
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of unsymmetrically tetrasubstituted pyrroles and studies of AIEE in pyrrolo[1,2-a]pyrimidine derivatives†
Taian
Li‡
ab,
Mong-Feng
Chiou‡
b,
Yajun
Li
b,
Changqing
Ye
b,
Min
Su
bc,
Mengyu
Xue
b,
Xiaobin
Yuan
ab,
Chuanchuan
Wang
b,
Wen-Ming
Wan
 b,
Daliang
Li
*a and
Hongli
Bao
*bc
b,
Daliang
Li
*a and
Hongli
Bao
*bc
aCollege of Chemistry and Material Science, Biomedical Research Center of South China, College of Life Sciences, Fujian Normal University, 1 Keji Road, Fuzhou, 350117, P. R. China. E-mail: daliangli@fjnu.edu.cn
bKey Laboratory of Coal to Ethylene Glycol and Its Related Technology, State Key Laboratory of Structural Chemistry, Center for Excellence in Molecular Synthesis, Chinese Academy of Sciences, 155 Yangqiao Road West, Fuzhou, Fujian 350002, P. R. China. E-mail: hlbao@fjirsm.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 20th April 2022
Abstract
Pyrroles are among the most important heterocycles in pharmaceuticals and agrochemicals. Construction of pyrrole scaffolds with different substituents and a free NH group, however, is challenging. Herein, a metal-free method for the synthesis of unsymmetrically tetrasubstituted NH-pyrroles using a consecutive chemoselective double cyanation is reported. The desired pyrroles were obtained with yields up to 99% and good functional group tolerance. Mechanistic studies identified a reaction mechanism that features a subtle sequence of first cyano-addition and migration, followed by cyano-addition and aromatization to afford the pyrrole skeleton. Pyrrolo[1,2-a]pyrimidines are synthesized as the synthetic applications of NH-pyrroles, and these pyrrolo[1,2-a]pyrimidines exhibit unpredicted time-dependent aggregation-induced emission enhancement (AIEE) properties.
Introduction
As one of the major challenges and driving forces for the development of innovative pharmaceuticals and agrochemicals, efficient construction of structurally diverse and highly functionalized heterocycles has long been a highly cited research field.1 As prevalent key motifs in bioactive natural molecules, pyrrole and its derivatives have diverse applications in therapeutically active compounds, such as the heavily used drugs, lipitor, sutent, and molindone (Fig. 1a).2 As core structures, these valuable polysubstituted pyrroles show that production of polysubstituted pyrrole scaffolds, especially with unsymmetrically disposed substituents and diverse functional groups, is highly sought. | ||
| Fig. 1 Pharmaceutically significant pyrroles and synthesis of heteroarenes from α,β-unsaturated sulfonimines. | ||
Classically, pyrroles can be synthesized by the Paal–Knorr reaction3 or the Hantzsch reaction.4 Many other syntheses of pyrroles have been established,5 including powerful coupling strategies realized by transition-metals5 and the van Leusen pyrrole synthesis.6 However, the major disadvantages of these protocols lie in their requirement for substrate pre-functionalization, substituent deficiency and formation of by-products. Synthesis of pyrrole scaffolds with unsymmetrical tetrasubstituents7 can be considerably difficult, and it is even more challenging to obtain polysubstituted pyrroles with an unprotected free NH group, so called NH-pyrroles.8 Examples such as the base-catalyzed cyclization of propargyl enamines by Wan's group,9 the rhodium-catalyzed tandem reaction of α-diazo oxime ethers by Park et al.,10 a visible-light-induced formal [3 + 2] cycloaddition by Xiao et al.11 and an electrochemical heterocoupling between two structurally diverse enamines reported by Roy and De Sarkar12 have disclosed excellent methods with which to synthesize polysubstituted NH-pyrroles. Nevertheless, only carbon substituents can be introduced into pyrroles with these methods, and procedures for the generation of NH-pyrroles with diverse functionalities are extremely rare.
On the basis of the widely used synthesis of commercially available chalcones,13 we have developed an efficient method for the synthesis of polysubstituted NH-pyrroles directly from α,β-unsaturated sulfonimines, which have been shown by Loh,14 Smith,15 and Yoshikai16 to be good precursors for the construction of multisubstituted pyridines (Fig. 1b). Judging by the proposed mechanism, an interesting consecutive chemo-selective double cyanation should be important in this strategy. This reaction provides access to unsymmetrically tetrasubstituted pyrroles and tolerates functional groups, such as a free NH group that can be further substituted (Fig. 1c). In addition, the pyrrolo[1,2-a]pyrimidine derivatives that were synthesized from NH-pyrroles exhibit unpredicted time-dependent aggregation-induced emission enhancement (AIEE) properties.
The initial screening and optimization of this pyrrole synthesis was carried out with an α,β-unsaturated sulfonimine (1s) as the model substrate and TMSCN. As shown in Table 1, the desired tetrasubstituted NH-pyrrole (1) can be easily generated when the reaction was performed in DMF at 80 °C with Cs2CO3 as the base (Table 1, entry 1). Screening of other alternative inorganic or organic bases showed that Cs2CO3 gave the best results (Table 1, entries 2 and 3 vs. entry 1). Decreasing or increasing the reaction temperature resulted in a lower yield (Table 1, entries 4 and 5). When anhydrous DMSO was used as the solvent, the best isolated yield of the NH-pyrrole (1) was 80% (Table 1, entry 6). While other solvents fail to improve the efficiency of the reaction, protic EtOH completely terminated the reaction (Table 1, entries 7 and 8). In the absence of a base, no reaction occurred (Table 1, entry 9), and finally, when the Bs (PhSO2) protecting group was replaced by a toluene sulfonyl group (Ts), the NH-pyrrole (1) was obtained in 73% yield (Table 1, entry 10). Other cyanide sources17 such as Zn(CN)2, K3[Fe(CN)6] and tBuCN have been studied, but only Zn(CN2) could afford the target molecule in 41% yield.
|
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Temp (°C) | Yieldb (%) |
| a Reaction conditions: sulfonimine 1s (0.2 mmol, 1 equiv.), TMSCN (2.2 equiv.), base (2 equiv.), anhydrous solvent (1.5 mL), 80 °C, 12 h, under a nitrogen atmosphere. b Yields were determined by GC-MS analysis of the crude product using 1,4-dimethoxybenzene as an internal standard. c Yields with K2CO3, Na2CO3, t-BuONa, and KF were 43%, 17%, 32%, and 37%, respectively. d Yields with TEA, DBU, DMAP, and DABCO were 4%, 29%, trace, and 12%, respectively. e Isolated yield in parentheses. f Yields with DMAc, MeCN, NMP, 1,4-dioxane, and THF were 49%, 57%, 29%, 18% and 45%, respectively. g N-((1E,2E)-1,3-Diphenylallylidene)-4-methylbenzenesulfonamide was used instead of 1s (Bs was replace by Ts). h ZnCN2 was used instead of TMSCN. i K3Fe(CN)6 was used instead of TMSCN. j t BuCN was used instead of TMSCN. | ||||
| 1 | Cs2CO3 | DMF | 80 | 68 |
| 2 | K2CO3, Na2CO3, t-BuONa, or KF | DMF | 80 | 17–43c |
| 3 | TEA, DBU, DMAP, or DABCO | DMF | 80 | 0–29d |
| 4 | Cs2CO3 | DMF | 70 | 47 |
| 5 | Cs2CO3 | DMF | 85 | 62 |
| 6 | Cs2CO3 | DMSO | 80 | 82 (80)e |
| 7 | Cs2CO3 | DMAc, MeCN, NMP, 1,4-dioxane, or THF | 80 | 18–57f |
| 8 | Cs2CO3 | EtOH | 80 | 0 |
| 9 | DMSO | 80 | 0 | |
| 10 | Cs2CO3 | DMSO | 80 | 73g |
| 11 | Cs2CO3 | DMSO | 80 | 41h |
| 12 | Cs2CO3 | DMSO | 80 | 0i |
| 13 | Cs2CO3 | DMSO | 80 | 0j |
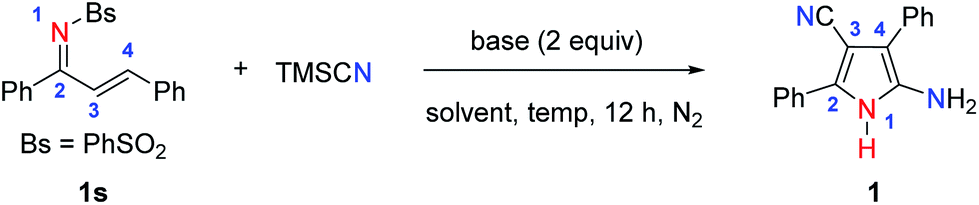
With the optimized conditions in hand, we explored the substrate scope and the limitations of the reaction. As shown by the results in Fig. 2, the reaction exhibits good functional group compatibility, and a variety of unsymmetrically tetrasubstituted NH-pyrroles can be obtained from α,β-unsaturated sulfonimines. Electron-donating substituents at the para-position of the aryl group (the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C side) are well-tolerated and give the corresponding products (1–4) in yields of 67–91%. The reaction proceeds smoothly with weak electron-withdrawing substituents, leading to the desired pyrroles (5–7, 10) in yields ranging from 72 to 96%. Compounds with ortho- and meta-methoxy substituents afford 9 and 11, respectively, in high yields. For substrates with a disubstituted aryl group, the reaction affords the corresponding pyrroles (12–14) in good to excellent yields. In addition to compounds with a phenyl ring, other aryl substituents are also tolerated. For example, naphthyl, pyrenyl, and thienyl groups are entirely compatible, leading to reactions which can convert the substrates into the corresponding NH-pyrroles (15–17) in excellent yields. Interestingly, when the C2-symmetric substrate, N-((1E,2E)-1,3-diphenylallylidene)benzene-sulfon-amide (18s) is employed in the reaction, both sides of the α,β-unsaturated sulfonimine moiety are involved in the formation of the pyrrole skeleton, and the reaction provides a C2-symmetric product (18) with two pyrrole substituents in 80% yield. However, when the R1 group is an alkyl group (Me and tBu), the desired NH-pyrrole (19, 20) was not detected. The structure of the NH-pyrrole (2) was confirmed by X-ray single crystal diffraction analysis.
C side) are well-tolerated and give the corresponding products (1–4) in yields of 67–91%. The reaction proceeds smoothly with weak electron-withdrawing substituents, leading to the desired pyrroles (5–7, 10) in yields ranging from 72 to 96%. Compounds with ortho- and meta-methoxy substituents afford 9 and 11, respectively, in high yields. For substrates with a disubstituted aryl group, the reaction affords the corresponding pyrroles (12–14) in good to excellent yields. In addition to compounds with a phenyl ring, other aryl substituents are also tolerated. For example, naphthyl, pyrenyl, and thienyl groups are entirely compatible, leading to reactions which can convert the substrates into the corresponding NH-pyrroles (15–17) in excellent yields. Interestingly, when the C2-symmetric substrate, N-((1E,2E)-1,3-diphenylallylidene)benzene-sulfon-amide (18s) is employed in the reaction, both sides of the α,β-unsaturated sulfonimine moiety are involved in the formation of the pyrrole skeleton, and the reaction provides a C2-symmetric product (18) with two pyrrole substituents in 80% yield. However, when the R1 group is an alkyl group (Me and tBu), the desired NH-pyrrole (19, 20) was not detected. The structure of the NH-pyrrole (2) was confirmed by X-ray single crystal diffraction analysis.
 | ||
| Fig. 2 Substrate scope of carbon substituents. Reaction conditions: sulfonimine (0.5 mmol, 1 equiv.), TMSCN (2.2 equiv.), Cs2CO3 (2 equiv.), anhydrous DMSO (4 mL), 80 °C, 12 h, under a nitrogen atmosphere. a TMSCN (4.4 equiv.) and Cs2CO3 (4 equiv.) were used instead. | ||
Subsequently, the scope of another substituent on the CN side of the α,β-unsaturated sulfonimines was explored (Fig. 3). Substrates with electron-rich substituents (21s–24s) and electron-deficient substituents (25s–29s) at the para-position of the aromatic ring afforded the corresponding unsymmetrically tetra-substituted NH-pyrroles (21–29) in yields of 75–99%. While substrates with a 3-chlorophenyl substituent (30s) afford the desired product in 69% yield, 3,5-dimethylphenyl (31s) and 4-fluoro-3-methylphenyl (32s) substituents are also well tolerated. Bis(4-(methylthio)phenyl (33)) and dinaphthyl (34) NH-pyrroles with the same substituents at the CC or the CN side could be obtained in good yield. When R2 is cyclopropyl, the corresponding product 35 can be obtained with 43% yield. However, when R2 is hydrogen and methyl, no trace of the corresponding NH-pyrrole (36, 37) is detected.
 | (1) |
 | ||
| Fig. 3 Substrate scope of substituents on the CN side. Reaction conditions: sulfonimine (0.5 mmol, 1 equiv.), TMSCN (2.2 equiv.), Cs2CO3 (2 equiv.), anhydrous DMSO (4 mL), 80 °C, 12 h, under a nitrogen atmosphere. | ||
Unexpectedly, distinct from the reaction with diaryl-substituted α,β-unsaturated sulfonimines, a substrate with a tert-butyl group next to the CN bond (38s) afforded only the 1,2-cyano addition product (38) in 73% yield (eqn (1)), presumably as a result of the considerable steric hindrance.18 This result suggested that this reaction might entail an initial 1,2-addition step.
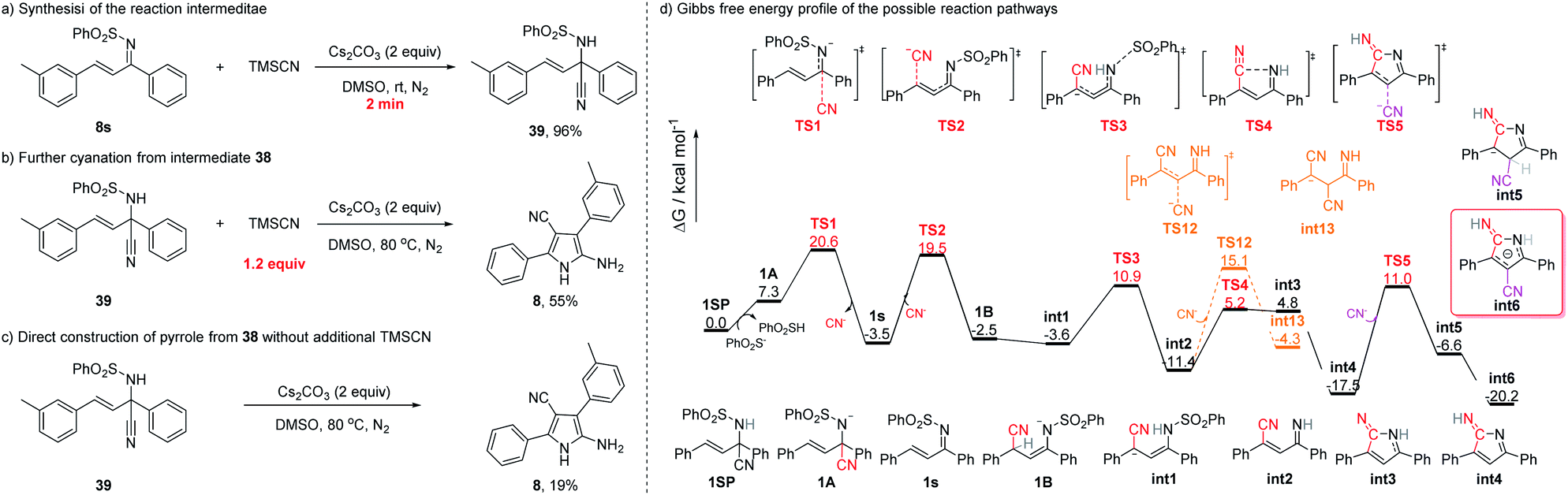
In order to gain some insight into the mechanism of the reaction, several control experiments were conducted (Fig. 4). First, when the reaction of 8s was conducted at rt, the 1,2-cyano addition product (39) was generated almost quantitatively in 2 min (Fig. 4a), a result similar to that obtained with substrate 38s.18 A reasonable hypothesis is that the reaction at rt of an α,β-unsaturated sulfonimine and TMSCN might undergo 1,2-addition to afford the corresponding cyano sulfonamide. Second, upon subjecting 39 to the standard conditions (with 1.2 equiv. of TMSCN), the corresponding NH-pyrrole (8) was isolated in 55% yield (Fig. 4b). Even in the absence of TMSCN, the cyano sulfonamide (39) can be converted into the NH-pyrrole (8) (Fig. 4c), implying that the 1,2-addition of the cyano group is reversible at higher temperatures.
 | ||
| Fig. 4 Experimental and theoretical studies on the reaction mechanism. | ||
Theoretical studies have also been conducted on the basis of the experimental results to investigate the reaction mechanisms of cyclization and double cyanation. Density functional theory (DFT) calculations were performed at the B3LYP-D3(SMD)/Def2-TZVP//B3LYP-D3/Def2-SVP level of theory. Since the reaction can be carried out starting from 39 without additional TMSCN but with much lower yields, the calculation of reversible CN elimination from 1SP was initially considered. Deprotonation of 1SP should be an easy process under the base reaction conditions, and the intermediate 1A can then eliminate the cyanide ion viaTS1 with a barrier of 13.3 kcal mol−1 to afford 1s (Fig. 4d). The isolated CN− ion therefore can add onto the carbon C4 of 1s to deliver the intermediate 1B through the transition state TS2, however, with a slightly higher barrier of 23.0 kcal mol−1, indicating that there may be a competition between 1,2- and 1,4-addition of 1s to reverse to the 1SP or forward to the pyrrole without external TMSCN at high temperature.
Several plausible pathways starting from intermediate 1B have been examined (see the ESI for more details), and the rearranged intermediate int1 with a relative free enthalpy −3.6 kcal mol−1 was found to be the more favorable conformation than 1B. Then, int1 can undergo the desulfurization rather than the second CN− addition to afford int2via the transition state TS3 with a barrier of 14.5 kcal mol−1. Next, the transition state of cyclization, TS4, was found to be a much lower state along the reaction pathway than that of TS12 (the second CN− addition).
Finally, the annulated intermediate int3 then should perform the proton transfer to afford intermediate int4 with a free enthalpy of −17.5 kcal mol−1. The second cyanide then can add onto int4via the transition state TS5 to deliver the intermediate int5 which will rearrange to the aromatic conformer int6 with a much stable free energy of −20.2 kcal mol−1.
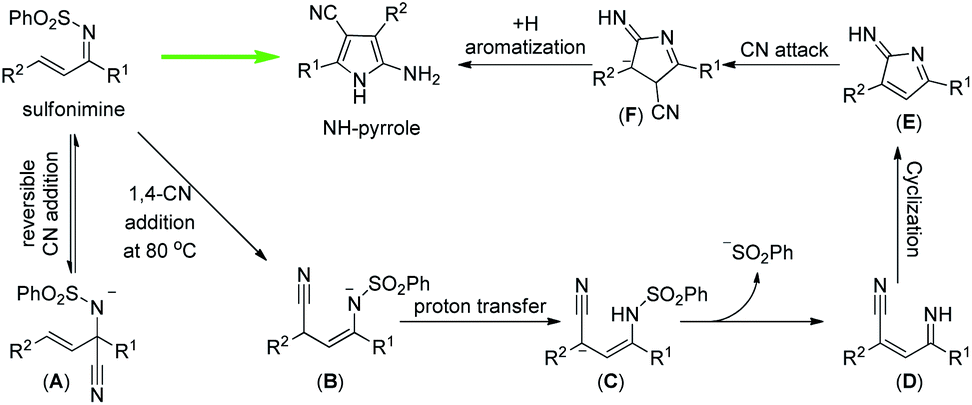
On the basic of these mechanistic studies, a plausible reaction mechanism was proposed and is shown in Fig. 5. Initially, a first cyanation occurs on the imine moiety to generate the cyano sulfonamide A.18 Upon heating, elimination of the cyano group from A takes place,19 regenerating the α,β-unsaturated sulfonimine, which will undergo 1,4-Michael addition to afford B. Intermediate B undergoes proton transfer and then deprotects the sulfone group to form intermediate D. Then, intramolecular 5-exo-dig cyclization provides an intermediate E which undergoes a second cyanation to offer intermediate F. Finally, the thermodynamically stable NH-pyrrole is formed by aromatization and protonation.
 | ||
| Fig. 5 Proposed mechanism. | ||
The synthetic value of the unsymmetrically tetrasubstituted NH-pyrroles was demonstrated by the construction of various multisubstituted pyrrolo[1,2-a]pyrimidines. As shown in Fig. 6, a wide range of fused aza-arenes (40–51) can be developed from the NH-pyrroles and diketones.
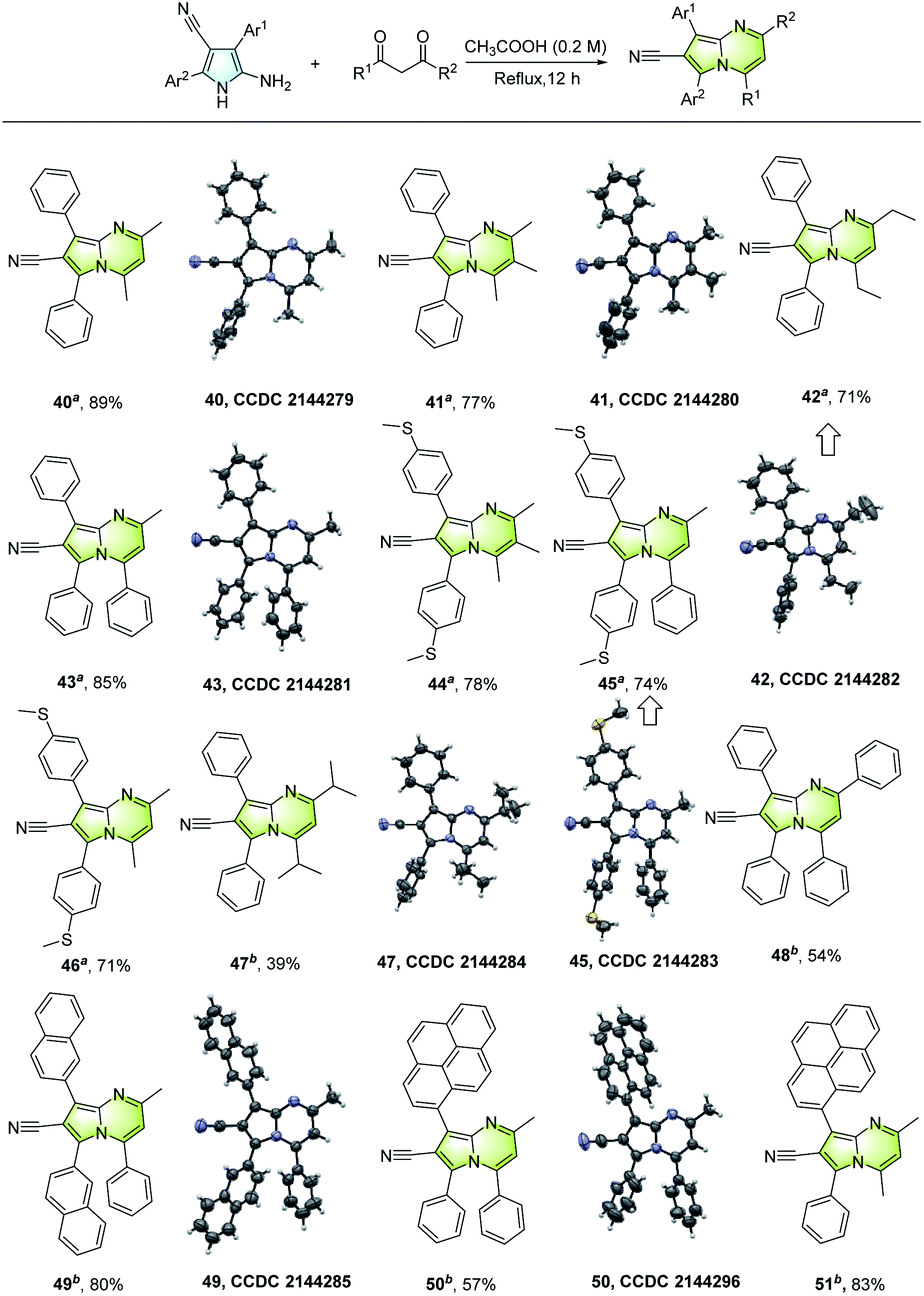 | ||
| Fig. 6 Construction of AIE-active multisubstituted pyrrolo[1,2-a]pyrimidines. aAIEE. bAIE. | ||
As known from the literature, pyrrole-containing fluorescent materials generally exhibit aggregation-caused quenching characteristics. Surprisingly, these pyrrole-containing materials exhibit AIE properties, and a remarkable fluorescence enhancement phenomenon was observed for most of these pyrrolo[1,2-a]pyrimidines in a mixed THF/water solvent (Fig. 7).20 For example, following the pioneering AIE studies by Tang et al.,21 the fluorescence intensity of compound 40 was measured and found to be slightly reduced before fw (water fraction) reached 60%. A sudden rise in fluorescence intensity occurred when fw increased from 80% to 95%, indicating an AIE behaviour. Unexpectedly, it was found that the fluorescence generated by aggregation continued to increase with time. In order to gain insight into this unusual phenomenon, several carefully designed experiments were performed. First, when the aggregate in THF/water (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90) solution was irradiated or kept in the dark for the same period of time, similar fluorescence intensities were obtained, indicating that an unexpected radiation time-dependent fluorescence enhancement had occurred. Further studies revealed that irradiation has very little effect on the intensity enhancement, and temperature was a minor factor. Second, upon photoactivation, the fluorescence lifetime of the THF/water (10:90) solution of 40 was prolonged from 3.529 ns to 9.966 ns. It was also shown that a solution in THF (0.1 mg mL−1) of compound 40 emitted green light with a maximum at 511 nm and a low fluorescence quantum yield,22 but the efficiencies of its solution in THF/water (10:90) and its crystal form reach 40% and 36%, respectively. Interestingly, the fluorescence of the THF/water solution of 47–51 is not time independent, which is thus different from 40. To investigate this difference, single crystal structures of these compounds were studied. As depicted in Fig. 7, a face-to-tail pyrrolo[1,2-a]pyrimidine stacking mode is present in crystals of 40, 41, 42, 43, and 45 and absent for 47, 49 and 50. Molecular packing in single crystals of 40 is shown in Fig. 7h as an example of face-to-tail packing of pyrrolo[1,2-a]pyrimidines. This trend is consistent with the AIE or AIEE properties. Interestingly, a π–π interaction of the naphthalene rings is found in crystalline 49. The different single crystal packing models of 40 and 49 might be responsible for the distinction in the fluorescence phenomena. So, the multisubstituted pyrrolo[1,2-a]pyrimidines represent a new kind of AIE luminogens with an unusual time-dependent fluorescence enhancement.
:90) solution was irradiated or kept in the dark for the same period of time, similar fluorescence intensities were obtained, indicating that an unexpected radiation time-dependent fluorescence enhancement had occurred. Further studies revealed that irradiation has very little effect on the intensity enhancement, and temperature was a minor factor. Second, upon photoactivation, the fluorescence lifetime of the THF/water (10:90) solution of 40 was prolonged from 3.529 ns to 9.966 ns. It was also shown that a solution in THF (0.1 mg mL−1) of compound 40 emitted green light with a maximum at 511 nm and a low fluorescence quantum yield,22 but the efficiencies of its solution in THF/water (10:90) and its crystal form reach 40% and 36%, respectively. Interestingly, the fluorescence of the THF/water solution of 47–51 is not time independent, which is thus different from 40. To investigate this difference, single crystal structures of these compounds were studied. As depicted in Fig. 7, a face-to-tail pyrrolo[1,2-a]pyrimidine stacking mode is present in crystals of 40, 41, 42, 43, and 45 and absent for 47, 49 and 50. Molecular packing in single crystals of 40 is shown in Fig. 7h as an example of face-to-tail packing of pyrrolo[1,2-a]pyrimidines. This trend is consistent with the AIE or AIEE properties. Interestingly, a π–π interaction of the naphthalene rings is found in crystalline 49. The different single crystal packing models of 40 and 49 might be responsible for the distinction in the fluorescence phenomena. So, the multisubstituted pyrrolo[1,2-a]pyrimidines represent a new kind of AIE luminogens with an unusual time-dependent fluorescence enhancement.
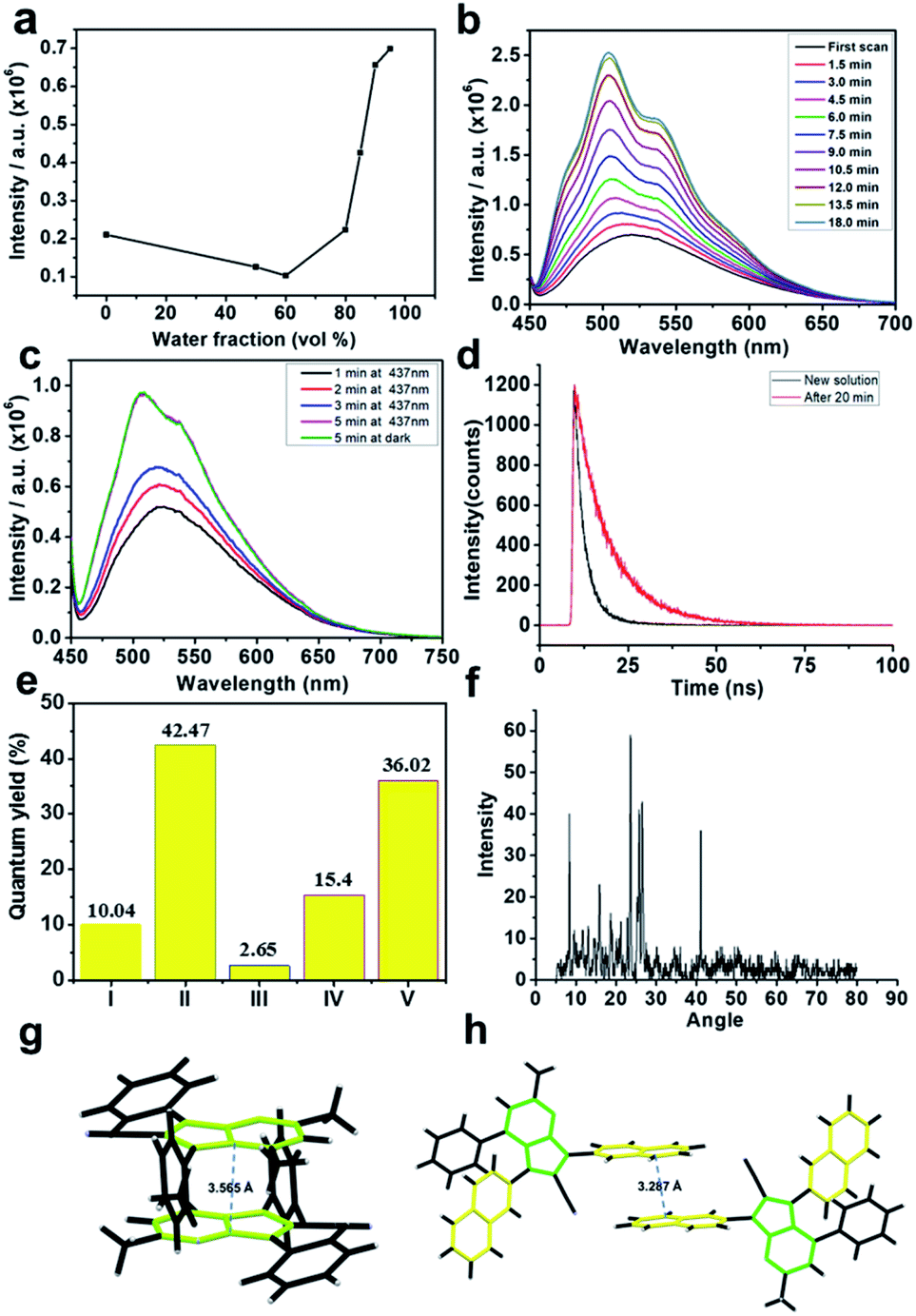 | ||
| Fig. 7 (a) The emission intensity at 518 nm of 40 in THF/water mixtures (0.1 mg mL−1) with different fw. (b) Time-dependent photoluminescence (PL) spectra in THF/water mixtures (fw = 90%, 0.2 mg mL−1). (c) Control experiment; fluorescence intensity at 437 nm light for 5 min (pink line) and in the dark for 5 min (green line); (d) transient photoluminescence decay spectra in THF and water mixtures (fw = 90%, 0.1 mg mL−1) freshly prepared (black line) and after 30 min (red line); λex = 437 nm. (e) Fluorescence quantum yield of THF and water mixtures (fw = 90%, 0.1 mg mL−1) (I) freshly prepared and (II) after 30 min, THF solution (0.1 mg mL−1) (III), solid powder (IV) and crystal particles (V). (f) XRD spectra of 40 powder, prepared by centrifugation of the THF and water mixtures (fw = 90%, 0.1 mg mL−1); (g) molecular packing in single crystals of 40, and (h) molecular packing in single crystals of 49. | ||
Conclusions
We have developed an efficient transition metal-free approach to the synthesis of a variety of unsymmetrically tetrasubstituted NH-pyrroles from chalcone derivatives. A consecutive double cyanation of α,β-unsaturated sulfonimines followed by aromatization is proposed as a plausible pathway. The feasible conversion of the kinetic intermediate into the corresponding thermodynamically stable NH-pyrrole might be the key to the success of this method. Pyrrolo[1,2-a]pyrimidines are synthesized as examples of synthetic applications of NH-pyrroles and these pyrrolo[1,2-a]pyrimidines exhibit unpredicted time-dependent aggregation-induced emission enhancement properties.Data availability
All experimental and characterization data, as well as DFT calculation data are available in the ESI.†Author contributions
H. Bao, D. Li and W.-M. Wan directed the investigations. H. Bao and Y. Li prepared the manuscript. T. Li and C. Ye performed the synthetic experiments and analyzed the experimental data. M.-F. Chiou completed the theoretical calculations. M. Su, M. Xue, X. Yuan and C. Wang performed the synthetic experiments. T. Li and M.-F. Chiou contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Professor Daqiang Yuan from our institute for X-ray crystallography analysis. This work was supported by the National Key R&D Program of China (Grant No. 2017YFA0700103), the NSFC (Grant No. 21871258 and 21922112), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000), and the Innovative Research Teams Program II of Fujian Normal University in China (IRTL1703).Notes and references
- (a) D. J. St Jean Jr and C. Fotsch, J. Med. Chem., 2012, 55, 6002 CrossRef PubMed; (b) O. Afzal, S. Kumar, M. R. Haider, M. R. Ali, R. Kumar, M. Jaggi and S. Bawa, Eur. J. Med. Chem., 2015, 97, 871 CrossRef CAS PubMed; (c) C. B. Mishra, S. Kumari and M. Tiwari, Eur. J. Med. Chem., 2015, 92, 1 CrossRef CAS PubMed; (d) S. S. Gholap, Eur. J. Med. Chem., 2016, 110, 13 CrossRef CAS PubMed; (e) J. Kilani and S. Fillinger, Front. Microbiol., 2016, 7, 1 Search PubMed; (f) K. Chand, Rajeshwari, A. Hiremathad, M. Singh, M. A. Santos and R. S. Keri, Pharmacol. Rep., 2017, 69, 281 CrossRef CAS PubMed; (g) S. Ahmad, O. Alam, M. J. Naim, M. Shaquiquzzaman, M. M. Alam and M. Iqbal, Eur. J. Med. Chem., 2018, 157, 527 CrossRef CAS PubMed.
- (a) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC; (b) C. Bailly, Curr. Med. Chem. Anticancer Agents, 2004, 4, 363 CrossRef CAS PubMed; (c) S. Agarwal, S. Cämmerer, S. Filali, W. Fröhner, J. Knöll, M. P. Krahl, K. R. Reddy and H.-J. Knölker, Curr. Org. Chem., 2005, 9, 1601 CrossRef CAS; (d) J. W. Huffman and L. W. Padgett, Curr. Med. Chem., 2005, 12, 1395 CrossRef CAS PubMed; (e) H. Fan, J. Peng, M. T. Hamann and J. F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS PubMed; (f) I. S. Young, P. D. Thornton and A. Thompson, Nat. Prod. Rep., 2010, 27, 1801 RSC; (g) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233 RSC; (h) J. Bracegirdle, L. P. Robertson, P. A. Hume, M. J. Page, A. V. Sharrock, D. F. Ackerley, A. R. Carroll and R. A. Keyzers, J. Nat. Prod., 2019, 82, 2000 CrossRef CAS PubMed; (i) I. Bauer and H.-J. Knölker, Top. Curr. Chem., 2012, 309, 203 CrossRef CAS PubMed.
- (a) L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 2863 CrossRef; (b) C. Paal, Ber. Dtsch. Chem. Ges., 1884, 17, 2756 CrossRef; (c) V. Amarnath, D. C. Anthony, K. Amarnath, W. M. Valentine, L. A. Wetterau and D. G. Graham, J. Org. Chem., 1991, 56, 6924 CrossRef CAS; (d) H. Cho, R. Madden, B. Nisanci and B. Török, Green Chem., 2015, 17, 1088 RSC.
- (a) A. Hantzsch, Ber. Dtsch. Chem. Ges., 1890, 23, 1474 CrossRef; (b) A. W. Trautwein, R. D. Sussmuth and G. Jung, Bioorg. Med. Chem. Lett., 1998, 8, 2381 CrossRef CAS PubMed; (c) J. Menéndez, M. Leonardi, V. Estévez and M. Villacampa, Synthesis, 2018, 51, 816 CrossRef.
- (a) S. Agarwal and H.-J. Knölker, Org. Biomol. Chem., 2004, 2, 3060 RSC; (b) S. Rakshit, F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585 CrossRef CAS PubMed; (c) B. Li, N. Wang, Y. Liang, S. Xu and B. Wang, Org. Lett., 2013, 15, 136 CrossRef CAS PubMed; (d) S. Michlik and R. Kempe, Nat. Chem., 2013, 5, 140 CrossRef CAS PubMed; (e) L. Wang and L. Ackermann, Org. Lett., 2013, 15, 176 CrossRef CAS PubMed; (f) M. Rostami Osanloo, A. Saadat, M. L. Van de Put, A. Laturia and W. G. Vandenberghe, Nanoscale, 2021, 14, 157 RSC; (g) W. Zhou, W. J. Pan, J. Chen, M. Zhang, J. H. Lin, W. Cao and J. C. Xiao, Chem. Commun., 2021, 57, 9316 RSC.
- (a) A. M. van Leusen, H. Siderius, B. E. Hoogenboom and D. van Leusen, Tetrahedron Lett., 1972, 13, 5337 CrossRef; (b) H. A. Houwing, J. Wildeman and A. M. v. Leusen, Tetrahedron Lett., 1976, 17, 143 CrossRef; (c) X. Zheng, W. Liu and D. Zhang, Molecules, 2020, 25, 1594 CrossRef CAS PubMed.
- (a) G. M. Torres, J. S. Quesnel, D. Bijou and B. A. Arndtsen, J. Am. Chem. Soc., 2016, 138, 7315 CrossRef CAS PubMed; (b) F. Kallmeier, B. Dudziec, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2017, 56, 7261 CrossRef CAS PubMed; (c) H. Xu, H. W. Liu, K. Chen and G. W. Wang, J. Org. Chem., 2018, 83, 6035 CrossRef CAS PubMed; (d) B. Kurpil, K. Otte, A. Mishchenko, P. Lamagni, W. Lipinski, N. Lock, M. Antonietti and A. Savateev, Nat. Commun., 2019, 10, 945 CrossRef PubMed; (e) J. K. Li, B. Zhou, Y. C. Tian, C. Jia, X. S. Xue, F. G. Zhang and J. A. Ma, Org. Lett., 2020, 22, 9585 CrossRef CAS PubMed; (f) L. Chen, J. Q. Huo, H. L. Si, X. Y. Xu, S. Kou, J. Mao and J. L. Zhang, Org. Lett., 2021, 23, 4348 CrossRef CAS PubMed; (g) W. Hu, Q. Zhan, H. Zhou, S. Cao and Z. Jiang, Chem. Sci., 2021, 12, 6543 RSC; (h) L. Zuo, Y. Yang and W. Guo, Org. Lett., 2021, 23, 2013 CrossRef CAS PubMed; (i) C. Ye, Y. Jiao, M. F. Chiou, Y. Li and H. Bao, Chem. Sci., 2021, 12, 9162 RSC.
- (a) I. Bergner, C. Wiebe, N. Meyer and T. Opatz, J. Org. Chem., 2009, 74, 8243 CrossRef CAS PubMed; (b) R. Suresh, S. Muthusubramanian, M. Nagaraj and G. Manickam, Tetrahedron Lett., 2013, 54, 1779 CrossRef CAS; (c) D. Imbri, N. Netz, M. Kucukdisli, L. M. Kammer, P. Jung, A. Kretzschmann and T. Opatz, J. Org. Chem., 2014, 79, 11750 CrossRef CAS PubMed; (d) H. P. Kalmode, K. S. Vadagaonkar, K. Murugan and A. C. Chaskar, New J. Chem., 2015, 39, 4631 RSC; (e) Z. Q. Lin, C. D. Li, Z. C. Zhou, S. Xue, J. R. Gao, Q. Ye and Y. J. Li, Synlett, 2019, 30, 1442 CrossRef CAS.
- X. Xin, D. Wang, X. Li and B. Wan, Angew. Chem., Int. Ed., 2012, 51, 1693 CrossRef CAS PubMed.
- Y. Jiang, W. C. Chan and C. M. Park, J. Am. Chem. Soc., 2012, 134, 4104 CrossRef CAS PubMed.
- J. Xuan, X. D. Xia, T. T. Zeng, Z. J. Feng, J. R. Chen, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 5653 CrossRef CAS PubMed.
- M. Baidya, D. Maiti, L. Roy and S. De Sarkar, Angew. Chem., Int. Ed., 2022, 61, e202111679 CrossRef CAS PubMed.
- (a) N. K. Sahu, S. S. Balbhadra, J. Choudhary and D. V. Kohli, Curr. Med. Chem., 2012, 19, 209 CrossRef CAS PubMed; (b) M. N. Gomes, E. N. Muratov, M. Pereira, J. C. Peixoto, L. P. Rosseto, P. V. L. Cravo, C. H. Andrade and B. J. Neves, Molecules, 2017, 22, 1210 CrossRef PubMed; (c) C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762 CrossRef CAS PubMed; (d) Z.-B. Dong and J.-Q. Chen, Synthesis, 2020, 52, 3714 CrossRef; (e) R. Luo, N. Luo, H. Shui, Y. Zhong and J. Huang, Synthesis, 2021, 53, 4516 CrossRef.
- Z. Shi and T. P. Loh, Angew. Chem., Int. Ed., 2013, 52, 8584 CrossRef CAS PubMed.
- D. G. Stark, L. C. Morrill, P. P. Yeh, A. M. Slawin, T. J. O'Riordan and A. D. Smith, Angew. Chem., Int. Ed., 2013, 52, 11642 CrossRef CAS PubMed.
- W. W. Tan, Y. J. Ong and N. Yoshikai, Angew. Chem., Int. Ed., 2017, 56, 8240 CrossRef CAS PubMed.
- A. M. Nauth and T. Opatz, Org. Biomol. Chem., 2018, 17, 11 RSC.
- Y. Fukuda, K. Kondo and T. Aoyama, Synthesis, 2006, 2006, 2649 CrossRef.
- (a) T. Zdrojewski and A. Jonczyk, Synthesis, 1990, 1990, 224 CrossRef; (b) J. Tao, T. T. Yang, Q. H. Li and T. L. Liu, Org. Chem. Front., 2021, 8, 3421 RSC.
- (a) Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (b) Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (c) J. Mei, Y. Hong, J. W. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed; (d) J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed; (e) J. Zhang, B. He, Y. Hu, P. Alam, H. Zhang, J. W. Y. Lam and B. Z. Tang, Adv. Mater., 2021, 33, e2008071 CrossRef PubMed.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- (a) J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956 CrossRef CAS PubMed; (b) M. Chen, L. Li, H. Nie, J. Tong, L. Yan, B. Xu, J. Z. Sun, W. Tian, Z. Zhao, A. Qin and B. Z. Tang, Chem. Sci., 2015, 6, 1932 RSC; (c) Y. Gong, L. Zhao, Q. Peng, D. Fan, W. Z. Yuan, Y. Zhang and B. Z. Tang, Chem. Sci., 2015, 6, 4438 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2133476, 2144279–2144285 and 2144296. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00837h |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |