
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickelII-catalyzed asymmetric photoenolization/Mannich reaction of (2-alkylphenyl) ketones†
Liangkun
Yang
 ,
Wang-Yuren
Li
,
Liuzhen
Hou
,
Tangyu
Zhan
,
Weidi
Cao
*,
Xiaohua
Liu
and
Xiaoming
Feng
*
,
Wang-Yuren
Li
,
Liuzhen
Hou
,
Tangyu
Zhan
,
Weidi
Cao
*,
Xiaohua
Liu
and
Xiaoming
Feng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, China. E-mail: wdcao@scu.edu.cn; xmfeng@scu.edu.cn
First published on 22nd June 2022
Abstract
A diastereo- and enantioselective photoenolization/Mannich (PEM) reaction of ortho-alkyl aromatic ketones with benzosulfonimides was established by utilizing a chiral N,N′-dioxide/Ni(OTf)2 complex as the Lewis acid catalyst. It afforded a series of benzosulfonamides and the corresponding ring-closure products, and a reversal of diastereoselectivity was observed through epimerization of the benzosulfonamide products under continuous irradiation. On the basis of the control experiments, the role of the additive LiNTf2 in achieving high stereoselectivity was elucidated. This PEM reaction was proposed to undergo a direct nucleophilic addition mechanism rather than a hetero-Diels–Alder/ring-opening sequence. A possible transition state model with a photoenolization process was proposed to explain the origin of the high level of stereoinduction.
Introduction
Photoinduced catalysis contributes in a significant way to the existing repertoire of carbon–carbon and carbon-hetero bond-forming reactions,1 allowing exceptional molecular transformations complementary to thermal catalytic reactions.2 Aromatic aldehydes and ketones represent an important subset of carbonyl compounds, which can undergo unique reaction pathways upon irradiation with UV light.3 As a kind of classic Norrish type II process, the photoenolization of 2-alkylphenyl containing carbonyls has been thoroughly studied since 1961.4 Therein, the photogenerated enols possess fascinating reactivities and have been regarded as versatile intermediates in diverse reactions.5 Nevertheless, the enantioselective versions were relatively limited. Early attempts focused on the asymmetric photoenolization/Diels–Alder (PEDA) reactions; Bach6 and Nicolaou7 obtained considerable results with the use of a stoichiometric catalyst. Later, Melchiorre creatively realized the enantioselective PEDA reaction with maleimides by using a cinchona-thiourea catalyst,8 and Gao's group took the PEDA reaction as a key step catalyzed by Ti–TADDOL complexes in asymmetric synthesis of natural products.9 In addition to dienophiles, other reaction partners to trap the ephemeral photoenols enantioselectively have been exploited, such as in the Michael reaction10 and aldol11 and allylic alkylation-type reactions.12 By contrast, the catalytic asymmetric photoenolization/Mannich reaction (PEM) seemed to encounter challenges such as: (1) the irrepressible background reaction and (2) the potential epimerization of the product due to its further photoenolization. Melchiorre first disclosed the enantioselective PEM reaction with N-protected benzaldimines promoted by a chiral (DHQ)2PHAL catalyst, affording the desired chiral amines with moderate enantioselectivity and low diastereoselectivity (Scheme 1a).13 Recently, He's group realized an organocatalytic enantioselective aldol reaction of photoenol with isatin, but isatin-derived ketimine transformed into the spirocyclic oxindole in an uncontrolled manner (Scheme 1a).11b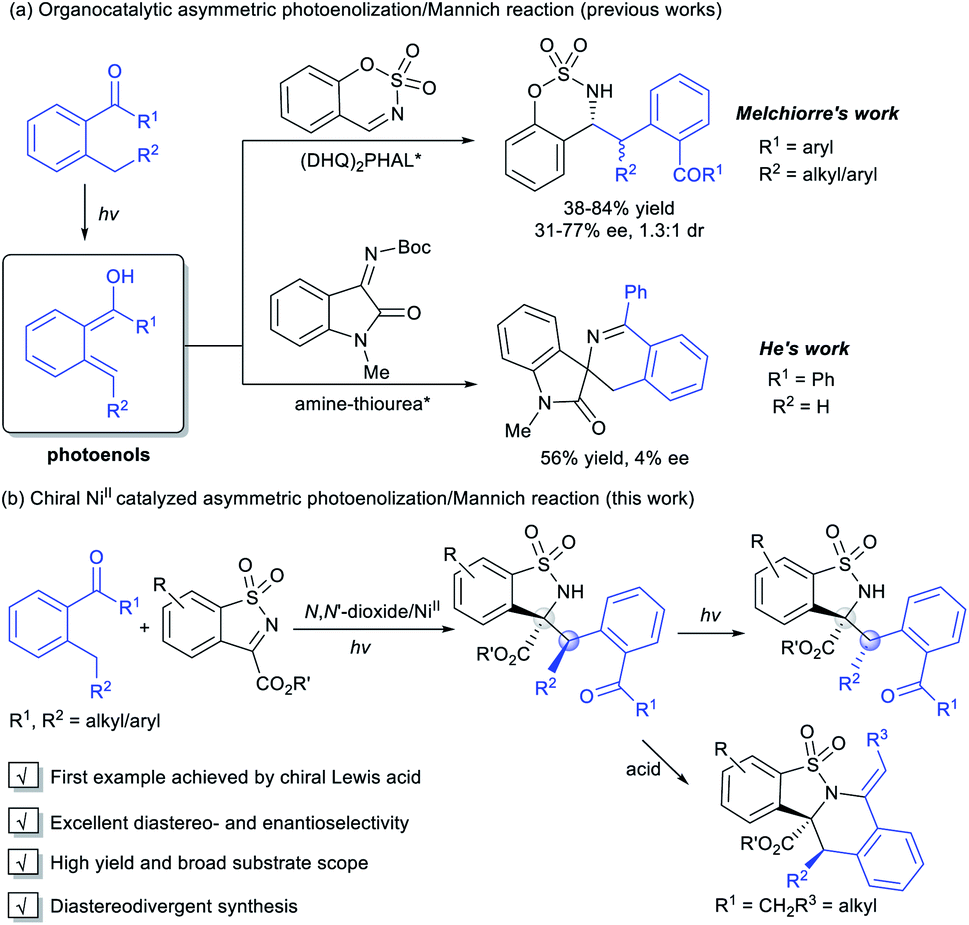 | ||
| Scheme 1 The catalytic asymmetric PEM reactions of 2-alkylphenyl ketones. | ||
Upon our long-term research, chiral N,N′-dioxide/metal complexes were demonstrated to exhibit excellent ability of chiral induction and delivery in asymmetric catalysis, including light-triggered reactions.14 We conceived that this kind of powerful Lewis acid catalyst could activate imines to trap the photoenol intermediate and suppress the background reaction. Herein, we disclosed the first NiII-mediated highly diastereo- and enantioselective PEM reaction of acyclic and cyclic 2-alkylphenyl ketones with N-sulfonyl ketimines, delivering various optically active α-amino acetate derivatives under mild conditions. The adducts could further undergo ring-closure to produce the tetrahydroisoquinoline derivatives under acidic conditions in one pot. The epimers could be obtained with high enantioselectivity under continuous stronger irradiation (Scheme 1b).
Results and discussion
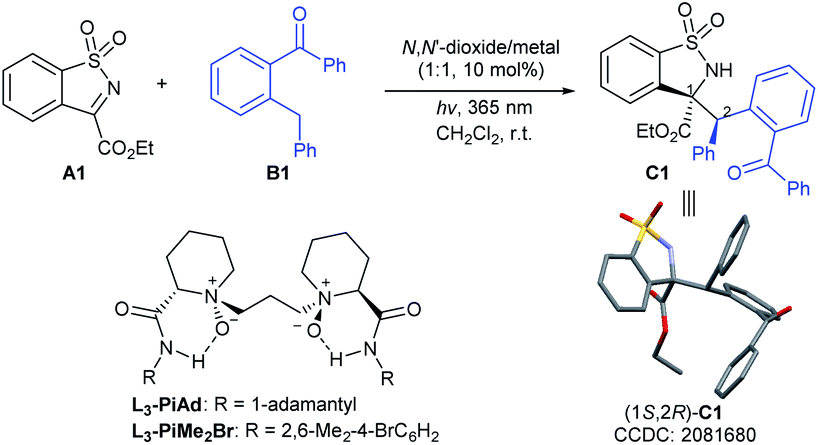
In our initial investigation, the N-sulfonyl cyclic ketimine A1 and 2-benzylbenzophenone B1 were applied as the model substrates to optimize the reaction conditions. The desired PEM reaction product C1 was formed in a yield of 67% with 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44 dr under 20 W UV LED irradiation in CH2Cl2, revealing the completion of a severe background reaction (Table 1, entry 1). To our delight, upon screening of metal salts (Table 1, entries 2–5) coordinated with chiral N,N′-dioxide L3-PiAd, we found that Ni(OTf)2 could provide promising enantioselectivity (68% yield, 58:42 dr, 57%/52% ee; entry 5). Other metal ions, such as ScIII, MgII, and ZnII yielded the product with no more than 5% ee. To improve the yield and stereoselectivity, additives and ligands were screened (see the ESI† for details). It was found that LiNTf2 played a crucial role in promoting diastereo- and enantioselectivity, and C1 could be isolated in 94% yield with 74:26 dr and 89%/85% ee by increasing its dosage to 30 mol% (Table 1, entry 6), which may benefit from exchanging the counterion with Ni(OTf)2.15 Switching the ligand from L3-PiAd to L3-PiMe2Br, the ee value of the major diastereomer was slightly improved to 92% (Table 1, entry 7). Considering that the occurrence of epimerization of the product C1 through the second photoenolization may provide the variation of diastereoselectivity, the intensity of the light source was reduced (entries 8 and 9). To our delight, a remarkable increase in diastereoselectivity was realized under 2 W UV LED irradiation albeit prolonged reaction time was necessary (8 h), and C1 was obtained in 90% yield, 91:9 dr and 93% ee (Table 1, entry 9). Increasing the amount of B1 and lowering the reaction concentration enhanced the yield to 96% with maintained stereoselectivity (entry 10). The exploration of wavelength showed that the reactivity reduced with the increase in the wavelength. Under the irradiation with a 385 nm UV LED, low diastereoselectivity (58:42) was obtained (entry 11). The PEM reaction could occur even with visible light (400 nm), but lower yield and stereoselectivity were obtained (entry 12). Other reaction parameters, including solvent, temperature and so on were also examined, but no better result was obtained (see the ESI† for details). The absolute configuration of C1 was determined to be (1S,2R) based on X-ray single crystal analysis.16a
:44 dr under 20 W UV LED irradiation in CH2Cl2, revealing the completion of a severe background reaction (Table 1, entry 1). To our delight, upon screening of metal salts (Table 1, entries 2–5) coordinated with chiral N,N′-dioxide L3-PiAd, we found that Ni(OTf)2 could provide promising enantioselectivity (68% yield, 58:42 dr, 57%/52% ee; entry 5). Other metal ions, such as ScIII, MgII, and ZnII yielded the product with no more than 5% ee. To improve the yield and stereoselectivity, additives and ligands were screened (see the ESI† for details). It was found that LiNTf2 played a crucial role in promoting diastereo- and enantioselectivity, and C1 could be isolated in 94% yield with 74:26 dr and 89%/85% ee by increasing its dosage to 30 mol% (Table 1, entry 6), which may benefit from exchanging the counterion with Ni(OTf)2.15 Switching the ligand from L3-PiAd to L3-PiMe2Br, the ee value of the major diastereomer was slightly improved to 92% (Table 1, entry 7). Considering that the occurrence of epimerization of the product C1 through the second photoenolization may provide the variation of diastereoselectivity, the intensity of the light source was reduced (entries 8 and 9). To our delight, a remarkable increase in diastereoselectivity was realized under 2 W UV LED irradiation albeit prolonged reaction time was necessary (8 h), and C1 was obtained in 90% yield, 91:9 dr and 93% ee (Table 1, entry 9). Increasing the amount of B1 and lowering the reaction concentration enhanced the yield to 96% with maintained stereoselectivity (entry 10). The exploration of wavelength showed that the reactivity reduced with the increase in the wavelength. Under the irradiation with a 385 nm UV LED, low diastereoselectivity (58:42) was obtained (entry 11). The PEM reaction could occur even with visible light (400 nm), but lower yield and stereoselectivity were obtained (entry 12). Other reaction parameters, including solvent, temperature and so on were also examined, but no better result was obtained (see the ESI† for details). The absolute configuration of C1 was determined to be (1S,2R) based on X-ray single crystal analysis.16a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal salt | Ligand | Yieldb (%) | drc | eed (%) |
|
a Unless otherwise noted, all reactions were carried out with Ni(OTf)2/ligand (1:1, 10 mol%), A1 (0.10 mmol), and B1 (0.15 mmol) in CH2Cl2 (1.5 mL) at room temperature under a N2 atmosphere and irradiation (20 W UV LED, λmax = 365 nm) for 2.5 h.
b Isolated yield.
c The dr value was determined by 1H NMR analysis.
d Determined by UPC2 on a chiral stationary phase.
e With LiNTf2 (30 mol%).
f Under irradiation with a 5 W UV LED for 5 h.
g Under irradiation with a 2 W UV LED for 8 h.
h In CH2Cl2 (2.5 mL) and B1 (0.2 mmol) were used.
i Under irradiation with a 2 W UV LED (λmax = 385 nm) for 8 h.
j Under irradiation with a 2 W blue LED (λmax = 400 nm) for 16 h.
|
|||||
| 1 | — | — | 67 | 56:44 |
— |
| 2 | Sc(OTf)3 | L3-PiAd | 67 | 54:46 |
0 |
| 3 | Mg(OTf)2 | L3-PiAd | 76 | 64:36 |
4/9 |
| 4 | Zn(OTf)2 | L3-PiAd | 67 | 53:47 |
5/5 |
| 5 | Ni(OTf)2 | L3-PiAd | 68 | 58:42 |
57/52 |
| 6e | Ni(OTf)2 | L3-PiAd | 94 | 74:26 |
89/85 |
| 7e | Ni(OTf)2 | L3-PiMe2Br | 91 | 72:28 |
92/84 |
| 8e,f | Ni(OTf)2 | L3-PiMe2Br | 89 | 85:15 |
93/76 |
| 9e,g | Ni(OTf)2 | L3-PiMe2Br | 90 | 91:9 |
93/64 |
| 10e,h | Ni(OTf)2 | L3-PiMe2Br | 96 | 92:8 |
93/65 |
| 11e,h,i | Ni(OTf)2 | L3-PiMe2Br | 92 | 58:42 |
91/71 |
| 12e,h,j | Ni(OTf)2 | L3-PiMe2Br | 87 | 90:10 |
91/52 |
With the optimized reaction conditions in hand, the substrate scope with respect to A was then evaluated (Table 2). Variations of the ester substituent of N-sulfonyl cyclic ketimines showed no obvious influence on the reactivities and stereoselectivities (Table 2, entries 1–4, 96–99% yields, 87:13–94:6 dr and 86–95% ee). Regardless of the position and electronic properties of the substituents on the phenyl ring, a wide range of substituted cyclic ketimines were converted into the corresponding products C5–C13 with good to excellent yields (Table 2, entries 5–13, 80–93%). In general, the imines bearing electron-donating groups, such as A5 (5-tBu), A6 (5-OMe), A9 (5,6-dimethoxy), and A10 (6-Me), exhibited relatively low reactivity and required prolonged reaction time (8–56 h) with consistently good diastereo- and enantioselectivities (Table 2, entries 5–6, 9–10; 84:16–90:10 dr and 91–94% ee). When electron-deficient imines were used, the corresponding products (C7, C8 and C10–C13) were obtained with decreased diastereo- and enantioselectivities, which may be attributed to the strong background reaction (Table 2, entries 7–8, 11–13, 80:20–90:10 dr and 67–87% ee). Imine with fused naphthyl could also be transformed into the desired product C14 but in 77% yield, 87:13 dr and 80% ee (Table 2, entry 14).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | R1/R2 | t (h) | Yieldb (%) | drc | eed (%) |
|
a Unless otherwise noted, all reactions were carried out with L3-PiMe2Br/Ni(OTf)2 (1:1, 10 mol%), A (0.10 mmol), B1 (0.20 mmol) and LiNTf2 (30 mol%) in CH2Cl2 (2.5 mL) at room temperature under a N2 atmosphere and 2 W LED (λmax = 365 nm) for a certain time.
b Isolated yield of C.
c Determined by 1H NMR analysis.
d Determined by UPC2 on a chiral stationary phase.
|
|||||
| 1 | Et/H | 8 | C1, 96 | 92:8 |
93 |
| 2 | Me/H | 8 | C2, 98 | 87:13 |
86 |
| 3 | i Pr/H | 8 | C3, 99 | 94:6 |
95 |
| 4 | Bn/H | 8 | C4, 99 | 88:12 |
88 |
| 5 | Et/5-tBu | 8 | C5, 93 | 90:10 |
91 |
| 6 | Et/5-OMe | 32 | C6, 80 | 84:16 |
93 |
| 7 | Et/5-OCF3 | 8 | C7, 87 | 80:20 |
75 |
| 8 | Et/5-F | 8 | C8, 87 | 85:15 |
87 |
| 9 | Et/5,6-(OMe)2 | 56 | C9, 93 | 85:15 |
94 |
| 10 | Et/6-Me | 20 | C10, 91 | 90:10 |
94 |
| 11 | Et/7-F | 8 | C11, 91 | 85:15 |
85 |
| 12 | Et/7-Cl | 8 | C12, 87 | 90:10 |
86 |
| 13 | Et/7-CF3 | 8 | C13, 92 | 81:19 |
67 |
| 14 |
|
72 | C14, 77 | 87:13 |
80 |

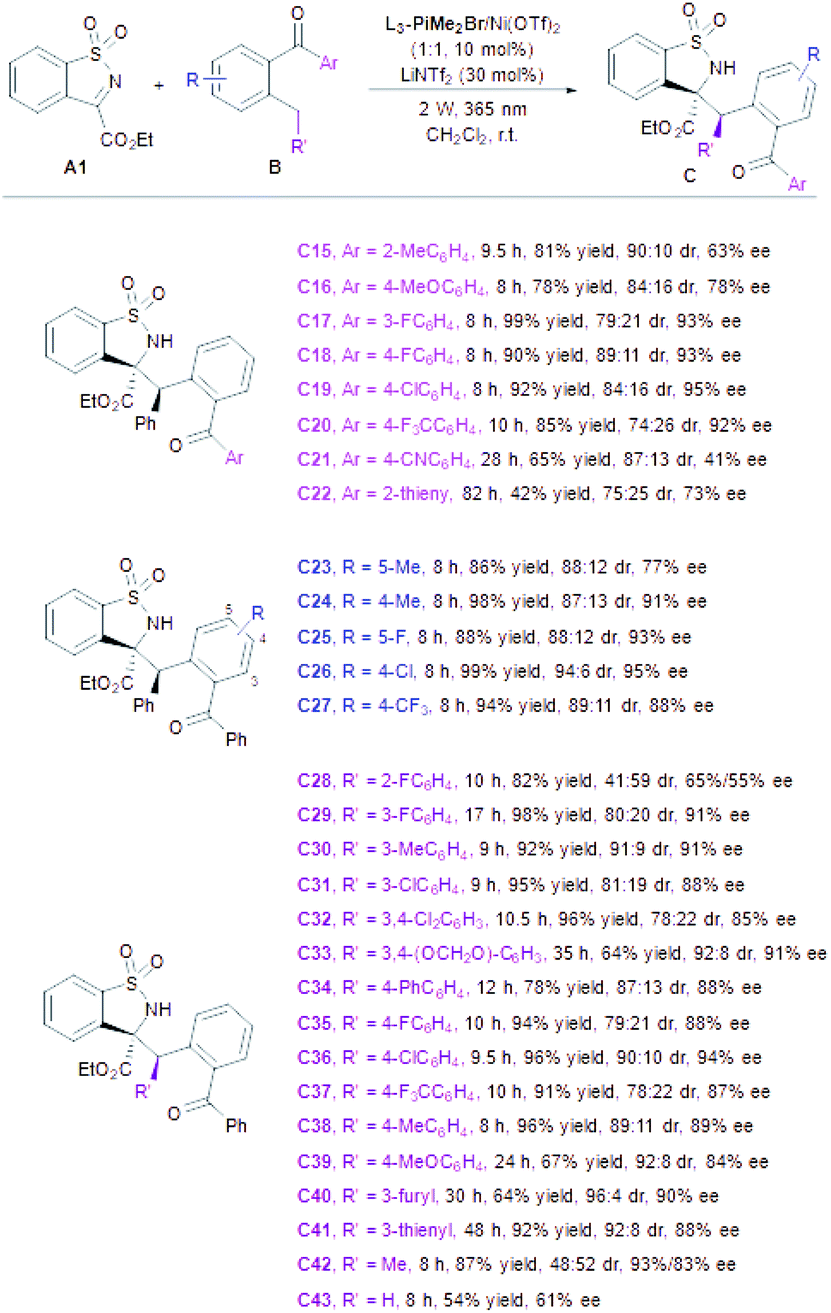
Next, the scope of 2-benzylbenzophenone derivatives was examined (Table 3). The 2-benzophenones B with electron-donating groups (2-Me, 4-OMe) on the Ar unit afforded the desired adducts (C15–C16) in lower yields (78–81%) and enantioselectivities (84:16–90:10 dr and 63–78% ee) than those containing electron-deficient aryl subunits (C17–C20, 85–99% yields, 74:26–89:11 dr and 92–95% ee). Nevertheless, the enantioselectivity dropped dramatically in the case of 4-CN and 2-thienyl substituted substrates (B21 and B22), probably due to the interferential coordination between the CN group or thienyl of ketone substrates with a central metal ion. Moreover, electron-withdrawing and electron-donating substituents (R) were well tolerated. 5-Methyl substituted substrate B23 gave relatively lower enantioselectivity than others, perhaps on account of the higher reactivity of photoenols that led to a strong background reaction. The steric hindrance of the R′ side-group has a pronounced effect on this transformation. The substrate bearing the ortho-fluorophenyl group was an exception to give the desired product (C28) smoothly (82% yield) with poor stereoselectivity (41:59 dr, 65%/55% ee). A broad array of benzophenones with electron-withdrawing and electron-donating substituents at the meta- and para-positions could be transformed to the corresponding products in good yields with moderate dr and high ee (C29–C39). To our delight, 3-furyl and 3-thienyl substituted substrates were also suitable under this catalytic system (C40–C41). However, low diastereoselectivity with maintained enantioselectivity was obtained when (2-ethylphenyl)(phenyl)methanone participated in (Me, C42), implying the high importance of an aryl substituent at the terminal of the photoenol intermediate to diastereocontrol. Furthermore, an obvious decrease in yield and the ee value of the desired Mannich product (C43) was obtained along with the formation of a retro-aza-vinylogous Michael product (18% yield) if phenyl(o-tolyl)methanone was employed in the reaction (see the ESI† for details).
|
a Unless otherwise noted, all reactions were carried out with L3-PiMe2Br/Ni(OTf)2 (1:1, 10 mol%), A1 (0.10 mmol), B (0.20 mmol) and LiNTf2 (30 mol%) in CH2Cl2 (2.5 mL) at room temperature under a N2 atmosphere and 2 W LED (λmax = 365 nm) for a certain time. The isolated yield of C is provided. The dr value was determined by 1H NMR analysis and the ee value was determined by UPC2 on a chiral stationary phase or chiral HPLC analysis.
|
|---|
|
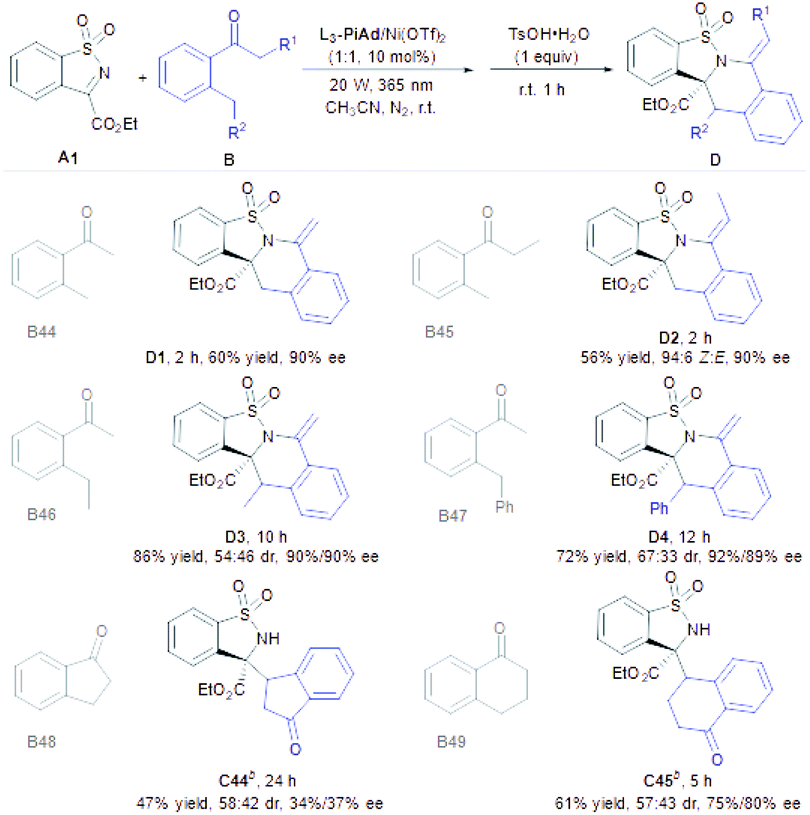
Subsequently, we turned our attention to the scope of aryl/alkyl-type of ketones (Table 4). Under the modified reaction conditions (see the ESI† for details), photoexcitation of ortho-methyl acetophenone B44 produced the photoenol followed by the Mannich reaction to provide the addition product smoothly, which was unstable and intended to undergo ring-closure for the construction of a tetracyclic skeleton (D1, 60% yield and 90% ee) by dealing with TsOH·H2O. This finding almost excluded the generally supposed cycloaddition pathway.13 As such, ortho-methyl propiophenone (B45) provided the tetrahydroisoquinoline derivative D2 in 56% yield with 90% ee. It was worth noting that the Z:E ratio of D2 increased to 94:6 with the extension of time, which indicated that the β-H elimination step is reversible (see the ESI† for details). 2-Ethyl acetophenone B46 and 2-benzyl acetophenone B47 were also effective to generate the desired cyclization products D3 and D4 in good yields with excellent enantioselectivities (72–86% yields and 90–92% ee), but the diastereoselectivities (54:46–67:33 dr) were poor even though the intensity of the light source was lowered. Benzannulated carbocyclic ketones, such as 1-indanone and 1-tetralone, were acceptable substrates as well to form the photoenols and incorporated into N-sulfonyl cyclic ketimine to provide C44 and C45 smoothly (47–71% yields, 58:42–57:43 dr and 34–75% ee), but no ring-closure products were observed with TsOH·H2O.
|
a Unless otherwise noted, all reactions were carried out with L3-PiAd/Ni(OTf)2 (1:1, 10 mol%), A (0.10 mmol) and B (0.20 mmol) in CH3CN (1.0 mL) at room temperature under a N2 atmosphere and 20 W LED (λmax = 365 nm) for a certain time, and then TsOH·H2O was added and stirred at room temperature for another 1 h under air. The isolated yield of D or C is provided. The dr value was determined by 1H NMR analysis and the ee value was determined by UPC2 on a chiral stationary phase or chiral HPLC analysis.
b Without further transformation in the presence of TsOH·H2O.
|
|---|
|
To show the synthetic potential of the current protocol, the gram-scale synthesis of C1 was conducted under slightly modified optimal conditions. Considering the difference in reaction vessels, four 3.75 W LEDs (λmax = 365 nm) were used (see the ESI† for details). As shown in Scheme 2a, N-sulfonyl cyclic ketimine A1 reacted with 2-benzylbenzophenone B1 smoothly, providing C1 (2.5 mmol scale, 1.23 g; 5 mmol scale, 2.36 g) with maintained yield, and diastereo- and enantioselectivity. Treatment of C1 with Pd/C and H2 afforded the reduction alcohol E1 in nearly equivalent yield, 87:13 dr and 92%/82% ee (Scheme 2b), and the absolute configuration of the major enantiomer was determined to be (S,R,R) by X-ray crystallographic analysis.16b The intramolecular Mitsunobu reaction of E1 proceeded well under the conventional reaction conditions,17 and afforded the tetrahydroisoquinoline derivatives F1 and F1′ with maintained enantioselectivities. The absolute configuration of F1′ was determined to be (S,R,R) by X-ray crystallographic analysis.16c
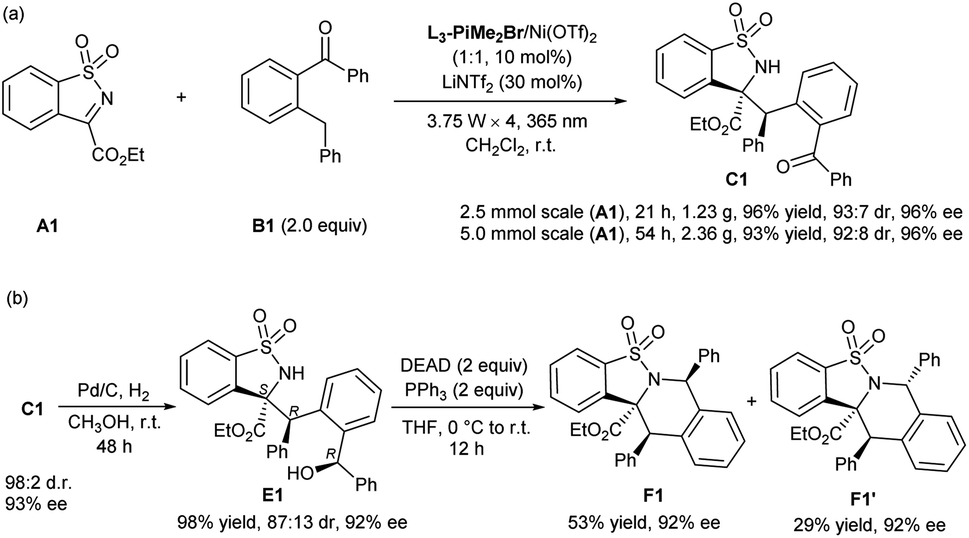 | ||
| Scheme 2 Gram-scale synthesis and further transformations. | ||
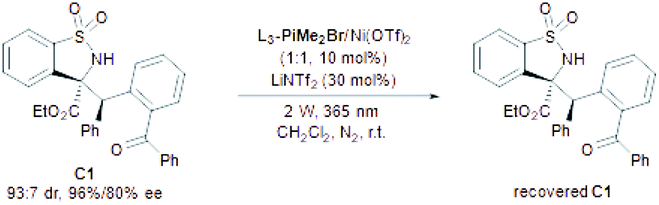
In order to gain insight into the mechanism of the PEM reaction, some control experiments were carried out. In the illumination experiments of the product (Table 5), C1 could be recovered with basically maintained stereoselectivity under the standard conditions for 10 h (Table 5, entry 1). On changing the wavelength to 385 nm, the diastereoselectivity dropped sharply due to the fast epimerization (Table 5, entry 2), which explained the low diastereoselectivity of the PEM reaction under a 385 nm UV LED (Table 1, entry 11). In the absence of the L3-PiMe2Br/Ni(OTf)2 complex, the stereoselectivity of C1 was nearly unchanged (Table 5, entry 3). However, the diastereoselectivity sharply decreased without LiNTf2 (Table 5, entry 4). These results suggested that the additive LiNTf2 may not only provide counterions for the chiral catalyst, but also coordinate with the carbonyl group of C1 to impede the epimerization. Interestingly, by increasing the illumination intensity to 20 W and extending the illumination time to 20 h without LiNTf2, the diastereoselectivity could be reversed through the second photoenolization of C1 albeit with partial decomposition (Table 5, entry 5), and the simple illumination with none of the catalysts gave a similar result (Table 5, entry 6), which provided a possibility for diastereodivergent synthesis.18 Additionally, no reaction occurred when replacing the benzoyl group with a nitrile group or hydrogen atom, but a self-coupling product of the imine was observed. It excluded the direct intermolecular hydrogen atom transfer process of excited imine19 with 2-benzyl benzophenone (Scheme 3a).
|
|
||||
|---|---|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) | drc | eed (%) |
|
a Unless otherwise noted, all reactions were carried out with L3-PiMe2Br/Ni(OTf)2 (1:1, 10 mol%), C1 (0.10 mmol) and LiNTf2 (30 mol%) in CH2Cl2 (2.5 mL) at room temperature under a N2 atmosphere and 2 W LED (λmax = 365 nm) for 10 h.
b Isolated yield of C.
c Determined by 1H NMR analysis.
d Determined by UPC2 on a chiral stationary phase.
|
||||
| 1 | — | 89 | 91:9 |
96/87 |
| 2 | 2 W, 385 nm | 74 | 37:63 |
95/93 |
| 3 | No L3-PiMe2Br/Ni(OTf)2 | 83 | 92:8 |
96/82 |
| 4 | No LiNTf2 | 92 | 79:21 |
96/91 |
| 5 | 20 W, no LiNTf2, 20 h | 51 | 10:90 |
76/95 |
| 6 | 20 W, no catalyst, 20 h | 52 | 11:89 |
78/95 |
 | ||
| Scheme 3 Control experiments and the proposed reaction process of the asymmetric PEM reaction. | ||
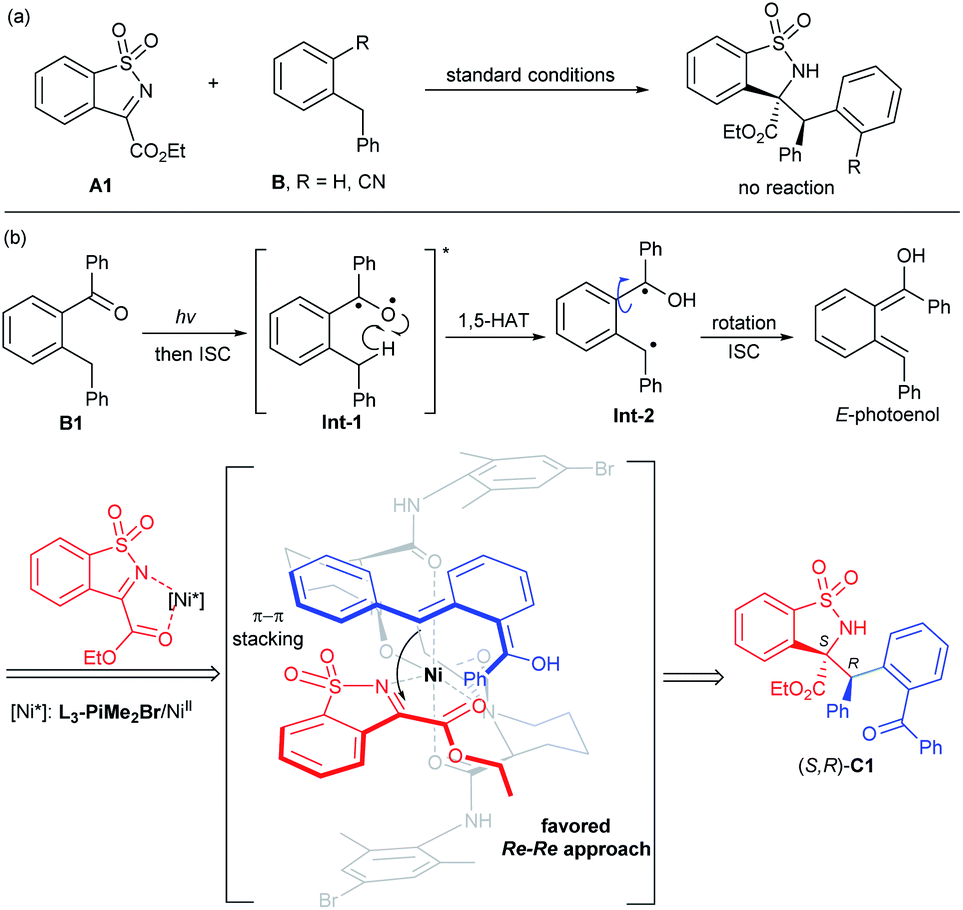
Based on the experimental results, the absolute configuration of the product C1 and our previous studies,20 a plausible transition state model was proposed (Scheme 3b). Under UV irradiation, the biradical Int-2 was obtained through 1,5-hydrogen atom transfer (HAT) of the triplet excited 2-benzylbenzophenone Int-1. Rotation of this biradical species and subsequent intersystem crossing (ISC) led to the reactive intermediate E-photoenol. According to the above experimental results, we conjectured that a π–π interaction between the aryl group at the benzyl position of the photoenol and the aryl group of N-sulfonyl cyclic ketimine exists. Meanwhile, the N,N′-dioxide/NiII complex activated the imine A1 in a bidentate coordination manner and the Si face of imine was shielded by the amide unit of the ligand. Thus, the Re–Re nucleophilic addition generated the Mannich product (S,R)-C1.
Conclusions
In summary, we have developed the first Lewis acid-catalyzed asymmetric PEM reaction. The combination of a chiral N,N′-dioxide/NiII complex catalyst and LiNTf2 could effectively suppress the background reaction and the epimerization of the products, offering a broad array of optically active α-amino acid ester derivatives with excellent stereoselectivities, and a reversal of diastereoselectivity was also achieved. The tetracyclic ring-closure products could be further yielded in one pot. In addition, the results of control experiments ruled out the general hetero-Diels–Alder/ring-opening sequence and the intermolecular direct HAT process. A plausible transition state model involving photo-mediated enol was also proposed to explain the origin of stereoselective control.Data availability
Further details of experimental procedure, 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, CD spectra and X-ray crystallographic data are available in the ESI.†Author contributions
L. K. Y. performed the experiments. W.-Y. L. repeated the data. L. Z. H. and T. Y. Z. participated in the synthesis of substrates and ligands. X. M. F., X. H. L. and W. D. C. supervised the project. X. H. L., W. D. C. and L. K. Y. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We appreciate the National Natural Science Foundation of China (No. 22071160 and 22188101) and the Science & Technology Department of Sichuan Province (No. 2021YJ0562) for financial support. We are grateful to Dr Yuqiao Zhou from the College of Chemistry, Sichuan University for the X-ray single crystal diffraction analysis.Notes and references
- For leading reviews, see: (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (d) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 RSC; (e) J. Xuan, L.-Q. Lu, J.-R. Chen and W.-J. Xiao, Eur. J. Org. Chem., 2013, 6755–6770 CrossRef CAS; (f) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355–360 CrossRef CAS; (g) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (h) R. A. Angnes, Z. Li, C. R. D. Correia and G. B. Hammond, Org. Biomol. Chem., 2015, 13, 9152–9167 RSC; (i) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872–3890 CrossRef CAS PubMed; (j) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC; (k) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed; (l) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (m) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 52 CrossRef CAS; (n) D. Wang, L. Zhang and S. Luo, Acta Chim. Sin., 2017, 75, 22–33 CrossRef CAS; (o) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41–49 CrossRef CAS PubMed; (p) B.-G. Cai, J. Xuan and W.-J. Xiao, Sci. Bull., 2019, 64, 337–350 CrossRef CAS; (q) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed; (r) W.-M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS; (s) H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem Catalysis, 2021, 1, 523–598 CrossRef; (t) T. Yasukawa and S. Kobayashi, ACS Cent. Sci., 2021, 7, 1099–1101 CrossRef CAS PubMed; (u) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (v) L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed; (w) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed; (x) M. J. Genzink, J. B. Kidd, W. B. Swords and T. P. Yoon, Chem. Rev., 2022, 122, 1654–1716 CrossRef CAS PubMed; (y) J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed; (z) N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2022, 122, 1925–2016 CrossRef CAS PubMed; (a a) P. Melchiorre, Chem. Rev., 2022, 122, 1483–1484 CrossRef CAS PubMed.
- For the reviews, see: (a) T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed; (b) S. Gao and Y. Qiu, Sci. China: Chem., 2016, 59, 1093–1108 CrossRef CAS; (c) M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (d) T. P. Nicholls, D. Leonori and A. C. Bissember, Nat. Prod. Rep., 2016, 33, 1248–1254 RSC; (e) M. K. Bogdos, E. Pinard and J. A. Murphy, Beilstein J. Org. Chem., 2018, 14, 2035–2064 CrossRef CAS PubMed; (f) B. Yang and S. Gao, Chem. Soc. Rev., 2018, 47, 7926–7953 RSC; (g) Y. Wei, Q.-Q. Zhou, F. Tan, L.-Q. Lu and W.-J. Xiao, Synthesis, 2019, 51, 3021–3054 CrossRef CAS; (h) C. M. Holden and P. Melchiorre, in Photochemistry, The Royal Society of Chemistry, 2020, vol. 47, pp. 344–378 Search PubMed; (i) J. B. Mateus-Ruiz and A. Cordero-Vargas, Synthesis, 2020, 52, 3111–3128 CrossRef CAS.
- For selected reviews and examples of photoreactivity of aromatic aldehydes and ketones, see: (a) R. G. W. Norrish and C. H. Bamford, Nature, 1937, 140, 195–196 CrossRef CAS; (b) N. C. Yang and D.-D. H. Yang, J. Am. Chem. Soc., 1958, 80, 2913–2914 CrossRef CAS; (c) A. Dalal, R. Khanna, D. Kumar, P. Jindal, A. Chaudhary and R. C. Kamboj, Curr. Org. Chem., 2015, 19, 2156–2195 CrossRef CAS; (d) C. Chen, Org. Biomol. Chem., 2016, 14, 8641–8647 RSC; (e) M. Oelgemöller and N. Hoffmann, Org. Biomol. Chem., 2016, 14, 7392–7442 RSC; (f) M. D'Auria, Photochem. Photobiol. Sci., 2019, 18, 2297–2362 CrossRef PubMed; (g) J. A. Dantas, J. T. M. Correia, M. W. Paixão and A. G. Corrêa, ChemPhotoChem, 2019, 3, 506–520 CrossRef CAS; (h) W. Zhang, Y. Li and S. Luo, J. Photochem. Photobiol., A, 2020, 396, 112553 CrossRef CAS; (i) J. Jurczyk, M. C. Lux, D. Adpressa, S. F. Kim, Y.-h. Lam, C. S. Yeung and R. Sarpong, Science, 2021, 373, 1004–1012 CrossRef CAS PubMed; (j) J. Mateos, S. Cuadros, A. Vega-Peñaloza and L. Dell'Amico, Synlett, 2022, 33, 116–128 CrossRef CAS.
- (a) N. C. Yang and C. Rivas, J. Am. Chem. Soc., 1961, 83, 2213 CrossRef CAS; (b) P. G. Sammes, Tetrahedron, 1976, 32, 405–422 CrossRef CAS; (c) J. L. Segura and N. Martín, Chem. Rev., 1999, 99, 3199–3246 CrossRef CAS PubMed; (d) A. Mavroskoufis, K. Rajes, P. Golz, A. Agrawal, V. Ruß, J. P. Götze and M. N. Hopkinson, Angew. Chem., Int. Ed., 2020, 59, 3190–3194 CrossRef CAS PubMed.
- For selected reviews and examples of reactions involving photoenols, see: (a) J. L. Charlton and M. M. Alauddin, Tetrahedron, 1987, 43, 2873–2889 CrossRef CAS; (b) J. L. Charlton and K. Koh, J. Org. Chem., 1992, 57, 1514–1516 CrossRef CAS; (c) R. Connors and T. Durst, Tetrahedron Lett., 1992, 33, 7277–7280 CrossRef CAS; (d) G. A. Kraus and Y. Wu, J. Org. Chem., 1992, 57, 2922–2925 CrossRef CAS; (e) K. C. Nicolaou, D. Gray and J. Tae, Angew. Chem., Int. Ed., 2001, 40, 3675–3678 CrossRef CAS; (f) K. C. Nicolaou, D. Gray and J. Tae, Angew. Chem., Int. Ed., 2001, 40, 3679–3683 CrossRef CAS; (g) K. Takaki, T. Fujii, H. Yonemitsu, M. Fujiwara, K. Komeyama and H. Yoshida, Tetrahedron Lett., 2012, 53, 3974–3976 CrossRef CAS; (h) Y. Masuda, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2015, 137, 14063–14066 CrossRef CAS PubMed; (i) T. Ide, S. Masuda, Y. Kawato, H. Egami and Y. Hamashima, Org. Lett., 2017, 19, 4452–4455 CrossRef CAS PubMed; (j) N. Ishida, T. Yano, T. Yuhki and M. Murakami, Chem.–Asian J., 2017, 12, 1905–1908 CrossRef CAS PubMed; (k) S. Cuadros and P. Melchiorre, Eur. J. Org. Chem., 2018, 2884–2891 CrossRef CAS; (l) J. Mateos, A. Cherubini-Celli, T. Carofiglio, M. Bonchio, N. Marino, X. Companyó and L. Dell'Amico, Chem. Commun., 2018, 54, 6820–6823 RSC; (m) Y. Masuda, N. Ishida and M. Murakami, Chem.–Asian J., 2019, 14, 403–406 CrossRef CAS PubMed; (n) J. Mateos, F. Rigodanza, A. Vega-Peñaloza, A. Sartorel, M. Natali, T. Bortolato, G. Pelosi, X. Companyó, M. Bonchio and L. Dell'Amico, Angew. Chem., Int. Ed., 2020, 59, 1302–1312 CrossRef CAS PubMed; (o) Y. Zhang, R. Jin, W. Kang and H. Guo, Org. Lett., 2020, 22, 5502–5505 CrossRef CAS PubMed; (p) Y. Zhang, R. Jin, G. Pan and H. Guo, Chem. Commun., 2020, 56, 11621–11624 RSC; (q) J. Y. J. Wang, M. T. Blyth, M. S. Sherburn and M. L. Coote, J. Am. Chem. Soc., 2022, 144, 1023–1033 CrossRef CAS PubMed; (r) Z.-L. Wang, L. Tang, W.-M. Zeng, Y.-H. He and Z. Guan, Tetrahedron Lett., 2022, 95, 153725 CrossRef CAS.
- B. Grosch, C. N. Orlebar, E. Herdtweck, W. Massa and T. Bach, Angew. Chem., Int. Ed., 2003, 42, 3693–3696 CrossRef CAS PubMed.
- K. C. Nicolaou, D. L. F. Gray and J. Tae, J. Am. Chem. Soc., 2004, 126, 613–627 CrossRef CAS PubMed.
- L. Dell'Amico, A. Vega-Peñaloza, S. Cuadros and P. Melchiorre, Angew. Chem., Int. Ed., 2016, 55, 3313–3317 CrossRef PubMed.
- (a) B. Yang, K. Lin, Y. Shi and S. Gao, Nat. Commun., 2017, 8, 622 CrossRef PubMed; (b) D. Xue, M. Xu, C. Zheng, B. Yang, M. Hou, H. He and S. Gao, Chin. J. Chem., 2019, 37, 135–139 CrossRef CAS; (c) D. Jiang, K. Xin, B. Yang, Y. Chen, Q. Zhang, H. He and S. Gao, CCS Chem., 2020, 2, 800–812 CrossRef CAS; (d) M. Hou, M. Xu, B. Yang, H. He and S. Gao, Chem. Sci., 2021, 12, 7575–7582 RSC; (e) M. Hou, M. Xu, B. Yang, H. He and S. Gao, Org. Lett., 2021, 23, 7487–7491 CrossRef CAS PubMed; (f) X.-L. Lu, Y. Qiu, B. Yang, H. He and S. Gao, Chem. Sci., 2021, 12, 4747–4752 RSC; (g) X.-L. Lu, B. Yang, H. He and S. Gao, Org. Chem. Front., 2021, 8, 1143–1148 RSC; (h) M. Xu, M. Hou, H. He and S. Gao, Angew. Chem., Int. Ed., 2021, 60, 16655–16660 CrossRef CAS PubMed; (i) B. Yang, G. Wen, Q. Zhang, M. Hou, H. He and S. Gao, J. Am. Chem. Soc., 2021, 143, 6370–6375 CrossRef CAS PubMed; (j) Q. Zhang, H. He and S. Gao, Chem. Commun., 2022, 58, 4239–4242 RSC.
- (a) L. Dell'Amico, V. M. Fernández-Alvarez, F. Maseras and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 3304–3308 CrossRef PubMed; (b) X. Yuan, S. Dong, Z. Liu, G. Wu, C. Zou and J. Ye, Org. Lett., 2017, 19, 2322–2325 CrossRef CAS PubMed.
- (a) S. Cuadros, L. Dell'Amico and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 11875–11879 CrossRef CAS PubMed; (b) S. Cao, J. Li, T. Yan, J. Han and Z. He, Org. Chem. Front., 2022, 9, 643–648 RSC.
- S. Paria, E. Carletti, M. Marcon, A. Cherubini-Celli, A. Mazzanti, M. Rancan, L. Dell'Amico, M. Bonchio and X. Companyó, J. Org. Chem., 2020, 85, 4463–4474 CrossRef CAS PubMed.
- H. B. Hepburn, G. Magagnano and P. Melchiorre, Synthesis, 2017, 49, 76–86 CAS.
- For reviews and selected examples on chiral N,N′-dioxides, see: (a) X. H. Liu, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed; (b) X. H. Liu, L. L. Lin and X. M. Feng, Org. Chem. Front., 2014, 1, 298–302 RSC; (c) X. H. Liu, H. F. Zheng, Y. Xia, L. L. Lin and X. M. Feng, Acc. Chem. Res., 2017, 50, 2621–2631 CrossRef CAS PubMed; (d) X. H. Liu, S. X. Dong, L. L. Lin and X. M. Feng, Chin. J. Chem., 2018, 36, 791–797 CrossRef CAS; (e) T. F. Kang, W. D. Cao, L. Z. Hou, Q. Tang, S. J. Zou, X. H. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2019, 58, 2464–2468 CrossRef CAS PubMed; (f) Z. Wang, X. H. Liu and X. M. Feng, Aldrichimica Acta, 2020, 53, 3–10 CAS; (g) M. Y. Wang and W. Li, Chin. J. Chem., 2021, 39, 969–984 CrossRef CAS; (h) X. B. Lin, Z. Tan, W. K. Yang, W. Yang, X. H. Liu and X. M. Feng, CCS Chem., 2021, 3, 1423–1433 CrossRef CAS; (i) G. H. Pan, C. L. He, M. Chen, Q. Xiong, W. D. Cao and X. M. Feng, CCS Chem., 2021, 3, 2012–2020 Search PubMed. For examples in photocatalysis: (j) H. Yu, S. X. Dong, Q. Yao, L. Chen, D. Zhang, X. H. Liu and X. M. Feng, Chem.–Eur. J., 2018, 24, 19361–19367 CrossRef CAS PubMed; (k) C.-X. Ye, Y. Y. Melcamu, H.-H. Li, J.-T. Cheng, T.-T. Zhang, Y.-P. Ruan, X. Zheng, X. Lu and P.-Q. Huang, Nat. Commun., 2018, 9, 410 CrossRef PubMed; (l) D. Zhang, Z. S. Su, Q. W. He, Z. K. Wu, Y. Q. Zhou, C. J. Pan, X. H. Liu and X. M. Feng, J. Am. Chem. Soc., 2020, 142, 15975–15985 CrossRef CAS PubMed; (m) H. Yu, T. Y. Zhan, Y. Q. Zhou, L. Chen, X. H. Liu and X. M. Feng, ACS Catal., 2022, 12, 5136–5144 CrossRef CAS; (n) Z. D. Tan, S. B. Zhu, Y. B. Liu and X. M. Feng, Angew. Chem., Int. Ed., 2022, e202203374 Search PubMed.
- Lithium salts with different counterions (LiCl, LiBF4, and LiOTf) and NaNTf2 were examined as well. The results showed that NTf2− plays an important role in enhancement of stereoselectivity (see the ESI† for details).
- (a) CCDC 2081680 (C1); †; (b) CCDC 2130299 (E1); †; (c) CCDC 2171982 (F1′).†.
- (a) M. Oyo and Y. Masaaki, Bull. Chem. Soc. Jpn., 1967, 40, 2380–2382 CrossRef; (b) M. Oyo, Y. Masaaki and M. Teruaki, Bull. Chem. Soc. Jpn., 1967, 40, 935–939 CrossRef; (c) T. Fukuyama, C.-K. Jow and M. Cheung, Tetrahedron Lett., 1995, 36, 6373–6374 CrossRef CAS; (d) N. García-Delgado, A. Riera and X. Verdaguer, Org. Lett., 2007, 9, 635–638 CrossRef PubMed.
- For selected reviews of diastereodivergent synthesis, see: (a) M. Bihani and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534–575 CrossRef CAS; (b) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627–5639 CrossRef CAS PubMed; (c) L. L. Lin and X. M. Feng, Chem.–Eur. J., 2017, 23, 6464–6482 CrossRef CAS PubMed; (d) G. Zhan, W. Du and Y.-C. Chen, Chem. Soc. Rev., 2017, 46, 1675–1692 RSC; (e) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080–5200 CrossRef CAS PubMed.
- For reviews of photoreactivity of imines, see: (a) A. Padwa, Chem. Rev., 1977, 77, 37–68 CrossRef CAS; (b) A. C. Pratt, Chem. Soc. Rev., 1977, 6, 63–81 RSC; (c) A. D. Richardson, M. R. Becker and C. S. Schindler, Chem. Sci., 2020, 11, 7553–7561 RSC; (d) D. Uraguchi, Y. Tsuchiya, T. Ohtani, T. Enomoto, S. Masaoka, D. Yokogawa and T. Ooi, Angew. Chem., Int. Ed., 2020, 59, 3665–3670 CrossRef CAS PubMed; (e) S. K. Kandappa, L. K. Valloli, S. Ahuja, J. Parthiban and J. Sivaguru, Chem. Soc. Rev., 2021, 50, 1617–1641 RSC.
- L. Z. Hou, T. F. Kang, L. K. Yang, W. D. Cao and X. M. Feng, Org. Lett., 2020, 22, 1390–1395 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H} and 19F{1H} NMR, HPLC spectra, and CD spectra. X-ray crystallographic data for C1, E1 and F1′. CCDC 2081680, 2130299 and 2171982. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02721f |
| This journal is © The Royal Society of Chemistry 2022 |