
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general electron donor–acceptor complex for photoactivation of arenes via thianthrenation†
Kai
Sun
 a,
Anzai
Shi
a,
Yan
Liu
b,
Xiaolan
Chen
*a,
Panjie
Xiang
a,
Xiaotong
Wang
a,
Lingbo
Qu
a and
Bing
Yu
*a
a,
Anzai
Shi
a,
Yan
Liu
b,
Xiaolan
Chen
*a,
Panjie
Xiang
a,
Xiaotong
Wang
a,
Lingbo
Qu
a and
Bing
Yu
*a
aGreen Catalysis Center, College of Chemistry, Zhengzhou University, Zhengzhou 450001, China. E-mail: chenxl@zzu.edu.cn; bingyu@zzu.edu.cn
bHenan International Joint Laboratory of Rare Earth Composite Material, College of Materials Engineering, Henan University of Engineering, Zhengzhou 451191, China
First published on 14th April 2022
Abstract
General photoactivation of electron donor–acceptor (EDA) complexes between arylsulfonium salts and 1,4-diazabicyclo[2.2.2]octane with visible light or natural sunlight was discovered. This practical and efficient mode enables the production of aryl radicals under mild conditions, providing an unrealized opportunity for two-step para-selective C–H functionalization of complex arenes. The novel mode for generating aryl radicals via an EDA complex was well supported by UV-vis absorbance measurements, nuclear magnetic resonance titration experiments, and density functional theory (DFT) calculations. The method was applied to the regio- and stereo-selective arylation of various N-heterocycles under mild conditions, yielding an assembly of challengingly linked heteroaryl–(hetero)aryl products. Remarkably, the meaningful couplings of bioactive molecules with structurally complex drugs or agricultural pharmaceuticals were achieved to display favorable in vitro antitumor activities, which will be of great value in academia or industry.
Introduction
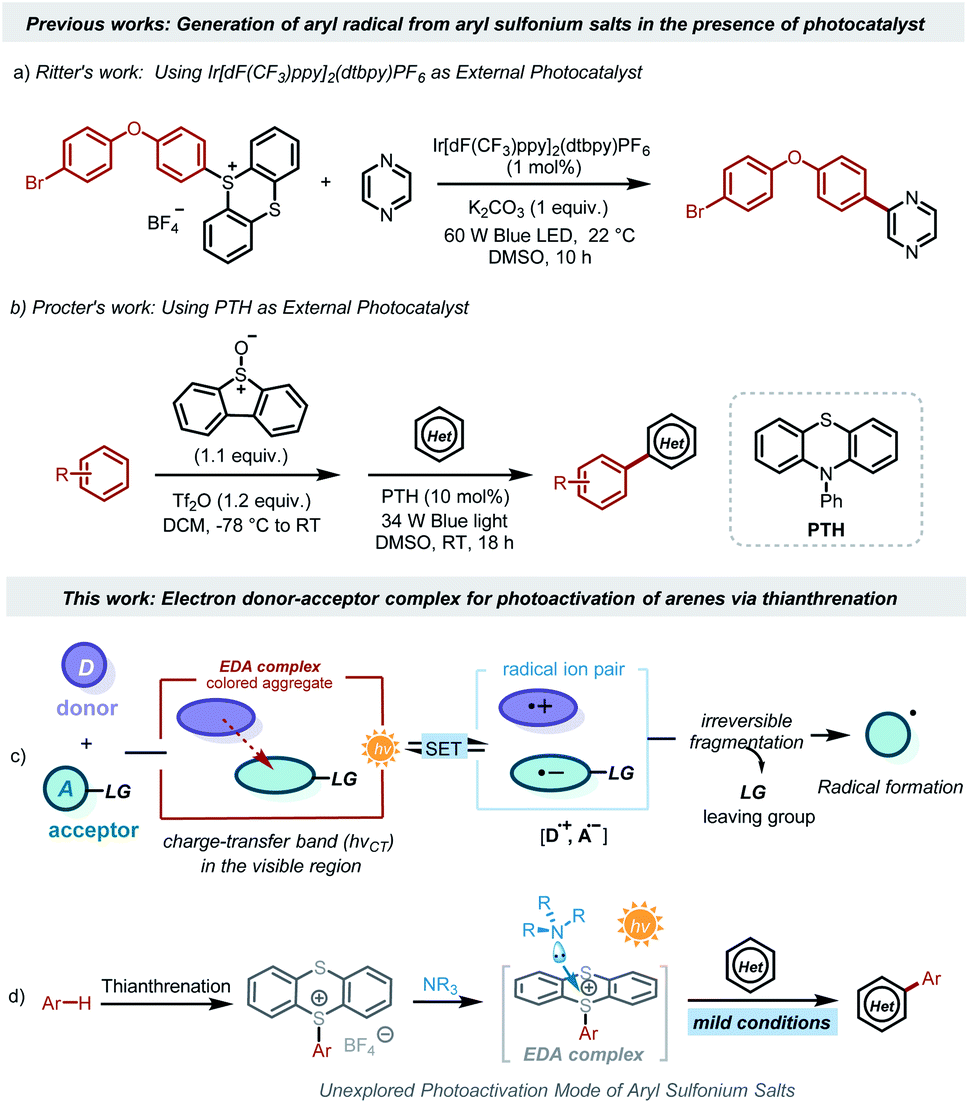
Aryl radicals have proven to be versatile synthetic intermediates in organic synthesis, displaying an important role in materials science, agricultural chemistry, and especially pharmaceutical chemistry.1 Classically, the generation of aryl radicals depends heavily on the reduction of aromatic halides by AIBN/n-Bu3SnH or energy-intensive diazonium salts by transition metal reductants.2 The oxidation of arylhydrazines and boronic acids emerged as an alternative to access aryl radicals in the presence of strong oxidants.3 However, site-selective synthesis of these active precursors from the corresponding arenes is challenging.4 The range of substrates is limited to simple arenes in most of the reported systems. Further disadvantages of these conventional reactions include the employment of toxic or expensive reagents and harsh oxidants (or reductants). Therefore, the development of sustainable approaches for the generation of aryl radicals that can exploit more general precursors without harsh oxidants or reductants would be highly desirable.Photocatalysis,5 which directly converts sustainable solar energy into chemical energy,6 now emerges as a promising technology to achieve diverse organic transformations in an environmentally friendly and energy-saving methodology.7 Some elegant methods for photocatalytic generation of aryl radicals have been reported.8 Very recently, it has been reported that sulfonium salts9 offer a feasible transformative platform for the late-stage functionalization of complex scaffolds. In particular, triarylsulfonium salts derived from aromatic C–H bonds are promising candidates for photocatalytic organic transformations. However, direct photochemical activation of triarylsulfonium salts requires strong energy such as ultraviolet light.10 The high energy of ultraviolet light will destroy more chemical bonds, resulting in low selectivity. Moreover, special light sources and equipment, such as high-pressure mercury lamps, lamp boxes, quartz reactors, etc., are usually required. Recently, some elegant visible-light mediated activations of triarylsulfonium salts have been reported.11 For example, the Ritter group disclosed an elegant arylation method of aryl sulfonium salts using the iridium complex as the photocatalyst (Scheme 1a).12 The Procter group reported a metal-free strategy for formal C–H/C–H cross-couplings using 10-phenylphenothiazine as the optimal photocatalyst (Scheme 1b).13 These strategies rely on the employment of an exogenous photocatalyst that harvests the energy of visible light to activate triarylsulfonium salts for the generation of aryl radicals under mild reaction conditions.
 | ||
| Scheme 1 Photocatalytic arylation of aryl sulfonium salts. | ||
Photoinduced intermolecular charge transfer through the association of an electron-donating substrate (D) and an electron acceptor molecule (A) via noncovalent interactions is a well-known process in photochemistry (Scheme 1c).14 Although each component itself (D or A) might not absorb visible light, the molecular aggregate formed between the donor and acceptor molecules in the ground state establishes a new charge-transfer band (hνCT) related to single electron transfer (SET) from the donor to the acceptor, enabling the ability for absorption of visible light.15 Upon light irradiation, the SET events in the electron-donor–accepter (EDA) complex can generate reactive open-shell intermediates, which act as valuable synthons for novel synthetic routes under mild conditions.16 In 2021, the Shi group disclosed the generation of alkyl radicals from EDA complexes between alkyl thianthrenium salts and the B2cat2·DMA adduct.17 During the preparation of our manuscript, the Procter group described the use of photoactive EDA complexes between arylsulfonium and triarylamines for the production of aryl radicals, leading to the formation of alkylation and cyanation of arenes.18 Such irradiation-induced intermolecular charge transfer would provide fresh opportunities for generating aryl radicals from arylthianthrenium salts. This virtually unexplored approach can significantly expand the synthetic toolbox of modern chemists.
Herein, we provide a practical approach for the photocatalytic generation of aryl radicals from EDA complexes between arylsulfonium salts and 1,4-diazabicyclo[2.2.2]octane (DABCO), enabling the preparation of high-value-added (hetero)biaryls under mild conditions (Scheme 1d). Through this simple strategy, bioactive molecules can be meaningfully coupled with structurally complex drugs or agricultural pharmaceuticals. The resulting compounds display favorable in vitro antitumor activities, providing great value in academia or industry. Moreover, this novel mode for generating aryl radicals via EDA complexes was comprehensively studied through UV−vis absorbance measurements, nuclear magnetic resonance (NMR) titration experiments, and density functional theory (DFT) calculations.
Results and discussion
As an azapyrimidinone analog of uracil, azauracil is a promising microbiological inhibitor that has been widely studied by chemists and pharmacists.19 In particular, the ribonucleosides of 6-azauracil display significant antiviral, antitumor, and antifungal activities. New synthetic methods that enrich the structural diversity of azauracils and nucleosides would significantly advance research in this area. We hypothesized that electron-deficient arylthianthrenium salts can serve as electron acceptors, forming EDA complexes with tertiary amines. This scheme provides an unrealized opportunity to access aryl radicals for the alkylation of azauracils and nucleosides. The feasibility of our hypothesis was evaluated by selecting toluene as a model substrate to produce aryl thianthrenium salt 2 to react with 2,4-dibenzyl-1,2,4-triazine-3,5(2H,4H)-dione (1) for the synthesis of 2,4-dibenzyl-6-(p-tolyl)-1,2,4-triazine-3,5(2H,4H)-dione (3) (Table 1). Initially, various commercially available tertiary amines, including N,N,N′,N′-tetramethylethylenediamine (TMEDA), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 4-dimethylaminopyridine (DMAP), Et3N, 1,1,3,3-tetramethylguanidine (TMG), and 1,4-diazabicyclo[2.2.2]octane (DABCO) (entries 1–6 in Table 1) were investigated. All of these tertiary amines effectively promoted the reaction, and the desired product 3 was obtained in 72% yield when DABCO was employed (entry 6). However, when triphenylamine or tri-p-tolylamine was employed as the electron-donating reagents, the yield of product 3 was significantly reduced (entries 7 and 8). Moreover, no desired product could be detected when PPh3 or tricyclohexylphosphane was used instead of tertiary amines (entries 9 and 10). Then, a series of other solvents, including dimethyl sulfoxide (DMSO), acetone, dichloromethane (DCM), dimethylformamide (DMF), ethanol (EtOH), 1,2-dichloroethane (DCE), and tetrahydrofuran (THF), were screened (entries 11–17). Unfortunately, these attempts failed to improve the yield, and no positive results were obtained. Next, the dosage of tertiary amine was optimized to further improve the reaction efficiency (entries 18 and 19). The best dosage was 3 equiv. of DABCO, which afforded the desired product 3 in 82% yield. The control experiments conducted in the absence of tertiary amines and visible light gave no desired product, indicating the important roles of tertiary amine and light in this transformation (entries 20 and 21). Furthermore, additional experiments showed that the donor loading could be decreased below 1 equivalent by adding other bases (for details, see Table S1†).|
|
|||
|---|---|---|---|
| Entry | Base | Solvent | Yield (%) |
| a Reaction conditions: 1 (0.1 mmol), 2 (0.2 mmol), base (2 equiv.), and an appropriate solvent (1.5 mL) were irradiated with a 10 W blue LED (430 nm) at room temperature under a N2 atmosphere for 12 h. Yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard based on 1. N.D = not detected. b DABCO (1 equiv.). c DABCO (3 equiv.). d No base. e In the dark. | |||
| 1 | TMEDA | MeCN | 42 |
| 2 | DBU | MeCN | 50 |
| 3 | DMAP | MeCN | 32 |
| 4 | Et3N | MeCN | 50 |
| 5 | TMG | MeCN | 59 |
| 6 | DABCO | MeCN | 72 |
| 7 | Triphenylamine | MeCN | Trace |
| 8 | Tri-p-tolylamine | MeCN | 21 |
| 9 | PPh3 | MeCN | N.D |
| 10 | Tricyclohexylphosphine | MeCN | N.D |
| 11 | DABCO | DMSO | 57 |
| 12 | DABCO | Acetone | 61 |
| 13 | DABCO | DCM | 32 |
| 14 | DABCO | DMF | Trace |
| 15 | DABCO | EtOH | Trace |
| 16 | DABCO | DCE | 40 |
| 17 | DABCO | THF | 21 |
| 18b | DABCO | MeCN | 52 |
| 19c | DABCO | MeCN | 82 |
| 20d | — | MeCN | N.D |
| 21e | DABCO | MeCN | N.D |
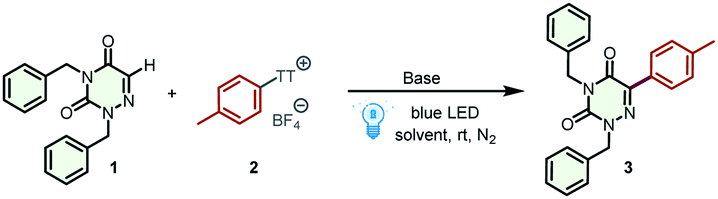
Having optimized the reaction conditions, we then turn our attention to examining the generality of our practical protocol. Initially, arenes bearing a wide range of functional groups were reacted with 2,4-dibenzyl-1,2,4-triazine-3,5(2H,4H)-dione (1) (Scheme 2). To our delight, all of these arenes were suitable substrates for the two-step site-selective C–H arylation reaction, obtaining the desired products 3–19 in moderate to good yields (44–82%). It is worth emphasizing that various functional groups were tolerated in these cases, and some sensitive functional groups such as –CHO, –CN, and –COCH3 were compatible with the transformation. The acid-sensitive free amino group was trifluoroacylated to product 19 in 68% yield. The straightforward and selective modification of biorelevant compounds are of importance in research and development campaigns.20 These conditions can be used in the late-stage diversification of drugs and agricultural pharmaceuticals, such as the multi-use insecticide pyriproxyphen, the fungicide boscalid, and atomoxetine for the treatment of attention deficit and hyperactivity disorder, affording the corresponding products 20–22 in satisfactory yields (53–71%). Flurbiprofen methyl ester, a drug derivative, was also amenable to this transformation, affording product 23 in 72% yield. Afterward, the scope of 6-azauracils was assessed. Gratifyingly, N,N′-disubstituted 6-azauracils were smoothly arylated with thianthrenium salt 2, affording the corresponding products 24–26 in satisfactory yields (57–73%). Moreover, the new anticoccidial drug diclazuril was also compatible with this efficient methodology, giving the product 27 in 53% yield. It is noteworthy that convenient C(sp2)–H arylation of 6-azauridine nucleosides was successfully achieved, furnishing the arylated 6-azauridine nucleosides 28–34 in moderate yields (47–71%). In these cases, the free secondary N–H group in the 6-azauridine nucleosides was tolerated with no significant effects on the reaction efficiency. Additionally, this protocol provides a feasible way for the effective coupling of 6-azauridine nucleosides and pharmaceuticals, leading to the corresponding nucleoside derivatives 35–39 in moderate yields (41–63%). Diclazuril was also coupled with boscalid to afford the corresponding product 40 in 38% yield. More importantly, in all of the above cases, thianthrene was recovered by separation and reused in subsequent cycles. Additionally, the model reaction efficiency of product 3 was investigated under different reaction parameters.21 In a comprehensively condition-based sensitivity assessment, this photocatalytic transformation was sensitive to low light intensity and high oxygen concentrations, which was generally tolerant toward the substrate concentration, reaction temperature, water, and scale (see the radar diagram in Scheme 2 and the ESI† for details).
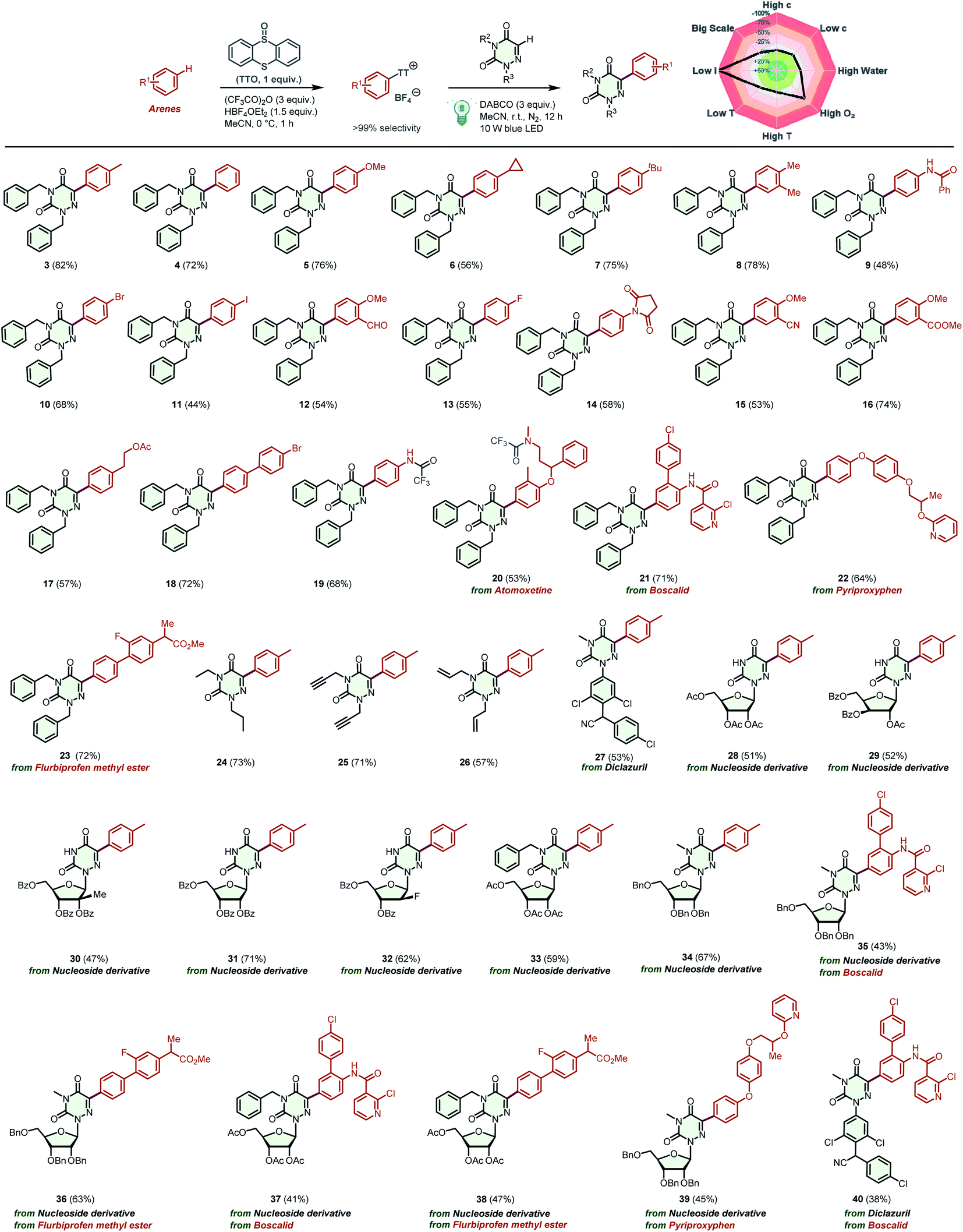 | ||
| Scheme 2 Substrate scope of the alkylation of azauracils and nucleosides. | ||
The proposed photoactivation of arenes via thianthrenation was then applied to the arylation of quinoxalin-2(1H)-ones (Scheme 3), which play a significant role in antimicrobial compounds, antitumor agents, and semiconductors.22 Through this protocol, various quinoxalin-2(1H)-ones bearing different functional groups afforded the corresponding products 41–57 with excellent site selectivity and reactivity. Owing to the mild reaction conditions, sensitive alkenyl, alkynyl, and hydroxy groups were well tolerated under the optimized conditions (44–46). Notably, N-unsubstituted quinoxalin-2(1H)-one was also a suitable substrate to furnish the desired product 58 in 34% yield. To explore the arene generality of the protocol, various electron-rich, neutral, and electron-poor arenes were reacted with N-methylquinoxalin-2(1H)-one under the optimized reaction conditions. Remarkably, the functional groups –Me, –Et, –Bn, –OAc, –Ph, –OMe, –OPh, –F, –Cl, –Br, –CN, –CHO, –COOMe, and –COCH3 were transformed into their corresponding products 59–82 in average to good yields, highlighting the compatibility of the protocol with a wide range of functional groups. Likewise, when aniline participated in this reaction, product 83 was generated in 65% yield, in which trifluoroacylation of the free amino group occurs. The scope of the protocol was further extended to facile late-stage functionalization of valuable scaffolds containing drug-like molecules and natural isolates. For example, quinoxalin-2(1H)-ones containing o-vanillin, p-vanillin, zingerone, ibuprofen, and isoxepac were selectively arylated to the desired products 84–88 in 48–81% yields. Nefiracetam, pyriproxyphen, flurbiprofen methyl ester, boscalid, and atomoxetine were all efficient arylation reagents in this two-step C–H alkylation reaction to produce products 89–93, demonstrating the utility of this transformation. Finally, the application of this methodology for the preparation of antimicrobial and antitumor agent 94 was achieved, and a 72% yield of the desired product was obtained. Likewise, in a condition-based sensitivity assessment, low light intensity, high oxygen concentrations, and the DABCO amount were the main factors influencing the preparation reproducibility of product 41 (for details, see the ESI†).
 | ||
| Scheme 3 Substrate scope for the arylation of quinoxalin-2(1H)-ones. | ||
Encouraged by the above results, we further examined the generality of the proposed protocol by screening other simple yet significant (hetero)aryl cycles (Scheme 4). To our delight, coumarin, 1-(fluoromethyl)cinnolin-4(1H)-one, and 2-phenylimidazo[1,2-a]pyridine were successfully arylated under the optimized reaction conditions, producing the corresponding products 95-97 in acceptable yields. Moreover, 3-methylene-1-phenylpyrrolidine-2,5-dione, which could be transformed into 3-methyl-1-phenyl-1H-pyrrole-2,5-dione in the presence of base,23 was a suitable substrate to access product 98 in 36% yield. Additionally, this method was also applied to the direct C–H arylation of tangeretin and 1,3,5-trimethoxybenzene, leading to the formation of desired products 99 and 100 in 36% and 61% yields, respectively. Unfortunately, some other (hetero)aromatic cycles, including pyridine, quinoline, benzothiazole, benzoxazole and indole were not suitable substrates for this transformation, and no desired products could be detected (for details, see Scheme S1†).
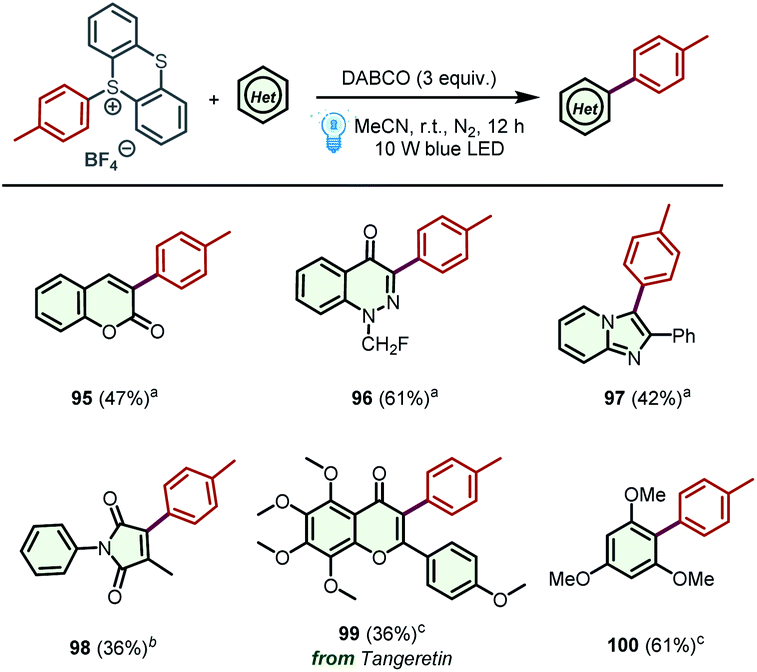 | ||
| Scheme 4 Screening of other (hetero)aromatic cycles. (a) Reaction conditions: (hetero)aromatic cycle (0.1 mmol) was reacted with thianthrenium salt (0.2 mmol) in the presence of DABCO (3 equiv.) under 10 W blue LED irradiation. (b) 3-Methylene-1-phenylpyrrolidine-2,5-dione (0.1 mmol) is used as a substrate to react with thianthrenium salt (0.2 mmol) in the presence of DABCO (3 equiv.) under 10 W blue LED irradiation. (c) Thianthrenium salt (0.1 mmol) was reacted with (hetero)aromatic cycle (10 equiv.) in the presence of DABCO (3 equiv.) under 10 W blue LED irradiation. | ||
To clarify the practicality of these transformations, the gram-scale synthesis of products 3 and 41 was performed on 4 mmol and 5 mmol scales, respectively. To our delight, the reactions proceeded smoothly to provide the desired products with no significant reduction in yields only by increasing the light intensity and reaction time (Scheme 5). Furthermore, natural sunlight-driven experiments were performed, leading to the formation of substantial amounts of products 3 and 41 in 79% and 76% yields, respectively. A one-pot sequence was also explored, and product 3 could be obtained in 37% yield (for details, see the ESI†).
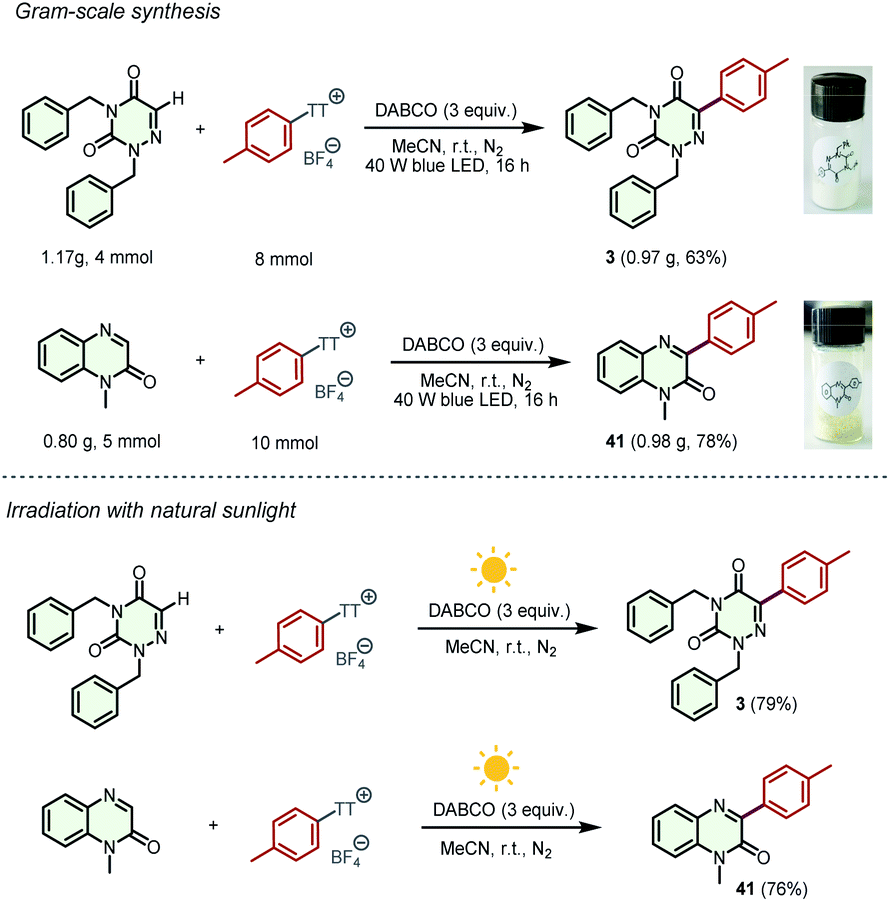 | ||
| Scheme 5 Synthetic applications. | ||
Importantly, the in vitro antitumor activities of the synthetic compounds 37 and 92 were evaluated in Ramos cells. The results indicated that compounds 37 and 92 exhibited excellent antitumor activities (Fig. S7†). In the aspect of anti-lymphoma activity, the IC50 values of compounds 37 and 92 against Ramos cells are slightly lower than that of the approved drug fluorouracil (5-FU, 13.7 μM), indicating the potential of our method in the development of novel drugs.
To get deep insight into the reaction mechanism, some radical trapping experiments were conducted (Scheme 6). When the radical scavenger (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl (TEMPO) or 1,1-diphenylethylene was added to the model reaction, the reactions were severely inhibited. Moreover, CuCl was also proven to be an effective inhibitor of this transformation. All of these results indicated the possible involvement of a radical pathway. Additionally, the reaction mixtures were analyzed by high-resolution mass spectrometry (HRMS), and the adducts 101 and 102 were successfully detected, respectively. These results strongly support the generation of aryl radicals in the photocatalytic transformation, which might be initiated by the photo-activated EDA complexes between arylsulfonium salts and DABCO. The preparation of product 41 was also severely inhibited by TEMPO, 1,1-diphenylethylene, or CuCl (for details, see the ESI†).
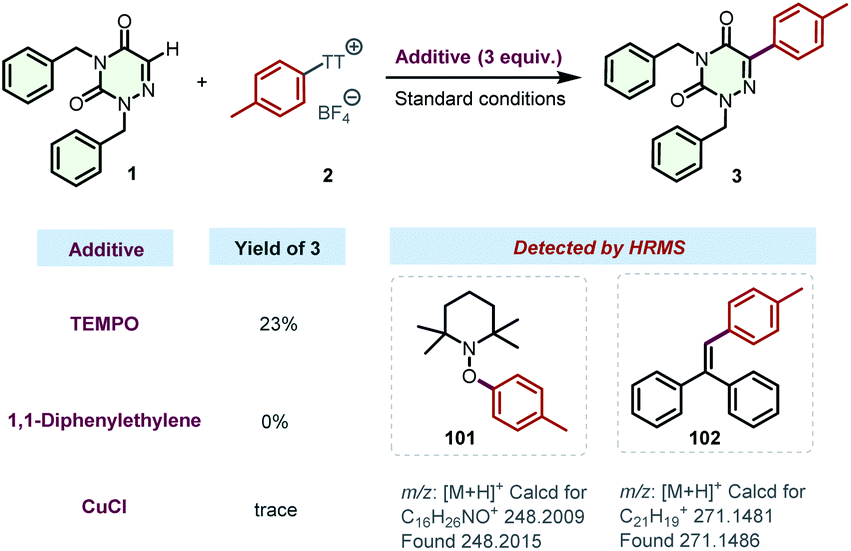 | ||
| Scheme 6 Control experiments. | ||
The formation of the EDA complex was confirmed in additional mechanism investigations (Fig. 1). When DABCO was added to a solution of arylthianthrenium salt 2 in CH3CN, the solution developed a marked yellow color. The UV-vis absorbance experimental results showed that the absorption peaks of the DABCO and arylthianthreniumsalt 2 mixture appeared at 430–460 nm, while those of control groups can only be observed in the near UV-region. This might be caused by the formation of a new EDA molecular aggregate. Moreover, 1H NMR titration experiments and a Job's plot analysis confirmed the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex between the arylthianthrenium salt 2 and DABCO. The binding constant Ka of complexation was 1.04 M−1 in CDCl3 (for details, see the ESI†). Additionally, DFT calculations were carried out to better understand the intermolecular charge transfer between arylthianthrenium salt 2 and DABCO. In the equilibrium structure, the distance d of the N/S interactions was 2.26 Å, shorter than the summed van der Waals radii of the two interacting atoms (3.78 Å).24 The binding energy was calculated to be 3.73 kcal mol−1, implying a feasible interaction between the arylthianthrenium salt 2 and DABCO. The low energy gap (0.64 eV) between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) indicated a favorable electron-transfer process under visible light irradiation.
:1 complex between the arylthianthrenium salt 2 and DABCO. The binding constant Ka of complexation was 1.04 M−1 in CDCl3 (for details, see the ESI†). Additionally, DFT calculations were carried out to better understand the intermolecular charge transfer between arylthianthrenium salt 2 and DABCO. In the equilibrium structure, the distance d of the N/S interactions was 2.26 Å, shorter than the summed van der Waals radii of the two interacting atoms (3.78 Å).24 The binding energy was calculated to be 3.73 kcal mol−1, implying a feasible interaction between the arylthianthrenium salt 2 and DABCO. The low energy gap (0.64 eV) between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) indicated a favorable electron-transfer process under visible light irradiation.
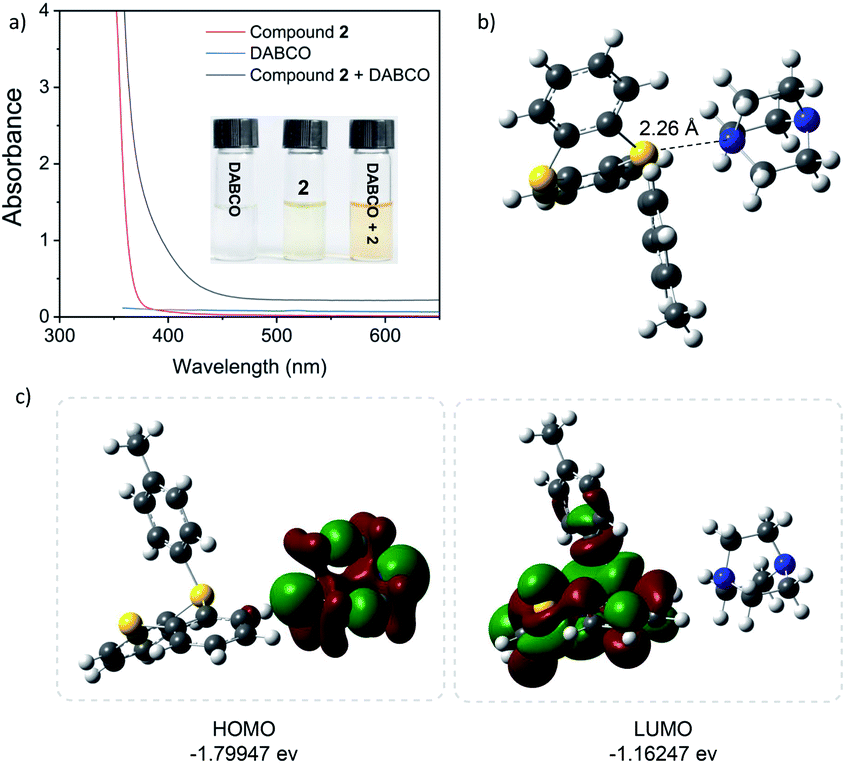 | ||
| Fig. 1 (a) UV-Vis absorption spectra of reactant mixtures recorded in CH3CN in 1 mm path-length quartz cuvettes, and visual appearance of the separate reaction components and the colored EDA complex between compound 2 and DABCO. (b) Calculated bond distances of arylsulfonium salt 2 and DABCO. (c) Calculated HOMO and LUMO of the EDA complex. | ||
Based on the above observations and previous reports,25 a plausible mechanism containing EDA complexes for the generation of aryl radicals was proposed (Scheme 7). Initially, DABCO combines with the aryl thianthrenium salt 2 to generate the EDA complex, which produces the aryl radical 103, thianthrene, and DABCO˙+ (106). Afterward, the addition of aryl radical 103 to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of 2,4-dibenzyl-1,2,4-triazine-3,5(2H,4H)-dione (1) affords radical 104, which undergoes a 1,2 H shift to furnish radical 105. Radical 105 can be oxidized by DABCO˙+ (106) to the carbon cation 107, which transforms into the desired product 3 in the presence of DABCO by deprotonation.
N bond of 2,4-dibenzyl-1,2,4-triazine-3,5(2H,4H)-dione (1) affords radical 104, which undergoes a 1,2 H shift to furnish radical 105. Radical 105 can be oxidized by DABCO˙+ (106) to the carbon cation 107, which transforms into the desired product 3 in the presence of DABCO by deprotonation.
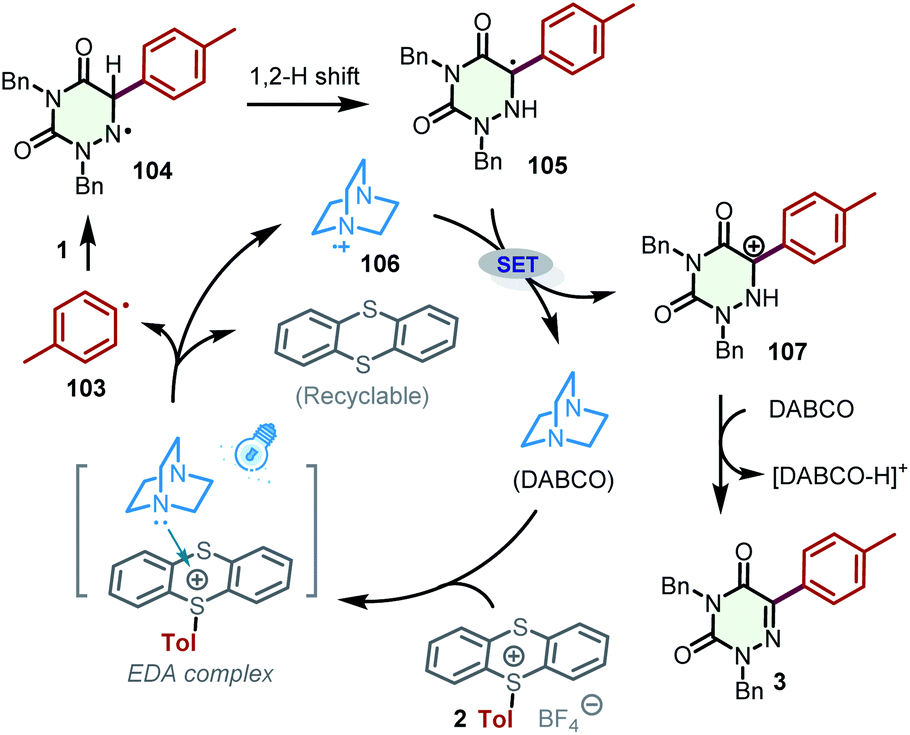 | ||
| Scheme 7 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a novel mode for the generation of aryl radicals by photoactivation of arenes via thianthrenation. When the EDA complex between arylsulfonium salts and DABCO is excited by weak visible light or natural sunlight, it forms aryl radicals that can be leveraged in synthetically useful transformations for the arylation of various N-heterocycles. The high versatility of the EDA complex facilitates an assembly of challengingly linked heteroaryl–(hetero)aryl products. More importantly, this practical protocol can efficiently and selectively combine bioactive molecules with structurally complex drugs or agricultural pharmaceuticals. The resulting compounds displayed favorable in vitro antitumor activities. The proposed methodology is promising for the development of novel synthetic methods and antitumor drugs in academia or industry. We believe that photoactivation of arenes to aryl radicals via thianthrenation will facilitate further arylation processes, providing meaningful compounds under mild reaction conditions.Data availability
The data that support the findings of this study are available in the ESI† or on request from the corresponding author.Author contributions
K. S., X. L. C. and B. Y. designed the subject and guided the experiments throughout. K. S., A. Z. S., Y. L., P. J. X. and X. T. W. conducted the organic synthesis and characterization. Y. L. conducted the DFT calculations, and K. S. performed the in vitro antitumor activity analysis. K. S., L. B. Qu and B. Y. completed the article writing together.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21971224, 22071222, and 22171249), the Key Research Projects of Universities in Henan Province (21A150053), the Natural Science Foundation of Henan Province (202300410375), China Postdoctoral Science Foundation (2021M692906) and Henan Postdoctoral Foundation (202003014).Notes and references
- (a) W. Liu, X. Yang, Y. Gao and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 8621 CrossRef CAS PubMed; (b) F. Mo, D. Qiu, L. Zhang and J. Wang, Chem. Rev., 2021, 121, 5741 CrossRef CAS PubMed; (c) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566 CrossRef CAS PubMed; (d) C. Zhou, T. Lei, X.-Z. Wei, C. Ye, Z. Liu, B. Chen, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2020, 142, 16805 CrossRef CAS PubMed; (e) H. Li, X. Tang, J. H. Pang, X. Wu, E. K. L. Yeow, J. Wu and S. Chiba, J. Am. Chem. Soc., 2021, 143, 481 CrossRef CAS PubMed.
- (a) N. Kvasovs and V. Gevorgyan, Chem. Soc. Rev., 2021, 50, 2244 RSC; (b) F. Mo, D. Qiu, Y. Zhang and J. Wang, Acc. Chem. Res., 2018, 51, 496 CrossRef CAS PubMed.
- (a) I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194 CrossRef CAS PubMed; (b) Y. Fujiwara, V. Domingo, I. B. Seiple, R. Gianatassio, M. Del Bel and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 3292 CrossRef CAS PubMed; (c) A. Hosseinian, R. Mohammadi, S. Ahmadi, A. Monfared and Z. Rahmani, RSC Adv., 2018, 8, 33828 RSC.
- B. Lansbergen, P. Granatino and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7909 CrossRef CAS PubMed.
- (a) P.-Z. Wang, X. Wu, Y. Cheng, M. Jiang, W.-J. Xiao and J.-R. Chen, Angew. Chem., Int. Ed., 2021, 60, 22956 CrossRef CAS PubMed; (b) W.-J. Zhou, Z.-H. Wang, L.-L. Liao, Y.-X. Jiang, K.-G. Cao, T. Ju, Y. Li, G.-M. Cao and D.-G. Yu, Nat. Commun., 2020, 11, 3263 CrossRef CAS PubMed; (c) J. Qi, F.-L. Zhang, J.-K. Jin, Q. Zhao, B. Li, L.-X. Liu and Y.-F. Wang, Angew. Chem., Int. Ed., 2020, 59, 12876 CrossRef CAS PubMed; (d) Z. Li, M. Wang and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 186 CrossRef CAS PubMed; (e) B.-G. Cai, S.-S. Luo, L. Li, L. Li, J. Xuan and W.-J. Xiao, CCS Chem., 2020, 3, 2764 CrossRef; (f) A.-Q. Xu, F.-L. Zhang, T. Ye, Z.-X. Yu and Y.-F. Wang, CCS Chem., 2019, 1, 504 CrossRef CAS; (g) C.-Y. Huang, J. Li, W. Liu and C.-J. Li, Chem. Sci., 2019, 10, 5018 RSC; (h) Y. Kuang, H. Cao, H. Tang, J. Chew, W. Chen, X. Shi and J. Wu, Chem. Sci., 2020, 11, 8912 RSC.
- (a) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506 CrossRef CAS PubMed; (b) L. Song, D.-M. Fu, L. Chen, Y.-X. Jiang, J.-H. Ye, L. Zhu, Y. Lan, Q. Fu and D.-G. Yu, Angew. Chem., Int. Ed., 2020, 59, 21121 CrossRef CAS PubMed; (c) J.-Q. Chen, X. Tu, Q. Tang, K. Li, L. Xu, S. Wang, M. Ji, Z. Li and J. Wu, Nat. Commun., 2021, 12, 5328 CrossRef CAS PubMed; (d) C.-Y. Huang, J. Li and C.-J. Li, Nat. Commun., 2021, 12, 4010 CrossRef CAS PubMed; (e) H.-H. Zhang, M. Tang, J.-J. Zhao, C. Song and S. Yu, J. Am. Chem. Soc., 2021, 143, 12836 CrossRef CAS PubMed.
- (a) J. Byun, W. Huang, D. Wang, R. Li and K. A. I. Zhang, Angew. Chem., Int. Ed., 2018, 57, 2967 CrossRef CAS PubMed; (b) X.-Y. Yu, Q.-Q. Zhao, J. Chen, W.-J. Xiao and J.-R. Chen, Acc. Chem. Res., 2020, 53, 1066 CrossRef CAS PubMed; (c) W.-Q. Liu, T. Lei, S. Zhou, X.-L. Yang, J. Li, B. Chen, J. Sivaguru, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 13941 CrossRef CAS PubMed; (d) X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620 CrossRef CAS PubMed; (e) X.-H. Ouyang, Y. Li, R.-J. Song, M. Hu, S. Luo and J.-H. Li, Sci. Adv., 2019, 5, eaav9839 CrossRef CAS PubMed; (f) Y. Ning, S. Wang, M. Li, J. Han, C. Zhu and J. Xie, Nat. Commun., 2021, 12, 4637 CrossRef CAS PubMed; (g) H. Huang, J.-H. Ye, L. Zhu, C.-K. Ran, M. Miao, W. Wang, H. Chen, W.-J. Zhou, Y. Lan, B. Yu and D.-G. Yu, CCS Chem., 2020, 2, 1746 Search PubMed; (h) H. Wang, W. Shi, Y. Li, M. Yu, Y. Gao and A. Lei, CCS Chem., 2020, 2, 1710 Search PubMed; (i) K. Muralirajan, R. Kancherla, J. A. Bau, M. R. Taksande, M. Qureshi, K. Takanabe and M. Rueping, ACS Catal., 2021, 11, 14772 CrossRef CAS; (j) T. Yuan, M. Zheng, M. Antonietti and X. Wang, Chem. Sci., 2021, 12, 6323 RSC.
- (a) B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616 CrossRef CAS PubMed; (b) A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 10882 CrossRef CAS PubMed; (c) C. M. Hendy, G. C. Smith, Z. Xu, T. Lian and N. T. Jui, J. Am. Chem. Soc., 2021, 143, 8987 CrossRef CAS PubMed.
- (a) R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161 CrossRef CAS PubMed; (b) F. Ye, F. Berger, H. Jia, J. Ford, A. Wortman, J. Börgel, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 14615 CrossRef CAS PubMed; (c) E. M. Alvarez, T. Karl, F. Berger, L. Torkowski and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 13609 CrossRef CAS PubMed; (d) J. Wu, Z. Wang, X.-Y. Chen, Y. Wu, D. Wang, Q. Peng and P. Wang, Sci. China: Chem., 2020, 63, 336 CrossRef CAS; (e) C. Chen, M. Wang, H. Lu, B. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 21756 CrossRef CAS.
- Y. Zhao, C. Yu, W. Liang and F. W. Patureau, Org. Lett., 2021, 23, 6232 CrossRef CAS PubMed.
- H. Wang, Y. Gao, C. Zhou and G. Li, J. Am. Chem. Soc., 2020, 142, 8122 CrossRef CAS PubMed.
- F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223 CrossRef CAS PubMed.
- M. H. Aukland, M. Šiaučiulis, A. West, G. J. P. Perry and D. J. Procter, Nat. Catal., 2020, 3, 163 CrossRef CAS.
- (a) Z.-Y. Cao, T. Ghosh and P. Melchiorre, Nat. Commun., 2018, 9, 3274 CrossRef PubMed; (b) C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS; (c) Y. Sumida and H. Ohmiya, Chem. Soc. Rev., 2021, 50, 6320 RSC; (d) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429 CrossRef CAS PubMed.
- (a) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS PubMed; (b) S. V. Rosokha and J. K. Kochi, Acc. Chem. Res., 2008, 41, 641 CrossRef CAS PubMed.
- (a) T. Li, K. Liang, J. Tang, Y. Ding, X. Tong and C. Xia, Chem. Sci., 2021, 12, 15655 RSC; (b) E. de Pedro Beato, D. Spinnato, W. Zhou and P. Melchiorre, J. Am. Chem. Soc., 2021, 143, 12304 CrossRef CAS PubMed; (c) M. Wang, C. Wang, Y. Huo, X. Dang, H. Xue, L. Liu, H. Chai, X. Xie, Z. Li, D. Lu and Z. Xu, Nat. Commun., 2021, 12, 6873 CrossRef CAS PubMed; (d) S. Jung, S. Shin, S. Park and S. Hong, J. Am. Chem. Soc., 2020, 142, 11370 CrossRef CAS PubMed.
- C. Chen, Z.-J. Wang, H. Lu, Y. Zhao and Z. Shi, Nat. Commun., 2021, 12, 4526 CrossRef CAS.
- A. Dewanji, L. van Dalsen, J. Rossi-Ashton, E. Gasson, G. Crisenza and D. Procter, ChemRxiv, 2021 DOI:10.26434/chemrxiv-2021-xbn7k.
- P. Ghosh, N. Y. Kwon, S. Kim, S. Han, S. H. Lee, W. An, N. K. Mishra, S. B. Han and I. S. Kim, Angew. Chem., Int. Ed., 2021, 60, 191 CrossRef CAS PubMed.
- (a) X. Xiao, J. Zeng, J. Fang, J. Sun, T. Li, Z. Song, L. Cai and Q. Wan, J. Am. Chem. Soc., 2020, 142, 5498 CrossRef CAS PubMed; (b) L. Meng, P. Wu, J. Fang, Y. Xiao, X. Xiao, G. Tu, X. Ma, S. Teng, J. Zeng and Q. Wan, J. Am. Chem. Soc., 2019, 141, 11775 CrossRef CAS PubMed; (c) J. Zeng, Y. Liu, W. Chen, X. Zhao, L. Meng and Q. Wan, Top. Curr. Chem., 2018, 376, 27 CrossRef PubMed; (d) P. Shu, X. Xiao, Y. Zhao, Y. Xu, W. Yao, J. Tao, H. Wang, G. Yao, Z. Lu, J. Zeng and Q. Wan, Angew. Chem., Int. Ed., 2015, 54, 14432 CrossRef CAS PubMed.
- L. Pitzer, F. Schäfers and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 8572 CrossRef CAS PubMed.
- (a) K. Sun, F. Xiao, B. Yu and W.-M. He, Chin. J. Catal., 2021, 42, 1921 CrossRef CAS; (b) K. Niu, L. Ding, P. Zhou, Y. Hao, Y. Liu, H. Song and Q. Wang, Green Chem., 2021, 23, 3246 RSC; (c) D. Zheng and A. Studer, Org. Lett., 2019, 21, 325 CrossRef CAS PubMed; (d) Q.-H. Teng, Y. Yao, W.-X. Wei, H.-T. Tang, J.-R. Li and Y.-M. Pan, Green Chem., 2019, 21, 6241 RSC; (e) W. Zhang, X.-X. Xiang, J. Chen, C. Yang, Y.-L. Pan, J.-P. Cheng, Q. Meng and X. Li, Nat. Commun., 2020, 11, 638 CrossRef CAS PubMed.
- S. Mangaleswaran and N. P. Argade, Synthesis, 2002, 2002, 0865 CrossRef.
- F. Zhou, J. Han, R. Liu, P. Li and H. Zhang, Comput. Theor. Chem., 2014, 1044, 80 CrossRef CAS.
- L. M. Kammer, S. O. Badir, R.-M. Hu and G. A. Molander, Chem. Sci., 2021, 12, 5450 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01241c |
| This journal is © The Royal Society of Chemistry 2022 |