
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced metal-free borylation of aryl halides catalysed by an in situ formed donor–acceptor complex†
Manhong
Li‡
a,
Siqi
Liu‡
 b,
Haoshi
Bao
a,
Qini
Li
a,
Yi-Hui
Deng
bc,
Tian-Yu
Sun
*b and
Leifeng
Wang
*a
b,
Haoshi
Bao
a,
Qini
Li
a,
Yi-Hui
Deng
bc,
Tian-Yu
Sun
*b and
Leifeng
Wang
*a
aSchool of Pharmaceutical Sciences (Shenzhen), Sun Yat-Sen University, No. 66, Gongchang Road, Shenzhen 518107, P. R. China. E-mail: wanglf33@mail.sysu.edu.cn
bShenzhen Bay Laboratory, Shenzhen 518132, P. R. China. E-mail: Tian-Yu_Sun@pku.edu.cn
cLab of Computational Chemistry and Drug Design, State Key Laboratory of Chemical Oncogenomics, Peking University Shenzhen Graduate School, Shenzhen 518055, P. R. China
First published on 4th April 2022
Abstract
Organoboron compounds are very important building blocks which can be applied in medicinal, biological and industrial fields. However, direct borylation in a metal free manner has been very rarely reported. Herein, we described the successful direct borylation of haloarenes under mild, operationally simple, catalyst-free conditions, promoted by irradiation with visible light. Mechanistic experiments and computational investigations indicate the formation of an excited donor–acceptor complex with a −3.12 V reduction potential, which is a highly active reductant and can facilitate single-electron-transfer (SET) with aryl halides to produce aryl radical intermediates. A two-step one-pot method was developed for site selective aromatic C–H bond borylation. The protocol's good functional group tolerance enables the functionalization of a variety of biologically relevant compounds, representing a new application of aryl radicals merged with photochemistry.
Introduction
Boronic esters are commonly encountered in pharmaceuticals, functional materials and agrochemicals.1 Aryl halides and related arenes are the most commonly employed precursors to produce them.2 Conventional routes for their synthesis include metalation (generally by Li or Mg reagents)3 followed by nucleophilic substitution and transition-metal catalysis,4 offering a broad substrate scope and good functional group compatibility if their disadvantages such as high cost, low reactivity and operational inconvenience could be ignored. Visible light catalysis5 has become a well-accepted and powerful method for the direct borylation of drug like molecules. Seminal studies by Larionov,6 Aggarwal,7 Wu,8 Schelter9 and others10 have established the viability and synthetic utility of this approach, circumventing the need for well recognized photocatalysts and metal additives. Studer11 developed radical borylation of aryl iodides with bis(catecholato)diboron (B2cat2) as the boron source under mild conditions. Recently, Jiao12 reported the borylation of aryl halides using 4-phenylpyridine as an efficient catalyst and sodium methanolate as a strong base with bis(pinacolato)diboron (B2pin2). Different from Jiao's work, Li,13 Larionov14 and Lin15 independently described simple and efficient methods to convert aryl triflates and aryl halides into aryl radicals under UV irradiation or reductive electrophotocatalysis. There is a constant quest for more efficient clean borylation strategies (e.g., metal-free visible light catalysis using an in situ formed photocatalyst) from simple and readily available starting materials.In recent years, in situ formed donor–acceptor complexes have been successfully used in the single-electron-transfer (SET) process to produce radical species by hybridization and modulation of the relevant energy levels.16,17 As a result, visible light excitation can occur favoured by a red shift of the absorption band, although both donor and acceptor components are individually insensitive to visible light irradiation. This novel protocol can provide a new approach for aryl radical intermediates through C–X bond activation in a metal-free manner (Scheme 1).
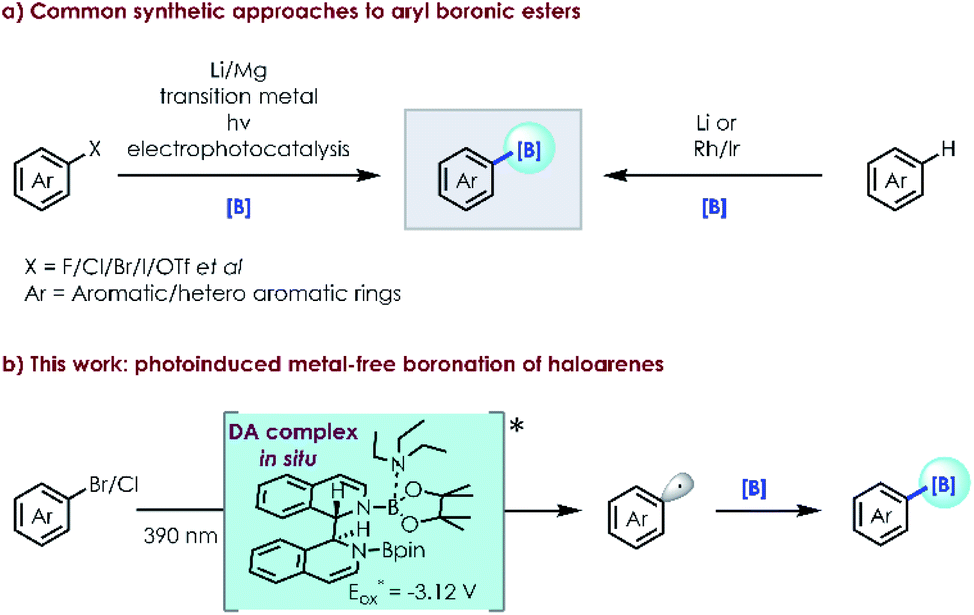 | ||
| Scheme 1 Synthetic approaches to aryl boronic esters. | ||
In 2020, we reported a photoinduced dehalogenation of aryl halides.18 Spectroscopic, computational and chemical studies indicate that the formation of a twisted intramolecular charge-transfer (TICT) species enables the population of higher-energy doublet states. König19 developed a versatile photocatalytic strategy for the ipso-borylation of substituted arenes by using thiolate as a catalyst, forming a donor–acceptor complex between thiolate/B2pin2 and boryl-anion-activated substrates. Inspired by our previous work and related reports,20 we questioned whether borylation of aryl halides could be achieved with the photocatalytic intermediate formed in situ employing simple available substrates (e.g., N-containing heterocycles/organic base/boron source). Herein, we described the successful direct borylation of haloarenes under mild, operationally simple, catalyst-free conditions, promoted by irradiation with visible light.
Results and discussion
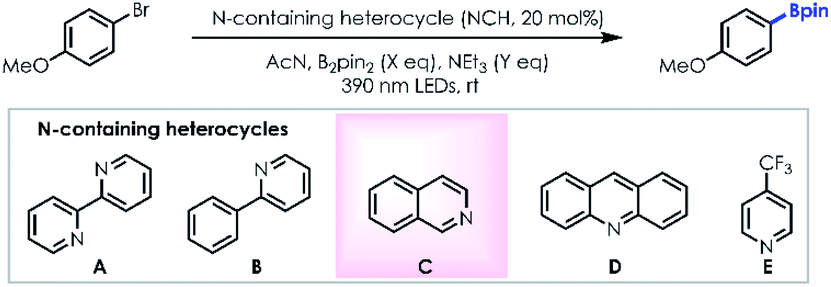
We made our initial discovery of the borylation reaction employing 4-bromoanisole as the model substrate (Table 1). First, under visible light photoirradiation, when the 2,2′-bipyridine (nitrogen containing heterocycle: NCH)/triethylamine (NEt3; final reductant)/B2pin2 (boron source) system was evaluated in acetonitrile, the desired product can be detected in 27% yield (entry 1). In order to generate the optimal ate-intermediate to facilitate the single-electron-transfer (SET) process, a series of NCHs were evaluated (entries 2–5). Only trace desired product was detected using the 2-phenyl pyridine/NEt3/B2pin2 system (entry 2) suggesting that the ate-intermediate formed with 2-phenyl pyridine couldn't deliver a single electron to the aromatic C–Br bond. To our delight, the yield was improved remarkably (49% yield) employing isoquinoline as the ate-intermediate precursor (entry 3). Neither acridine (entry 4) nor 4-trifluoromethyl pyridine (entry 5) could benefit the borylation reaction, indicating that steric resistance and electron-deficient factors of NCHs can decrease the reductive ability of the ate-intermediate. A brief investigation of the solvent effect demonstrates that DMSO (entry 6), DMF (entry 7) and DCE (entry 8) are not as efficient as acetonitrile. Gratifyingly, the desired product could be isolated with 81% yield via increasing the loading of B2pin2/NEt3 and prolonging the photoirradiation process (entry 9). Not surprisingly, control experiments without NCH (entry 10) and in the dark (entry 11) couldn't afford the desired product effectively.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | B2pin2 (X eq.) | NEt3 (Y eq.) | NCH | Time (h) | Yield (%)b |
| a Reactions were performed with 0.2 mmol 4-bromoanisole, 1.0 mmol NEt3, 20 mol% NCH, 0.5 ml solvent and suitable B2pin2 for the indicated period of time under 2 × 390 nm LED (40 W) irradiation. b NMR yields using pyrazine or hexamethyl disiloxane as the internal standard. c Isolated yields. d Without irradiation. | ||||||
| 1 | AcN | X = 2 | Y = 3 | A | 12 | 27 |
| 2 | AcN | X = 2 | Y = 3 | B | 12 | Trace |
| 3 | AcN | X = 2 | Y = 3 | C | 12 | 49 |
| 4 | AcN | X = 2 | Y = 3 | D | 12 | 17 |
| 5 | AcN | X = 2 | Y = 3 | E | 12 | Trace |
| 6 | DMSO | X = 2 | Y = 3 | C | 12 | Trace |
| 7 | DMF | X = 2 | Y = 3 | C | 12 | NR |
| 8 | DCE | X = 2 | Y = 3 | C | 12 | 16 |
| 9 | AcN | X = 4 | Y = 5 | C | 36 | 87 (81) |
| 10 | AcN | X = 4 | Y = 5 | — | 36 | 14 |
| 11 | AcN | X = 4 | Y = 5 | C | 36 | NRd |
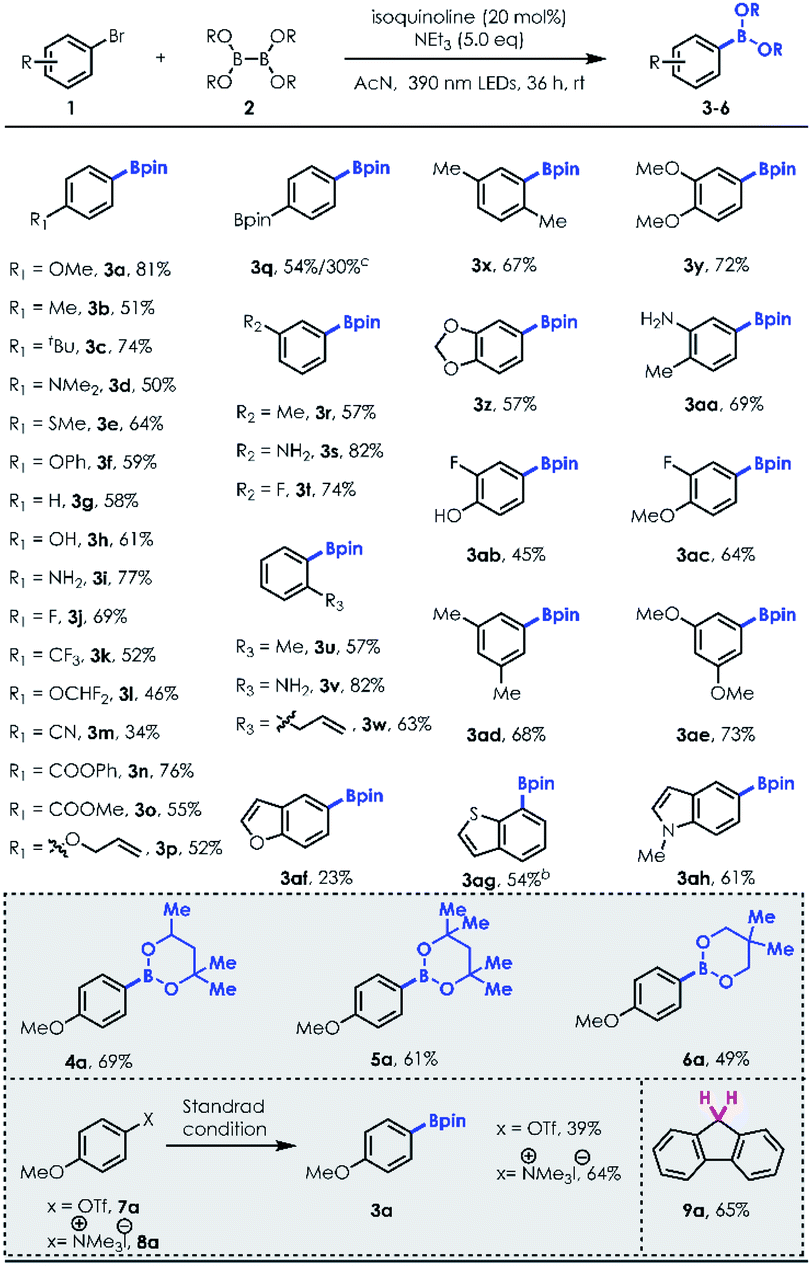
With the optimized reaction conditions in hand, we first examined the substrate scope of aryl bromides (Table 2). Various para-substituted aryl bromides, including electron donating group substituted (3a–3f), nonsubstituted (3g), unprotected –OH (3h)/–NH2 (3i), electron withdrawing group substituted (3j–3o) and even simple alkene moieties (3o), could be well tolerated in this system, delivering the corresponding arylboronates in good to excellent yields. However, bromo-styrene was not tolerated. A dual borylation product (3q) could be synthesized using dihaloarene or haloaryl boronic ester. Meta-(3r–3t) and ortho-(3u–3w) substituted arylboronates can both be obtained in good yields (e.g., 3w, 1-allyl-2-bromobenzene, 63% yield). Moreover, 2,5-(3x)/3,4-(3y–3ac)/3,5-disubstituted (3ad–3ae) aryl bromides produced borylation products in good yields (3aa and 3ac, 69% and 64%, respectively). Heteroaromatic bromides, such as benzothiophene (3ag) and indole (3ah) derivatives, can be employed to access the desired borylation products with good efficiencies other than brominated benzofuran (3af, acceptable 23% yield). This methodology also worked well with a series of diboron reagents to produce arylboronates 4a–6a with useful efficiencies (e.g., 4a, 69% yield). In contrast, B2(OH)4 was not a good borylation candidate with a poor solubility in acetonitrile. Notably, except for aryl halides, 3a could also be prepared through more challenging Ar–O (7a)/Ar–N (8a) bond cleavage under standard conditions. Surprisingly, reductive deoxygenation of fluorenone was accomplished with high efficiency (9a, 65%).
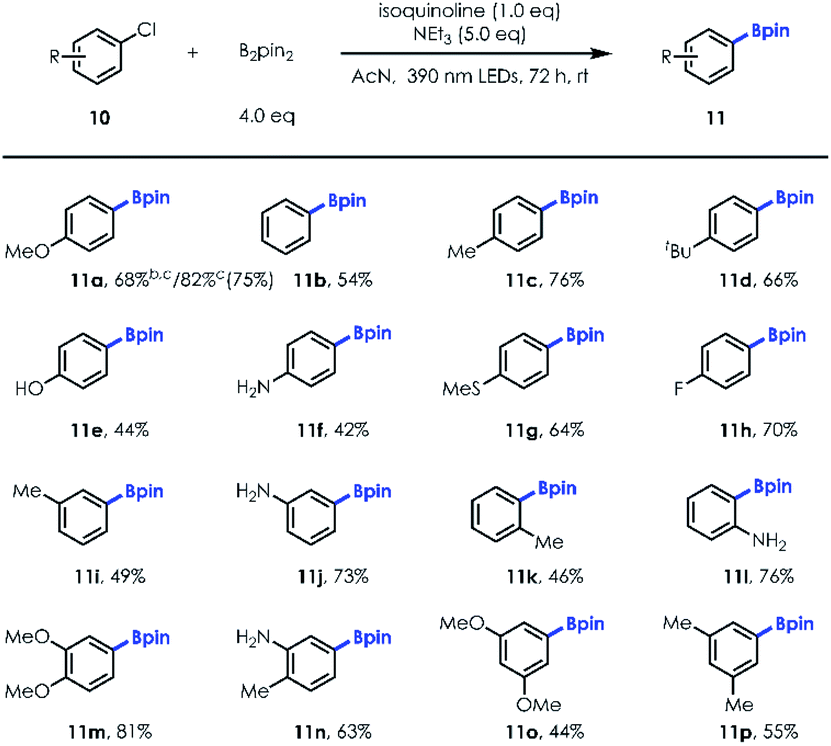
Next, we turned our attention to exploring the substrate scope of the borylation of unactivated aryl chlorides, since cleavage of the C–Cl bond in a metal-free manner remains quite challenging [e.g., for PhCl, Ered = −3.04 V vs. SCE].21 Our investigations revealed that both electron-rich and electron-deficient chlorobenzene derivatives could be employed to provide the corresponding arylboronates in good yields with a little more loading of NCH and longer irradiation time (Table 3; 68% NMR yield for 20 mol% NCH loading and 36 h irradiation vs. 82% NMR yield for 100 mol% NCH loading and 72 h irradiation). It's worth noting that the reaction was allowed for production of fluorinated boronic ester (11h). In addition, unprotected para, meta, and ortho –OH/–NH2 substituted aryl chlorides (10e–10f; 10j; 10l) were well tolerated, illustrating the immediate utility of this approach in preparing value-added boronic ester in a protecting group-free manner. Similar to aryl bromide substrates, more electron-rich and sterically hindered aryl chlorides (10m–10p) could also be transformed into desired products with good yields using this new protocol (Table 4).
| a Reactions were run on a 0.2 mmol scale; isolated yield. |
|---|
|
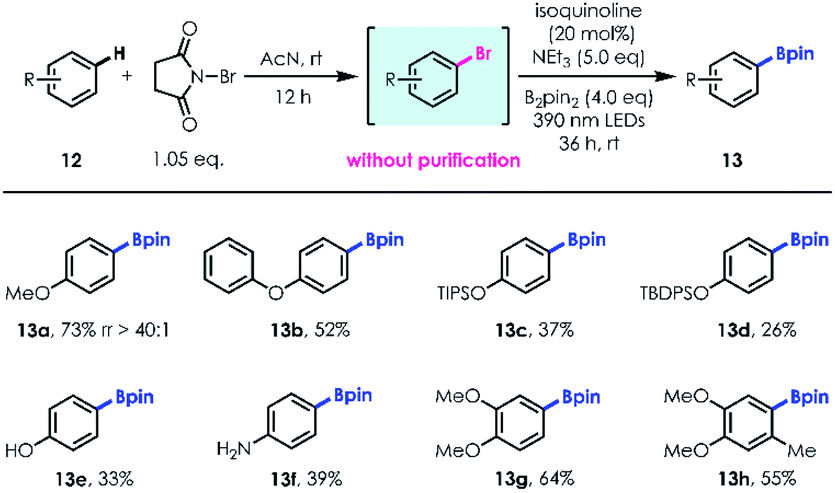
Site selective aromatic C–H functionalization provides rapid access to useful structural and functional molecular complexity.22 Based on the optimized reaction conditions, we developed a two-step one-pot method for simple aromatic C–H borylation. In the first step, almost equivalent regioselective bromination is accomplished using N-bromosuccinimide (NBS) as the bromination reagent, controlled by the electrophilicity and steric hindrance effect of the aromatic ring.23 Subsequently, a series of aryl bromides without further purification were transformed into the corresponding boronic esters with high regioselectivities and efficiencies (e.g., 13a, 73% yield; rr up to 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Aromatic silyl-ethers (13c–d), unprotected phenol (13e), aniline (13f) and polysubstituted benzenes (13g–h) were well tolerated, delivering site selective products with moderate to good yields (Table 5).
:1). Aromatic silyl-ethers (13c–d), unprotected phenol (13e), aniline (13f) and polysubstituted benzenes (13g–h) were well tolerated, delivering site selective products with moderate to good yields (Table 5).
| a Reactions were run on a 0.2 mmol scale; isolated yield. |
|---|
|
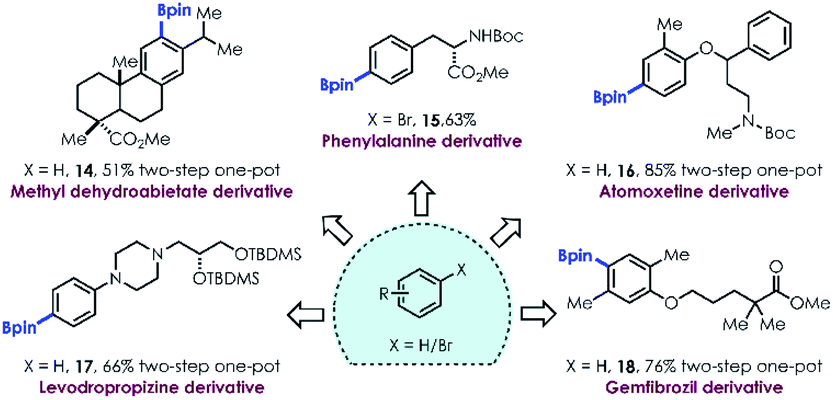
The broad functional group tolerance and two-step one-pot method for site selective aromatic C–H borylation encouraged us to test this mild strategy for late stage functionalization of complex molecules, such as natural products and active pharmaceutical ingredients (APIs). Boronates derived from methyl dehydroabietate (14), amino acid tyrosine (15), and a variety of APIs (16–18) containing polar groups, sterically hindered substituents and polycyclic frameworks were still readily produced under the optimized conditions, highlighting the utility of this methodology in the late-stage functionalization of biologically relevant compounds.
To obtain insights into the reaction mechanism, an N,N-diboronate complex24 (dimer A) was prepared through reductive coupling of isoquinoline and B2pin2 (Scheme 2a). Under photoirradiation, no reaction of 4-bromoanisole and dimer A was detected without addition of NEt3 (see the ESI†). In contrast, the potential excited donor–acceptor complex was able to slowly reduce 4-bromoanisole into anisole in the presence of NEt3; when additional B2pin2 (2.0 eq.) was applied as the trapping reagent without loading of NEt3, no dehalogenation product except boronic ester product was detected; furthermore, the reduction of 4-bromoanisole took place more efficiently in the presence of NEt3 and B2pin2, delivering the corresponding boronic ester as the major product (Scheme 2b). These observations indicated that the reduction ability of the coordination complex of acceptor dimer A and donor NEt3 has indeed been enhanced after photoexcitation. To obtain a clearer picture of the complex of acceptor dimer A and donor NEt3, density functional theory (DFT) calculations were carried out. First, the computational UV-Vis spectrum was employed to compare with the experimental UV-Vis spectrum (Scheme 2c). Without NEt3, there exist two smooth peaks in the computational UV-Vis spectrum of dimer A and the peak at 227 nm is higher than that at 301 nm, which is in agreement with the experiments (Scheme 2c(I) and d(III)). When one NEt3 molecule binds to dimer Ain silico, the curve between the two peaks becomes sharp, which is also consistent with the experimental UV-Vis spectrum (Scheme 2c(II) and d(IV)). However, when two NEt3 bind to dimer A, the computational UV-Vis spectrum reveals that the peak around 220 nm becomes lower than the peak around 300 nm, which is quite different from experiments (Scheme 2d(V)). Therefore, the computational results suggest that only one NEt3 binds to dimer A under the optimized reaction conditions. In addition, the binding energy between dimer A and one NEt3 is 11.6 kcal mol−1 by calculation. Considering that the ratio of dimer A (20 mol% isoquinoline → 10 mol% dimer A) to NEt3 (5.0 eq.) is 1:50 under the optimized reaction conditions, dimer A and complex B can equilibrate (Scheme 2d). After B is determined to be the complex combining acceptor dimer A and donor NEt3, time dependent TD-DFT calculations were conducted for the excited-state of B. The excited-state reduction potential of B* is −3.12 V, indicating that B* is a highly active reductant and can facilitate SET with aryl halides to produce aryl radical intermediates.
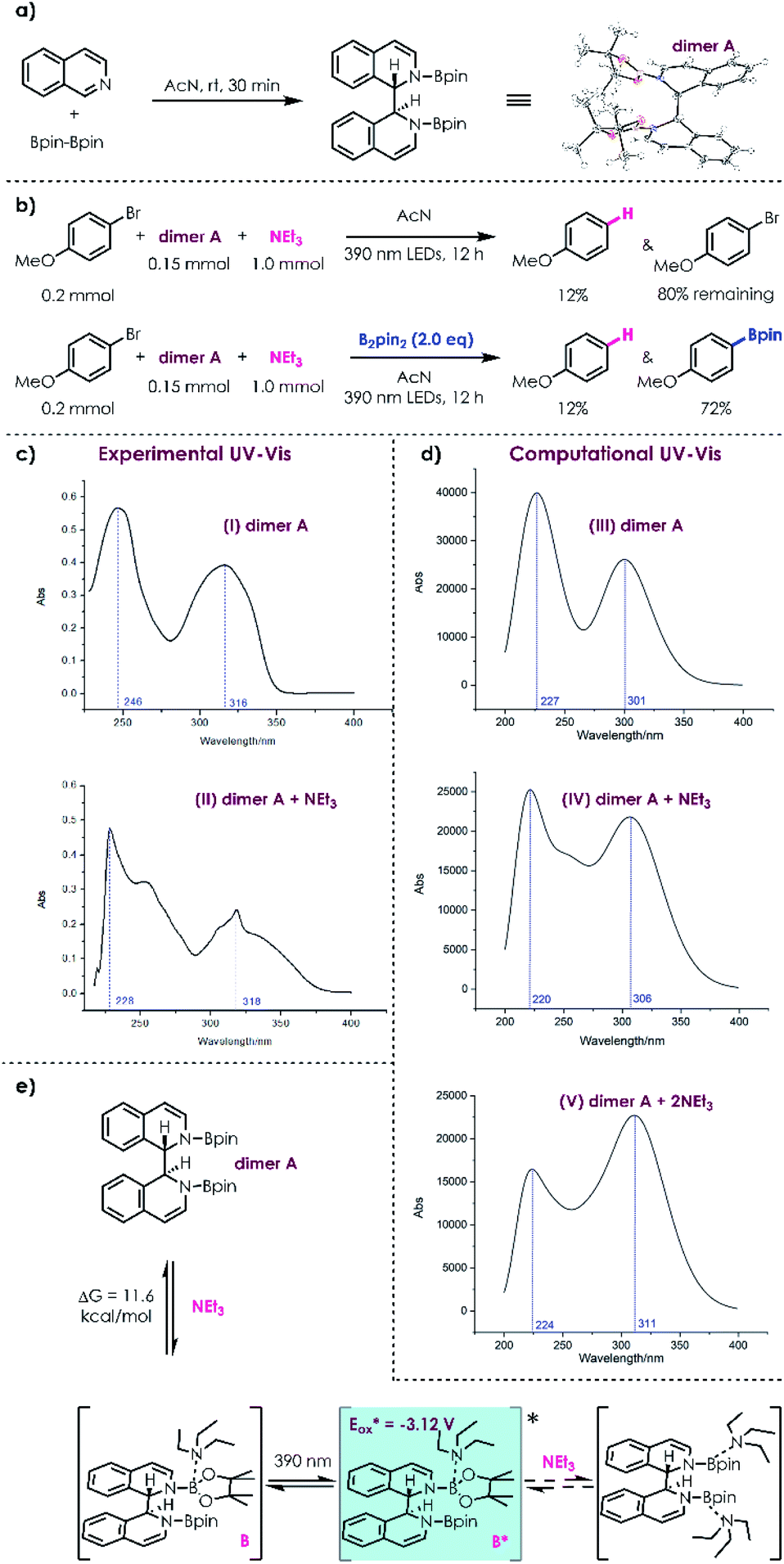 | ||
| Scheme 2 Mechanistic and computational experiments: (a) preparation of dimer A; (b) mechanistic experiments using dimer A (0.15 mmol)/4-bromoanisole (0.2 mmol)/NEt3 (1.0 mmol) with or without B2pin2 under 2 × 390 nm LEDs (40 W) irradiation for 12 h; (c) experimental UV-Vis spectra (I: dimer A/II: dimer A + NEt3); (d) computational UV-Vis spectra (III: dimer A; IV: dimer A + NEt3; V: dimer A + 2NEt3); (e) binding between dimer A and one NEt3 and then photoexcitation of D–A complex B by 390 nm light. | ||
With this mechanistic insight, a plausible mechanism for the conversion of aryl halides to their corresponding boronic esters is proposed in Scheme 3. Reductive coupling of isoquinoline and B2pin2 generates dimer A, which can coordinate with NEt3in situ to afford intermediate B. Donor–acceptor complex B is then excited by 390 nm light, populating a highly reducing excited state B*, which can undergo a single electron transfer process with an electronically matched aryl halide to generate a radical cation species C, aryl radical D, and regenerate isoquinoline/B2pin2. Radical cation C could be transformed into enamine E followed by hydrogen atom transfer and deprotonation processes. Finally, radical D is trapped by B2pin2 immediately, yielding the desired boronic ester.
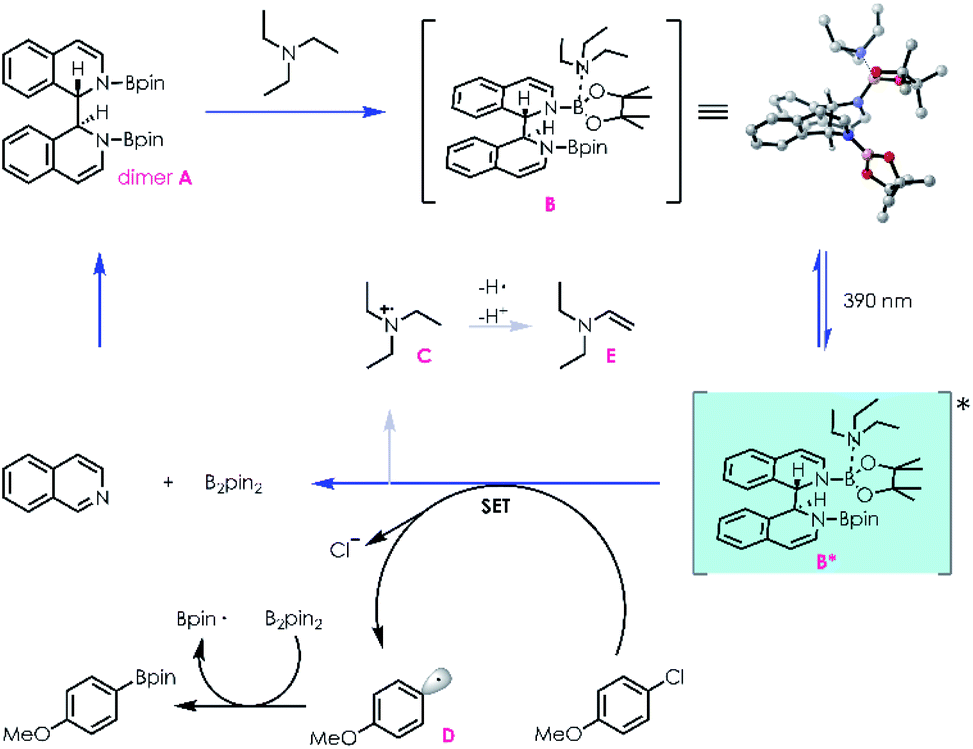 | ||
| Scheme 3 The plausible mechanism. | ||
Conclusions
In summary, a facile transition-metal-free conversion of aryl halides to the corresponding boronic esters through a radical process has been developed. A series of aryl boronic esters were synthesized with good functional group tolerance in moderate to excellent yields under mild conditions. Mechanistic experiments and computational investigations indicate the formation of an excited donor–acceptor complex, serving as the super single electron reductant. Furthermore, the key role of the donor–acceptor complex formed in situ was confirmed by control experiments and DFT calculations. The protocol's functional group tolerance and two-step one-pot method for site selective aromatic C–H bond borylation enabled the functionalization of a variety of biologically relevant compounds, representing a new application of aryl radical merged with photochemistry.Data availability
Data for all compounds in this manuscript are available in the ESI,† which includes experimental details, characterisation data, computational data and copies of 1H, 13C, 19F, 11B NMR spectra.Author contributions
M.-H. L. performed the experiments with contributions from H.-S. B and Q.-N. L., S.-Q. L. and Y.-H. D. performed DFT calculations. T.-Y. S. and L.-F. W. supervised the project. All the authors analyzed the data, discussed results and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support of this research by the National Natural Science Foundation of China (No. 21933004), the Key-Area Research and Development Program of Guangdong Province (No. 2020B010188001), the Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515011994), the Technology & Innovation Commission of Shenzhen Municipality (No. 75110-42100033), the Fundamental Research Funds for the Central Universities (No. 75110-30610023) and the Sun Yat-sen University Startup Fund (No. 75110-1841230 & 75110-18841290). The calculations were carried out at the SZBL supercomputing center.Notes and references
- (a) )D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis Medicine and Materials, Wiley-VCH, Weinheim, Germany, 2nd edn, 2011 CrossRef; (b) A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415–430 RSC; (c) Y.-M. Tian, X.-N. Guo, H. Braunschweig, U. Radius and T. B. Marder, Chem. Rev., 2021, 121, 3561–3597 CrossRef CAS PubMed.
- (a) F. W. Friese and A. Studer, Chem. Sci., 2019, 10, 8503–8518 RSC; (b) Z.-H. Qiu and C.-J. Li, Chem. Rev., 2020, 120, 10454–10515 CrossRef CAS PubMed; (c) S. K. Bose, L.-J. Mao, L. Kuehn, U. Radius, J. Nekvinda, W. L. Santos, S. A. Westcott, P. G. Steel and T. B. Marder, Chem. Rev., 2021, 121, 13238–13341 CrossRef CAS PubMed; (d) E. M. Larin, J. Loup, I. Polishchuk, R. J. Ross, A. Whyte and M. Lautens, Chem. Sci., 2020, 11, 5716–5723 RSC; (e) T. Lim, J. Y. Ryoo and M. S. Han, J. Org. Chem., 2020, 85, 10966–10972 CrossRef CAS PubMed; (f) G.-L. Gao, J.-X. Yan, K. Yang, F.-E. Chen and Q.-L. Song, Green Chem., 2017, 19, 3997–4001 RSC; (g) X.-H. Pu, J.-F. Hu, Y. Zhao and Z.-Z. Shi, ACS Catal., 2016, 6, 6692–6698 CrossRef CAS; (h) A. Verma, K. Tomar and P. K. Bharadwaj, Inorg. Chem., 2019, 58, 1003–1006 CrossRef CAS PubMed.
- (a) R. L. Letsinger and I. H. Skoog, J. Org. Chem., 1953, 18, 895–897 CrossRef CAS; (b) M. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef PubMed; (c) J. A. Knöller, G.-Y. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 3156–3160 CrossRef PubMed; (d) D.-T. Yang, J. Zheng, J.-B. Peng, X. Wang and S. Wang, J. Org. Chem., 2021, 86, 829–836 CrossRef CAS PubMed.
- For reviews, see: (a) D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC; (b) F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078–7115 RSC; (c) Y.-J. Quan and Z. W. Xie, Chem. Soc. Rev., 2019, 48, 3660–3673 RSC; (d) H.-M. Huang, P. Bellotti and F. Glorius, Chem. Soc. Rev., 2020, 49, 6186–6197 RSC; (e) H. Lu, T.-Y. Yu, P.-F. Xu and H. Wei, Chem. Rev., 2021, 121, 365–411 CrossRef CAS PubMed; (f) U. B. Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S. Lee, Chem. Rev., 2020, 120, 13382–13433 CrossRef CAS PubMed; (g) J. Trouvé and R. Gramage-Doria, Chem. Soc. Rev., 2021, 50, 3565–3584 RSC; (h) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B.-X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- For reviews, see: (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed; (b) C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed; (c) L. Troian-Gautier, M. D. Turlington, S. A. M. Wehlin, A. B. Maurer, M. D. Brady, W. B. Swords and G. J. Meyer, Chem. Rev., 2019, 119, 4628–4683 CrossRef CAS PubMed; (d) H.-M. Huang, P. Bellotti and F. Glorius, Chem. Soc. Rev., 2020, 49, 6186–6197 RSC; (e) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (f) N. L. Reed and T. P. Yoon, Chem. Soc. Rev., 2021, 50, 2954–2967 RSC; (g) J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC; (h) M. S. Galliher, B. J. Roldan and C. R. J. Stephenson, Chem. Soc. Rev., 2021, 50, 10044–10057 RSC; (i) F.-D. Lu, J. Chen, X. Jiang, J.-R. Chen, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2021, 50, 12808–12827 RSC; (j) M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC; (k) N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed; (l) S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed; (m) K. Kwon, R. T. Simons, M. Nandakumar and J. L. Roizen, Chem. Rev., 2022, 122, 2353–2428 CrossRef CAS PubMed; (n) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- S. Jin, H. T. Dang, G. C. Haug, R. He, V. D. Nguyen, V. T. Nguyen, H. D. Arman, K. S. Schanze and O. V. Larionov, J. Am. Chem. Soc., 2020, 142, 1603–1613 CrossRef CAS PubMed.
- A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS PubMed.
- J.-H. Xu, J.-L. Cao, X.-Y. Wu, H. Wang, X.-N. Yang, X.-X. Tang, R.-W. Toh, R. Zhou, E. K. L. Yeow and J. Wu, J. Am. Chem. Soc., 2021, 143, 13266–13273 CrossRef CAS PubMed.
- Y.-S. Qiao, Q.-M. Yang and E. J. Schelter, Angew. Chem., Int. Ed., 2018, 57, 10999–11003 CrossRef CAS PubMed.
- (a) Y.-M. Tian, X.-N. Guo, I. Krummenacher, Z. Wu, J. Nitsch, H. Braunschweig, U. Radius and T. B. Marder, J. Am. Chem. Soc., 2020, 142, 18231–18242 CrossRef CAS PubMed; (b) J. H. Docherty, K. Nicholson, A. P. Dominey and S. P. A. Thomas, ACS Catal., 2020, 10, 4686–4691 CrossRef CAS; (c) Z.-J. Kuang, H.-H. Chen, J. Qiu, Z.-L. Ou, Y. Lan and Q.-L. Song, Chem, 2020, 6, 2347–2363 CrossRef CAS; (d) G. Zhang, M.-Y. Li, W.-B. Ye, Z.-T. He, C.-G. Feng and G.-Q. Lin, Org. Lett., 2021, 23, 2948–2953 CrossRef CAS PubMed.
- Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS PubMed.
- (a) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607–610 CrossRef CAS PubMed; (b) L. Zhang and L. Jiao, J. Am. Chem. Soc., 2019, 141, 9124–9128 CrossRef PubMed; (c) H. Yang, L. Zhang, F.-Y. Zhou and L. Jiao, Chem. Sci., 2020, 11, 742–747 RSC.
- (a) W.-B. Liu, X.-B. Yang, Y. Gao and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 8621–8627 CrossRef CAS PubMed; (b) W. Liu, J. Li, C.-Y. Huang and C.-J. Li, Angew. Chem., Int. Ed., 2020, 59, 1786–1796 CrossRef PubMed.
- (a) A. M. Mfuh, J. D. Doyle, B. Chhetri, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 139, 2985–2988 CrossRef PubMed; (b) S.-F. Jin, H. T. Dang, G. C. Haug, R. He, V. D. Nguyen, V. T. Nguyen, H. D. Arman, K. S. Schanze and O. V. Larionov, J. Am. Chem. Soc., 2020, 142, 1603–1613 CrossRef CAS PubMed.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed.
- (a) S. Takeuchi, T. Nakagawa and Y. Yokoyama, Chem. Commun., 2020, 56, 6492–6494 RSC; (b) M. Madhu, R. Ramakrishnan, V. Vijay and M. Hariharan, Chem. Rev., 2021, 121, 8234–8284 CrossRef CAS PubMed; (c) K. Kohara, A. Trowbridge, M. A. Smith and M. J. Gaunt, J. Am. Chem. Soc., 2021, 143, 19268–19274 CrossRef CAS PubMed.
- (a) Y. Wang, W. Zhu, W. Du, X. Liu, X. Zhang, H. Dong and W. Hu, Angew. Chem., Int. Ed., 2018, 57, 3963–3967 CrossRef CAS PubMed; (b) T. J. Whittemore, A. Millet, H. J. Sayre, C. Xue, B. S. Dolinar, E. G. White, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2018, 140, 5161–5170 CrossRef CAS PubMed.
- I. A. MacKenzie, L.-F. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- S. Wang, H. Wang and B. König, Chem, 2021, 7, 1653–1665 CAS.
- (a) A. B. Grommet, L. M. Lee and R. Klajn, Acc. Chem. Res., 2020, 53, 2600–2610 CrossRef CAS PubMed; (b) M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed; (c) Si. Witzel, A. S. K. Hashmi and J. Xie, Chem. Rev., 2021, 121, 8868–8925 CrossRef CAS PubMed; (d) W.-S. Guo, Q. Wang and J.-P. Zhu, Chem. Soc. Rev., 2021, 50, 7359–7377 RSC; (e) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed; (f) L. M. Moreau, E. Lapsheva, J. I. Amaro-Estrada, M. R. Gau, P. J. Carroll, B. C. Manor, Y. Qiao, Q. Yang, W. W. Lukens, D. Sokaras, E. J. Schelter, L. Maron and C. H. Booth, Chem. Sci., 2022, 13, 1759 RSC; (g) H.-Y. Li, X.-X. Tang, J.-H. Pang, X.-Y. Wu, E. K. L. Yeow, J. Wu and S. Chiba, J. Am. Chem. Soc., 2021, 143, 481–487 CrossRef CAS PubMed; (h) L. F. T. Novaes, J.-J. Liu, Y.-F. Shen, L. X. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
- (a) C. B. Kelly and R. Padilla-Salinas, Chem. Sci., 2020, 11, 10047–10060 RSC; (b) L. Zhang and T. Ritter, J. Am. Chem. Soc., 2022, 144, 2399–2414 CrossRef CAS PubMed; (c) F. Berger and T. Ritter, Synlett, 2022, 33, 339–345 CrossRef CAS; (d) R. Cannalire, S. Pelliccia, L. Sancineto, E. Novellino, G. C. Tron and M. Giustiniano, Chem. Soc. Rev., 2021, 50, 766–897 RSC; (e) B. Prabagar, Y.-Q. Yang and Z.-Z. Shi, Chem. Soc. Rev., 2021, 50, 11249–11269 RSC.
- (a) X. Xiong, F. Tan and Y.-Y. Yeung, Org. Lett., 2017, 19, 4243–4246 CrossRef CAS PubMed; (b) D. R. Motati, D. Uredi and E. B. Watkins, Chem. Sci., 2018, 9, 1782–1788 RSC; (c) R.-J. Tang, T. Milcent and B. Crousse, J. Org. Chem., 2018, 83, 930–938 CrossRef CAS PubMed; (d) W.-J. Wang, X.-Y. Li, X.-X. Yang, L.-S. Ai, Z.-W. Gong, N. Jiao and S. Song, Nat. Commun., 2021, 12, 3873–3882 CrossRef CAS PubMed.
- D.-P. Chen, G.-Q. Xu, Q.-H. Zhou, L. W. Chung and W.-J. Tang, J. Am. Chem. Soc., 2017, 139, 9767–9770 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2144922. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00552b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |