
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperative B–H bond activation: dual site borane activation by redox active κ2-N,S-chelated complexes†
Mohammad
Zafar‡
a,
Asif
Ahmad‡
a,
Suvam
Saha
a,
Rongala
Ramalakshmi
a,
Thierry
Roisnel
 b and
Sundargopal
Ghosh
*a
b and
Sundargopal
Ghosh
*a
aDepartment of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: sghosh@iitm.ac.in
bUniv of Rennes, CNRS, Institut des Sciences Chimiques de Rennes, UMR 6226, F-35042 Rennes, France
First published on 23rd June 2022
Abstract
Cooperative dual site activation of boranes by redox-active 1,3-N,S-chelated ruthenium species, mer-[PR3{κ2-N,S-(L)}2Ru{κ1-S-(L)}], (mer-2a: R = Cy, mer-2b: R = Ph; L = NC7H4S2), generated from the aerial oxidation of borate complexes, [PR3{κ2-N,S-(L)}Ru{κ3-H,S,S′-BH2(L)2}] (trans–mer-1a: R = Cy, trans–mer-1b: R = Ph; L = NC7H4S2), has been investigated. Utilizing the rich electronic behaviour of these 1,3-N,S-chelated ruthenium species, we have established that a combination of redox-active ligands and metal–ligand cooperativity has a big influence on the multisite borane activation. For example, treatment of mer-2a–b with BH3·THF led to the isolation of fac-[PR3Ru{κ3-H,S,S′-(NH2BSBH2N)(S2C7H4)2}] (fac-3a: R = Cy and fac-3b: R = Ph) that captured boranes at both sites of the κ2-N,S-chelated ruthenacycles. The core structure of fac-3a and fac-3b consists of two five-membered ruthenacycles [RuBNCS] which are fused by one butterfly moiety [RuB2S]. Analogous fac-3c, [PPh3Ru{κ3-H,S,S′-(NH2BSBH2N)(SC5H4)2}], can also be synthesized from the reaction of BH3·THF with [PPh3{κ2-N,S-(SNC5H4)}{κ3-H,S,S′-BH2(SNH4C5)2}Ru], cis–fac-1c. In stark contrast, when mer-2b was treated with BH2Mes (Mes = 2,4,6-trimethyl phenyl) it led to the formation of trans- and cis-bis(dihydroborate) complexes [{κ3-S,H,H-(NH2BMes)Ru(S2C7H4)}2], (trans-4 and cis-4). Both the complexes have two five-membered [Ru–(H)2–B–NCS] ruthenacycles with κ2-H–H coordination modes. Density functional theory (DFT) calculations suggest that the activation of boranes across the dual Ru–N site is more facile than the Ru–S one.
Introduction
Metal–ligand cooperation (MLC) is an important model in catalytic reactions that generates new reactivity patterns in many inorganic/organic transformations.1,2 Unlike the sole participation of the metal center in classical catalysis, MLC involves a reactive ligand bound to metal that can activate small molecules across the metal–ligand bond, such as, H2, CO2, boranes, silanes, alcohols, etc.3–6 Among them, the activation of the B–H bond of boranes along with their catalytic applications in hydroboration became of interest.7 Metal complexes with polarized M–L bonds (L = O, N, S or C) proved to be very effective for B–H bond activation to yield M–H–B–L species. For example, the M–O bond cooperation (M = Ru or Rh and Ir) can capture H2BMes or HBCy2 across the metal–oxygen bond.4c,7b,8 Transition metal complexes with M–S (M = Ru and Fe) or redox non-innocent ligands in combination with MLC can activate BH3 and 9-BBN molecules.9,10 For example, ruthenium complexes containing o-(N-arylamino) thiophenol derivatives show oxidative aromatic ring cleavage in the presence of the superoxide ion (I, Chart 1).11 Reactions such as hydrogen atom transfer and proton-coupled electron transfer (PCET) reactions that determine the reactivity of H2 noticeably occurred at the redox-active sites of complexes (II–IV, Chart 1).12,13 On the other hand, for the B–H activation the engagement of MLC with multifunctional reactive sites with redox-active ligands is more useful for accessing key organic transformations.14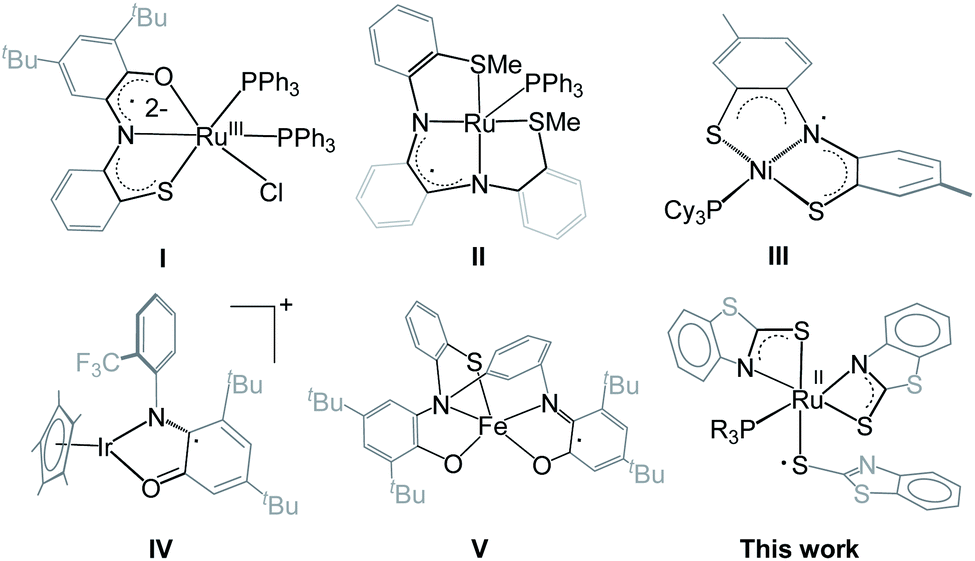 | ||
| Chart 1 Non-innocent redox-active ligand complexes of various transition metals (I–V). | ||
As part of our current interest in activation of boranes utilizing cooperative reactivity,15 we have synthesized a number of molybdenum(II) hydroborate species, in which BH3 is stabilized through Mo–H–B(H2)–E molybdacycles (E = S, Se or Te).16 Further, very recently, we have established cooperative Si–H and B–H bond activations by a κ2-N,S-chelated borate complex, trans–mer-1b that led to the formation of six-membered ruthenahetero-cycles through hemilabile ring-opening of Ru–N bonds.17 While working on κ2-N,S-chelated species trans–mer-1a–b, we have observed that aerial oxidation of these Ru(II) complexes unusually generates redox-active Ru(III) species, mer-2a–b that contain dual reactive sites. As a result, we have explored the reactivity of these redox-active Ru(III) species with different types of boranes that demonstrate the synergetic effect of metal and redox active ligands for the activation of boranes.
Results and discussion
As shown in Scheme 1, the room temperature aerial oxidation of Ru(II) borate complexes, trans–mer-1a–b in CDCl3 yielded green Ru(III) complexes, mer–2a–b. In order to get insight into these reactions, we have monitored the aerial oxidation of one of the molecules 1a in CDCl3 by 11B{1H} NMR that converted to trans–mer-2a after 48 h. The 11B{1H} NMR spectrum shows two peaks at δ = 19.4 and −43 ppm that correspond to boric acid [B(OH)3] and the borane adduct [BH3·PCy3]. We believe that boric acid (H3BO3) has been generated from the aerial oxidation of BH3 and the [BH3·PCy3] adduct has been generated from the reaction of released BH3 and PCy3, believed to be produced during the course of the reaction. The 1H and 31P{1H} NMR spectra show broad resonances that suggest the presence of paramagnetic species. The ESI-MS spectra show peaks at m/z 881.0789 (mer-2a) and 862.9324 (mer-2b) with isotopic distribution patterns.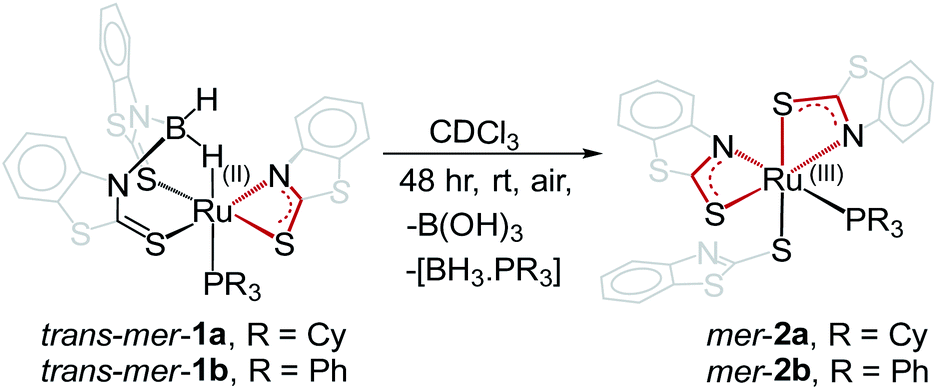 | ||
| Scheme 1 Conversion of ruthenium(II) borate complexes, trans–mer-1a–b to ruthenium(III) N,S-chelating mercapto-benzothiazole complexes, mer-2a–b. | ||
Single crystals for one of these species (mer-2a) appropriate for XRD analyses were grown from the slow diffusion of a CH2Cl2–hexane solution. The geometry of mer-2a around the Ru center is octahedral (Ma3b2c type) comprising two κ2-1,3-N,S-chelated rings along with one dangling mercaptobenzo-thiazole and phosphine ligand (Fig. 1a). In the meridional form, the PPh3 ligand is trans to the N atom of one of the κ2-N,S-mercaptobenzothiazole donor ligands. The asymmetric unit of mer-2a possesses a π···π interaction arising from the overlap of one of the κ2-N,S-heterodentate ligands and the dangling one (Fig. S29†). The torsion angles of the four-membered [SCNRu] rings (1.6(6)° and −0.4(6)°) match well with that of trans–mer-1a (0.9(4)°). The Ru–S distances of 2.491(2) and 2.342(2) Å in the four-membered chelate rings are longer as compared to the pendant one (2.278(2) Å).
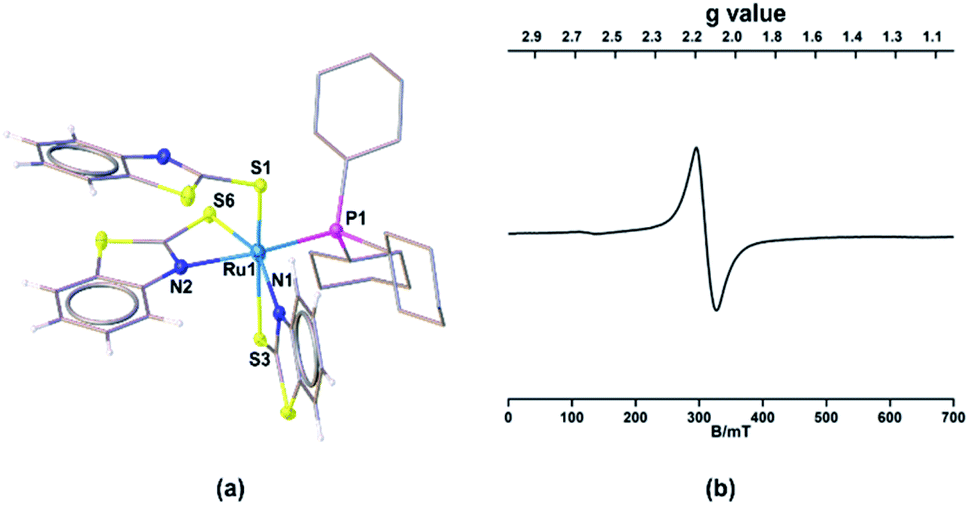 | ||
| Fig. 1 Molecular structures of mer-2a. (a) Selected bond lengths (Å) and angles (°): Ru1–P1 2.347(2), Ru1–S1 2.278(2), Ru1–S3 2.491(2), Ru1–S6 2.342(2), Ru1–N1 2.138(6), Ru1–N2 2.164(6); N1–Ru1–S3 66.88(18), N2–Ru1–S6 68.49(18); (b) EPR spectrum of mer-2a. | ||
Further, to understand the nature of these species, the gas phase geometry of mer-2a was optimized with a doublet spin state by the DFT method with the X-ray coordinates. The computed Ru–P and Ru–S bond lengths were found to be slightly longer than the experimental values (Table S1†). The molecular orbital analysis shows that the unpaired electron typically lies on the orbital with a significant ruthenium dyz-character and a smaller py-character of the sulfur atom of the dangling as well as κ2-N,S-benzothiazole ligand (SOMO, Fig. 2a). Indeed, the Mulliken spin density for the unpaired electron in mer-2a is located mostly on the Ru atom (+0.64) with a smaller contribution from the ligated sulfur (+0.20) atom, which is consistent with the spin density plot (Fig. 2b). The Wiberg bond indices (WBI) for Ru–S and Ru–N of 0.771 and 0.340 respectively, suggest two different types of interactions (Table S1†).
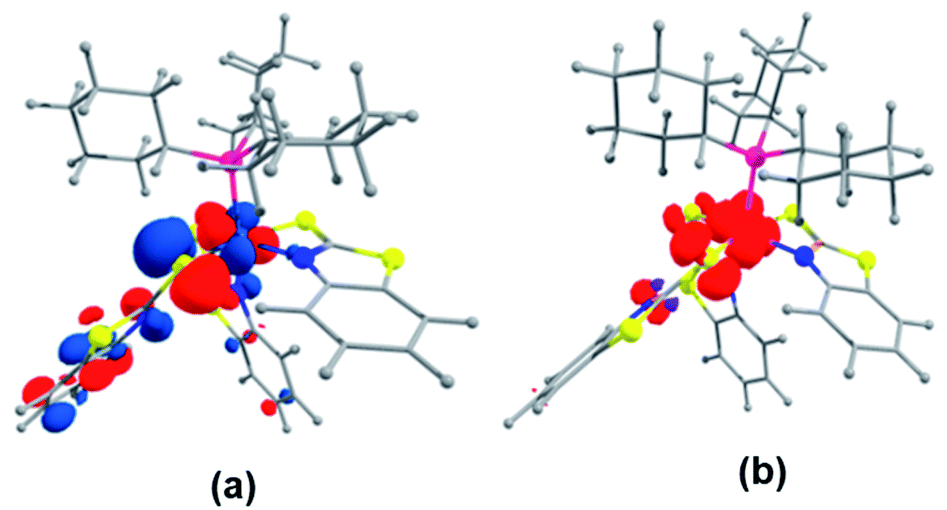 | ||
| Fig. 2 (a) Calculated SOMO of mer-2a (isovalue ±0.04 [e bohr−3]1/2). (b) A spin density plot for mer-2a (isovalue 0.004 [e bohr−3]1/2). | ||
The solid-state EPR spectra of mer-2a and mer-2b at 195 K display signals with the g values of 2.116 and 2.118, respectively that confirm the presence of spin delocalization over Ru and the ligand (Fig. 1b and S7†). Although the EPR study confirms the contribution of both [(L−)RuIII] and [(L˙)RuII] components to the ground state, the g values of 2.116 (mer-2a) and 2.118 (mer-2b) differ from that of the organic radical. Note that the typical range of g values for RuIII complexes is 2.033–2.205. Thus, this may be due to the distinctive contribution of [(LNCS−)RuIII] species.14,18 Although the EPR spectra of any open-shell metal based paramagnetic species typically exhibit rhombohedral signals at low temperature, no observable hyperfine splitting was observed for these species due to the 14N (I = 1) nucleus.19,20a
The UV-Vis spectra of mer-2a–b show a strong absorption band at 240 nm due to the π → π* transitions and a weak absorption at 430 nm (Fig. S30†). By comparing the UV-Vis spectra of trans–mer-1a and mer-2a–b, shown in Fig. S9,† we presume that the broad absorption at 720 nm for mer-2a and 740 nm for mer-2b is due to the contribution of the (LNCS˙)RuII organic radical in which the ligand has been reduced to the 2-mercaptobenzothiazolate form [(LNCS−)RuIII ↔ (LNCS˙)RuII]. Note that similar systems describing amido and aminyl radical complexes of Ru(II) have recently been reported by Ghosh and co-workers.20a The TD-DFT calculations further suggest that the low energy absorption band for mer-2a corresponds to the SOMO–LUMO(β) transition (Table S5†). The redox behaviour of both trans–mer-1a and mer-2a–b species has further been supported by cyclic voltammetry (CV) studies. The cyclic voltammograms of mer-2a and mer-2b in acetonitrile show reversible waves at E1/2 = −0.57 V, (Ipc/Ipa = 0.97) for mer-2a and −0.25 V (Ipc/Ipa = 0.98) for mer-2b (Fig. S8†), which are assigned to the [RuII(L˙)/RuII(L−)] redox couple.20a In addition, the quasi-reversible waves at 0.79 V (mer-2a) and 0.87 V (mer-2b) arised due to the RuIII/RuII redox centered couple.20
The redox behaviours of mer-2a and mer-2b species were compared with those of other ruthenium complexes containing N2P2 or N2S2 ligands derived from o-phenylenediamine. For example, Mascharak and co-workers reported the cyclic voltammogram of [cis-(dppQ)RuCl2] (dppQ = 1,2-bis-N-[2′(diphenylphosphanyl) benzoyl]benzoquinonediimine) that shows two reversible redox events at −0.28 and 0.90 V versus Fc+/Fc and an irreversible event at −1.35 V.20c The irreversible feature at −1.35 V was assigned to the ligand-centered reduction of the o-diiminosemi-quinone radical to a fully reduced o-phenylenediamine unit. The reversible wave at −0.28 V was assigned to the second ligand-centered redox event, and the reversible wave at 0.90 V was assigned to the RuIII/RuII redox couple. Similarly, Daly and co-workers reported two reversible redox events at −0.78 V and −0.28 V versus Fc+/Fc (RuII/RuI and RuIII/RuII redox couples) and irreversible features at −2.46 V as the ligand centered redox event.12 Therefore, based on the above results, the additional irreversible half-wave potentials, appearing at 0.46 V (mer-2a) and 0.54 V (mer-2b), have been assigned to the oxidation of the second N,S-donor mercapto-benzothiazolyl ligand.20e Nonetheless, to gain further insight into the nature of the half-wave potentials of these complexes, we performed the molecular orbital analysis of the oxidised form of mer-2a and mer-2b that shows mixing of orbitals between Ru–metal and the heterocyclic ligand (Fig. S39†). Thus, based on the DFT calculations, the assignments of the redox couples as ligand or metal-centered oxidation are unclear. We have also recorded the current vs. square root of scan rate for one of the molecules mer-2a and the corresponding plot (current vs. square root of scan rate) is provided in the revised ESI (Fig. S31–S33†). Note that the redox couple observed at −0.57 V shows reversible wave linearity with increasing scan rate (Fig. S32†). However, the peak current at 0.79 V is not proportional to the square root of scan rate and thus, it may be considered as a quasi-reversible wave. The peak current at 0.46 V shows an irreversible half wave.
The presence of dual reactive sites and the redox active hemilabile κ2-1,3-N,S-bidentate chelate ligands in mer-2a–b encouraged us to study their reactivity with various boranes. As a result, we treated these species with an excess of BH3·THF that resulted in the formation of yellow 3a and 3b with 38% and 42% yields, respectively (Scheme 2). Both the complexes were characterized by 1H and 13C{1H} NMR, IR spectroscopy, and single crystal X-ray diffraction studies. The 11B{1H} NMR spectra of both the complexes feature a single resonance at δ −11.5 and δ −10.1 ppm, respectively for 3a and 3b. The 1H NMR spectra show the presence of the mercaptobenzothiazole ligand in the region of δ 7.87–7.26 ppm. In addition, two broad resonances, appearing at δ 2.97 and −14.43 ppm for 3a and 3.01 and −13.24 ppm for 3b, have been assigned to B–H and Ru–H–B hydrides. The broad 1H chemical shifts were resolved into a doublet and an apparent triplet upon 11B decoupling (2JHP = 14.1 Hz, 2JHH = 14.1 Hz (3a); and 2JHP = 12.0 Hz, 2JHH = 12.0 Hz (3b). These NMR signatures indirectly ensure that the hydride is cis oriented to the phosphine ligand. The 31P{1H} NMR revealed a singlet at δ 71.0 for 3a and 60.9 ppm for 3b. The mass spectra showed a molecular ion peak at m/z 772.1165 for 3a and 776.9636 for 3b. Based on all the spectroscopic data along with mass spectrometric data, it was clear that both the species are diamagnetic. However, a clear explanation eluded us until the single-crystal X-ray diffraction analysis of one of these species 3a was performed.
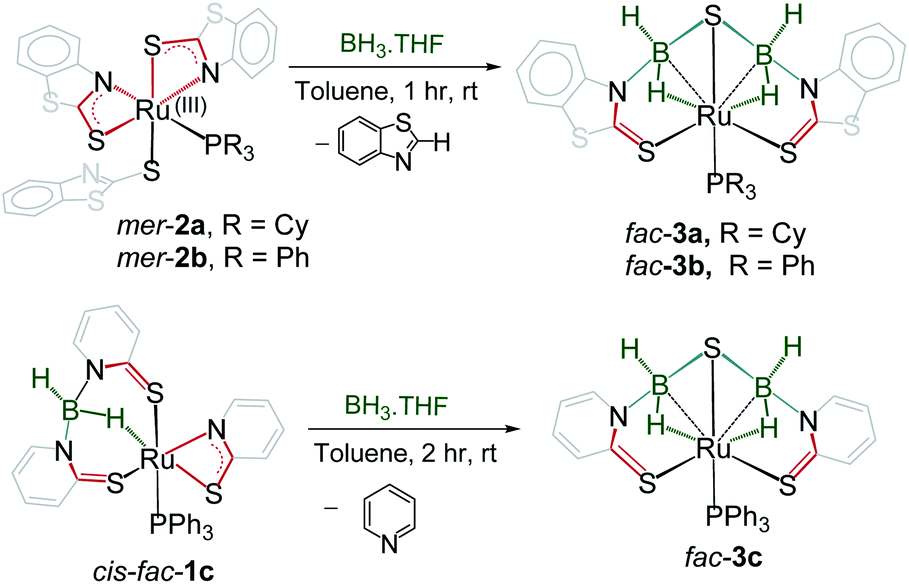 | ||
| Scheme 2 Reactivity of mer-2a–b and cis–fac-1c with BH3·THF. | ||
The solid-state X-ray structure of 3a, shown in Fig. 3a, displays a symmetrical structure with Cs symmetry, wherein the mirror plane of symmetry passed through the S5–Ru1–P1. The ruthenium center adopts an octahedral geometry in the facial form in which the phosphine ligand is present in trans to the sulfur atom. Thus, we believe that complex 3a, now fac-3a, is generated from the insertion of two BH2 moieties across the initial Ru–N bonds of κ2-N,S-chelated heterocycles. The S atom in the butterfly core is presumably generated from the C–S bond cleavage of the pendant mercaptobenzothiazole ligand. To validate this concept, we have monitored the reaction of mer-2a with BH3·THF by 13C{1H} and 1H NMR spectroscopy in toluene-d8. The 13C{1H} NMR spectrum (after 2 h) shows chemical shifts in the range of δ = 149–134 ppm that correspond to the benzothiazole ligand. On the other hand, the 1H chemical shift at δ = 9.4 ppm corresponds to the C–H proton of the benzothiazole ligand.21 Thus, we firmly believe that the source of sulphide for the formation of 3a or 3b is the mercapto-benzothiazole ligand. Note that recently Wang and co-workers reported a similar type of reaction that yielded the Mo(II)hydride complex, [Cp*MoH(1,2-Ph2PC6H4SBH2)] in which a BH2 moiety is coordinated with the Mo–S bond.22 The BH2 moieties in fac-3a and fac-3b are further stabilized by one sulfur atom (S5, fac-3a), generating two unique five-membered RuBNCS ruthenacycles, which are fused by one {RuB2S} butterfly unit. The average Ru–B distance (2.284 Å) in fac-3a is comparable with the bond lengths of 2.266(8) Å and 2.216(6) Å observed in [RuH(PCy3)2{(μ-H)2BMeCH2SMe}]23 and [Cp*Ru(μ-H)2B(NC7H4S2)],24 respectively. Similarly, the average B–S bond distance of 1.917 Å is consistent with borane–thiolate species that typically fall in the range of 1.949–1.911 Å.25 The DFT analysis further established that the borane activation across the Ru–N bond is more favourable than the Ru–S bond with a lower energy of 50.2 kcal mol−1. This has also been supported by NBO analysis, where the Wiberg bond index of Ru–S (0.771) is significantly stronger than that of Ru–N (0.340) (Table S1†).
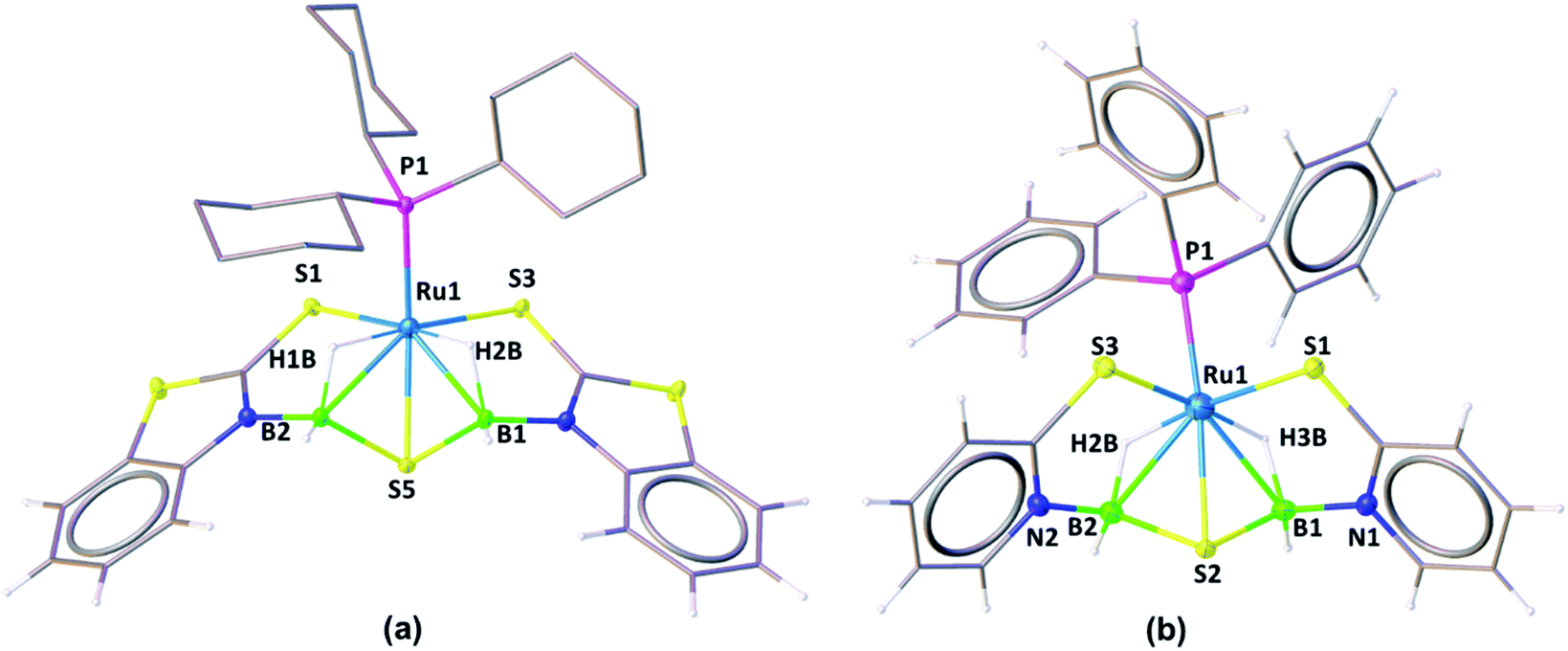 | ||
| Fig. 3 Molecular structures of fac-3a (a) and fac-3c (b). Selected bond lengths (Å) and angles (°): fac-3a (a) Ru1–B2 2.276(2), Ru1–B1 2.293(3), Ru1–P1 2.307(6), Ru1–S1 2.384(6), Ru1–S3 2.383(6), Ru1–S5 2.4184(6), Ru1–H1B 1.69(3), Ru1–H2B 1.69(3), B2–Ru1–B1 75.66(9), B2–Ru1–S5 48.08(7); fac-3c (b) Ru1–B2 2.285(4), Ru1–B1 2.281(4), Ru1–P1 2.2830(9), Ru1–S1 2.3611(8), Ru1–S3 2.3535(9), Ru1–S2 2.4090(9), Ru1–H2B 1.63(3), Ru1–H3B 1.66(3), B2–Ru1–B1 77.74(14), B2–Ru1–S5 48.17(10). | ||
The meridional and facial orientations of all the complexes have been assigned largely based on the coordination of hydride and phosphine ligands to metal. The coordination modes were established in comparison with the JPH coupling constants of similar molecules.14,23,26a For example, the 1H NMR spectrum of trans–mer-1b shows a broad hydride peak at δ = −3.71 ppm that converted to a doublet of doublet upon 11B decoupling (2JHP = 33.1 Hz, 2JHH = 16.4 Hz). This indirectly suggests that the hydride is trans to the phosphine ligand, and all mercaptobenzothiazole sulfurs are arranged in the meridional fashion (Fig. S10†). However, in facial-orientation, the broad hydride peak at δ = −13.24 ppm transformed to an apparent triplet with coupling constants 2JHP = 12.0 Hz and 2JHH = 12.0 Hz. This is due to the presence of the adjacent cis-oriented phosphine group in which the mercaptobenzothiazolyl sulfur coordinated facially.26b,c
Note that analogous fac-3c, [PPh3Ru{κ3-H,S,S′-(NH2BSBH2 N)(SC5H4)2}] was synthesized as a yellow crystalline solid from the room temperature reaction of [{κ3-H,S,S′-H2B(SNC5H4)2}Ru{κ2-N,S-(SNC5H4)}PPh3],27cis–fac-1c and BH3·THF (Scheme 2). Complex fac-3c was characterized by comparing its spectroscopic data with the mass spectrometric data of fac-3a–b that show a molecular ion peak at 642.0321, and a solid-state X-ray diffraction analysis. The 11B{1H} NMR spectrum shows a sharp peak in the upfield region δ = −3.8 ppm. The 1H NMR spectrum of fac-3c displayed an upfield resonance at δ = −12.27 ppm due to the Ru–H–B proton. This broad 1H chemical shift was resolved into an apparent triplet upon 11B decoupling with coupling constants 2JHP = 13.1 Hz and 2JHH = 13.1 Hz. The solid-state X-ray structure of fac-3c, shown in Fig. 3b, shows a butterfly core containing the S atom similar to that of fac-3a. The S atom in the butterfly core has presumably been generated from the C–S bond cleavage of the pendant mercaptopyridyl ligand. Further, to get some insight into the reaction intermediates, we have monitored the reaction of cis–fac-1c with BH3·THF using 1H and 13C NMR in toluene-d8. The 13C{1H} NMR spectrum (after 2 h) shows chemical shifts in the range of δ = 134–148.6 ppm that correspond to free pyridine. On the other hand, the 1H chemical shift at δ = 8.68 ppm corresponds to the C–H proton of the pyridine ligand.28
To have a further understanding of the MLC binding effect in 1,3-N,S-chelated ruthenium complexes, the electrochemistry of fac-3a–b was studied (Fig. S8†). The single quasi-reversible redox observed at E1/2 = 0.59 V (fac-3a) and 0.62 V (fac-3b) assigned to the RuIII/RuII redox couple indicates that the MLC reduces the electrochemical activity of fac-3a–b when compared with trans–mer-1a–b and mer-2a–b species. As there exists no equilibrium between fac-3a–b and mer-2a–b under an applied potential (loss of BH3), we believe that the cooperativity with two BH3 units in fac-3a–b is stronger as compared to trans–mer-1a–b.
The NBO and QTAIM analyses show the coordination of BH2 with S and Ru atoms. As shown in Fig. 4a–d, it is evident that one of the B–H bonds donates electron density to the Ru center and the S donates a lone pair of electrons to Ru. This was further supported by natural charge analysis that indicates positive natural charges both at S and B atoms (qB = 0.029, qS = 0.026). Thus, it is apparent that S and B act as donors and the Ru center (qRu = −0.429) can be considered as an acceptor. The Wiberg bond indices (WBI) of 0.984 and 0.979 with regard to B1–S5 and B2–S5 bonds support strong bonding interactions. The HOMO−2 of fac-3a, shown in Fig. 4c, suggests that the electron density is mostly localized on the B–S–B moiety and Ru center. Further, topology analysis of fac-3a reveals the presence of B–S, B–H, Ru–H and Ru–S bond critical points (BCPs) and ring critical points (RCPs). The topological features at BCPs of Ru–H, Ru–S and Ru–P bonds are characterized as dative interactions (Fig. 4d and Table S4†).
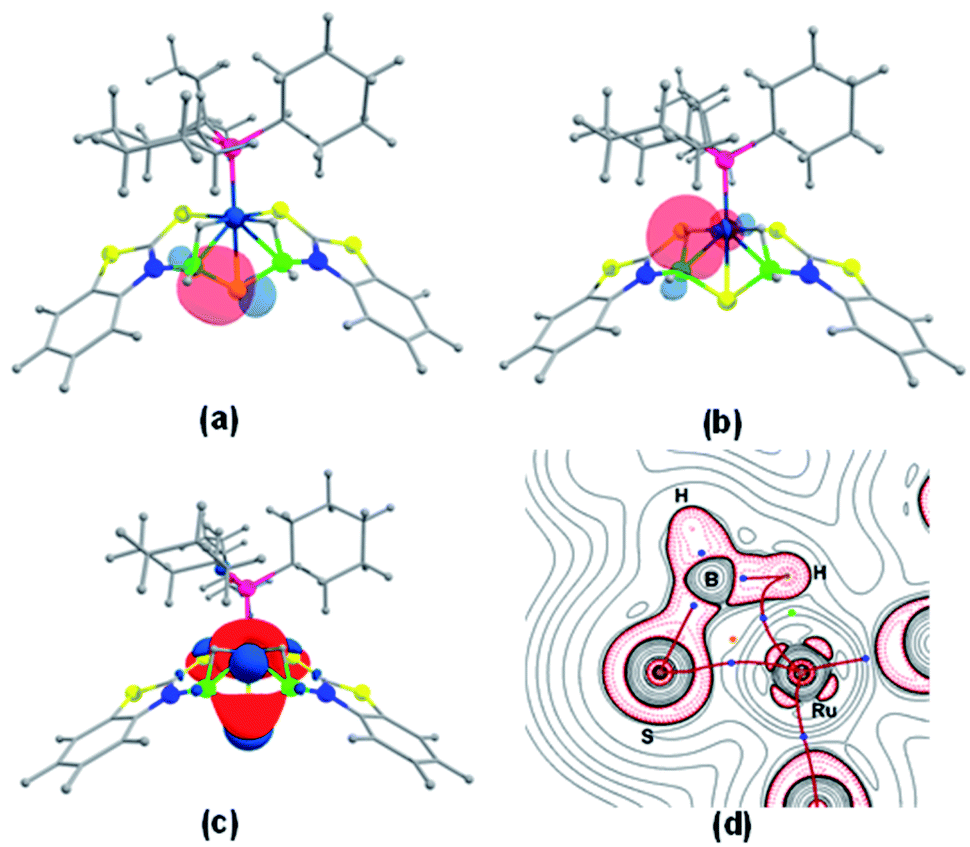 | ||
| Fig. 4 Natural bond orbital interaction between the B–S bond (a) and the B–H–Ru bond (b) in fac-3a (isovalue ±0.04 [e bohr−3]1/2); (c) HOMO−2 of fac-3a; (d) contour-line map of the Laplacian of the electron density in the Ru–S–H plane of fac-3a. BCPs and RCPs correspond to blue and orange dots and red lines indicate bond paths. | ||
With the conditions for the formation of fac-3a–c in hand, reactivity of mer-2a–b with bulky borane such as mesityl borane (H2BMes) became of interest. Although the reaction of mer-2a with H2BMes resulted in decomposition of the starting material over time, mild thermolysis of mer-2b with a stoichiometric amount of mesityl borane in toluene resulted in the formation of complex 4. Thin-layer chromatographic workup allowed us to isolate pure 4 as a yellow crystalline solid in 45% yield, which was characterized by multinuclear NMR and IR spectroscopic methods (Scheme 3). The 11B{1H} NMR spectrum of 4 shows two broad resonances at δ 39.4 and 31.2 ppm. In addition to 1H chemical shifts for the mercaptobenzo-thiazolyl ligand, the 1H NMR spectrum of 4 shows three up-field chemical shifts at δ −11.42, −10.41 and −10.01 ppm. The mass spectrum shows a molecular ion peak at m/z 698.0799. The 31P{1H} NMR shows the presence of no 31P chemical shift. All the spectroscopic data along with mass spectrometric data suggest 4 as a mixture of two Ru−borate species. However, the identity was unclear until an X-ray crystallographic analysis was carried out for one of them.
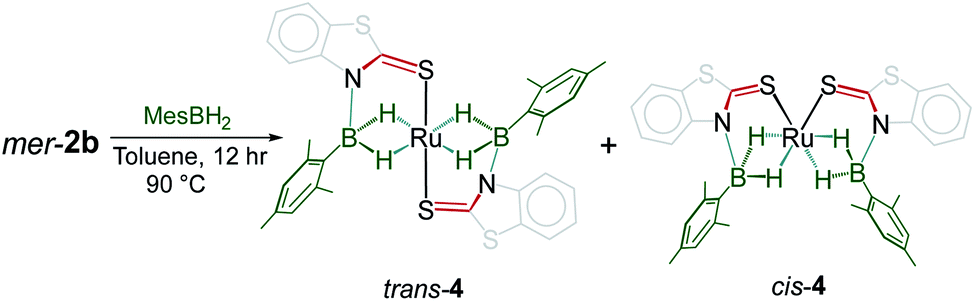 | ||
| Scheme 3 Reactivity of mer-2b with mesityl borane. | ||
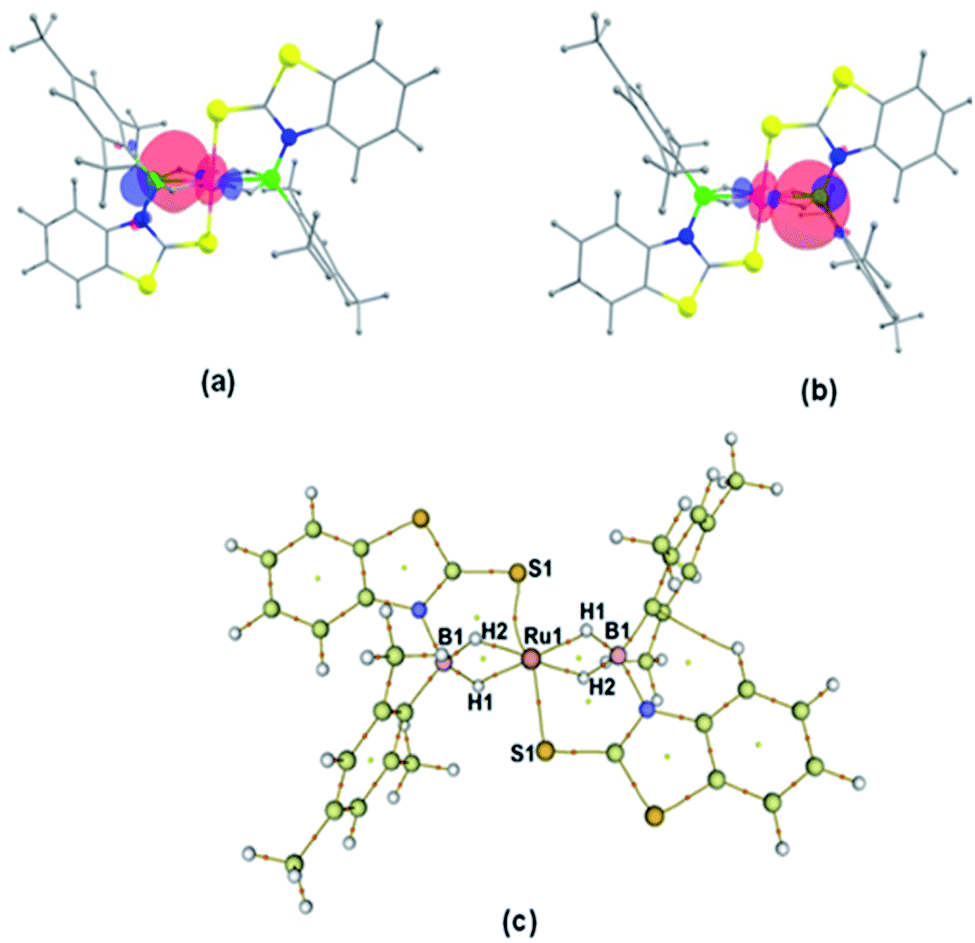
A slow evaporation of a CH2Cl2/hexane solution of 4 at −5 °C yielded two different types of crystals. The X-ray diffraction analysis was performed on a yellow crystal which was manually picked up from the Schlenk tube. The solid-state X-ray structure of this yellow crystal, shown in Fig. 5, shows a distorted-octahedral geometry in which four hydrogen atoms are placed in a square plane and two S atoms occupy the axial positions. This clearly shows that two units of H2BMes have been inserted into two Ru–N bonds of κ2-N,S-chelated ligands of mer-2b resulting in the formation of two five-membered Ru–B–NCS ruthenacycles. Interestingly, the geometry of this crystal has an inversion center on Ru. Thus, this molecule can be defined as trans-[Ru{κ3-S,H,H-(NBH2Mes)(S2C7H4)}2] (trans-4).
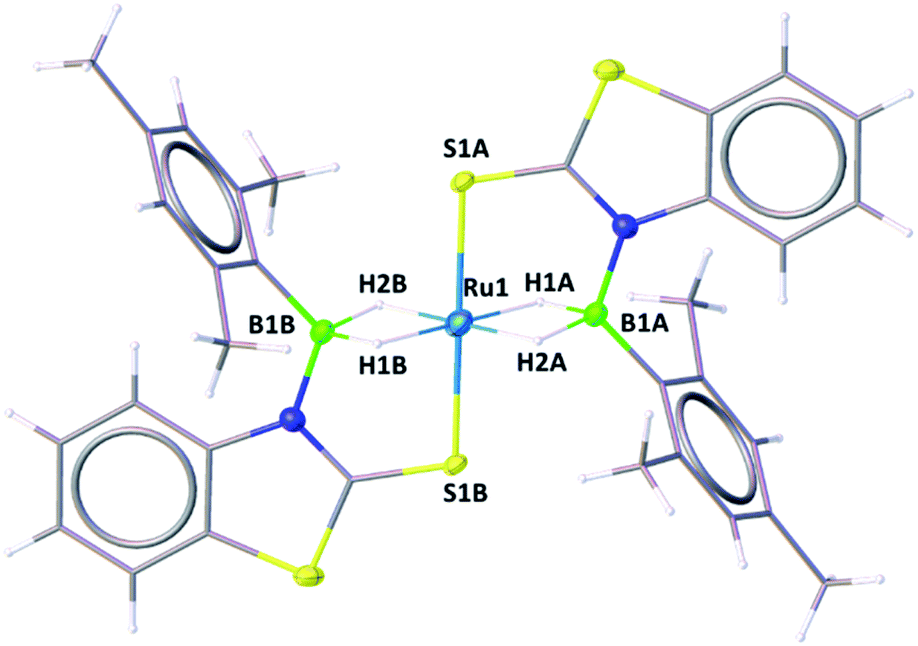 | ||
| Fig. 5 Molecular structures of trans-4. Selected bond lengths (Å) and angles (°): Ru1–B1A 2.172(4), Ru1–S1A 2.3221(10), B1–N1 1.542(5), B1–C1 1.580(5), B1–Ru1–B1 180.0(2), B1–Ru1–S1 95.86(11), S1–Ru1–S1 180.0, C1–B1–Ru1 127.2(3). | ||
Although we failed to get single crystals for the other species, all the spectroscopic data and mass spectrometric data clearly suggest the presence of both trans-4 and cis-4 species. For the trans-4 isomer (C2h symmetry) the 11B chemical shift at δ = 39.4 ppm has been assigned to two equivalents of boron atoms and the 1H chemical shift at δ = −11.42 ppm has been assigned to four Ru–H–B protons which are in an equivalent environment. However, for the cis-4 (C2V symmetry), the 11B chemical shift at δ = 31.2 ppm is due to the presence of two equivalents of boron. The two broad 1H chemical shifts at δ = −10.41 and δ = −10.01 ppm for the Ru–H–B protons emanate from the presence of two different groups opposite to the hydrogen atom. Two Ru–H–B protons are trans to mercaptobenzothiazolyl sulphur and other two Ru–H–B protons are trans to each other (Fig. S26†).
As listed in Table 1, the Ru–B distance of 2.172(4) Å for trans-4 is consistent with that of the bis(dihydroborate) complex, Ru[(μ-H)2 BC8H14]2(PCy3)29 (2.160(2) Å) and other reported dihydroborate species.29–33 The Ru–B distances in trans-4 fall in the range of 2.103(2)–2.266(8) Å,15 which are significantly longer as compared to those of σ-borane complexes, for example, [Ru(PCy3)2(H)2(BH2Mes)]29b (1.938(4) Å) and [Cp*Ru(PiPr3)(BH2Mes)]B(C6F5)4 (ref. 34) (1.921(2) Å). The B1–Ru1–B1 angle of 180.0(2)° shows a perfect symmetrical coordination of borane to the metal center, unlike tetrahydroborate species, [PBP](μ-H)2Ru(η2-BH4)]32 (177.4(4)°) and Ru[(μ-H)2BC8H14]2(PCy3)30 (147.68(8)°). The WBI of 0.369 for the Ru–B bonds of trans-4 also supports symmetrical interaction (Table S1†). The analysis further suggests that the trans-isomer is thermodynamically more stable in which the relative total energy for the trans-4 isomer is 0.46 kcal mol−1 lower than that of the cis-4 isomer. The donor–acceptor interaction between the B–H bond and Ru is confirmed by second-order perturbation analysis with a stabilization energy of 11.34 kcal mol−1 (Fig. 6a and b). Although the QTAIM analysis shows all the BCPs in trans-4, it can't identify the interaction between Ru and B (Fig. 6c).35
| Bis-(dihydroborate) | Spectroscopic parameters (ppm) | Structural parameters (Å) | Ref. | ||
|---|---|---|---|---|---|
| 1H(Ru–H) | 11B{1H} | d[M–B] | d[M–H] | ||
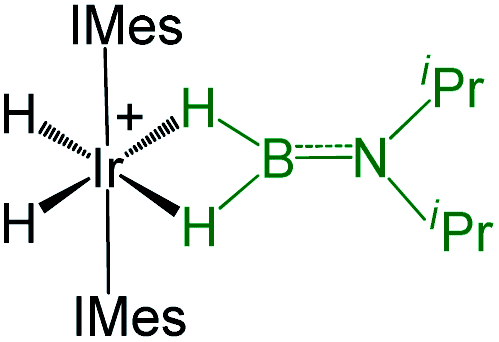
|
−15.50, −5.83 | 37.9 | 2.088(5) | 1.85(12) | 29a |
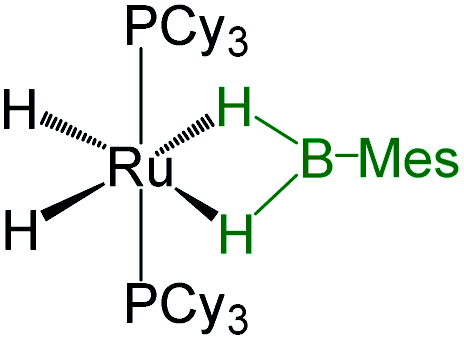
|
−11.26, −6.10 | 58.0 | 1.938(4) | 1.61(3), 1.59(3), 1.73(3), 1.77(3) | 29b |
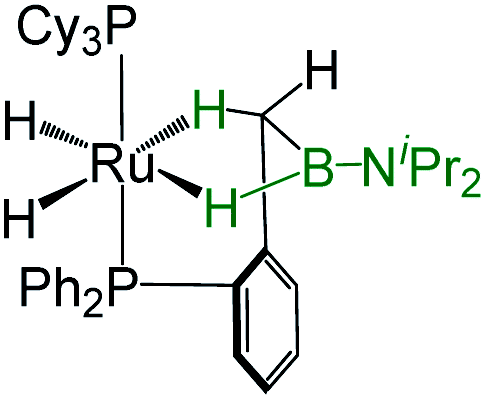
|
−13.22, −8.58, −6.71, −1.00 | 35.5 | 2.173(3) | 1.59(3), 1.69(3), 1.49(3), 1.97(3) | 33 |

|
−11.58, −6.29 | 46.0 | — | — | 33 |
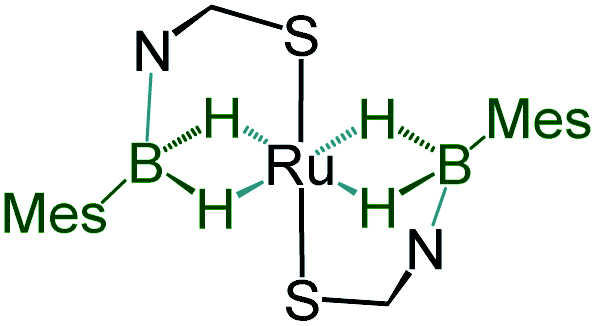
|
−11.42 | 39.4 | 2.172(4) | 1.60(4), 1.62(4) | This work |
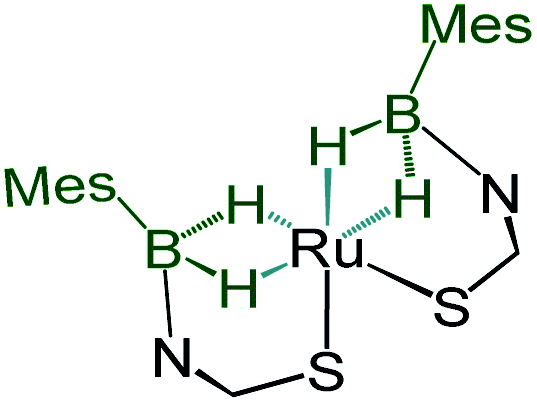
|
−10.41, −10.01 | 31.2 | — | — | This work |
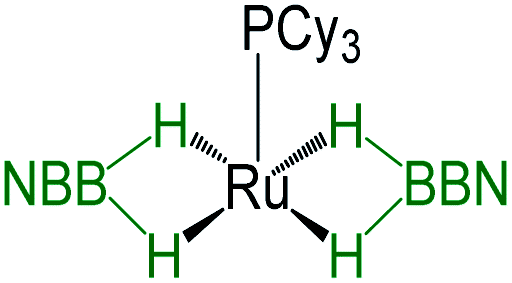
|
−12.43 | 58.8 | 2.160(2), 2.085(2) | 1.63(2), 1.66(2), 1.60(2), 1.63(2) | 30 |
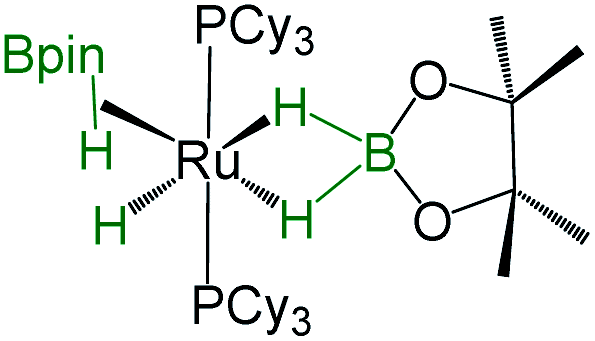
|
−11.4, −8.03, −7.13 | 37.3 | 2.157(5), 2.188(5) | 1.48(3), 1.58(3), 1.49(3), 1.55(3) | 31 |
|
−14.57, −5.78 | 11.9, 40.8 | 2.048(9), 2.333(9) | 1.615, 1.629, 1.843, 1.860 | 32 |
 | ||
| Fig. 6 (a and b) Donor–acceptor interaction between B–H and the Ru atom obtained by NBO analysis of trans-4. (c) QTAIM analysis of trans-4. Bond paths are indicated by yellow lines and BCPs and RCPs are represented by orange and yellow dots respectively. | ||
Although mesityl borane, H2BMes, has been accessible since 1994,36 structurally characterized species, other than borylene complexes, are very limited.37 For example, the first ruthenium terminal borylene complex, [Ru(BMes)(PCy3)2], was synthesized by double B–H activation of H2BMes.37a Recently, Hayes and co-workers have isolated and structurally characterized the Rh–borylene complex37b from the rhodium pincer complex, [(iPrNNN)Rh(CO)] (iPrNNN = 2,5-[iPr2P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(4-iPrC6H4)]2N(C4H2)−]) and H2B–Mes by reversible dehydrogenation of H2BMes. Interestingly, not many examples are known where the addition of H2BMes occurred in a κ2-coordination fashion into the polar M–L bonds.7b,17,38 Some of the dihydroborate species, for example, [Cp*Ru(κ3-P,H,H-(iPr)P(C9H6O–H2BMes)]38 and [M{κ3-N,H,H-Xyl(N)P(OH2 BMes)(OEt)2}(η4-COD)] (M = Rh and Ir),7b have been isolated from the insertion of H2BMes into the corresponding M–O bonds. The first bis(σ-borane) species, [Ru(PCy3)2(H)2 (η2:η2-BH2Mes)], was reported by Sabo-Etienne and Alcaraz, isolated from the reaction of H2BMes and [RuH2 (PCy3)2(η2-H2)2].29b
N(4-iPrC6H4)]2N(C4H2)−]) and H2B–Mes by reversible dehydrogenation of H2BMes. Interestingly, not many examples are known where the addition of H2BMes occurred in a κ2-coordination fashion into the polar M–L bonds.7b,17,38 Some of the dihydroborate species, for example, [Cp*Ru(κ3-P,H,H-(iPr)P(C9H6O–H2BMes)]38 and [M{κ3-N,H,H-Xyl(N)P(OH2 BMes)(OEt)2}(η4-COD)] (M = Rh and Ir),7b have been isolated from the insertion of H2BMes into the corresponding M–O bonds. The first bis(σ-borane) species, [Ru(PCy3)2(H)2 (η2:η2-BH2Mes)], was reported by Sabo-Etienne and Alcaraz, isolated from the reaction of H2BMes and [RuH2 (PCy3)2(η2-H2)2].29b
Conclusions
In summary, we have developed some redox-active complexes supported by hemilabile κ2-N,S-chelated ruthena-cycles that undergo unusual dual site B–H bond activation with free and bulky boranes. When the reaction was carried out with free borane, one of the B–H bonds of the BH3 unit cleaved and the rest of the BH2 moiety was captured by the Ru–N bond that led to two five-membered (RuBNCS) ruthenacycles. In contrast, bulky borane mesitylborane generated trans and cis species, in which two H2BMes units are coordinated to the Ru center. A combined experimental and theoretical study shows that a combination of redox-active ligands and metal–ligand cooperativity has a major role in multisite borane activation for smaller and bulky boranes.Data availability
Crystallographic data have been deposited with the Cambridge Crystallographic Data Center as supplementary publication no CCDC-1875697 (mer-2a), CCDC-1984218 (fac-3a), CCDC-2126931 (fac-3c), CCDC-2040802 (trans-4). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttps://www.ccdc.cam.ac.uk/data_request/cif.Author contributions
M. Zafar, A. Ahmad and S. Saha have executed the experimental synthesis, characterization, and analysis of the data. R. Ramalakshmi has conducted the theoretical calculations. All authors have contributed to the preparation of the manuscript. S. Ghosh has supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by CEFIPRA, grant number 5905-1. DST-FIST, India. M. Z. thanks IIT Madras for a research fellowship. A. A. thanks the Council of Scientific & Industrial Research (CSIR) for a research fellowship. S. S. thanks INSPIRE for a research fellowship. R. R. thanks the University Grant Commission (UGC), India, for the research fellowship.Notes and references
- (a) R. Peters, Cooperative Catalysis, Wiley-VCH, Weinheim, 2015 CrossRef; (b) L. Omann, C. D. F. Konigs, H. F. T. Klare and M. Oestreich, Acc. Chem. Res., 2017, 50, 1258–1269 CrossRef CAS PubMed; (c) K.-S. Feichtner and V. H. Gessner, Chem. Commun., 2018, 54, 6540–6553 RSC.
- (a) T. Higashi, S. Kusumoto and K. Nozaki, Chem. Rev., 2019, 119, 10393–10402 CrossRef CAS PubMed; (b) H. Li, T. P. Goncalves, D. Lupp and K.-W. Huang, ACS Catal., 2019, 9, 1619–1629 CrossRef CAS; (c) E. R. M. Habraken, A. R. Jupp, M. B. Brands, M. Nieger, A. W. Ehlers and J. C. Slootweg, Eur. J. Inorg. Chem., 2019, 2019, 2436–2442 CrossRef CAS PubMed; (d) M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- (a) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (b) R. H. Morris, Acc. Chem. Res., 2015, 48, 1494–1502 CrossRef CAS PubMed.
- (a) C. C. Comanescu and V. M. Iluc, Chem. Commun., 2016, 52, 9048–9051 RSC; (b) J. Y. Corey, Chem. Rev., 2016, 116, 11291–11435 CrossRef CAS PubMed; (c) L. T. Scharf, J. Weismann, K.-S. Feichtner, F. Lindl and V. H. Gessner, Chem.–Eur. J., 2018, 24, 3439–3443 CrossRef CAS PubMed.
- (a) R. Stichauer, A. Helmers, J. Bremer, M. Rohdenburg, A. Wark, E. Lork and M. Vogt, Organometallics, 2017, 36, 839–848 CrossRef CAS; (b) C. Erken, A. Kaithal, S. Sen, T. Weyhermüller, M. Hölscher, C. Werle and W. Leitner, Nat. Commun., 2018, 9, 4521 CrossRef PubMed; (c) I. Heuermann, B. Heitmann, R. Stichauer, D. Duvinage and M. Vogt, Organometallics, 2019, 38, 1787–1799 CrossRef CAS.
- (a) S. P. Cronin, J. M. Strain, M. S. Mashuta, J. M. Spurgeon, R. M. Buchanan and C. A. Grapperhaus, Inorg. Chem., 2020, 59, 4835–4841 CrossRef CAS PubMed; (b) R. Stichauer and M. Vogt, Organometallics, 2018, 37, 3639–3643 CrossRef CAS.
- (a) J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS PubMed; (b) M. W. Drover, L. L. Schafer and J. A. Love, Angew. Chem., 2016, 128, 3233–3238 ( Angew. Chem., Int. Ed. , 2016 , 55 , 3181–3186 ) CrossRef; (c) M. Ito, M. Itazaki and H. Nakazawa, Inorg. Chem., 2017, 56, 13709–13714 CrossRef CAS PubMed; (d) J. W. Nugent, M. G. Melchor and A. R. Fout, Organometallics, 2020, 39, 2917–2927 CrossRef CAS; (e) X. Zhai, M. Pang, L. Feng, J. Jia, C.-H. Tung and W. Wang, Chem. Sci., 2021, 12, 2885–2889 RSC.
- M. A. Rankin, K. D. Hesp, G. Schatte, R. McDonald and M. Stradiotto, Dalton Trans., 2009, 4756–4765 RSC.
- (a) T. Stahl, K. Mether, Y. Ohki, K. Tatsumi and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 10978–10981 CrossRef CAS PubMed; (b) H. Song, K. Ye, P. Geng, X. Han, R.-Z. Liao, C.-H. Tung and W. Wang, ACS Catal., 2017, 7, 7709–7717 CrossRef CAS.
- V. Lyaskovskyy and B. Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- S. Maity, S. Kundu, S. Mondal, S. Bera and P. Ghosh, Inorg. Chem., 2017, 56, 3363–3376 CrossRef CAS PubMed.
- (a) G. Durgaprasad, J. A. Luna, K. D. Spielvogel, C. Haas, S. K. Shaw and S. R. Daly, Organometallics, 2017, 36, 4020–4031 CrossRef CAS; (b) K. E. Rosenkoetter, M. K. Wojnar, B. J. Charette, J. W. Ziller and A. F. Heyduk, Inorg. Chem., 2018, 57, 9728–9737 CrossRef CAS PubMed; (c) M. R. Ringenberg, S. L. Kokatam, Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2008, 130, 788–789 CrossRef CAS PubMed.
- (a) J. I. van der Vlugt, Chem.–Eur. J., 2019, 25, 2651–2662 CrossRef CAS PubMed; (b) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed; (c) D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- K. D. Spielvogel, J. A. Luna, S. M. Loria, L. P. Weisburn, N. C. Stumme, M. R. Ringenberg, G. Durgaprasad, J. M. Keith, S. K. Shaw and S. R. Daly, Inorg. Chem., 2020, 59, 10845–10853 CrossRef CAS PubMed.
- K. Saha, D. K. Roy, R. D. Dewhurst, S. Ghosh and H. Braunschweig, Acc. Chem. Res., 2021, 54, 1260–1273 CrossRef CAS PubMed.
- R. Ramalakshmi, K. Saha, D. K. Roy, B. Varghese, A. K. Phukan and S. Ghosh, Chem.–Eur. J., 2015, 21, 17191–17195 CrossRef CAS PubMed.
- M. Zafar, R. Ramalakshmi, A. Ahmad, P. K. S. Antharjanam, S. Bontemps, S. Sabo-Etienne and S. Ghosh, Inorg. Chem., 2021, 60, 1183–1194 CrossRef CAS PubMed.
- M. M. T. Khan, D. Srinivas, R. I. Kureshy and N. H. Khan, Inorg. Chem., 1990, 29, 2320–2326 CrossRef CAS.
- M. Chatterjee, S. Mondal, P. Ghosh, W. Kaim and G. K. Lahiri, Inorg. Chem., 2018, 57, 12187–12194 CrossRef CAS PubMed.
- (a) S. Kundu, D. Dutta, S. Maity, T. Weyhermüller and P. Ghosh, Inorg. Chem., 2018, 57, 11948–11960 CrossRef CAS PubMed; (b) N. P. V. Leest, M. A. Tepaske, J. H. Oudsen, B. Venderbosch, N. R. Rietdijk, M. A. Siegler, M. Tromp, J. I. V. D. Vlugt and B. D. Bruin, J. Am. Chem. Soc., 2020, 142, 552–563 CrossRef PubMed; (c) N. L. Fry, M. J. Rose, C. Nyitray and P. K. Mascharak, Inorg. Chem., 2008, 47, 11604–11610 CrossRef CAS PubMed; (d) H. Masui, A. B. P. Lever and P. R. Auburn, Inorg. Chem., 1991, 30, 2402–2410 CrossRef CAS; (e) S. D. J. McKinnon, B. O. Patrick, A. B. P. Lever and R. G. Hicks, Chem. Commun., 2010, 46, 773–775 RSC.
- (a) D. A. Lomov, Y. M. Yutilov and N. N. Smolyar, Russ. J. Org. Chem., 2006, 42, 241–242 CrossRef CAS; (b) T. Itoh and T. Mase, Org. Lett., 2007, 9(18), 3687–3689 CrossRef CAS PubMed.
- S.-F. Hou, J.-Y. Chen, M. Xue, M. Jia, X. Zhai, R.-Z. Liao, C.-H. Tung and W. Wang, ACS Catal., 2020, 10, 380–390 CrossRef CAS.
- Y. Gloaguen, G. Alcaraz, A.-F. Péharman, E. Clot, L. Vendier and S. Sabo-Etienne, Angew. Chem., 2009, 121, 3008–3012 ( Angew. Chem., Int. Ed. , 2009 , 48 , 2964–2968 ) CrossRef.
- R. S. Anju, D. K. Roy, B. Mondal, K. Yuvaraj, C. Arivazhagan, K. Saha, B. Varghese and S. Ghosh, Angew. Chem., 2014, 126, 2917–2921 ( Angew. Chem., Int. Ed. , 2014 , 53 , 2873–2877 ) CrossRef.
- K. Saha, U. Kaur, S. Kar, B. Mondal, B. Joseph, P. K. S. Antharjanam and S. Ghosh, Inorg. Chem., 2019, 58, 2346–2353 CrossRef CAS PubMed.
- (a) G. Alcaraz, L. Vendier, E. Clot and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2010, 49, 918–920 CrossRef CAS PubMed; (b) R. Adam, E. Alberico, W. Baumann, H. Drexler, R. Jackstell, H.-J. Junge and M. Beller, Chem.–Eur. J., 2016, 22, 4991–5002 CrossRef CAS PubMed; (c) D. Y. Shopov, B. Rudshteyn, J. Campos, V. S. Batista, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2015, 137, 7243–7250 CrossRef CAS PubMed.
- M. Zafar, R. Ramalakshmi, K. Pathak, A. Ahmad, T. Roisnel and S. Ghosh, Chem.–Eur. J., 2019, 25, 13537–13546 CrossRef CAS PubMed.
- B. Jiang, J. Jia, Y. Sun, Y. Wang, J. Zeng, X. Bu, L. Shi and X. Sun Xiaobo Yang, Chem. Commun., 2020, 56, 13389–13392 RSC.
- (a) C. Y. Tang, A. L. Thompson and S. Aldridge, Angew. Chem., 2010, 122, 933–937 ( Angew. Chem., Int. Ed. , 2010 , 49 , 921–925 ) CrossRef; (b) G. Alcaraz, E. Clot, U. Hemstedt, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2007, 129, 8704–8705 CrossRef CAS PubMed.
- K. Essalah, J.-C. Barthelat, V. Montiel, S. Lachaize, B. Donnadieu, B. Chaudret and S. Sabo-Etienne, J. Organomet. Chem., 2003, 680, 182–187 CrossRef CAS.
- V. Montiel-Palma, M. Lumbierres, B. Donnadieu, S. Sabo-Etienne and B. Chaudret, J. Am. Chem. Soc., 2002, 124, 5624–5625 CrossRef CAS PubMed.
- T. Miyada, E. H. Kwan and M. Yamashita, Organometallics, 2014, 33, 6760–6770 CrossRef CAS.
- A. Cassen, Y. Gloaguen, L. Vendier, C. Duhayon, A. Poblador-Bahamonde, C. Raynaud, E. Clot, G. Alcaraz and S. Sabo-Etienne, Angew. Chem., 2014, 126, 7699–7703 ( Angew. Chem., Int. Ed. , 2014 , 53 , 7569–7573 ) CrossRef.
- K. D. Hesp, F. O. Kannemann, M. A. Rankin, R. McDonald, M. J. Ferguson and M. Stradiotto, Inorg. Chem., 2011, 50, 2431–2444 CrossRef CAS PubMed.
- (a) J. Brugos, J. A. Cabeza, P. García-Álvarez, A. R. Kennedy, E. Pérez-Carreño and J. F. Van der Maelen, Inorg. Chem., 2016, 55, 8905–8912 CrossRef CAS PubMed; (b) J. Brugos, J. A. Cabeza, P. García-Álvarez, E. Pérez-Carreño and J. F. Van der Maelen, Dalton Trans., 2017, 46, 4009–4017 RSC; (c) J. F. Van der Maelen, J. Brugos, P. García-Álvarez and J. A. Cabeza, J. Mol. Struct., 2020, 1201, 12717 CrossRef.
- K. Smith, A. Pelter and Z. Jin, Angew. Chem., 1994, 106, 913–914 ( Angew. Chem. Int. Ed. Engl. , 1994 , 33 , 851–853 ) CrossRef CAS.
- (a) G. Alcaraz, U. Helmstedt, E. Clot, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2008, 130, 12878–12879 CrossRef CAS PubMed; (b) C. S. MacNeil, S.-J. Hsiang and P. G. Hayes, Chem. Commun., 2020, 56, 12323–12326 RSC.
- M. A. Rankin, K. D. Hesp, G. Schatte, R. McDonald and M. Stradiotto, Dalton Trans., 2009, 4756–4765 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1875697, 1984218, 2126931 and 2040802. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00907b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |