
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective radical cascade (4+2) annulation with olefins towards the synthesis of chroman derivatives via organo-photoredox catalysis†
Zhipeng
Guan‡
,
Xingxing
Zhong‡
,
Yayu
Ye
,
Xiangwei
Li
,
Hengjiang
Cong
 ,
Hong
Yi
,
Heng
Zhang
,
Zhiliang
Huang
* and
Aiwen
Lei
*
,
Hong
Yi
,
Heng
Zhang
,
Zhiliang
Huang
* and
Aiwen
Lei
*
The Institute for Advanced Studies (IAS), College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, Hubei, People's Republic of China. E-mail: aiwenlei@whu.edu.cn; zlhuang@whu.edu.cn
First published on 21st April 2022
Abstract
Due to the importance of chroman frameworks in medicinal chemistry, the development of novel synthetic methods for these structures is gaining increasing interest of chemists. Reported here is a new (4 + 2) radical annulation approach for the construction of these functional six-membered frameworks via photocatalysis. Featuring mild reaction conditions, the protocol allows readily available N-hydroxyphthalimide esters and electron-deficient olefins to be converted into a wide range of valuable chromans in a highly selective manner. Moreover, the present strategy can be used in the late-stage functionalization of natural product derivatives and biologically active compounds, which demonstrated the potential application. This method is complementary to the traditional Diels–Alder [4 + 2] cycloaddition reaction of ortho-quinone methides and electron-rich dienophiles, since electron-deficient dienophiles were smoothly transformed into the desired chromans.
Chroman moieties frequently exist as the key subunit in a wide array of natural products, pharmaceuticals, and bioactive molecules.1 For example, vitamin E,2 centchroman,2 cromakalim3 and rubioncolin B4 are well-known active pharmaceutical ingredients in various therapeutic areas (Scheme 1a). Due to their significant importance in medicinal chemistry, developing new methods towards the synthesis of chromans and the installation of a variety of the functional groups in chroman frameworks are gaining increasing attention of the chemical community.5
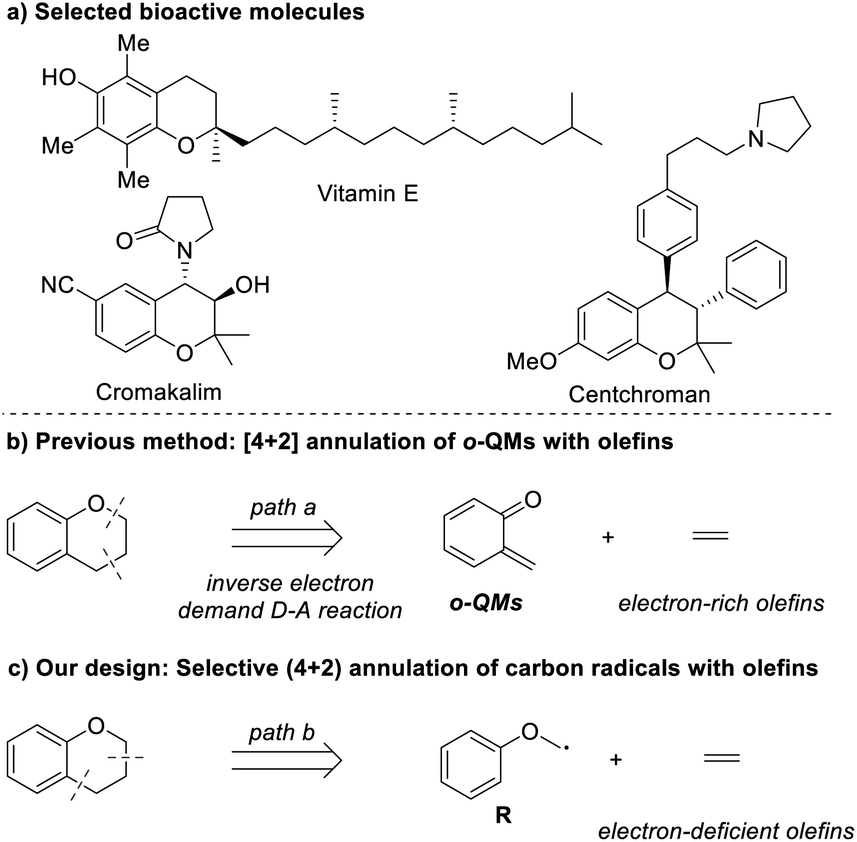 | ||
| Scheme 1 Selected bioactive molecules and the synthetic methods of chromans. | ||
In the past few decades, a great deal of methods have been developed for the assembly of substituted chromans, and among them, the Diels–Alder [4 + 2] cycloaddition reaction provides a highly efficient synthetic platform in the construction of these functional six-membered frameworks.6 Extensive work has been done with this strategy, resulting in a lot of significant progress. The ortho-quinone methides (o-QMs) are generally essential dienes for the Diels–Alder reaction towards the synthesis of chromans, as they are highly reactive for rapid rearomatization via Michael addition of nucleophiles, cycloaddition with a dienophile of 2π partners or 6π-electrocyclization (Scheme 1b).7 Herein, although various valuable chromans have been successfully synthesized with the Diels–Alder [4 + 2] cycloaddition reaction, the use of o-QMs may lead to several potential limitations in some cases. One of the potential limitations is that o-QMs are used mainly as Michael acceptor and electron-deficient dienes to react only with nucleophiles and electron-rich dienophiles. In these considerations, the evolution of synthetic methods for chromans is very important and highly desirable. In particular, novel (4 + 2) cycloaddition strategies capable of synthesizing chromans with the use of easily available materials and electron-deficient dienophiles are of utmost interest.
On the basis of retrosynthetic analysis of chroman shown in Scheme 1c, (4 + 2) radical annulation of the corresponding carbon-centered radical R with olefin would be an alternative route, which is able to overcome the above-mentioned potential limitations. Considering that radical species R is normally nucleophilic, thus, it could react with electron-deficient olefins affording chroman products that generally can't be synthesized by the traditional Diels–Alder [4 + 2] cycloaddition reaction involving o-QMs. Herein, we reported a highly selective (4 + 2) radical–annulation reaction to construct the chroman framework with the use of easily available NHPI ester as the radical precursor and olefin as the radical acceptor under mild conditions.
Compared with other alkyl radical precursors, the redox-active N-(acyloxy)phthalimides (NHPI esters) come to the fore, since they are cheap, stable, readily available, and non-toxic.8 Bearing above hypothesis in mind, we commenced to investigate the (4 + 2) annulation reaction by utilizing readily available N-hydroxyphthalimide ester A′ and commercially available ethyl acrylate as model substrates. After a great deal of screening on the reaction parameters, only a trace amount of the target product was detected by GC-MS. In contrast, the main product is anisole, which may result from a rapid hydrogen abstraction reaction of the unstable primary alkyl radical intermediate. To restrain the formation of this by-product, we designed N-hydroxyphthalimide esters A and A′′, which could produce more stable tertiary radicals, for the target (4 + 2) annulation reaction instead of A′ (Table 1). Pleasantly, with Eosin Y as the photosensitizer,9 74% yield of ethyl-2,2-dimethylchromane-4-carboxylate upon 1 was selectively obtained after irradiation of the reaction system under blue LEDs at room temperature for 12 h, despite a little by-product (Table 1, entry 1). Control experiments showed that both Eosin Y and light are essential for the annulation reaction (Table 1, entries 2 and 3). Further investigation of the photosensitizer revealed that 4-CzIPN and Ru(bpy)3(PF6)2 are either ineffective or inferior in this transformation (Table 1, entries 4 and 5). Other solvents were also evaluated. A poor yield was observed when MeCN was used instead of DMAc, meanwhile, the target product was not detected in DCE (Table 1, entries 6 and 7). This radical annulation reaction was sensitive to air, and dramatically decreased yield was obtained when the reaction was carried out under air (Table 1, entry 8).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yield/% |
| a Standard conditions: A (0.2 mmol), ethyl acrylate (0.5 mmol), Eosin Y (2 mol%), DMAc (2.0 mL), blue light, N2, rt, 12 h, isolated yield; n.d. = not detected. | ||
| 1 | None | 74 |
| 2 | No light | n.d |
| 3 | No EY | n.d |
| 4 | 4-CzIPN | n.d |
| 5 | Ru(bpy)3(PF6)2 | 36 |
| 6 | MeCN | 34 |
| 7 | DCE | n.d |
| 8 | Air | 39 |
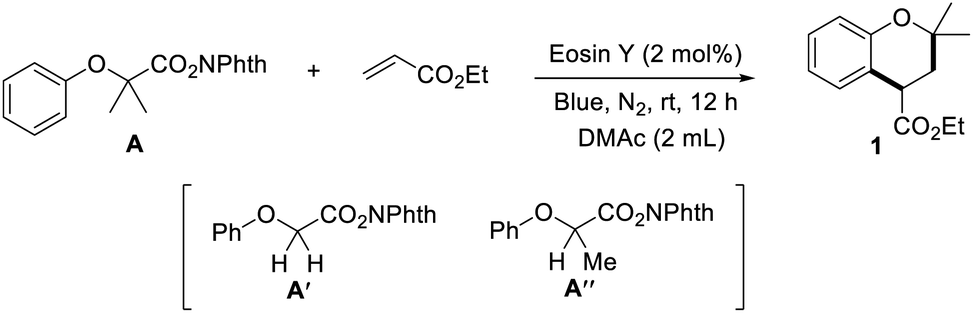
In order to explore the substrate scope of the (4 + 2) annulation reaction, we commenced to scrutinize the generality and selectivity with respect to N-hydroxyphthalimide esters. The functional group applicability of N-hydroxyphthalimide esters was investigated by the examination of various electron donating/withdrawing substituents at the varying positions, as illustrated in Scheme 2. Gratifyingly, we found that substances bearing electron-donating substituents (Me, OMe, tBu, SMe, OPh, OBn, and Ph) at the para-position could smoothly be transformed into the corresponding chromans with satisfactory yields (2–8). N-Hydroxyphthalimide esters with halogen substituents, such as fluoride, chloride, bromide and iodide are suitable to produce the corresponding chromans in satisfactory yields, which enable potential application in further functionalization (9–12). Surprisingly, electron-withdrawing substituents, such as MeCO, OCF3, and CF3, were also tolerated under standard conditions (13–15). This reaction could proceed effectively with N-hydroxyphthalimide esters containing one group or multiple groups in different positions, which delivered a variety of chroman compounds in moderate to good yields (16–19, 21–23). The annulation reaction is not limited to the construction of benzene compounds, as ethyl-3,3-dimethyl-2,3-dihydro-1H-benzo[f]chromene-1-carboxylate was also obtained in 68% yield (20). After the simple esterification, drug molecules, such as clofibric acid, fenofibric acid and ciprofibrate, could be transformed into the corresponding N-hydroxyphthalimide esters, further engaging with ethyl acrylate (10 and 24–25), which highlighted the synthetic applicability of this protocol.
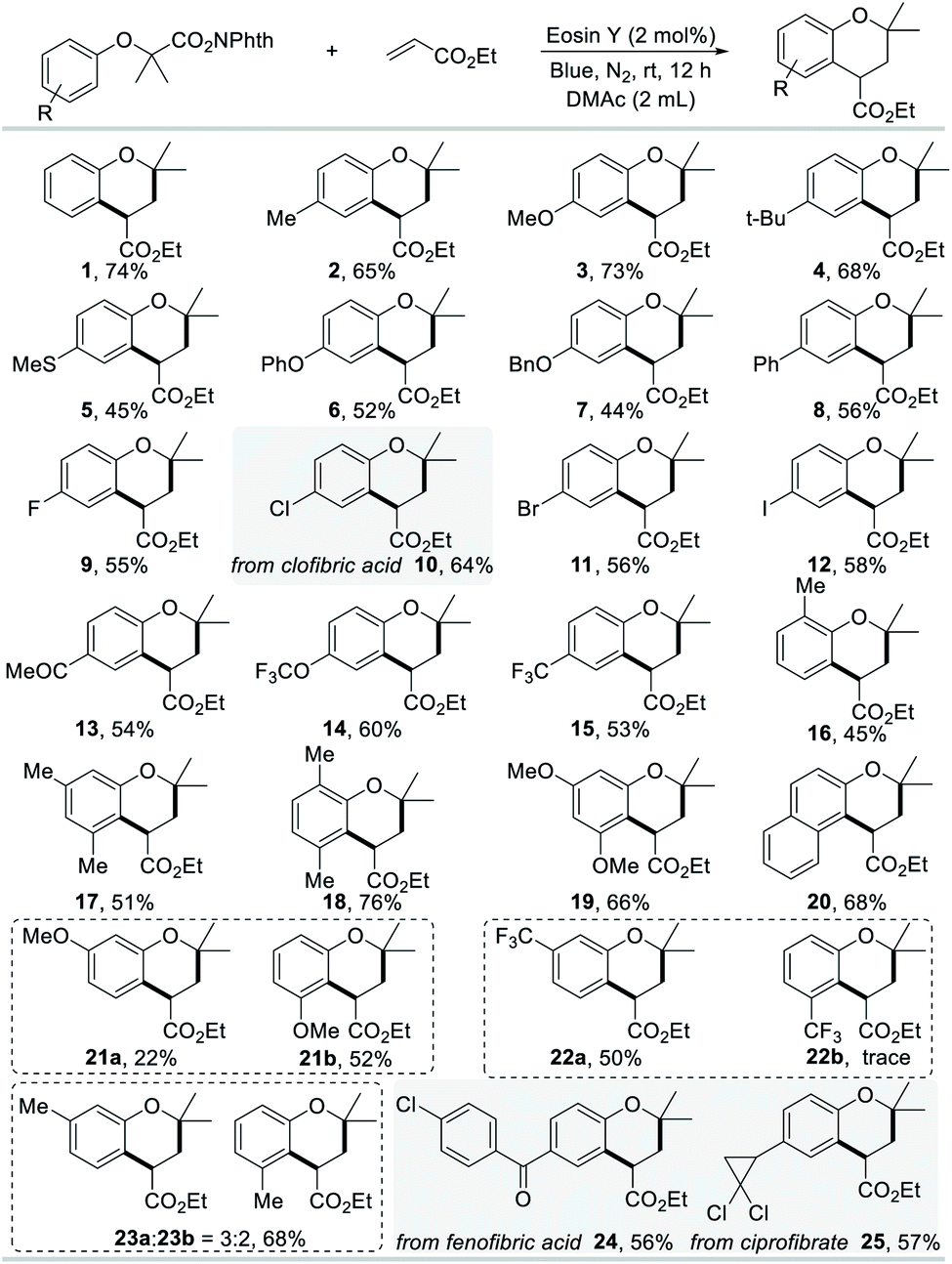 | ||
| Scheme 2 Reactions of NHPI esters with ethyl acrylate. Standard conditions: NHPI ester (0.2 mmol), ethyl acrylate (0.5 mmol), Eosin Y (2 mol%), DMAc (2.0 mL), blue light, N2, rt, 12 h, isolated yield. | ||
Next, we shifted attention to the scope with respect to a wide range of acrylates, as shown in Scheme 3. Methyl acrylate and butyl acrylate were well amenable with N-hydroxyphthalimide esters (26–27). Other acrylates, such as cyclohexyl, tert-butyl and phenyl, were also competent reaction partners with a satisfactory efficiency (28–30). Ethyl (E)-but-2-enoate was tolerant to afford the desired chroman product, albeit in 29% yield (31). It is particularly noteworthy that dimethyl maleate was demonstrated to be a suitable substrate, leading to the formation of sterically hindered product (32). The sensitive benzylic C–H bond and the fragile furan and thiophene moieties could be retained in the radical cascade reaction, providing a series of functionalized chromans (33–35). Alkoxy and aligned alkoxy on substances did not reduce the reaction efficacy (36–37). Chromans possessing various subtle trimethylsilyl, hydroxyl, primary/secondary bromoalkene, cyano and thiomethylene were accessed in reasonable yields, which provided the basis for late-stage derivatization of products (38–41, 43). Owing to the superiority of lipophilicity, permeability and metabolism, we tried to introduce trifluoromethyl into chroman skeletons. To our delight, 2,2,2-trifluoroethyl acrylate gave rise to the corresponding chromans with 52% yield (42). The unactivated alkynyl moiety and alkenyl moiety survived in the photoredox catalysis (44–46).
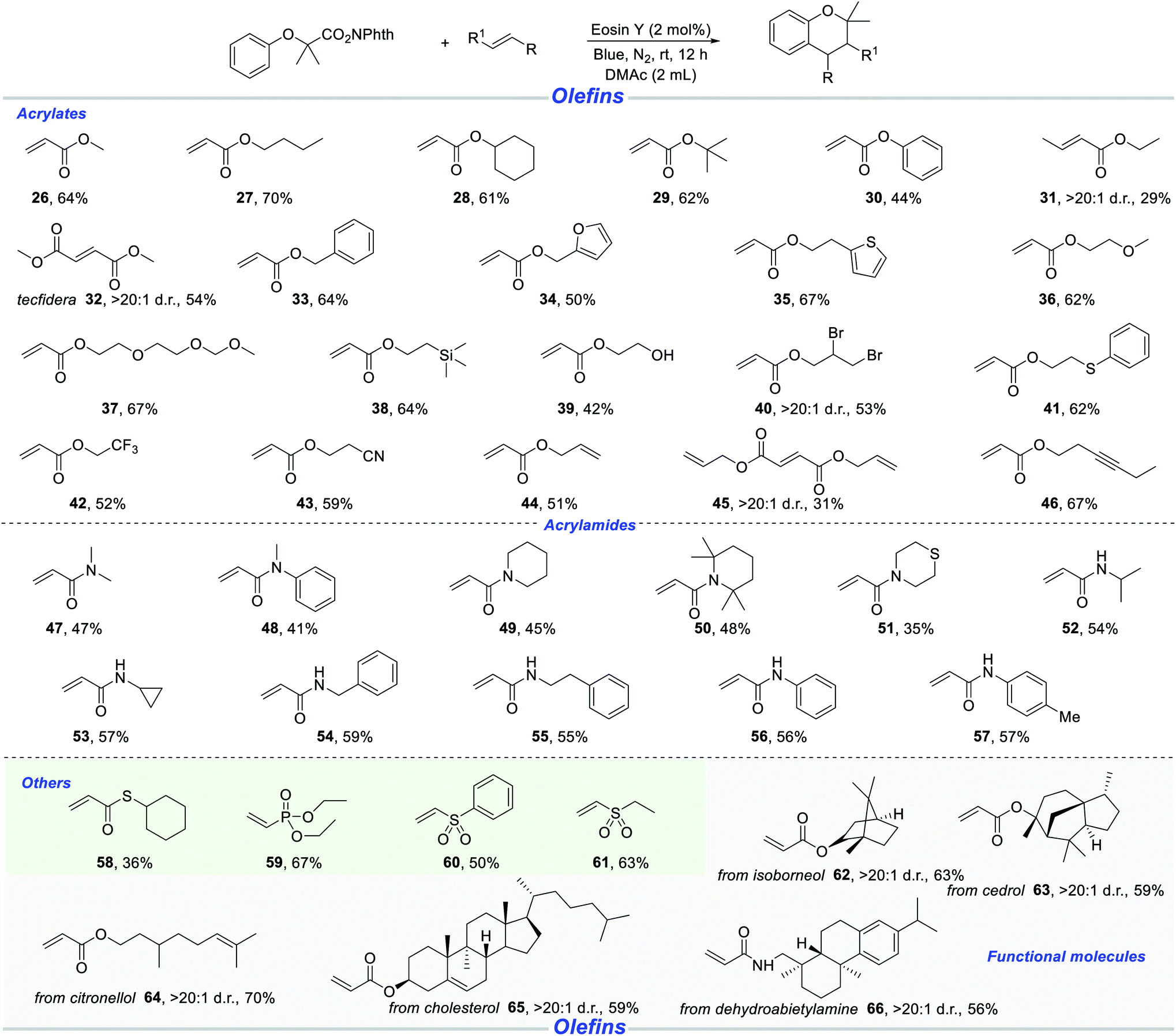 | ||
| Scheme 3 Reactions of A with various olefins. Standard conditions: A (0.2 mmol), olefin (0.5 mmol), Eosin Y (2 mol%), DMAc (2.0 mL), blue light, N2, rt, 12 h, isolated yield. | ||
It is well-known notorious that compounds possessing nitrogen atoms are a very important class of biologically active and functional molecules. Thus, we turned our attention from acrylates to acrylamide derivatives. We were delighted to find that N,N-dimethylacrylamide was a suitable radical receptor to give the target molecule in moderate yield (47). Similarly, a series of chroman products were obtained with cyclic and acyclic acrylamides (48–51). Subsequently, we continued to investigate the reaction of different secondary acrylamides with N-hydroxyphthalimide ester A. These secondary acrylamides bearing NH-isopropyl, -cylopropyl, -benzyl, -phenylethyl and -aryl functionalities, could smoothly be transformed into the desired (4 + 2) annulation products under standard conditions (52–57). Besides acrylates and acrylamides, this method was successfully applied to other Michael acceptors resulting in the synthesis of various functionalized chromans (58–61). In order to demonstrate the potential applicability of this methodology, a variety of natural products, their derivatives and functional molecules, such as isoborneol (62), cedrol (63), citronellol (64), cholesterol (65), and dehydroabietylamine (66), were examined, and all these structures could be embedded into target products in 56–70% yields.
The (4 + 2) annulation protocol is not limited to the synthesis of chromans. Under standard conditions, the thiochromane derivative could be formed, although less efficiently (Scheme 4a). With curiosity, we tried to use the commercially available pinacol vinylboronate instead of acrylates for this transformation because of the widespread use of organoboron compounds in organic synthesis. The target compound 2-(2,2-dimethylchroman-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, which is a versatile building block in functionalization of chromans, was obtained in 48% yield under the slightly revised conditions (Scheme 4b). It is noting that the reaction could be conducted smoothly to afford 60% yield under sunlight irradiation, showing the potential of industrial application (Scheme 4c). Furthermore, the versatility of chroman 1 was also explored. The oxidative dehydrogenation process of 1 led to the formation of value-added ethyl 2,2-dimethyl-2H-chromene-4-carboxylate 69 by using DDQ as the oxidant (Scheme 4d). 1 could also be reduced to (2,2-dimethylchroman-4-yl)methanol 70 with lithium aluminum hydride in ethyl ether (Scheme 4d).
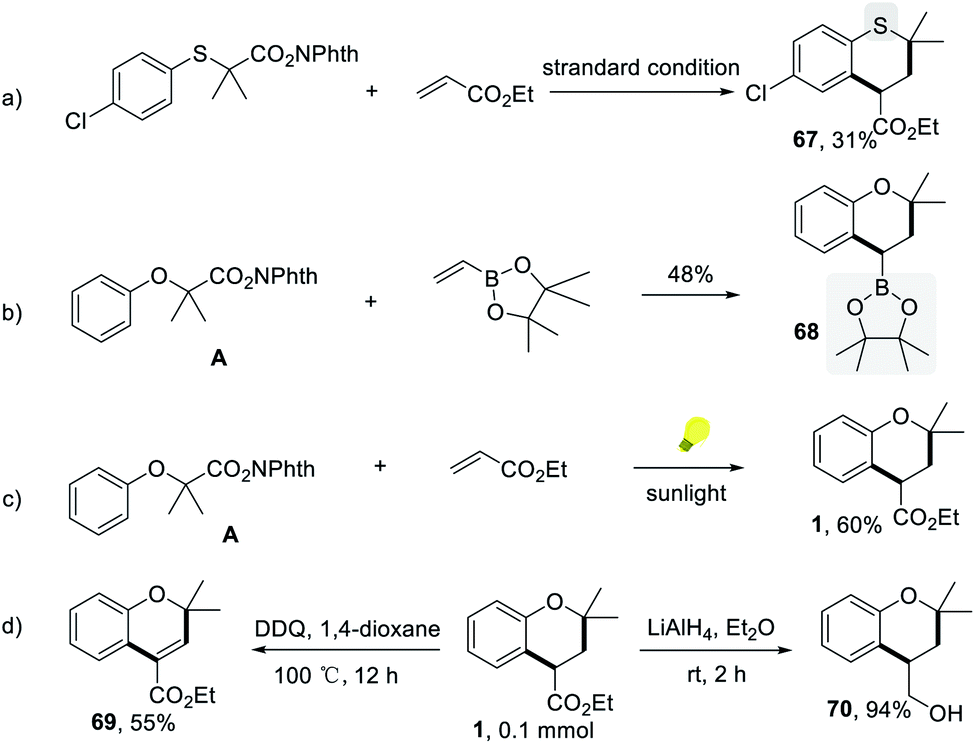 | ||
| Scheme 4 The synthetic applications. (a) The synthesis of thiochromane. (b) Pinacol vinylboronate as a substrate. (c) Sunlight condition. (d) The derivatization of products. | ||
To further gain mechanistic insights into this process, a series of experiments were conducted. When the model reaction was performed under standard conditions but in the presence of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as a radical scavenger, the target product was not detected (Scheme 5a). Notably, when butylated hydroxytoluene (BHT) was added to this reaction system, the annulation reaction was significantly suppressed, meanwhile, a coupling product was detected by GC-MS and HRMS (Scheme 5b). These results indicated a radical-involved pathway for this transformation. Subsequently, the carbon radical was captured by an intramolecular aromatic ring, giving the cyclization product 69 in excellent yield (Scheme 5c). Moreover, the intermolecular kinetic isotope effect (KIE) experiment was carried out by using A and A-d5 as competitive substrates. Under standard conditions, a KIE value of 1.05 was observed, indicating that the cleavage of the aromatic C–H bond might not be the rate-determining step in the transformation (Scheme 5d).
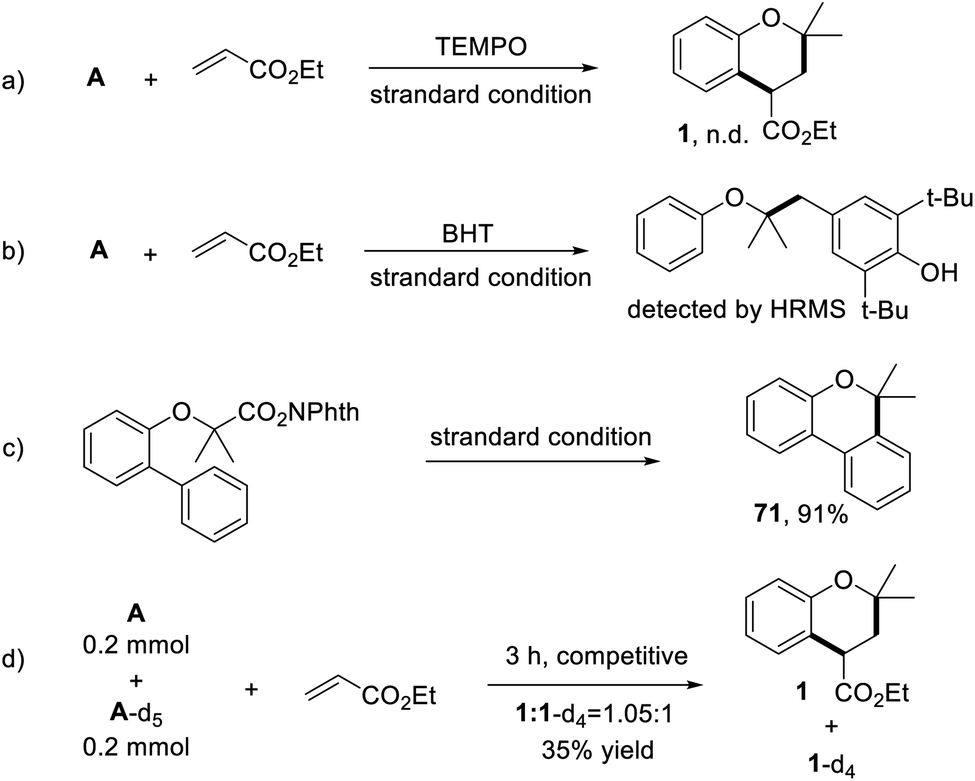 | ||
| Scheme 5 The control experiments. (a) The addition of TEMPO. (b) The addition of BHT. (c) Intramolecular reaction. (d) KIE experiment. | ||
On the basis of the above experimental results, we proposed a possible mechanism cycle for the reaction, as shown in Scheme 6. Initially, the photocatalyst Eosin Y (EY) was transformed into the excited species EY* (E1/2[EY˙+/EY*] = −1.1 V vs. SCE) under the irradiation with visible light. As a redox-active species, EY* was able to reduce N-hydroxyphthalimide ester (E1/2[A/I] = −0.8 V, see the CV in the ESI†) via a single-electron-transfer (SET) process, generating the EY˙+ radical cation and the corresponding N-hydroxyphthalimide ester radical anion I. The intermediate I underwent rapid homolytic fragmentation to generate carbon-centered nucleophilic radical II by releasing the phthalimide anion and carbon dioxide. Subsequently, the carbon radical II was captured by ethyl acrylate to form the electrophilic radical III, which underwent rapid intramolecular radical cyclization to afford aryl radical IV. Then, the intermediate IV was converted into cation Vvia a SET oxidation. On the other hand, the EY˙+ radical cation was transformed into Eosin Y to accomplish the photocatalytic cycle. The rapid deprotonation of V leads to the formation of the product 1.
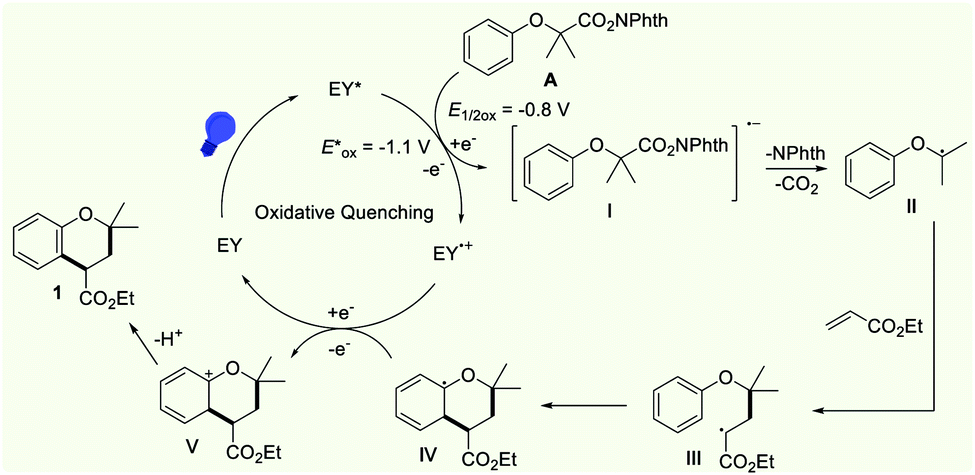 | ||
| Scheme 6 Proposed reaction mechanism. | ||
Conclusions
In summary, a novel (4 + 2) radical annulation approach has been established for the synthesis of diverse chromans from readily available redox-active esters and olefins. This method operates under mild conditions and provides reactivity with a broad range of N-hydroxyphthalimide esters and electron-deficient olefins. The demonstrated late-stage utility of this protocol makes it particularly promising as a versatile tool to generate diverse valuable chroman structures for drug discovery and chemical biology. Compared with the traditional Diels–Alder [4 + 2] cycloaddition reaction of ortho-quinone methides with electron-rich dienophiles, this protocol opens a new pathway for the synthesis of chromans that generally can't be accessed by the use of ortho-quinone methides. We anticipate that the (4 + 2) radical annulation reaction will continue to be developed and deployed for the synthesis of highly functionalized six-membered rings, ideally providing synthetic chemists access to diverse drug and natural product derivatives by the utilization of readily available starting materials.Data availability
The datasets supporting this article have been uploaded as part of the ESI† material.Author contributions
Z. G. and A. L. conceived the project. Z. G., X. Z., Y. Y., and X. L. conducted and analyzed the experiments. H. C. performed the crystallographic data. Z. G., A. L., Z. H., X. Z., H. Z. and H. Y. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the China Postdoctoral Science Foundation (2021M702516), National Key R&D Program of China (No. 2021YFA1500104), National Natural Science Foundation of China (22031008) and Science Foundation of Wuhan (2020010601012192). The numerical calculations in this paper have been done on the supercomputing system in the Supercomputing Center of Wuhan University.Notes and references
- (a) E. E. Schweizer and O. Meeder-Nycz, Chromenes, Chromanes, and Chromones, Wiley-Interscience, New York, 1977 Search PubMed; (b) G. P. Ellis, in Chemistry of Heterocyclic Compounds, 1977, pp. 1–10 Search PubMed; (c) H. C. Shen, Tetrahedron, 2009, 65, 3931–3952 CrossRef CAS; (d) K. C. Nicolaou, J. A. Pfefferkorn, S. Barluenga, H. J. Mitchell, A. J. Roecker and G. Q. Cao, J. Am. Chem. Soc., 2000, 122, 9968–9976 CrossRef CAS; (e) G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285–2310 CrossRef CAS PubMed.
- (a) V. W. Bowry and R. Stocker, J. Am. Chem. Soc., 1993, 115, 6029–6044 CrossRef CAS; (b) M. Uyanik, H. Hayashi and K. Ishihara, Science, 2014, 345, 291–294 CrossRef CAS PubMed.
- (a) J. De Crée, H. Geukens, J. Leempoels and H. Verhaegen, Drug Dev. Res., 1986, 8, 109–117 CrossRef; (b) A. Van de Water, W. Janssens, J. Van Neuten, R. Xhonneux, J. De Cree, H. Verhaegen, R. S. Reneman and P. A. J. Janssen, J. Cardiovasc. Pharmacol., 1988, 11, 552–563 CrossRef CAS PubMed.
- J.-P. Lumb, K. C. Choong and D. Trauner, J. Am. Chem. Soc., 2008, 130, 9230–9231 CrossRef CAS PubMed.
- (a) P.-S. Wang, P. Liu, Y.-J. Zhai, H.-C. Lin, Z.-Y. Han and L.-Z. Gong, J. Am. Chem. Soc., 2015, 137, 12732–12735 CrossRef CAS PubMed; (b) B. J. Nachtsheim, Science, 2014, 345, 270–271 CrossRef CAS PubMed; (c) S. E. Ammann, G. T. Rice and M. C. White, J. Am. Chem. Soc., 2014, 136, 10834–10837 CrossRef CAS PubMed; (d) J. Barluenga, M. Trincado, E. Rubio and J. M. González, J. Am. Chem. Soc., 2004, 126, 3416–3417 CrossRef CAS PubMed; (e) A. F. Ward, Y. Xu and J. P. Wolfe, Chem. Commun., 2012, 48, 609–611 RSC; (f) S. W. Youn and J. I. Eom, J. Org. Chem., 2006, 71, 6705–6707 CrossRef CAS PubMed; (g) R. T. Thornbury, V. Saini, T. d. A. Fernandes, C. B. Santiago, E. P. A. Talbot, M. S. Sigman, J. M. McKenna and F. D. Toste, Chem. Sci., 2017, 8, 2890–2897 RSC; (h) D. W. Manley, R. T. McBurney, P. Miller, R. F. Howe, S. Rhydderch and J. C. Walton, J. Am. Chem. Soc., 2012, 134, 13580–13583 CrossRef CAS PubMed; (i) J. D. Hepworth and B. M. Heron, in Progress in Heterocyclic Chemistry, ed. G. W. Gribble and T. L. Gilchrist, Elsevier, 2002, vol. 14, pp. 332–355 Search PubMed; (j) X.-F. Wang, Q.-L. Hua, Y. Cheng, X.-L. An, Q.-Q. Yang, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2010, 49, 8379–8383 CrossRef CAS PubMed; (k) Y. K. Chung and G. C. Fu, Angew. Chem., Int. Ed., 2009, 48, 2225–2227 CrossRef CAS PubMed; (l) B. M. Trost, H. C. Shen, L. Dong and J.-P. Surivet, J. Am. Chem. Soc., 2003, 125, 9276–9277 CrossRef CAS PubMed; (m) M. Wollenburg, J. Bajohr, A. D. Marchese, A. Whyte, F. Glorius and M. Lautens, Org. Lett., 2020, 22, 3679–3683 CrossRef CAS PubMed; (n) K. Fukamizu, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2008, 130, 10498–10499 CrossRef CAS PubMed; (o) H. Ishibashi, K. Ishihara and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 11122–11123 CrossRef CAS PubMed.
- (a) W. Oppolzer, Angew. Chem., Int. Ed., 1977, 16, 10–23 CrossRef; (b) B.-s. Jeon, S.-A. Wang, M. W. Ruszczycky and H.-w. Liu, Chem. Rev., 2017, 117, 5367–5388 CrossRef CAS PubMed; (c) A. G. Fallis, Acc. Chem. Res., 1999, 32, 464–474 CrossRef CAS; (d) W.-J. Bai, J. G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and T. R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655–3664 CrossRef CAS PubMed; (e) B. Liu, S. Fu and C. Zhou, Nat. Prod. Rep., 2020, 37, 1627–1660 RSC.
- (a) C.-C. Hsiao, S. Raja, H.-H. Liao, I. Atodiresei and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 5762–5765 CrossRef CAS PubMed; (b) J.-J. Zhao, S.-B. Sun, S.-H. He, Q. Wu and F. Shi, Angew. Chem., Int. Ed., 2015, 54, 5460–5464 CrossRef CAS PubMed; (c) S. K. Alamsetti, M. Spanka and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 2392–2396 CrossRef CAS PubMed; (d) K. Zhao, Y. Zhi, T. Shu, A. Valkonen, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2016, 55, 12104–12108 CrossRef CAS PubMed; (e) M. Uyanik, K. Nishioka, R. Kondo and K. Ishihara, Nat. Chem., 2020, 12, 353–362 CrossRef CAS PubMed; (f) X. Lin, X. Liu, K. Wang, Q. Li, Y. Liu and C. Li, Nat. Commun., 2021, 12, 4958 CrossRef CAS PubMed; (g) M. Xiang, C.-Y. Li, X.-J. Song, Y. Zou, Z.-C. Huang, X. Li, F. Tian and L.-X. Wang, Chem. Commun., 2020, 56, 14825–14828 RSC; (h) G. Desimoni, G. Faita and P. Quadrelli, Chem. Rev., 2018, 118, 2080–2248 CrossRef CAS PubMed; (i) B. Yang and S. Gao, Chem. Soc. Rev., 2018, 47, 7926–7953 RSC; (j) Y. Xie and B. List, Angew. Chem., Int. Ed., 2017, 56, 4936–4940 CrossRef CAS PubMed; (k) F. Göricke and C. Schneider, Angew. Chem., Int. Ed., 2018, 57, 14736–14741 CrossRef PubMed; (l) J. Zhang, L. Lin, C. He, Q. Xiong, X. Liu and X. Feng, Chem. Commun., 2018, 54, 74–77 RSC; (m) T. J. Doyon, J. C. Perkins, S. A. Baker Dockrey, E. O. Romero, K. C. Skinner, P. M. Zimmerman and A. R. H. Narayan, J. Am. Chem. Soc., 2019, 141, 20269–20277 CrossRef CAS PubMed.
- (a) W. Zhao, R. P. Wurz, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2017, 139, 12153–12156 CrossRef CAS PubMed; (b) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283 CrossRef CAS PubMed; (c) L. Candish, M. Teders and F. Glorius, J. Am. Chem. Soc., 2017, 139, 7440–7443 CrossRef CAS PubMed; (d) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed; (e) H.-M. Huang, M. Koy, E. Serrano, P. M. Pflüger, J. L. Schwarz and F. Glorius, Nat. Catal., 2020, 3, 393–400 CrossRef CAS; (f) A. Tlahuext-Aca, R. A. Garza-Sanchez and F. Glorius, Angew. Chem., Int. Ed., 2017, 56, 3708–3711 CrossRef CAS PubMed; (g) R. Mao, A. Frey, J. Balon and X. Hu, Nat. Catal., 2018, 1, 120–126 CrossRef CAS; (h) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS PubMed; (i) X. Yi, R. Mao, L. Lavrencic and X. Hu, Angew. Chem., Int. Ed., 2021, 60, 23557–23563 CrossRef CAS PubMed; (j) R. Mao, J. Balon and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 9501–9504 CrossRef CAS PubMed; (k) R. Mao, J. Balon and X. Hu, Angew. Chem., Int. Ed., 2018, 57, 13624–13628 CrossRef CAS PubMed; (l) C. Zhu, H. Yue, L. Chu and M. Rueping, Chem. Sci., 2020, 11, 4051–4064 RSC.
- D. P. Hari and B. Konig, Chem. Commun., 2014, 50, 6688–6699 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2089433. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00903j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |