
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion-coordination-driven single–double helix switching and chiroptical molecular switching based on oligoureas†
Hongfei
Li‡
a,
Lei
Kou‡
a,
Lin
Liang
b,
Boyang
Li
a,
Wei
Zhao
 *b,
Xiao-Juan
Yang
b and
Biao
Wu
*ab
*b,
Xiao-Juan
Yang
b and
Biao
Wu
*ab
aKey Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education, College of Chemistry and Materials Science, Northwest University, Xi'an 710069, China
bKey Laboratory of Medical Molecule Science and Pharmaceutics Engineering, Ministry of Industry and Information Technology, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 102488, China. E-mail: zhaochem@bit.edu.cn; wubiao@bit.edu.cn
First published on 7th April 2022
Abstract
Synthetic foldamers with helical conformation are widely seen, but controllable interconversion amongst different geometries (helical structure and sense) is challenging. Here, a family of oligourea (tetra-, penta-, and hexa-) ligands bearing stereocenters at both ends are designed and shown to switch between single and double helices with concomitant inversion of helical senses upon anion coordination. The tetraurea ligand forms a right-handed single helix upon chloride anion (Cl−) binding and is converted into a left-handed double helix when phosphate anion (PO43−) is coordinated. The helical senses of the single and double helices are opposite, and the conversion is further found to be dependent on the stoichiometry of the ligand and phosphate anion. In contrast, only a single helix is formed for the hexaurea ligand with the phosphate anion. This distinction is attributed to the fact that the characteristic phosphate anion coordination geometry is satisfied by six urea moieties with twelve H-bonds. Our study revealed unusual single–double helix interconversion accompanied by unexpected chiroptical switching of helical senses.
Introduction
Allosteric regulation of DNA helical geometries (helical senses and structures) is tightly associated with gene expression and other cellular processes in biological systems.1 For example, right-handed B-DNA converts into left-handed Z-DNA during transcription by binding to spermine, spermidine, or metal cations.2 To mimic these biological processes, considerable advances have been made to regulate single and double helical forms of synthetic foldamers.3–6 Pioneering work from Lehn and co-workers showed temperature-dependent single–double helix switching based on oligoamides while retaining their left-handed helical sense.7 Other external stimuli, e.g., solvent,8–10 light,11–15 and guest molecules16–20 have also been shown to regulate single–double helix switching. However, most of these studies have focused on the interconversion between single and double helical forms, and controlling changes in the helical sense along with these processes remains a challenge.Synthetic foldamers with stimuli-responsive helical sense are promising candidates for chiroptical molecular switches, which are amenable with fabrication of functional materials for data storage.21–23 As an example, the seminal report on chiroptical molecular switches from Feringa showed the conversion between left- and right-handed configurations of overcrowded chiral alkenes.24 Triggered by a light source, the chirality of the stereocenter was inversed, thus reversing the helical sense of the pendant foldamers. Besides, anions have also been utilized to regulate helical structures and conformations.25–30 For example, chiroptical molecular switches based on oligo-indolocarbazole foldamers have been shown to alter their helical senses by anion binding and the solvent effect.25–27 Very recently, anions were demonstrated to be able to simultaneously regulate single–double helix switching and reversion of helical senses for aryl-triazole based foldamers.28–30 To the best of our knowledge, this aryl-triazole based foldamer is the only reported example showing the anion-mediated two-in-one switching process involving changes in both the helical structure and helical sense.12
In our previous work, we found that ortho-phenylene spaced oligoureas display strong coordination affinity toward anions, including chloride and oxoanions.31–33 Through multiple hydrogen bonds between urea moieties (NH) and anions, the backbones of oligoureas readily wrap around the bound anions in the centers. As the chain length of oligourea increases, single-strand helical structures are formed by coordinating to two chloride anions.34 By comparison, oxoanions with higher charges prefer more urea moieties (and therefore hydrogen bonds) in order to stabilize the coordination geometry. For instance, the phosphate anion exhibits a characteristic coordination geometry with six urea moieties through 12 hydrogen bonds, which is easily satisfied in a double helix. Thus, we hypothesize that ortho-phenylene spaced oligourea foldamers with a suitable chain length allow for switching of their helical forms by varying the identity of coordinated anions. Yet so far, most of these helical structures are found to be racemic with no preferential helical senses.35 By introducing a pair of chiral centers, the switching of double-single helical forms and helical senses can be envisioned by coordinating to the phosphate anion.36–39
To test this hypothesis, we designed a family of oligourea ligands that incorporate a chiral α-methylbenzyl group at each end to induce single helices with a preferred helical sense.40–43 It was found that the tetraurea L1 shows unexpected, different helical structures and helical senses when coordinating to chloride or phosphate anions (Fig. 1). The single-crystal X-ray diffraction and circular dichroism (CD) results unambiguously support a single, right-handed helix (P) formed with two chloride anions (LA2, L: ligand, A: anion) and double, left-handed helices (MM) driven by phosphate coordination (L2A), respectively. In addition, the latter double, left-handed helices can be further switched into a single, right-handed helix (L1A1 complex) by interacting with extra phosphate anions.
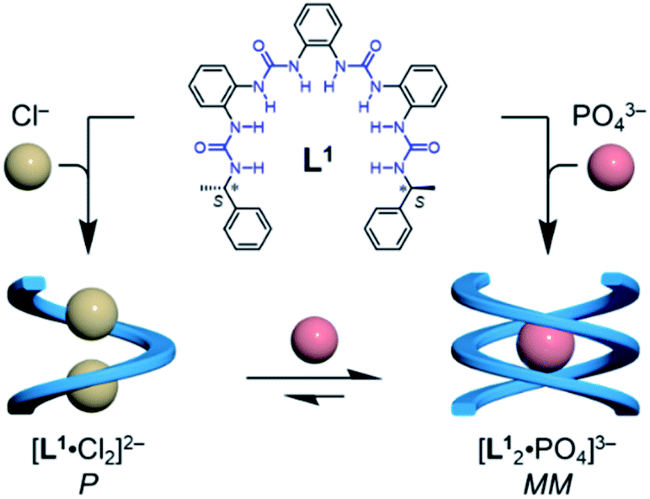 | ||
| Fig. 1 Formation of single and double helices of the tetraurea ligand L1 mediated by chloride and phosphate anion binding. | ||
Results and discussion
Single right-handed helix induced by chloride anion coordination
The details of the syntheses of oligourea ligands L1, L2 and L3 are shown in the ESI.† All the three ligands bear the same α-methylbenzyl chiral group at each end, yet consisting of various numbers of ortho-phenylene spaced urea moieties (Fig. 2a). The formation of a folded structure of these ligands upon chloride coordination was first investigated in solution (Fig. 2b). Strong CD signals with a positive Cotton effect were observed for all the three ligands. The structures of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (ligand to chloride) complexes were confirmed by single-crystal X-ray diffraction (Fig. 2c, d and S2, ESI†), which are consistent with our previous report.35 As expected, only right-handed conformation was found in the solid state on account of the presence of chiral ending groups. The 1:2 complexes were also supported by mass spectrometry analysis (Fig. S19, ESI†).
:2 (ligand to chloride) complexes were confirmed by single-crystal X-ray diffraction (Fig. 2c, d and S2, ESI†), which are consistent with our previous report.35 As expected, only right-handed conformation was found in the solid state on account of the presence of chiral ending groups. The 1:2 complexes were also supported by mass spectrometry analysis (Fig. S19, ESI†).
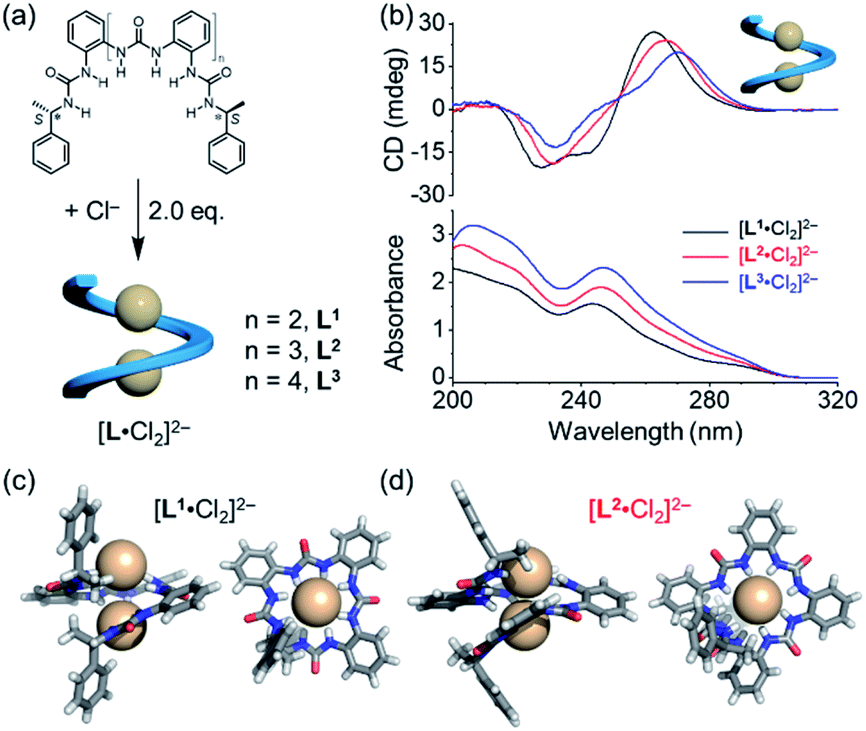 | ||
| Fig. 2 (a) Structures of ligands L1–L3 and their chloride-coordinated single helices [L·Cl2]2−. (b) CD and UV-vis spectra of [L·Cl2]2− complexes in acetonitrile ([L] = 30 μM). Crystal structures for the single helices of (c) [L1·Cl2]2− and (d) [L2·Cl2]2− showing right-handed helical conformation in the solid state (front and top views). Countercations and solvent molecules were omitted for clarity. | ||
Consistent with the crystal structures of [L·Cl2]2− complexes, CD spectra in acetonitrile also support the formation of right-handed helices (Fig. 2b). According to the exciton chirality method,44–46 the observed pattern of CD signals (positive at long wavelength) is a signature for the formation of right-handed (P) helical conformation. This observation suggests that the stereo information of the (S) α-methylbenzyl group at both ends is transferred to the folded structures. In addition, the attenuated intensities of CD signals in different oligourea ligands could be attributed to a decrease in the anisotropy factors (g-factor) of folded conformations (Fig. S3, ESI†).47–49 It is worth noting that both CD and UV absorptions are slightly red shifted for [L3·Cl2]2− as compared to [L1·Cl2]2−, which may be attributed to the increased coupling through the oligomer backbone in the longer oligomer.50
Double helices induced by phosphate anion coordination and single–double helix switching
Based on the characteristic coordination geometry between ortho-phenylene spaced urea moieties and phosphate anions seen in our previous work,51 we expect to see a different helical structure with phosphate from that with chloride anions. Indeed, instead of forming a single helix as seen with chloride anions, phosphate coordination was found to drive the formation of a double helix with the tetraurea ligand L1. A similar anion-mediated interconversion of single and double helices has been reported in a previous study of aryl-triazole-based foldamers.12 In addition to this, we also observed that the helical sense is also inverted from being right-handed for the single helix to being left-handed for the double helices, which was usual in previous studies.52–58Inversion of the helical sense from right to left was first shown by CD spectra. We observed a negative Cotton effect in the CD spectrum (negative in a long wavelength region) for the complex of L1 and phosphate anion (Fig. 3a), indicative of the formation of the left-handed helix as compared to the right-handed helix found with chloride.59 The CD intensity of the phosphate complex (ΔCD = +145 mdeg at 259 nm) is ca. seven times stronger than that of the chloride complex (ΔCD = +22 mdeg at 259 nm), suggesting the formation of a more compact foldamer-anion complex with phosphate than that with the chloride anion.
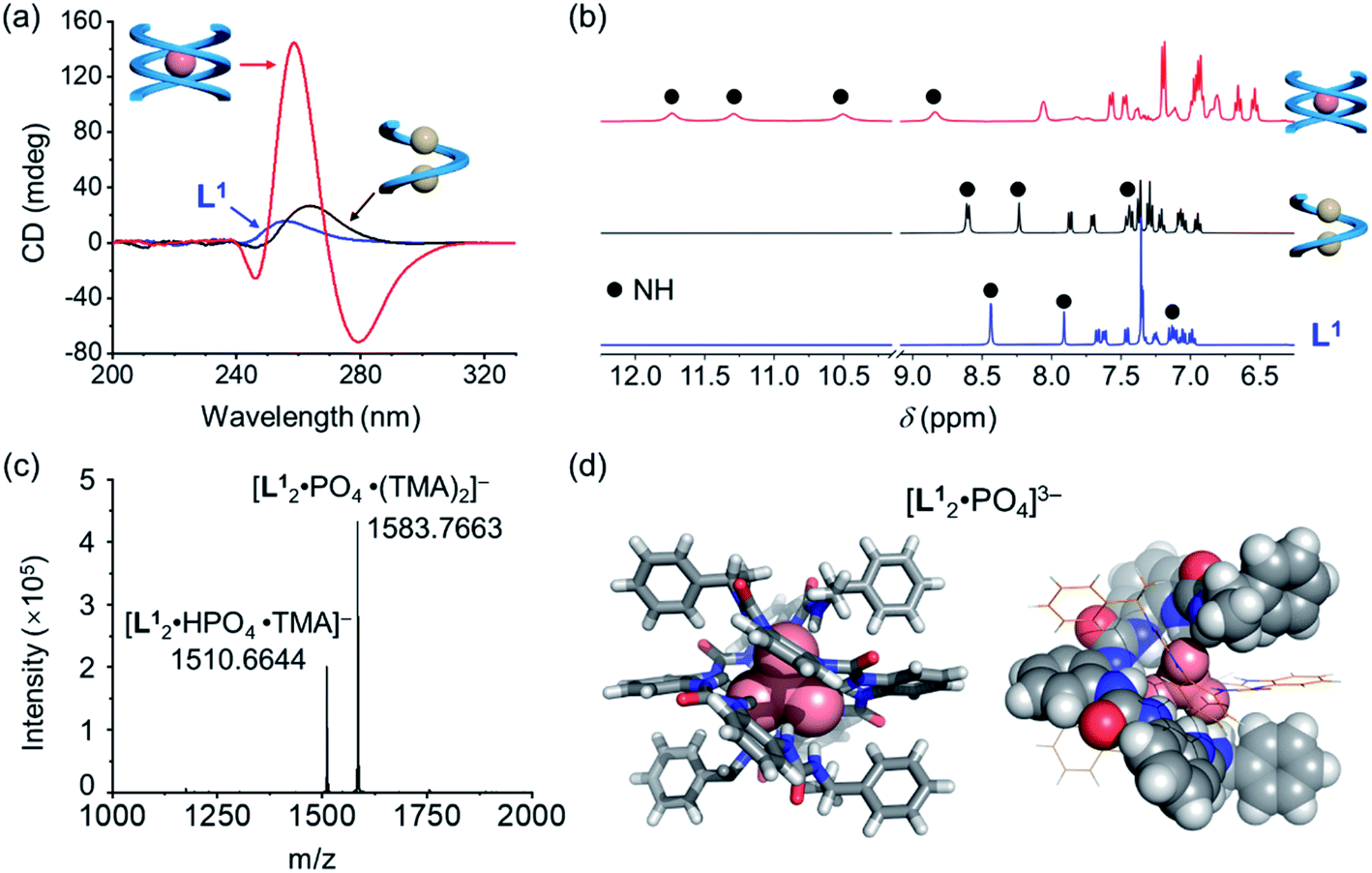 | ||
| Fig. 3 (a) CD spectra ([L1] = 30 μM, 5% v/v DMSO/CH3CN) and (b) stacked partial 1H NMR spectra ([L1] = 0.6 mM, 400 MHz, DMSO-d6) of the tetraurea ligand L1 in its single-helix chloride complex and double-helix phosphate complex. (c) ESI-MS spectrum and (d) crystal structure of the double helical complex [L12·PO4]3−, tetramethylammonium countercations and solvent molecules were omitted for clarity. | ||
Considering that the phosphate ion (PO43−) with three negative charges is a much stronger hydrogen-bond acceptor than chloride,60 NMR spectroscopy was used to study these interactions. Larger downfield shifts of N–H (urea) groups were observed compared to the chloride complex (Fig. 3b and S18, ESI†). Specifically, the largest chemical shift change for the N–H peak is about 3.30 ppm (from 8.40 ppm to 11.70 ppm) for the phosphate complex, while only less than 0.33 ppm (7.87 ppm to 8.20 ppm) was seen for the chloride complex (Tables S9 and S10, ESI†). This is consistent with the stronger interactions between the ligand and phosphate than the chloride ion.61
To further confirm the stoichiometry of the phosphate complex, we performed mass spectrometry analysis. The major peak in the ESI-MS spectrum is assigned to the L2A complex, [L12·PO4·(TMA)2]− (TMA = tetramethylammonium, Fig. 3c). The single-crystal X-ray diffraction structure confirms the formation of a double, left-handed helix (Fig. 3d and S1, ESI†), in accordance with the strong CD intensities and negative Cotton effect for the tetraurea ligand L1 with the phosphate anion (Fig. 3a). As shown in the crystal structure, all 16 urea hydrogens (N–H) from the two intertwining ligands form hydrogen bonds with the bound phosphate anion. This coordination number of 16 for phosphate is unusually high in comparison with the typical coordination number of 12 seen in our previous studies.32
Addition of 0.5 equivalents of phosphate anions into the solution of [L1·Cl2]2− drives a complete conversion from a single helix to a double helix of [L12·PO4]3− (Fig. 4a and S5, ESI†). According to the CD spectra in acetonitrile (Fig. 4b), the positive Cotton effect gradually shifts to the negative pattern by adding phosphate anions. The final CD spectrum is very similar to that by adding 0.5 equivalents of phosphate into the solution of the free tetraurea ligand (Fig. 3a), suggesting the formation of left-handed double helices of the [L12·PO4]3− complex. Along with change in helical forms, the helical sense is also inverted from being right-handed to being left-handed. This simultaneous dual conversion in both the helical structure and helical sense was rarely seen in previous studies.62–64 Note that only 0.5 equivalents of phosphate anions are sufficient to fully drive the conversion from the [L1·Cl2]2− complex to [L12·PO4]3−, inferring once again that the binding affinity of tetraurea ligand L1 with phosphate is much stronger than that with chloride anions.
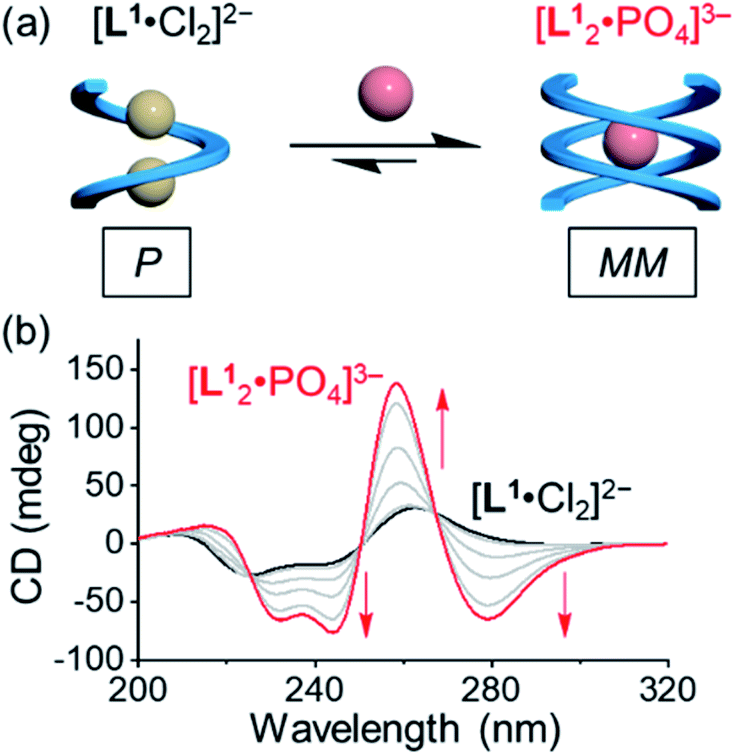 | ||
| Fig. 4 (a) Schematic representation of the anion-mediated conversion from a single helix to a double helix and (b) the corresponding changes in CD spectra. ([L1] = 30 μM, CH3CN). | ||
Quantitative NMR and UV-vis titration confirms that all oligourea ligands bind to phosphate much strongly than chloride anion (Fig. S6–S17, ESI†). Using 1H NMR titration in DMSO-d6 (Fig. S12–S14, ESI†), the binding constants of oligoureas L1, L2, and L3 with the chloride ion were determined to be (2.47 ± 0.03) × 103 M−1, (3.17 ± 0.06) × 103 M−1, and (2.11 ± 0.04) × 103 M−1, respectively. In such a polar solvent, 1:1 binding mode was applied for chloride coordination. The hexaurea L3 shows slightly weaker binding affinity compared to the other two ligands, perhaps due to its larger and/or less preorganized cavity for chloride binding. In the case of phosphate, a binding mode of 2:1 (L2A1) was observed in the titration of oligourea ligands with the phosphate ion (Fig. S15, ESI†). We saw the formation of two distinct complexes (L2A1 and L1A1), which were supported by the slow exchange between NMR signals during titrations in DMSO-d6. The binding affinity of phosphate is too high for quantitative determination using NMR measurements. Yet based on changes in the absorbance of UV-vis spectra (Fig. S9–S11, ESI†), we believe that the overall binding affinities of these oligoureas with the phosphate anion are much stronger than those with the chloride anion.
The inversion of the helical sense from a single helix to a double helix is unexpected and unusual. The underlying driving force involved in the transition is not yet clear.12,26,27,65,66 Flood and co-workers found that the helicity bias between the racemic single helix and chiral double helices for aryl-triazole foldamers is dependent on the anion size.12 Huc and co-workers found that extra contacts (H-bonds or π–π stacks) among double strands could contribute to the inversion of the helical sense.66 In our case, based on the crystal structure of [L1·Cl2]2− and [L12·PO4]3−, we see that the terminal phenyl rings are aligned parallel to the radial direction in the single helix and vertical to the radial direction for the double helix. In the double helix, extra C–H⋯π contacts (Fig. S1, ESI†) between the terminal phenyl ring and mid-chain phenyl ring are observed, and may help to stabilize the M-helicity thus forming left-handed double helices (MM). Computational studies (Fig. S23 and S24, ESI†) on the double helix with the opposite right-handed helical sense (PP) do not result in similar C–H⋯π contacts, and so the right-handed structure is suggested to be less stable than the left-handed one. Thus, we postulate that these extra C–H⋯π contacts in the left-handed double helix are one driving force for the inversion of helicity (Fig. S24, ESI†).
Stoichiometry dependent helix switching induced by phosphate anion coordination
In addition to the anion-mediated interconversion of single and double helices, stoichiometry-controlled helix switching is also observed by further adding phosphate anions (Fig. 5a and S4, ESI†). First, when compared to the mass spectrum of double helices of the phosphate complex, [L12·PO4·(TMA)2]− (Fig. 3c and S20a, ESI†), the major peaks were assigned to a 1:1 complex of [L1·PO4·(TMA)2]− when more than one equivalent of phosphate anions were mixed with the tetraurea ligand (Fig. 5b and S20b, ESI†). This is indicative of the conversion from double helices to a single helix. According to the CD signal changes during the titration of double helices with extra phosphate anions in acetonitrile (Fig. 5c and d), we observed consistent shifting of the Cotton effect from being negative to being positive when 0.5 equivalents of phosphate anions were added. The inverted CD signals suggest that the helical sense changed to right-handed. Moreover, the decreased CD intensity also suggests the formation of a less folded structure that is similar to a single helical conformation. The corresponding 1H NMR titration also supports this transformation in solution (Fig. S15, ESI†). When ligand L1 was mixed with less than 0.5 equivalents of phosphate anions, only one set of peaks were observed, which were assigned to the double helices of the [L12·PO4]3− complex. By adding more phosphate anions (up to one equivalent), we observed a decrease in the peaks of the double helix and emergence of a new set of peaks (Fig. S15, ESI†). The 1H NMR spectroscopy results and the CD spectra support stoichiometry-controlled conversion from double helices to a single helix with inversed helical senses.
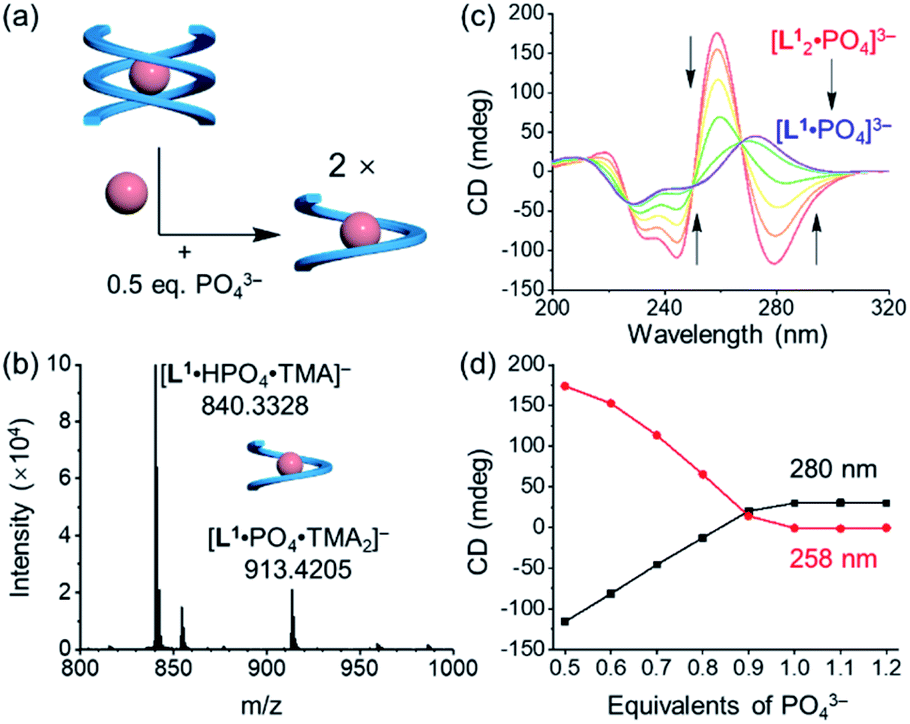 | ||
| Fig. 5 (a) Schematic representation of the stoichiometry-controlled conversion from double helices to a single helix and conversion of the helical sense from M to P. (b) ESI-MS spectrum showing the formation of the single helix. (c) CD spectra of [L12·PO4]3− by adding 0.5 equivalents of phosphate anions gradually showing conversion in the helical sense. (d) Changes in CD spectra at a wavelength of 258 nm and 280 nm. ([L1] = 30 μM, CH3CN). | ||
Given that the phosphate anion typically requires six urea moieties (twelve hydrogen bonds) to satisfy the coordination sphere, we expect to see different helical structures induced by phosphate coordination by increasing the chain length from tetra-urea to penta- and hexa-urea. In particular, the hexaurea ligand L3 with twelve NH moieties may be able to wrap around one phosphate anion and form a single-strand helix instead of double helices. Indeed, unlike the tetraurea ligand L1, treating the other two ligands L2 and L3 with phosphate anions did not clearly show the transition from a double helix to a single helix (Fig. S16 and S17, ESI†).
For the pentaurea ligand L2, we observed a set of new peaks when 0.5 equivalents of phosphates were added based on 1H NMR spectra (Fig. S16, ESI†), which is similar to the peaks seen in the case of ligand L1. However, we did not see a comparable Cotton effect for the double helices (Fig. 3a). Presumably, an intermediate species of an irregular helical structure for the L2A complex was formed, which readily converted into the single helix [L2·PO4]3− when more phosphate ions were added. The CD spectrum of the [L2·PO4]3− complex in acetonitrile shows a similar Cotton effect to that of the [L1·PO4]3− complex that is assigned to be the right-handed single helix (Fig. 6a).
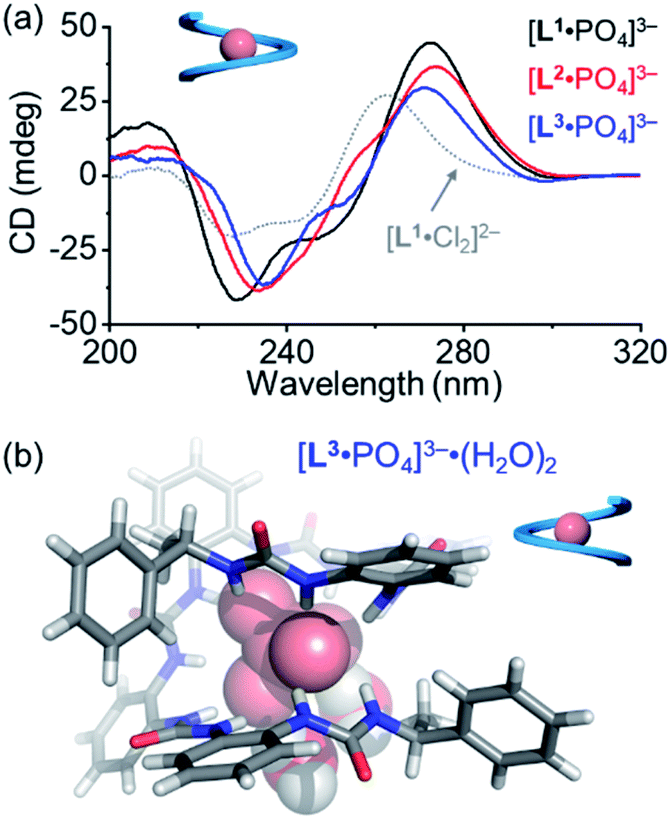 | ||
| Fig. 6 (a) CD spectra of the single helix of ligands with 1.0 equivalent of phosphate anions. ([L] = 30 μM, CH3CN) (b) X-ray diffraction crystal structures of the single helix of the [L3·PO4]3− complex. The 1:1 complex is shown, and two water molecules are found to interact with the phosphate anion through hydrogen bonding. The tetramethylammonium countercations and solvent molecules were omitted for clarity. | ||
During the titration of the hexaurea ligand L3 with the phosphate anion, we only observed the formation of the [L3·PO4]3− complex based on 1H NMR spectra, mass spectrum, and CD spectra (Fig. 6a and S22, ESI†) as a single helix. In addition, the CD signals of the [L·PO4]3− complex in acetonitrile also show a similar decreasing tendency to those of the right-handed single helix [L1·Cl2]2−, and of the ligands (L1–L3) (Fig. 6a). This indicates the formation of the right-handed single helix for all the three [L·PO4]3− complexes in solution.
We obtained the crystal structure of the [L3·PO4]3− complex, where to our surprise only a left-handed helix is seen in the solid state (Fig. 6b), which is not consistent with the CD results. This difference could be attributed to specific solvation, and similar solvation-driven inversion of helical senses has been observed before for the oligo-indolocarbazole based foldamers.26,27,65 In the solid state, two water molecules are seen to interact with the phosphate anion through two hydrogen bonds. This specific solvation affects the orientation of the terminal chiral groups, which is more folded around the phosphate anion than the other oligourea ligands, and thus is more sensitive to what solvates the anions in the solid state. In solution, this coordination geometry with water molecules may not survive, and the right-handed helical conformation is more stabilized just as the other shorter foldamers.
Conclusions
In summary, we report the interconversion of single–double helix structures with simultaneous helical sense switching that is regulated by anion coordination. Based on the characteristic anion coordination ability of ortho-phenylene spaced urea moieties, the chloride anion (Cl−) drives the tetraurea ligand to form a right-handed (P) single helix (LA2 complex) while phosphate (PO43−) binding drives the formation of a left-handed (MM) double helix (L2A complex). One equivalent of phosphate anions further converts the double helix into a single helix (LA complex) suggesting stoichiometry dependent single-to-double helix switching. Our study shows a new anion-coordination-mediated approach to the regulation of helical conformations with specific chirality.Data availability
All experimental and computational data associated with this article are included in the main text and ESI.†Author contributions
Conceptualization: B. Wu, and H. Li. Data curation: H. Li, B. Li, and L. Kou. Formal analysis: B. Wu, W. Zhao, and H. Li. Investigation: H. Li, L. Kou, and B. Li. Methodology: H. Li, B. Li., and L. Liang. Project administration: H. Li, B. Wu, X.-J. Yang, and W. Zhao. Supervision & funding acquisition: B. Wu, X.-J. Yang, and W. Zhao. Writing (original draft, reviewing & editing): W. Zhao, H. Li, B. Wu, X.-J. Yang, and L. Liang.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (91856102, 21772154, 22171023 and 22101024). W. Z. acknowledges the support of the Beijing Institute of Technology Research Fund Program for Young Scholars. We thank the staff of the BL17B beamline at the National Centre for Protein Sciences Shanghai and Shanghai Synchrotron Radiation Facility, Shanghai, People's Republic of China, for assistance during data collection. We also thank Dr Yun Liu and Dr Xiaoyan Zheng for helpful discussions.Notes and references
- T. Pawson and P. Nash, Science, 2003, 300, 445–452 CrossRef CAS PubMed
.
- A. Rich and S. Zhang, Nat. Rev. Genet., 2003, 4, 566–572 CrossRef CAS PubMed
- Y. Ferrand, N. Chandramouli, A. M. Kendhale, C. Aube, B. Kauffmann, A. Grélard, M. Laguerre, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2012, 134, 11282–11288 CrossRef CAS PubMed
- N. Saito, H. Kobayashia and M. Yamaguchi, Chem. Sci., 2016, 7, 3574–3580 RSC
- L. R. Holloway, H. H. McGarraugh, M. C. Young, W. Sontising, G. J. O. Beran and R. J. Hooley, Chem. Sci., 2016, 7, 4423–4427 RSC
- G. Iadevaia, J. A. Swain, D. Núñez-Villanueva, A. D. Bond and C. A. Hunter, Chem. Sci., 2021, 12, 10218–10226 RSC
- V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720–723 CrossRef CAS PubMed
- A. Acocella, A. Venturini and F. Zerbetto, J. Am. Chem. Soc., 2004, 126, 2362–2367 CrossRef CAS PubMed
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Ihara and I. Huc, Chem. Commun., 2012, 48, 6337–6339 RSC
- M. Umetani, T. Tanaka and A. Osuka, Chem. Sci., 2018, 9, 6853–6859 RSC
- W. Miao, S. Wang and M. Liu, Adv. Funct. Mater., 2017, 27, 1701368 CrossRef
- F. C. Parks, Y. Liu, S. Debnath, S. R. Stutsman, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 17711–17723 CrossRef CAS PubMed
- B. Gole, B. Kauffmann, V. Maurizot, I. Huc and Y. Ferrand, Angew. Chem., Int. Ed., 2019, 58, 8063–8067 CrossRef CAS PubMed
- S. Pramanik, B. Kauffmann, S. Hecht, Y. Ferrand and I. Huc, Chem. Commun., 2021, 57, 93–96 RSC
- N. A. Simeth, S. Kobayashi, P. Kobauri, S. Crespi, W. Szymanski, K. Nakatani, C. Dohno and B. L. Feringa, Chem. Sci., 2021, 12, 9207–9220 RSC
- M. Barboiu, G. Vaughan, N. Kyritsakas and J.-M. Lehn, Chem.–Eur. J., 2003, 9, 763–769 CrossRef CAS PubMed
- N. Chandramouli, Y. Ferrand, B. Kauffmanncde and I. Huc, Chem. Commun., 2016, 52, 3939–3942 RSC
- Q. Gan, X. Wang, B. Kauffmann, F. Rosu, Y. Ferrand and I. Huc, Nat. Nanotechnol., 2017, 12, 447–452 CrossRef CAS PubMed
- P. Mateus, N. Chandramouli, C. D. Mackereth, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2020, 59, 5797–5805 CrossRef CAS PubMed
- T. Kinoshita, Y. Haketa, H. Maeda and G. Fukuhara, Chem. Sci., 2021, 12, 6691–6698 RSC
- B. L. Feringa, R. A. v. Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789–1816 CrossRef CAS PubMed
- D. Zhao, T. v. Leeuwen, J. Cheng and B. L. Feringa, Nat. Chem., 2017, 9, 250–256 CrossRef CAS PubMed
- N. Ousaka, Y. Takeyama, H. Iida and E. Yashima, Nat. Chem., 2011, 3, 856–861 CrossRef CAS PubMed
- B. L. Feringa, N. P. M. Huck and A. M. Schoevaars, Adv. Mater., 1996, 8, 681–684 CrossRef CAS
- J.-m. Suk and K.-S. Jeong, J. Am. Chem. Soc., 2008, 130, 11868–11869 CrossRef CAS PubMed
- J.-m. Suk, V. R. Naidu, X. Liu, M. S. Lah and K.-S. Jeong, J. Am. Chem. Soc., 2011, 133, 13938–13941 CrossRef CAS PubMed
- D. A. Kim, P. Kang, M.-G. Choi and K.-S. Jeong, Chem. Commun., 2013, 49, 9743–9745 RSC
- H. Juwarker, J. M. Lenhardt, D. M. Pham and S. L. Craig, Angew. Chem., Int. Ed., 2008, 47, 3740–3743 CrossRef CAS PubMed
- S. Wei, X. Li, Z. Yang, J. Lan, G. Gao, Y. Xue and J. You, Chem. Sci., 2012, 3, 359–363 RSC
- Y. Liu, F. C. Parks, W. Zhao and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 15477–15486 CrossRef CAS PubMed
- S. Li, M. Wei, X. Huang, X.-J. Yang and B. Wu, Chem. Commun., 2012, 48, 3097–3099 RSC
- B. Wu, F. Cui, Y. Lei, S. Li, N. d. S. Amadeu, C. Janiak, Y.-J. Lin, L.-H. Weng, Y.-Y. Wang and X.-J. Yang, Angew. Chem., Int. Ed., 2013, 52, 5096–5100 CrossRef CAS PubMed
- X. Zhao, H. Wang, B. Li, W. Zhang, X. Li, W. Zhao, C. Janiak, A. W. Heard, X.-J. Yang and B. Wu, Angew. Chem., Int. Ed., 2022, 61, e202115042 CAS
- P. Yang, J. Wang, C. Jia, X.-J. Yang and B. Wu, Eur. J. Org. Chem., 2013, 2013, 3446–3454 CrossRef CAS
- B. Wu, C. Jia, X. Wang, S. Li, X. Huang and X.-J. Yang, Org. Lett., 2012, 14, 684–687 CrossRef CAS PubMed
- C. A. Hunter, P. S. Jones, P. M. N. Tigera and S. Tomas, Chem. Commun., 2003, 1642–1643 RSC
- Y. Wang, J. Xiang and H. Jiang, Chem.–Eur. J., 2011, 17, 613–619 CrossRef CAS PubMed
- X. Jin, J. Jiang and M. Liu, ACS Nano, 2016, 10, 11179–11186 CrossRef CAS PubMed
- W. Zheng, T. Ikai and E. Yashima, Angew. Chem., Int. Ed., 2021, 60, 11294–11299 CrossRef CAS PubMed
- E. Yashima, Nat. Chem., 2011, 3, 12–14 CrossRef CAS PubMed
- D. Zheng, L. Zheng, C. Yu, Y. Zhan, Y. Wang and H. Jiang, Org. Lett., 2019, 21, 2555–2559 CrossRef CAS PubMed
- G. N. Vemuri, R. R. Pandian, B. J. Spinello, E. B. Stopler, Z. J. Kinney and C. S. Hartley, Chem. Sci., 2018, 9, 8260–8270 RSC
- C. A. Hunter, A. Spitaleri and S. Tomas, Chem. Commun., 2005, 29, 3691–3693 RSC
- D. Gargiulo, N. Ikemoto, J. Odingo, N. Bozhkova, T. Iwashita, N. Berova and K. Nakanishi, J. Am. Chem. Soc., 1994, 116, 3760–3767 CrossRef CAS
- T. Taniguchi and K. Monde, J. Am. Chem. Soc., 2012, 134, 3695–3698 CrossRef CAS PubMed
- J. Chen, A. J. Ferreira and C. M. Beaudry, Angew. Chem., Int. Ed., 2014, 53, 11931–11934 CrossRef CAS
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed
- J. Yao, W. Wu, W. Liang, Y. Feng, D. Zhou, J. J. Chruma, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, Angew. Chem., Int. Ed., 2017, 56, 6869–6873 CrossRef CAS PubMed
- J. Hao, H. Lu, L. Mao, X. Chen, M. C. Beard and J. L. Blackburn, ACS Nano, 2021, 15, 7608–7617 CrossRef CAS PubMed
- B. Li, B. Zheng, W. Zhang, D. Zhang, X.-J. Yang and B. Wu, Cryst. Growth Des., 2019, 19, 6527–6533 CrossRef CAS
- R. Li, Y. Zhao, S. Li, P. Yang, X. Huang, X.-J. Yang and B. Wu, Inorg. Chem., 2013, 52, 5851–5860 CrossRef CAS PubMed
- C. Dolain, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2003, 42, 2738–2740 CrossRef CAS PubMed
- T. Miyagawa, A. Furuko, K. Maeda, H. Katagiri, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2005, 127, 5018–5019 CrossRef CAS PubMed
- K. Okoshi, S.-i. Sakurai, S. Ohsawa, J. Kumaki and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 8173–8176 CrossRef CAS PubMed
- S.-i. Sakurai, K. Okoshi, J. Kumaki and E. Yashima, J. Am. Chem. Soc., 2006, 128, 5650–5651 CrossRef CAS PubMed
- D. Pijper and B. L. Feringa, Angew. Chem., Int. Ed., 2007, 46, 3693–3696 CrossRef CAS PubMed
- I. Louzao, J. M. Seco, E. Quiñoá and R. Riguera, Angew. Chem., Int. Ed., 2010, 49, 1430–1433 CrossRef CAS PubMed
- F. Freire, J. M. Seco, E. Quiñoá and R. Riguera, Angew. Chem., Int. Ed., 2011, 50, 11692–11696 CrossRef CAS PubMed
- N. Berova, L. D. Bari and G. Pescitellib, Chem. Soc. Rev., 2007, 36, 914–931 RSC
- C. Jia, B. Wu, S. Li, Z. Yang, Q. Zhao, J. Liang, Q.-S. Li and X.-J. Yang, Chem. Commun., 2010, 46, 5376–5378 RSC
- J. Zhao, D. Yang, Y. Zhao, X.-J. Yang, Y.-Y. Wang and B. Wu, Angew. Chem., Int. Ed., 2014, 53, 6632–6636 CrossRef CAS
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. D. Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed
- H. Liang, B. Hua, F. Xu, L.-S. Gan, L. Shao and F. Huang, J. Am. Chem. Soc., 2020, 142, 19772–19778 CrossRef CAS PubMed
- J. S. S. K. Formen and C. Wolf, Angew. Chem., Int. Ed., 2021, 60, 27031–27038 CrossRef CAS PubMed
- H. J. Lee, H.-G. Jeon and K.-S. Jeong, Supramol. Chem., 2014, 27, 378–385 CrossRef
- Q. Gan, Y. Ferrand, N. Chandramouli, B. Kauffmann, C. Aube, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2012, 134, 15656–15659 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Details of syntheses, CD, NMR, mass spectroscopy results and crystal structure. CCDC 2130743–2130746. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00876a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |