedures and analytical data are provided as the ESI.†Author contributions
PL performed the experiments and wrote the manuscript. HL and CH conceptualized the project and revised the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22077040 to HL and No. 91853117 and 22077036 to CH), the Fundamental Research Funds for the Central Universities of SCUT (No. 2020ZYGXZR056) and the Shenzhen Fundamental Research Program (No. JCYJ20200109105836705). The technical assistance in ICP-MS from Ms Shaomin Lin and Mr Zhipeng Zhao was gratefully acknowledged. We also thank Dr Yuqi Zhang and Mr Zeyuan Mo for helpful discussion.Notes and references
- V. N. Gladyshev, K.-T. Jeang, J. C. Wootton and D. L. Hatfield, J. Biol. Chem., 1998, 273, 8910–8915 CrossRef CAS PubMed.
- B. Ren, M. Liu, J. Ni and J. Tian, Nutrients, 2018, 10, 1619 CrossRef PubMed.
- V. M. Labunskyy, A. D. Ferguson, D. E. Fomenko, Y. Chelliah, D. L. Hatfield and V. N. Gladyshev, J. Biol. Chem., 2005, 280, 37839–37845 CrossRef CAS PubMed.
- A. D. Ferguson, V. M. Labunskyy, D. E. Fomenko, D. Araç, Y. Chelliah, C. A. Amezcua, J. Rizo, V. N. Gladyshev and J. Deisenhofer, J. Biol. Chem., 2006, 281, 3536–3543 CrossRef CAS PubMed.
- V. M. Labunskyy, D. L. Hatfield and V. N. Gladyshev, IUBMB Life, 2007, 59, 1–5 CrossRef CAS PubMed.
- V. M. Labunskyy, M.-H. Yoo, D. L. Hatfield and V. N. Gladyshev, Biochemistry, 2009, 48, 8458–8465 CrossRef CAS PubMed.
- P. Liao and C. He, Front. Chem., 2021, 9, 735149 CrossRef CAS PubMed.
- T. Mukai, A. Sevostyanova, T. Suzuki, X. Fu and D. Söll, Angew. Chem., Int. Ed., 2018, 57, 7215–7219 CrossRef CAS PubMed.
- R. Rakauskaitė, G. Urbanavičiūtė, A. Rukšėnaitė, Z. Liutkevičiūtė, R. Juškėnas, V. Masevičius and S. Klimašauskas, Chem. Commun., 2015, 51, 8245–8248 RSC.
- A. P. Welegedara, L. A. Adams, T. Huber, B. Graham and G. Otting, Bioconjugate Chem., 2018, 29, 2257–2264 CrossRef CAS PubMed.
- J. C. Peeler, J. A. Falco, R. E. Kelemen, M. Abo, B. V. Chartier, L. C. Edinger, J. Chen, A. Chatterjee and E. Weerapana, ACS Chem. Biol., 2020, 15, 1535–1540 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- S. B. H. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS PubMed.
- Y. Tan, H. Wu, T. Wei and X. Li, J. Am. Chem. Soc., 2020, 142, 20288–20298 CrossRef CAS PubMed.
- T. W. Muir, D. Sondhi and P. A. Cole, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6705 CrossRef CAS PubMed.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef CAS PubMed.
- L. Dery, P. S. Reddy, S. Dery, R. Mousa, O. Ktorza, A. Talhami and N. Metanis, Chem. Sci., 2017, 8, 1922–1926 RSC.
- N. J. Mitchell, J. Sayers, S. S. Kulkarni, D. Clayton, A. M. Goldys, J. Ripoll-Rozada, P. J. Barbosa Pereira, B. Chan, L. Radom and R. J. Payne, Chem, 2017, 2, 703–715 CAS.
- B. Eckenroth, K. Harris, A. A. Turanov, V. N. Gladyshev, R. T. Raines and R. J. Hondal, Biochemistry, 2006, 45, 5158–5170 CrossRef CAS PubMed.
- N. Metanis, E. Keinan and P. E. Dawson, J. Am. Chem. Soc., 2006, 128, 16684–16691 CrossRef CAS PubMed.
- G. Casi, G. Roelfes and D. Hilvert, ChemBioChem, 2008, 9, 1623–1631 CrossRef CAS PubMed.
- R. J. Hondal, B. L. Nilsson and R. T. Raines, J. Am. Chem. Soc., 2001, 123, 5140–5141 CrossRef CAS PubMed.
- K. Medini, P. W. R. Harris, A. Menorca, K. Hards, G. M. Cook and M. A. Brimble, Chem. Sci., 2016, 7, 2005–2010 RSC.
- R. Quaderer, A. Sewing and D. Hilvert, Helv. Chim. Acta, 2001, 84, 1197–1206 CrossRef CAS.
- N. Metanis and D. Hilvert, Chem. Sci., 2015, 6, 322–325 RSC.
- S. M. Berry, M. D. Gieselman, M. J. Nilges, W. A. van der Donk and Y. Lu, J. Am. Chem. Soc., 2002, 124, 2084–2085 CrossRef CAS PubMed.
- Z. Zhao, D. Shimon and N. Metanis, J. Am. Chem. Soc., 2021, 143, 12817–12824 CrossRef CAS PubMed.
- Z. Zhao and N. Metanis, Angew. Chem., Int. Ed., 2019, 58, 14610–14614 CrossRef CAS PubMed.
- T. Takei, T. Ando, T. Takao, Y. Ohnishi, G. Kurisu, M. Iwaoka and H. Hojo, Chem. Commun., 2020, 56, 14239–14242 RSC.
- N. Metanis, E. Keinan and P. E. Dawson, Angew. Chem., Int. Ed., 2010, 49, 7049–7053 CrossRef CAS PubMed.
- S. S. Kulkarni, E. E. Watson, B. Premdjee, K. W. Conde-Frieboes and R. J. Payne, Nat. Protoc., 2019, 14, 2229–2257 CrossRef CAS PubMed.
- M. P. Malakhov, M. R. Mattern, O. A. Malakhova, M. Drinker, S. D. Weeks and T. R. Butt, J. Struct. Funct. Genomics, 2004, 5, 75–86 CrossRef CAS PubMed.
- A. Reif, S. Siebenhaar, A. Tröster, M. Schmälzlein, C. Lechner, P. Velisetty, K. Gottwald, C. Pöhner, I. Boos, V. Schubert, S. Rose-John and C. Unverzagt, Angew. Chem., Int. Ed., 2014, 53, 12125–12131 CrossRef CAS PubMed.
- J. L. Lauer, C. G. Fields and G. B. Fields, Lett. Pept. Sci., 1995, 1, 197–205 CrossRef CAS.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248–9252 CrossRef CAS PubMed.
- As shown in Fig. S21,† the desulfurization of 4 works the best at 30 min in our hands, because the substrate 4 can still be observed at a shorter reaction time, e.g., 15 min, and the deselenized side-product 5′ can be observed when incubate longer, e.g., 1 h..
- S. K. Maity, M. Jbara, S. Laps and A. Brik, Angew. Chem., Int. Ed., 2016, 55, 8108–8112 CrossRef CAS PubMed.
- M. Jbara, S. Laps, M. Morgan, G. Kamnesky, G. Mann, C. Wolberger and A. Brik, Nat. Commun., 2018, 9, 3154 CrossRef PubMed.
- J. Liu, F. Zheng, R. Cheng, S. Li, S. Rozovsky, Q. Wang and L. Wang, J. Am. Chem. Soc., 2018, 140, 8807–8816 CrossRef CAS PubMed.
- M. Muttenthaler and P. F. Alewood, J. Pept. Sci., 2008, 14, 1223–1239 CrossRef CAS PubMed.
- Z. Zhao and N. Metanis, J. Org. Chem., 2020, 85, 1731–1739 CrossRef CAS PubMed.
- T. Koide, H. Itoh, A. Otaka, H. Yasui, M. Kuroda, N. Esaki, K. Soda and N. Fujii, Chem. Pharm. Bull., 1993, 41, 502–506 CrossRef CAS PubMed.
- K. M. Harris, S. Flemer Jr and R. J. Hondal, J. Pept. Sci., 2007, 13, 81–93 CrossRef CAS PubMed.
- A. L. Schroll, R. J. Hondal and S. Flemer Jr, J. Pept. Sci., 2012, 18, 155–162 CrossRef CAS PubMed.
- D. Lu, H. Yin, S. Wang, F. Tang, W. Huang and P. Wang, J. Org. Chem., 2020, 85, 1652–1660 CrossRef PubMed.
- H. Rohde, J. Schmalisch, Z. Harpaz, F. Diezmann and O. Seitz, ChemBioChem, 2011, 12, 1396–1400 CrossRef CAS PubMed.
- N. Ollivier, A. Blanpain, E. Boll, L. Raibaut, H. Drobecq and O. Melnyk, Org. Lett., 2014, 16, 4032–4035 CrossRef CAS PubMed.
- S. Dery, P. S. Reddy, L. Dery, R. Mousa, R. N. Dardashti and N. Metanis, Chem. Sci., 2015, 6, 6207–6212 RSC.
- F. Åslund, K. D. Berndt and A. Holmgren, J. Biol. Chem., 1997, 272, 30780–30786 CrossRef PubMed.
- J. Liu, F. Li and S. Rozovsky, Biochemistry, 2013, 52, 3051–3061 CrossRef CAS PubMed.
- A. Holmgren, J. Biol. Chem., 1979, 254, 9627–9632 CrossRef CAS.
- J. Lundstroem and A. Holmgren, Biochemistry, 1993, 32, 6649–6655 CrossRef CAS PubMed.
- E. Mössner, M. Huber-Wunderlich and R. Glockshuber, Protein Sci., 1998, 7, 1233–1244 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc00492e |
| This journal is © The Royal Society of Chemistry 2022 |