
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence“Click handle”-modified 2′-deoxy-2′-fluoroarabino nucleic acid as a synthetic genetic polymer capable of post-polymerization functionalization†
Kevin B.
Wu
,
Christopher J. A.
Skrodzki‡
,
Qiwen
Su
,
Jennifer
Lin
and
Jia
Niu
 *
*
Department of Chemistry, Boston College, 2609 Beacon Street, Chestnut Hill, MA 20467, USA. E-mail: jia.niu@bc.edu
First published on 17th May 2022
Abstract
The functions of natural nucleic acids such as DNA and RNA have transcended genetic information carriers and now encompass affinity reagents, molecular catalysts, nanostructures, data storage, and many others. However, the vulnerability of natural nucleic acids to nuclease degradation and the lack of chemical functionality have imposed a significant constraint on their ever-expanding applications. Herein, we report the synthesis and polymerase recognition of a 5-(octa-1,7-diynyl)uracil 2′-deoxy-2′-fluoroarabinonucleic acid (FANA) triphosphate. The DNA-templated, polymerase-mediated primer extension using this “click handle”-modified FANA (cmFANA) triphosphate and other FANA nucleotide triphosphates consisting of canonical nucleobases efficiently generated full-length products. The resulting cmFANA polymers exhibited excellent nuclease resistance and the ability to undergo efficient click conjugation with azide-functionalized molecules, thereby becoming a promising platform for serving as a programmable and evolvable synthetic genetic polymer capable of post-polymerization functionalization.
Introduction
Natural nucleic acids, such as DNA and RNA, have emerged as a new class of functional materials in a wide range of diagnostic and therapeutic applications beyond carrying genetic information.1 As sequence-defined functional polymers, nucleic acids possess unique advantages such as ease of amplification, predictable thermal properties, programmable three-dimensional folding, and well-established technologies for their intracellular trafficking.2,3 Despite the rapid growth of nucleic acid-based technologies in recent years, two key limitations have posed significant constraints on the development of this field: the susceptibility of natural nucleic acids toward nuclease-mediated degradation, and the lack of chemical diversity compared to proteins.The advent of xenobiotic nucleic acids (XNAs) that consist of non-natural sugar backbones has provided a promising solution to overcome nuclease susceptibility.4–11 To date, aptamers, XNAzymes, and nanostructures based on several different XNA systems have been reported, including 2′-deoxy-2′-fluoroarabino nucleic acid (FANA),12–15 hexitol nucleic acid (HNA),16 locked nucleic acid (LNA),17,18 and threose nucleic acid (TNA).19–22 To enable the efficient uptake of XNA building blocks in the enzymatic polymerization reactions, engineered polymerases with altered substrate specificity were discovered through rational mutagenesis and directed evolution that can tolerate unnatural XNA nucleotide triphosphates (NTPs). For example, Holliger and coworkers evolved DNA polymerases that could recognize six classes of XNA NTPs including FANA, HNA, LNA, and TNA.16 Chaput and coworkers engineered Kod23 and Tgo12 DNA polymerases that showed superior activities in synthesizing TNA and FANA, respectively. However, the majority of these XNA structures generated to date still incorporate the same nucleobases, adenine (A), cytosine (C), guanine (G), and thymine (T) or uracil (U), as the natural nucleic acids.
On the other hand, installing functional groups to the nucleobases has been demonstrated to be an effective strategy to expand the chemical repertoire and enhance the bioactive functions in natural nucleic acids.24–27 An array of hydrophobic groups have been incorporated in both DNA and RNA aptamers, with notable examples including slow off-rate modified aptamers (SOMAmers)28 and highly functionalized nucleic acid polymers (HFNAPs).29 Impressively, in 2018 Chaput and coworkers reported a TNA aptamer uniformly incorporating a 7-deaza-7-phenyl guanidine against HIV reverse transcriptase with nanomolar affinity.30 They further expanded the nucleobase modification of TNA to 10 different hydrophobic groups.22,31 While this approach ensures quantitative incorporation of base-modified nucleotides, bulky modifications often become challenging to incorporate and necessitate re-engineering of the polymerase enzymes. An alternative strategy to introduce nucleobase modifications is through the incorporation of nucleotides consisting of click handle-modified bases followed by efficient conjugation of a variety of chemical motifs via click chemistry. Mayer32 and Krauss33 pioneered this technology and demonstrated its utility in introducing hydrophobic groups and carbohydrates onto DNA aptamers. Soh and Niu further combined the click handle-modified DNA structures with the particle display platform to enable the discovery of highly specific DNA aptamers.34
To expand the chemical repertoire of XNA systems and incorporate a wide range of functional side chains, herein we developed a “click handle”-modified FANA (cmFANA) strategy centered on a 5-octa-1,7-diynyl FANA uracil triphosphate (C8-alkyne-FANA UTP), in which an alkyne modification at the 5-position of the FANA uridine is used to conjugate with a variety of chemical motifs bearing an azido group via the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction (Fig. 1). We demonstrated that C8-alkyne-FANA UTP can be efficiently incorporated into cmFANA polymers in DNA-templated primer extension reactions. The high efficiency of the CuAAC reaction enabled near quantitative post-polymerization conjugation of several carbohydrate structures ranging from monosaccharides to oligosaccharides that are otherwise challenging to incorporate by using engineered polymerases. Finally, a DNA-display strategy was developed to covalently link the carbohydrate-conjugated cmFANA polymer with its encoding DNA template, laying the foundation for the in vitro selection of these FANA-based synthetic genetic polymers.
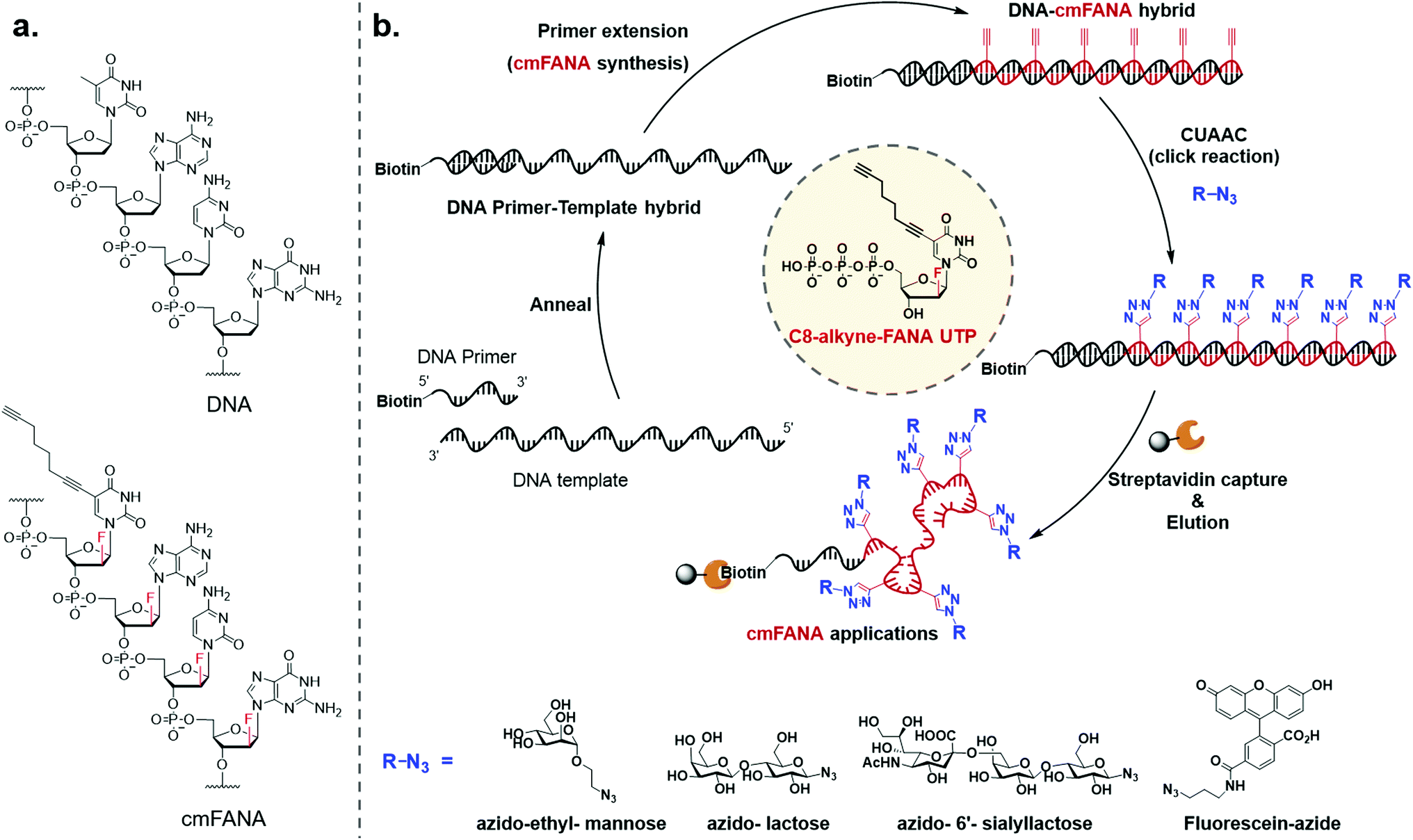 | ||
| Fig. 1 An overview of the cmFANA technology. (a) Structural comparison between DNA and cmFANA. (b) Transcription of a DNA template into cmFANA, conjugation by CuAAC, and the DNA-display strategy to covalently link the modified FANA with its encoding DNA template. The structure of cmFANA uridine triphosphate, C8-alkyne-FANA UTP, is presented in the middle. Examples of azido-modified molecules conjugated to cmFANA are shown below. | ||
Results and discussion
Synthesis of C8-alkyne-FANA UTP
We began our investigation by developing a facile synthetic route for C8-alkyne-FANA UTP from readily available building blocks (Scheme 1). Fluorination of commercially available 1,3,5-tri-O-benzoyl-α-(D)-ribofuranose by diethylaminosulfur trifluoride (DAST) produced 2-fluoroarabanose 1. Bromination of 1 afforded the glycosyl donor 2, which was subjected to glycosylation with 5-iodouracil to yield a crude product with an α/β isomer ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. The desired β-isomer 4 was isolated by recrystallization. Next, Sonogashira coupling of 4 and 1,7-octadiyne afforded 3′,5′-benzoyl protected nucleoside 5. It is noteworthy that an excessive amount of 1,7-octadiyne (12 equiv.) was used to ensure monofunctionalization and avoid crosslinking. Subsequently, deprotection of 5 by sodium methoxide yielded C8-alkyne-FANA uridine 6.
:3. The desired β-isomer 4 was isolated by recrystallization. Next, Sonogashira coupling of 4 and 1,7-octadiyne afforded 3′,5′-benzoyl protected nucleoside 5. It is noteworthy that an excessive amount of 1,7-octadiyne (12 equiv.) was used to ensure monofunctionalization and avoid crosslinking. Subsequently, deprotection of 5 by sodium methoxide yielded C8-alkyne-FANA uridine 6.
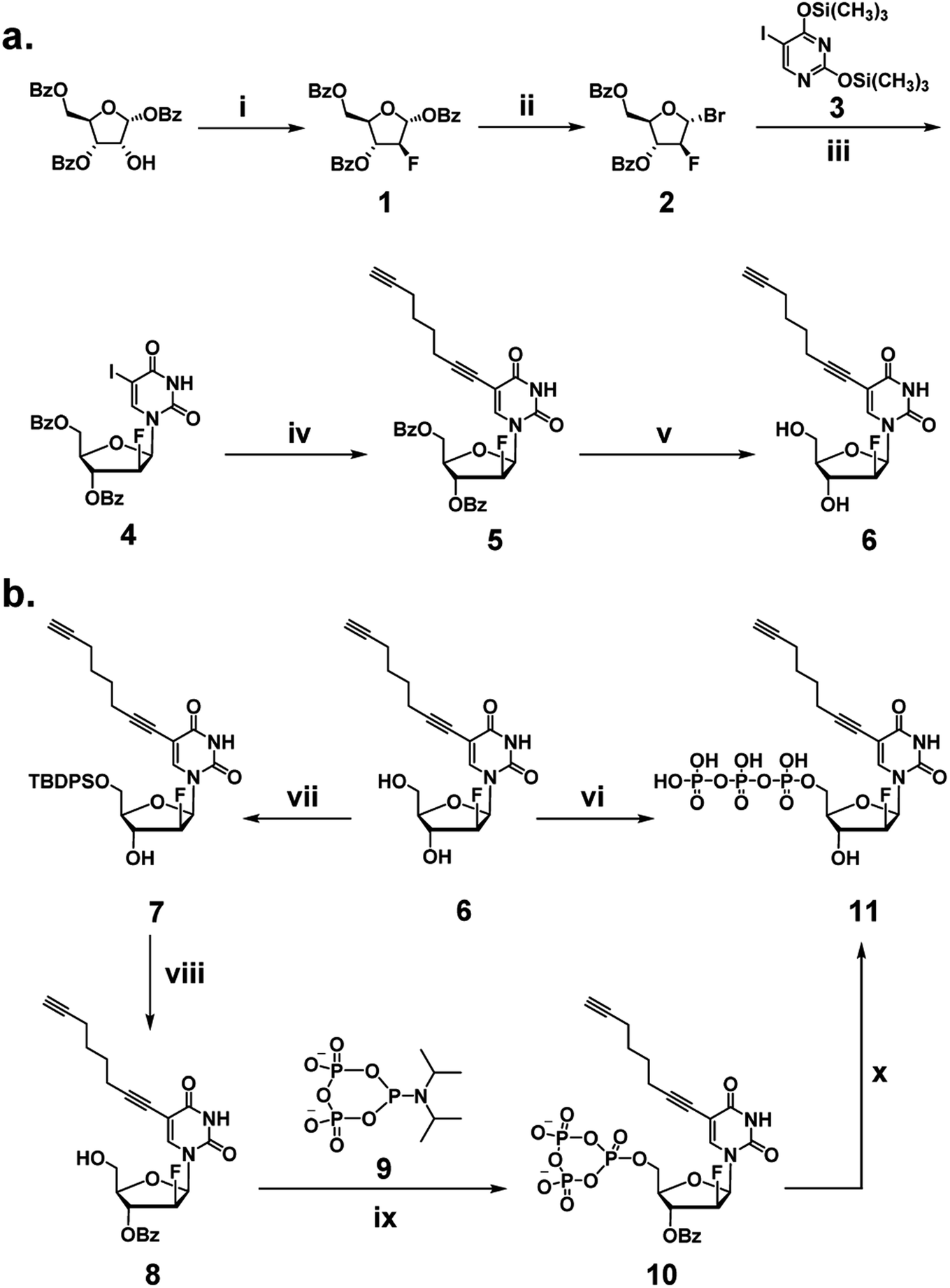 | ||
| Scheme 1 Synthesis of C8-alkyne-FANA UTP. (a) Synthetic route to C8-alkyne-FANA nucleoside 6. (i) DAST, DCM, 40 °C, 16 h, 85%; (ii) HBr, AcOH, DCM, r.t., 24 h, 80%; (iii) 3, NaI, DCM/ACN, r.t., 7 d, 48%; (iv) 1,7-octadiyne, PdCl2(PPh3)2, CuI, Et3N, DMF, r.t., 16 h, 76%; (v) NaOMe/MeOH, r.t., 2 h, 84%. (b) Conventional triphosphorylation strategy to yield C8-alkyne-FANA UTP 11, (vi) (1) POCl3, PO(OMe)3, 0 °C, 8 h; (2) (NHBu3)2H2P2O7, Bu3N, DMF, 0 °C, 1.5 h. An alternative triphosphorylation strategy using a cyclic pyrophosphoryl phosphoramidite (cPyPA) for coupling/ring-opening (steps vii–x) to give 11. (vii) TBDPSCl, imidazole, DMF, r.t., 18 h, 85%; (viii) (1) BzCl, pyridine, DCM, 0 °C, 2 h and (2) TBAF, THF, r.t., 1 h, 90%; (ix) (1) 9, 5-(ethylthio)-1H-tetrazole (ETT), MeCN, r.t., 3 h and (2) mCPBA, 0 °C, 5 min; (x) (1) D2O, r.t., 3 h and (2) NH4OH, r.t., 18 h, 60%. | ||
Triphosphorylation of 6 was initially attempted using POCl3 and pyrophosphate following the conventional Yoshikawa phosphorylation protocol (Scheme 1b, vi).35 This reaction resulted in a complex mixture, which required preparative HPLC for separation and only produced the desired C8-alkyne-FANA UTP 11 in poor yield (Fig. 2a). After examining several different triphosphorylation strategies, we found that the method using a cyclic pyrophosphoryl phosphoramidite (cPyPA) reported by Singh et al.36 enabled the preparation of 11 in high yield and good purity (Scheme 1b, vii–x and Fig. 2a). 1H-NMR, 31P-NMR, and mass spectrometry characterization confirmed the structure of 11 (Fig. 2b and c). Furthermore, accurate quantitation of 11 in the lyophilized product was achieved by 31P-NMR using a known amount of the sodium phosphate monobasic salt as the internal standard (Fig. S1†).
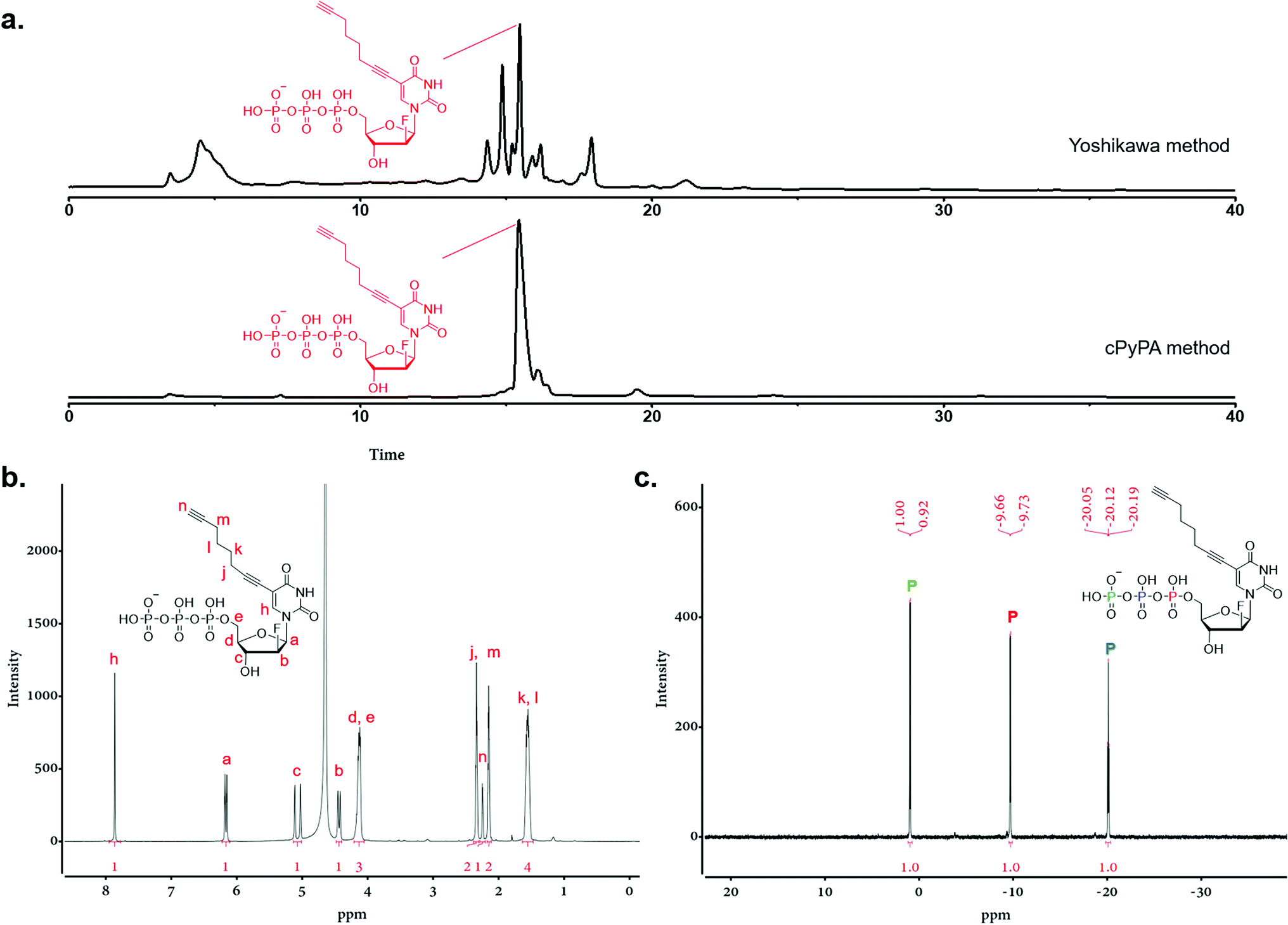 | ||
| Fig. 2 Characterization of C8-alkyne-FANA UTP (11). (a) The HPLC traces of triphosphorylation products using the Yoshikawa method (up) and the cPyPA method (down). (b) 1H-NMR and (c) 31P-NMR characterization of 11. | ||
Polymerase recognition of C8-alkyne-FANA UTP
The 5-1,7-octadiyne modification on uracil has previously been found to be well-tolerated by several distinct wild-type and engineered DNA polymerases.34 Based upon these findings, we reasoned that this modification could also be tolerated by Thermococcus gorgonarius (Tgo) DNA polymerase, the enzyme that has shown good activities in incorporating FANA nucleotides.12 To investigate this hypothesis, a primer extension experiment was performed, in which a DNA primer–template complex was enzymatically extended by Tgo DNA polymerase using commercially available FANA nucleotide triphosphates (A, C, and G) and 11. The DNA template was prepared to contain a 40-nucleotide (nt) random region to avoid sequence bias, and a 24-nt poly-AAC tail at the 3′-end, which would not be transcribed, to provide a mobility difference between the extension product (76 nt) and DNA template (100 nt) in denaturing polyacrylamide gel electrophoresis (PAGE). The DNA primer was labeled by a 5′-Cy5 fluorophore to allow easy detection of the extended product. A control experiment in which all FANA nucleotide triphosphates consisting of natural bases (A, C, G, and T) was also conducted to determine the impact of the 5-1,7-octadiyne modification of uracil on the primer extension reactivity. Tgo DNA polymerase was added in various amounts to confirm that the generation of the extended products was mediated by the Tgo DNA polymerase in a dose-dependent fashion. To our delight, both the primer extension reaction involving FANA NTPs of all natural bases and the one involving FANA ATP, CTP, GTP, and 11 produced the full-length products in a polymerase dose-dependent fashion (Fig. 3).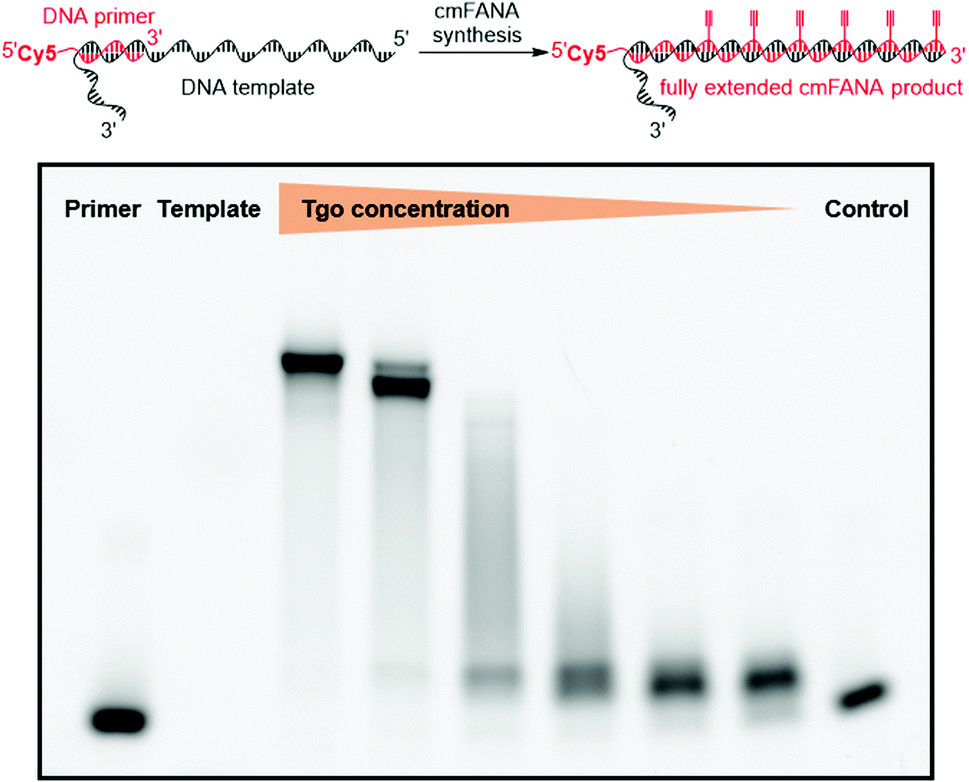 | ||
| Fig. 3 Tgo polymerase activity assay suggested that C8-alkyne-FANA UTP can successfully serve as a substrate. Various amounts of the polymerase were added to the reaction. The extended product was detected via a Cy5 fluorescent tag at the 5′-end of the primer. | ||
To unambiguously confirm the primer extension product, a cmFANA sequence was synthesized using a DNA template (T-ConA-XL, 98 nt including an 18-nt non-transcribed AAC repeating sequence at the 3′-end) encoding an aptamer targeting concanavalin A (ConA),34 and a 5′-biotinylated DNA primer. After the primer extension reaction, the DNA-cmFANA heteroduplex was captured by streptavidin magnetic particles, and the cmFANA polymer was separated from the DNA template strand by treating the captured heteroduplex with sodium hydroxide. The cmFANA polymer was then eluted from the magnetic particles by incubation with concentrated ammonium hydroxide at 70 °C. Electrospray ionization mass spectrometry (ESI-MS) analysis of the strand-separated cmFANA polymer suggested that the observed molecular weight of the cmFANA polymer (27632.5 Da) was consistent with the expected value (27632.5 Da). Taken together, these results indicated that the 5-1,7-octadiyne modification of uracil is well-tolerated by Tgo DNA polymerase and that C8-alkyne-FANA UTP could be quantitatively incorporated in a DNA-templated primer extension reaction mediated by this enzyme.
cmFANA exhibits superior nuclease resistance to DNA
To demonstrate cmFANA as a functionalizable synthetic genetic polymer for in vivo applications, we investigated its stability in the human serum, which contains a variety of nucleases and mimics the typical environment for in vivo applications. To allow facile quantitation of the cmFANA after human serum treatment, we conjugated azido-fluorescein to the cmFANA via CuAAC (Fig. 4a). A single-stranded DNA oligonucleotide fluorescently labeled by the Cy5 dye was used as a control. Both DNA and cmFANA were incubated in human serum for up to 48 hours, and the remaining nucleic acid species were analyzed by denaturing PAGE. We observed that while DNA started to degrade after one hour and was almost fully degraded after six hours (Fig. 4b), cmFANA was completely stable after 24 hours of incubation in human serum and could last even longer than 48 hours (Fig. 4c). The short ssDNA primer sequence that remained at the 5′-end of cmFANA seemed to withstand degradation as long as the cmFANA region, as the mobility of the product did not change throughout the incubation time. The superior nuclease resistance of cmFANA suggests that it is a promising synthetic genetic polymer for in vivo applications.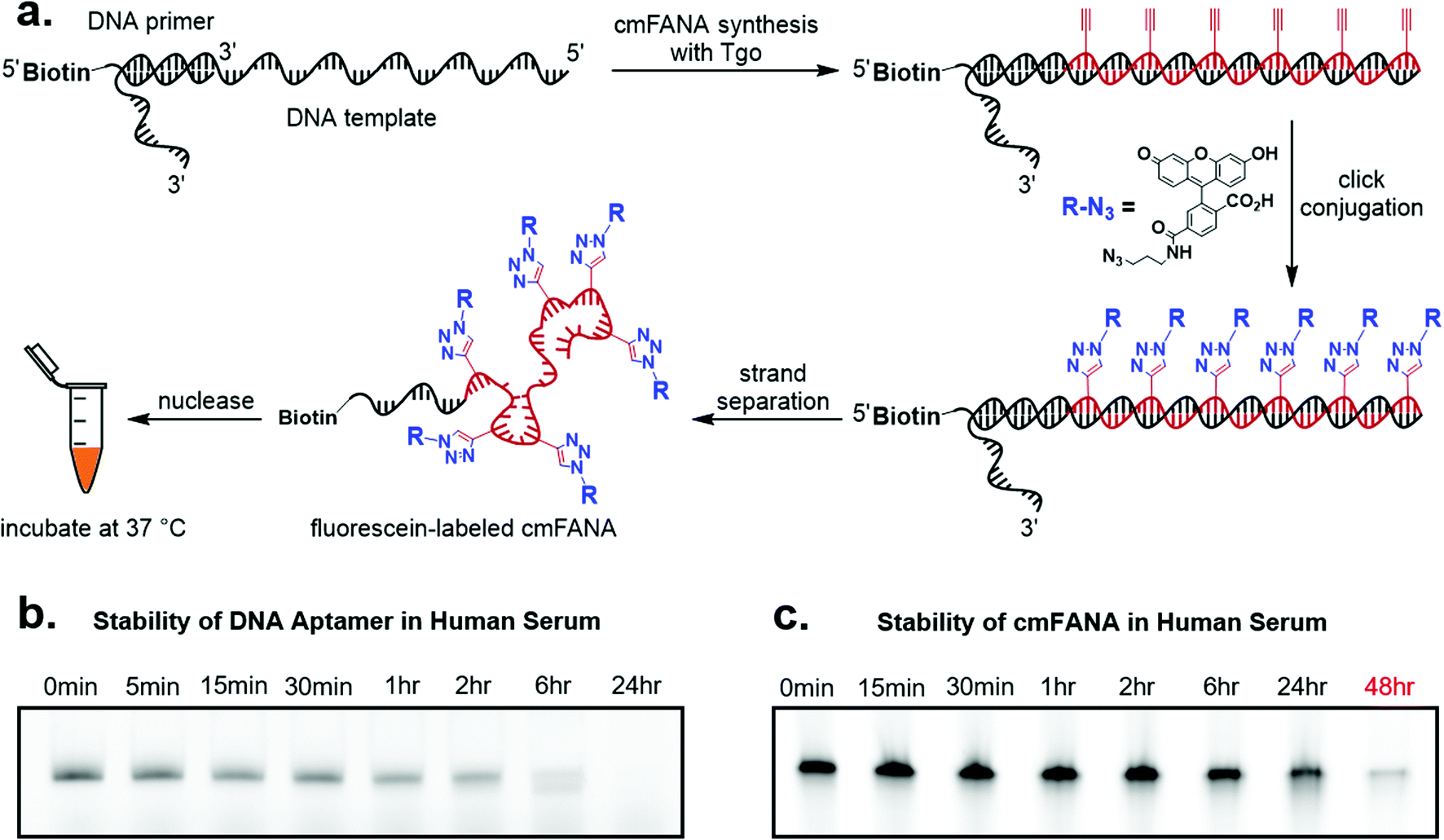 | ||
| Fig. 4 cmFANA stability assay. (a) Reaction scheme for generating cmFANA with fluorescein labeling. (b) Denaturing PAGE analysis of the stability status in the presence of human serum for single-stranded DNA and (c) fluorescein-labeled cmFANA. The gels were imaged in the Cy5 channel for DNA and the fluorescein channel for cmFANA. | ||
Click conjugation of carbohydrates to the cmFANA polymer
Carbohydrate-modified nucleic acids have emerged as a promising technology to develop affinity reagents for glycan-binding proteins (GBPs) and vaccine epitopes.33,37 To demonstrate that cmFANA can serve as a general platform for carbohydrate-modified XNAs, we investigated the click conjugation of azido-functionalized glycans with cmFANA (Fig. 5a). To this end, a cmFANA sequence (cmFANA1) was generated from the primer extension reaction using the DNA template T-ConA-XL and a 5′-biotinylated primer. The DNA-cmFANA1 heteroduplex was then coupled with a commercially available monosaccharide azidoethyl-penta-O-acetyl-mannose (Man-N3) by CuAAC. The conjugate was subsequently captured by streptavidin magnetic particles, strand-separated by the sodium hydroxide treatment, and deprotected and eluted from the magnetic particles by incubation with concentrated ammonium hydroxide for three hours. Denaturing PAGE analysis confirmed successful click conjugation with Man-N3, in which a clear reduction of the mobility of the band was observed after the click reaction (Fig. 5c). ESI-MS analysis of the product of the conjugation reaction suggested the quantitative coupling of the mannose residue to all alkyne groups in cmFANA1 (Fig. S2†). Notably, click conjugation of Man-N3 to cmFANA2, which has the same nucleotide composition as cmFANA1 but is rearranged to consist a string of consecutive alkynes in the sequence, did not reduce the conjugation efficiency (Fig. S3†).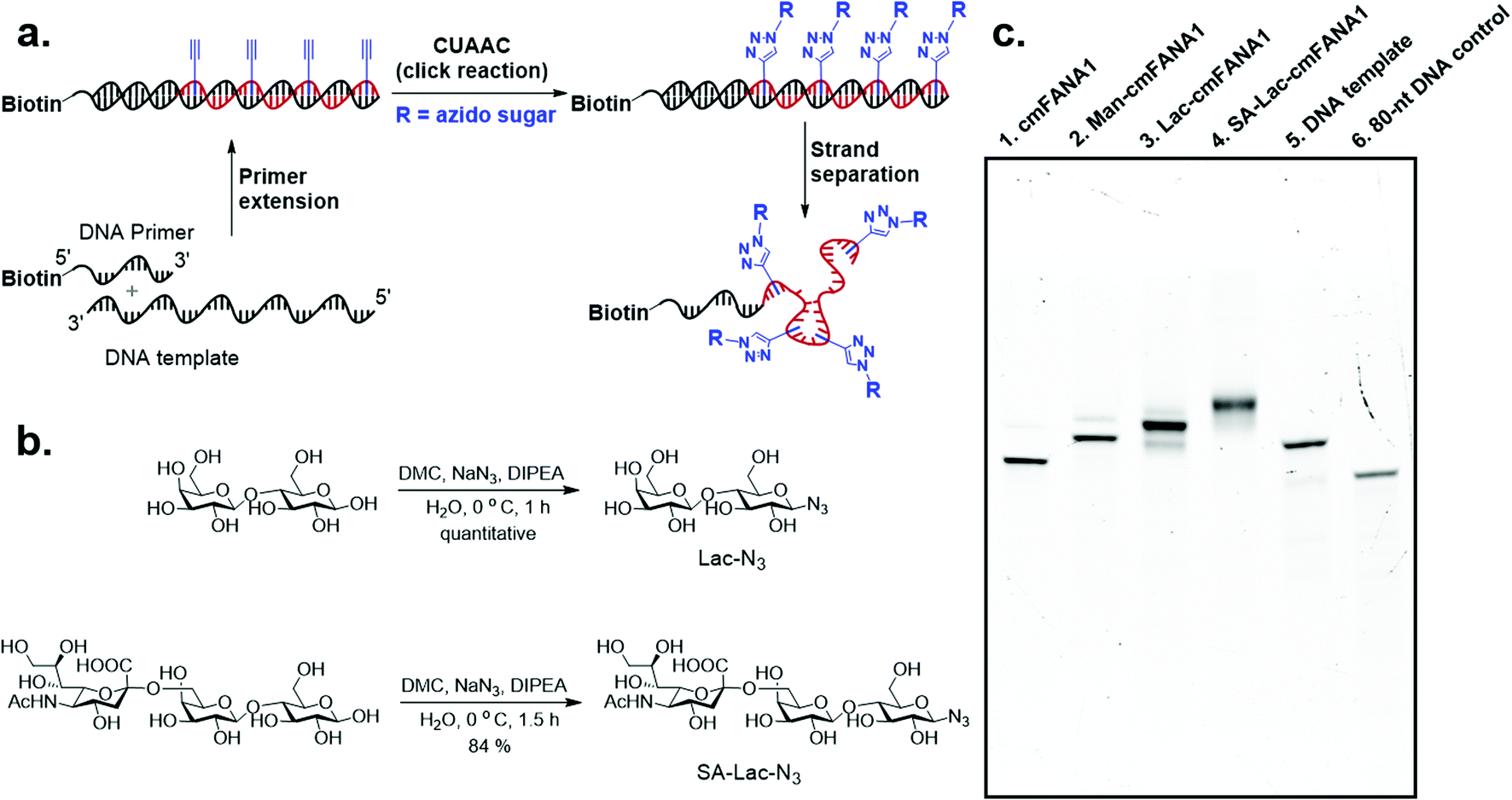 | ||
| Fig. 5 Click conjugation of azide-functionalized carbohydrates to the cmFANA polymer. (a) Reaction scheme of generation of single-stranded cmFANA conjugates. (b) Synthesis of sugar mimetics: Lac-N3 (up) and SA-Lac-N3 (down). (c) Gel analysis of cmFANA and carbohydrate-conjugated cmFANA. 10% TBE-urea gel, stained with ethidium bromide. Lane 1: cmFANA1, 80 nt; lane 2: Man-N3-conjugated cmFANA1 (Man-cmFANA1, 80 nt); lane 3: Lac-N3-conjugated cmFANA1 (Lac-cmFANA1, 80 nt); lane 4: SA-Lac-N3-conjugated cmFANA1 (SA-Lac-cmFANA1, 80 nt); lane 5: DNA template (T-ConA-XL, 98 nt); lane 6: a 80 nt DNA control. Trace amounts of the cmFANA-DNA template hybrids resulting from incomplete strand separation were observed above the major bands. | ||
Next, we attempted to conjugate a disaccharide lactose and a trisaccharide 2,6-sialyllactose to cmFANA. First, lactose and 2,6-sialyllactose from commercial sources were furnished with azido groups at the reducing end by reacting with 2-chloro-1,3-dimethylimidazolinium chloride (DMC) and sodium azide in water, following a method reported by Tanaka et al.38 (Fig. 5b). The resulting 1-azido-lactose (Lac-N3) and 1-azido-2,6-sialyllactose (SA-Lac-N3) were conjugated to the biotinylated DNA-cmFANA heteroduplex, before being subjected to strand separation using streptavidin magnetic particles as described above. Denaturing PAGE analysis showed one major band in each well which indicated a single major product in each conjugation (Fig. 5c). While all the cmFANA and carbohydrate-cmFANA conjugates consist of a cmFANA backbone with a length of 80 nt, we observed notable differences in gel mobility of the bands, which was attributed to the different molecular weights of conjugated carbohydrate substrates. ESI-MS analysis suggested that the fully conjugated cmFANA with all alkynes functionalized by Lac-N3 or SA-Lac-N3 were the major products, although byproducts with 1–3 unreacted alkynes were also observed (Fig. S4 and S5†). The slightly lower conjugation efficiency of Lac-N3 or SA-Lac-N3 compared to Man-N3 was likely a result of the large sizes of these substrates.
A DNA-display approach to link the carbohydrate-conjugated cmFANA to its encoding DNA template
Directed evolution of modified nucleic acids has become a powerful approach to discover affinity reagents and catalysts with enhanced functions.12,39 A critical step during the laboratory evolution process is the amplification of the genetic information carried by the evolving population.40–43 Base-modified nucleic acids often cause a challenging amplification step due to the inability to be reverse-transcribed back into native DNA. In particular, the bulky carbohydrate-conjugated cmFANA structure raised the question of whether it could be directly reverse-transcribed back to DNA. To this end, we tested a series of polymerases, including Taq, Bst LF*, Bst2.0, Bst3.0, Kod, Deep Vent, Tgo, and Q5, for the reverse transcriptase activity of cmFANA and Man-N3-conjugated cmFANA. Unfortunately, none of the polymerases tested could reverse-transcribe cmFANA or Man-N3-conjugated cmFANA (Fig. S7†).To overcome the challenging reverse transcription, an alternative strategy for amplifying the genetic information of the carbohydrate-conjugated cmFANA based on DNA-display44–46 was devised. We adopted the DNA-display strategy developed by Krauss et al.47 in their selection with modified aptamer (SELMA) method, which involves using a hairpin structure to establish the critical phenotype-genotype linkage (Fig. 6). To validate that the genetic information of a library of diverse carbohydrate-conjugated cmFANA sequences could be amplified using DNA-display, the cycle started from a hairpin regeneration process where a biotinylated hairpin-regenerating primer annealed to a library template (94 nt, with the N40 random region) and was subsequently extended into a double-stranded DNA library (134 bp) through bidirectional extension (Fig. 6, lane i). Streptavidin magnetic particle-enabled strand separation removed the unwanted biotinylated strand, yielding a single-stranded DNA template with a 40 nt random region (the genotype) that self-hybridizes into a hairpin structure. Due to the presence of the random region, this library appeared as a smear in the native PAGE (Fig. 6, lane ii). Then, Tgo DNA polymerase extended the hairpin from the 3′-end by using FANA ATP, CTP, GTP, and 11 (Fig. 6, lane iii), followed by the conjugation of Man-N3 with the cmFANA library via CuAAC (Fig. 6, lane iv). Next, strand displacement was affected by the addition of a primer that annealed in the loop region and was extended by DNA dNTPs using Bst2.0 WarmStart DNA polymerase. This primer extension step formed a dsDNA and displaced the Man-N3-conjugated cmFANA polymers (the phenotype, also referred to as Man-cmFANA hereafter ) from the DNA template (Fig. 6, lane v). After strand displacement, the Man-cmFANA-dsDNA hybrid structure was confirmed by exonuclease I digestion, which cleaves the 3′-terminal ssDNA portion along with the displaced Man-cmFANA phenotype, leaving the genotype dsDNA intact (Fig. 6, lane v-2). This genotype dsDNA contains a copy of the same sequence of the Man-cmFANA phenotype in the native DNA form, which could be readily amplified by PCR using a 5′-biotinylated primer. The PCR product was confirmed to be of the same length (94 bp) as the genotype dsDNA (Fig. 6, lane vi, comparable to lane v-2). Finally, capturing the PCR product by streptavidin magnetic particles enables strand separation to generate a single-stranded DNA coding sequence, which was subjected to the bidirectional extension again along with the hairpin-regenerating primer (Fig. 6, lane vii). The regenerated DNA template could be extended once again to produce a second-generation cmFANA polymer library (Fig. 6, lane viii, comparable to lane iii). Next-generation sequencing of the initial and regenerated DNA templates indicate similar numbers of unique sequences in the library region, suggesting the efficient recovery of the library diversity and that no significant sequence bias was introduced during the DNA-display cycle (Fig. S8†).
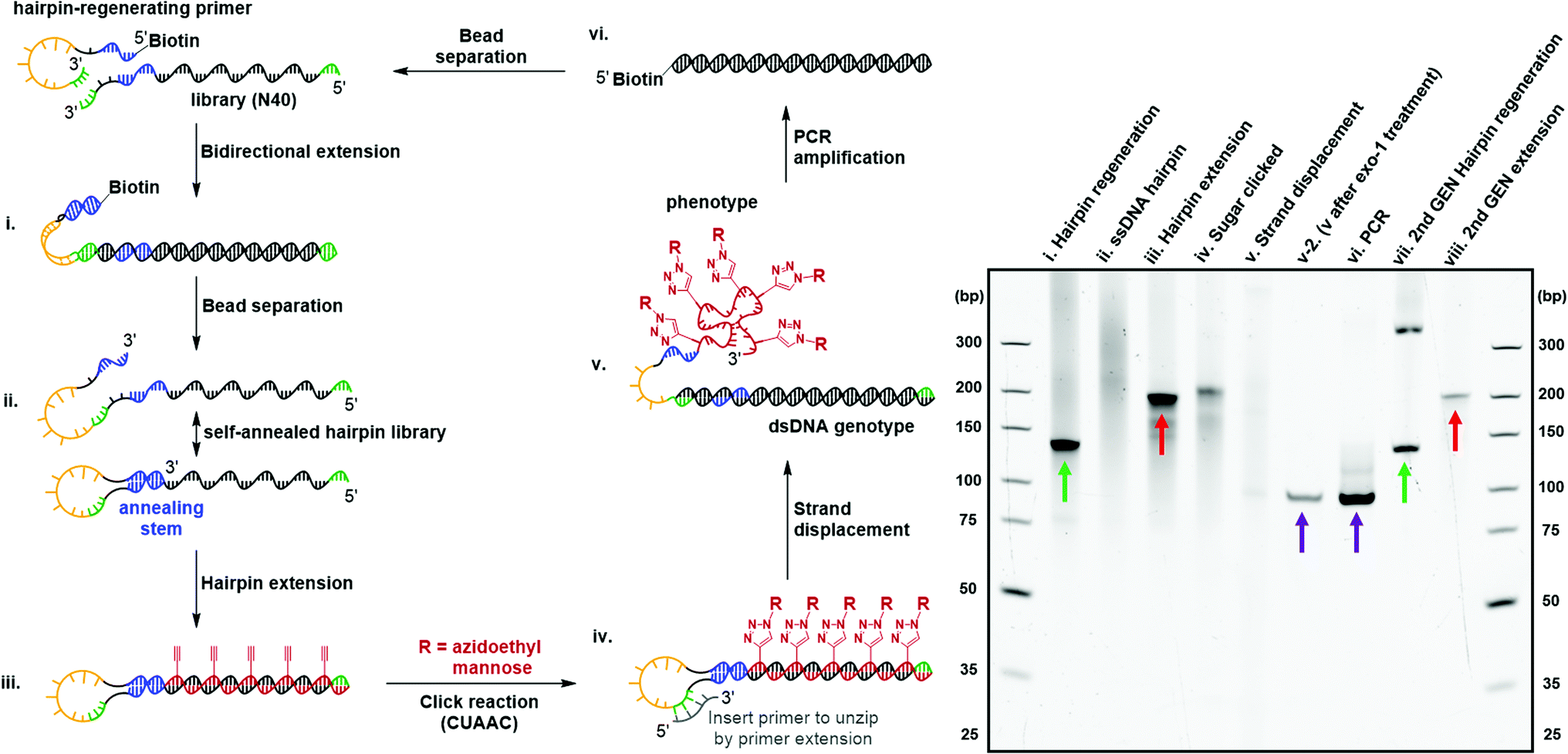 | ||
| Fig. 6 DNA-display and native PAGE analysis of the carbohydrate-conjugated cmFANA library. The genetic information of the mannose-conjugated cmFANA library can be amplified and reproduced through a complete DNA-display cycle. Products of the same mobility are indicated by red, green, and purple arrows, respectively, with each color representing a pair of products of the same mobility. A side product was observed above the targeted product in the template regeneration step in lane vii, which was attributed to the over-extension in the bidirectional extension. The amount of this byproduct was greatly reduced after the subsequent strand separation step and did not affect the second round of cmFANA synthesis (lane viii). | ||
The feasibility of the carbohydrate-conjugated cmFANA polymers to be evolved by in vitro selection was further validated by a mock selection experiment (Fig. 7). A positive control DNA template TConA-hairpin was transcribed into a cmFANA sequence and conjugated with an azide-functionalized biotin via CuAAC to form a biotinylated cmFANA-DNA hybrid structure. This biotinylated positive control was then added to a Man-cmFANA-DNA hybrid library at a ratio of library:control = 1000:1. The selection began with strand displacement to form the dsDNA genotype and the conjugated cmFANA phenotype. Following capture by streptavidin magnetic particles and six cycles of buffer wash, the dsDNA genotype was eluted by the exonuclease-I treatment and was subjected to PCR amplification. The enrichment of the positive control sequence after a full cycle of mock selection was confirmed by restriction enzyme digestion and next-generation sequencing. First, independent digestion by two restriction enzymes, BsrI and SmlI, both of which cut the positive control sequence but not the library, showed that the positive control has become the predominant constituent of the post-selection library. Furthermore, next-generation sequencing of the post-selection library indicated that 55% of the total reads have become the positive control sequence. Collectively, these results support the potential of the DNA-display strategy to be applied to the in vitro selection of functionality-conjugated cmFANA.
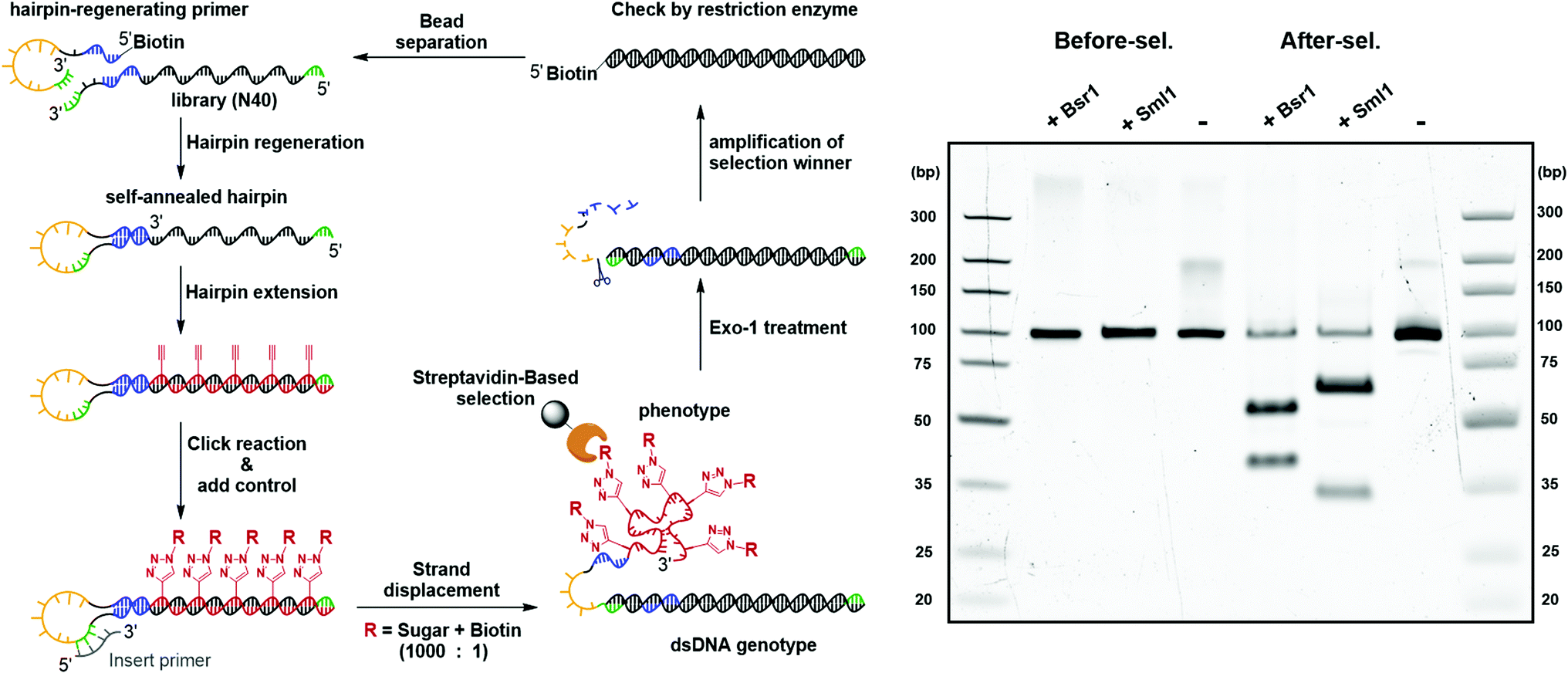 | ||
| Fig. 7 Mock selection via DNA-display. Two restriction enzymes with distinct cut sites, BsrI and SmlI, were implemented to independently cut the positive control sequence, but not the library, in the coding region before and after the mock selection. | ||
Conclusions
We have demonstrated the development of a cmFANA triphosphate that could be recognized by Tgo DNA polymerase and efficiently incorporated in DNA-templated primer extension reactions. The introduction of a C8 extended alkyne linker enabled efficient conjugation of azido-functionalized compounds without compromising the polymerase recognition. Furthermore, the cmFANA polymer exhibited excellent resistance to nuclease degradation. Although direct reverse-transcription of the cmFANA polymer or the carbohydrate-conjugated cmFANA by polymerases was challenging, an alternative method based on DNA-display was demonstrated to enable the critical genotype-phenotype linkage for amplifying the genetic information of the cmFANA polymer, making it suitable for further evolution of cmFANA-based aptamers. The cmFANA technology provides a valuable platform for presenting a variety of chemical functionalities including carbohydrates, fluorophores, and hydrophobic or charged moieties.Data availability
All other data supporting this article, including all experimental procedures, characterization details, and copies of NMR spectra for all compounds, have been uploaded as part of the ESI.†Author contributions
J. N. and K. B. W. designed all the experiments and analyzed the results. J. N. mentored the research. K. B. W. performed the experiments. C. S. participated in the initial design and execution of this study. Q. S. performed the expression and purification of the Bst-LF* polymerase. J. L. participated in the synthesis and validation of the Man-cmFANA. J. N. and K. B. W. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a start-up fund from Boston College to J. N. and the NIH through a Director's New Innovator Award to J. N. (1DP2HG011027-01). Q. S. acknowledges the support from the NSF graduate research fellowship. We graciously thank Prof. John Chaput for providing the early inspiration of the project and for providing the plasmid of the Tgo polymerase. We also thank Dr Yajun Wang for the helpful discussion.Notes and references
- A. Condon, Nat. Rev. Genet., 2006, 7, 565–575 CrossRef CAS PubMed.
- Y. Zhang, B. S. Lai and M. Juhas, Molecules, 2019, 24, 941 CrossRef PubMed.
- C. Yang, K. B. Wu, Y. Deng, J. Yuan and J. Niu, ACS Macro Lett., 2021, 10, 243–257 CrossRef CAS PubMed.
- J. K. Watts and M. J. Damha, Can. J. Chem., 2008, 86, 641–656 CrossRef.
- I. Anosova, E. A. Kowal, M. R. Dunn, J. C. Chaput, W. D. Van Horn and M. Egli, Nucleic Acids Res., 2015, 44, 1007–1021 CrossRef PubMed.
- M. R. Dunn, R. M. Jimenez and J. C. Chaput, Nat. Rev. Chem., 2017, 1, 1–16 CrossRef.
- A. W. Feldman and F. E. Romesberg, Acc. Chem. Res., 2018, 51, 394–403 CrossRef CAS PubMed.
- V. T. Dien, S. E. Morris, R. J. Karadeema and F. E. Romesberg, Curr. Opin. Chem. Biol., 2018, 46, 196–202 CrossRef CAS PubMed.
- V. B. Pinheiro and P. Holliger, Trends Biotechnol., 2014, 32, 321–328 CrossRef CAS PubMed.
- K. Duffy, S. Arangundy-Franklin and P. Holliger, BMC Biol., 2020, 18, 1–14 CrossRef PubMed.
- J. C. Chaput, H. Yu and S. Zhang, Chem. Biol., 2012, 19, 1360–1371 CrossRef CAS PubMed.
- Y. Wang, A. K. Ngor, A. Nikoomanzar and J. C. Chaput, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- C. G. Peng and M. J. Damha, Nucleic Acids Res., 2007, 35, 4977–4988 CrossRef CAS PubMed.
- I. Alves Ferreira-Bravo, C. Cozens, P. Holliger and J. J. DeStefano, Nucleic Acids Res., 2015, 43, 9587–9599 Search PubMed.
- K. M. Rose, I. Alves Ferreira-Bravo, M. Li, R. Craigie, M. A. Ditzler, P. Holliger and J. J. DeStefano, ACS Chem. Biol., 2019, 14, 2166–2175 CAS.
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S.-Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS PubMed.
- H. Shi, X. He, W. Cui, K. Wang, K. Deng, D. Li and F. Xu, Anal. Chim. Acta, 2014, 812, 138–144 CrossRef CAS PubMed.
- A. S. Jørgensen, L. H. Hansen, B. Vester and J. Wengel, Bioorg. Med. Chem. Lett., 2014, 24, 2273–2277 CrossRef PubMed.
- H. Yu, S. Zhang and J. C. Chaput, Nat. Chem., 2012, 4, 183–187 CrossRef CAS PubMed.
- M. R. Dunn, C. M. McCloskey, P. Buckley, K. Rhea and J. C. Chaput, J. Am. Chem. Soc., 2020, 142, 7721–7724 CrossRef CAS PubMed.
- J. C. Chaput, Acc. Chem. Res., 2021, 54, 1056–1065 CrossRef CAS PubMed.
- C. M. McCloskey, Q. Li, E. J. Yik, N. Chim, A. K. Ngor, E. Medina, I. Grubisic, L. Co Ting Keh, R. Poplin and J. C. Chaput, ACS Synth. Biol., 2021, 10, 3190–3199 CrossRef CAS PubMed.
- A. Nikoomanzar, M. R. Dunn and J. C. Chaput, Curr. Protoc. Nucleic Acid Chem., 2017, 69, 4.75.1–4.75.20 CAS.
- A. P. Mehta, H. Li, S. A. Reed, L. Supekova, T. Javahishvili and P. G. Schultz, J. Am. Chem. Soc., 2016, 138, 14230–14233 CrossRef CAS PubMed.
- D. A. Malyshev, K. Dhami, T. Lavergne, T. Chen, N. Dai, J. M. Foster, I. R. Corrêa and F. E. Romesberg, Nature, 2014, 509, 385–388 CrossRef CAS PubMed.
- A. W. Feldman, V. T. Dien, R. J. Karadeema, E. C. Fischer, Y. You, B. A. Anderson, R. Krishnamurthy, J. S. Chen, L. Li and F. E. Romesberg, J. Am. Chem. Soc., 2019, 141, 10644–10653 CrossRef CAS PubMed.
- R. L. Redman and I. J. Krauss, J. Am. Chem. Soc., 2021, 143, 8565–8571 CrossRef CAS PubMed.
- S. Strauss, P. C. Nickels, M. T. Strauss, V. J. Sabinina, J. Ellenberg, J. D. Carter, S. Gupta, N. Janjic and R. Jungmann, Nat. Methods, 2018, 15, 685–688 CrossRef CAS PubMed.
- Z. Chen, P. A. Lichtor, A. P. Berliner, J. C. Chen and D. R. Liu, Nat. Chem., 2018, 10, 420–427 CrossRef CAS PubMed.
- H. Mei, J.-Y. Liao, R. M. Jimenez, Y. Wang, S. Bala, C. McCloskey, C. Switzer and J. C. Chaput, J. Am. Chem. Soc., 2018, 140, 5706–5713 CrossRef CAS PubMed.
- Q. Li, V. A. Maola, N. Chim, J. Hussain, A. Lozoya-Colinas and J. C. Chaput, J. Am. Chem. Soc., 2021, 143, 17761–17768 CrossRef CAS PubMed.
- F. Tolle, G. M. Brändle, D. Matzner and G. Mayer, Angew. Chem., Int. Ed., 2015, 54, 10971–10974 CrossRef CAS PubMed.
- I. S. MacPherson, J. S. Temme, S. Habeshian, K. Felczak, K. Pankiewicz, L. Hedstrom and I. J. Krauss, Angew. Chem., Int. Ed., 2011, 50, 11238–11242 CrossRef CAS PubMed.
- C. K. Gordon, D. Wu, A. Pusuluri, T. A. Feagin, A. T. Csordas, M. S. Eisenstein, C. J. Hawker, J. Niu and H. T. Soh, ACS Chem. Biol., 2019, 14, 2652–2662 CrossRef CAS PubMed.
- M. Yoshikawa, T. Kato and T. Takenishi, Tetrahedron Lett., 1967, 8, 5065–5068 CrossRef.
- J. Singh, A. Ripp, T. M. Haas, D. Qiu, M. Keller, P. A. Wender, J. S. Siegel, K. K. Baldridge and H. J. Jessen, J. Am. Chem. Soc., 2019, 141, 15013–15017 CrossRef CAS PubMed.
- R. L. Redman and I. J. Krauss, J. Am. Chem. Soc., 2021, 143, 8565–8571 CrossRef CAS PubMed.
- T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S.-i. Shoda, Chem. Commun., 2009, 3378–3379 RSC.
- G. Houlihan, S. Arangundy-Franklin, B. T. Porebski, N. Subramanian, A. I. Taylor and P. Holliger, Nat. Chem., 2020, 12, 683–690 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1992, 355, 850–852 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, science, 1990, 249, 505–510 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Biomol. Eng., 2007, 24, 381–403 CrossRef CAS PubMed.
- M. Yonezawa, N. Doi, Y. Kawahashi, T. Higashinakagawa and H. Yanagawa, Nucleic Acids Res., 2003, 31, e118 CrossRef PubMed.
- R. W. Roberts and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12297–12302 CrossRef CAS PubMed.
- J. K. Ichida, K. Zou, A. Horhota, B. Yu, L. W. McLaughlin and J. W. Szostak, J. Am. Chem. Soc., 2005, 127, 2802–2803 CrossRef CAS PubMed.
- J. S. Temme and I. J. Krauss, Curr. Protoc. Chem. Biol., 2015, 7, 73–92 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc00679k |
| ‡ Current address: Moderna, Inc., 200 Technology Square, Cambridge, MA 02139, USA. |
| This journal is © The Royal Society of Chemistry 2022 |