
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater molecular bridge-induced selective dual polarization in crystals for stable multi-emitters†
Yi
Xing
a,
Zhongyu
Li
a,
Glib V.
Baryshnikov
 b,
Shen
Shen
a,
Danfeng
Ye
a,
Hans
Ågren
c and
Liangliang
Zhu
*a
b,
Shen
Shen
a,
Danfeng
Ye
a,
Hans
Ågren
c and
Liangliang
Zhu
*a
aState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200438, China. E-mail: zhuliangliang@fudan.edu.cn
bLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, 60174 Norrköping, Sweden
cDepartment of Physics and Astronomy, Uppsala University, Box 516, SE-751 20 Uppsala, Sweden
First published on 26th April 2022
Abstract
In the solid state, the molecular polarization of donor–acceptor (D–A) molecules can be implemented in a simple way via the use of an external polarizing source (e.g., an electric field). However, internal chemical polarization approaches are less studied due to difficulties related to controlling the charge-separation orientation in the solid state. Herein, a series of D–A molecules with both a proton donor and an acceptor were designed. Water-based molecular bridges were then established in their crystal structures, which firmly and alternately connected the proton donor of one molecule and the acceptor of another via an intermolecular H-bond network. In this way, the selective dual polarization of a phenolic hydroxyl group and a pyridinyl group could be achieved, owing to the strengthening of the charge-separation orientation upon the simultaneous deprotonation and protonation of the D–A molecules. This effect led to a 3–5-fold amplification of the molecular dipole moment in the crystal form relative to the monomeric state. On this basis, multi-excitation and multi-emission characteristics were achieved in these charge-separated crystals, endowing them with the ability to visually detect the energy of a light source, covering a wide range of the UV-Vis spectral region. This work provides a practical chemical approach for developing intrinsically polarized systems that can exhibit stable but distinct molecular photophysical properties.
Introduction
Donor–acceptor (D–A) molecules have received much attention due to their fascinating applications in modern optoelectronics.1 They can be easily polarized to regulate the optoelectronic properties, particularly the molecular photoluminescence and other photophysical behavior.2 A common polarization case in solution involves the internal charge-transfer mechanism, which is generally connected with a wavelength shift and/or multiple emission from D–A molecules in polar solvents.1g,3 However, solution states generally suffer from solvent loss4 and recycling-based challenges5 when used. In crystals or solid films, external stimuli, like an electronic field, polarized/circular polarized light, magnetic control, or an additional energy source, are usually employed for polarizing molecules and tuning their corresponding photophysical properties.6 Developing internal chemical polarization approaches that are smart and low cost remains challenging, but they are desirable for achieving stable and distinct molecular photophysical properties.In the process of molecular polarization, the molecular dipole moment is normally used to measure the sum effects of charge separation.7 Therefore, the internal chemical-based control of charge-separation orientation in a solid material can be a key factor for addressing the above-mentioned issue. ortho-Pyridinil phenols, a class of D–A molecular structures with both a proton donor and acceptor, can readily form an intermolecular H-bond dimeric structure through the proton donor and acceptor.8 Inspired by the selective bonding mode in these structures, we expect that a stable H-bond network can be constructed formed from ortho-pyridinil phenols together with polar chemicals (like water) in an ordered and alternating fashion, so as to create an oriented polarization effect induced by a perfect H-bond environment.
Here, we designed and synthesized a series of new ortho-pyridinil phenol molecules with a phenolic hydroxyl group (proton donor) and a pyridinyl group (proton acceptor) (as illustrated in the chemical structures of DPP-F, DPP, and PP-F in Fig. 1a). The introduction of a strongly electronegative fluorine atom that may make an extra contribution to molecular polarization is also considered.9 Then, via the hydrated crystal growth of these molecules, water-based molecular bridges were constructed, which firmly and alternately connected the proton donor of one molecule and the acceptor of another through an intermolecular H-bond network. This ordered and alternating H-bond superstructure led to water-induced intermolecular charge separation, followed by the strong selective dual polarization of the phenolic hydroxyl group and the pyridinyl group. The strengthened charge-separation orientation relied on the simultaneous deprotonation and protonation of the D–A molecules in the inherent crystal system (see the illustration in Fig. 1b), where enlarged dipole moments were obtained (a 3–5-fold amplification of the molecular dipole moments in the crystal form relative to the monomeric state). Based on this characteristic, multi-excitation and multi-emission properties can be obtained using these crystal platforms, originating from these charge-separated species, and these properties can be further bathochromically or hypochromatically regulated via J-aggregation or H-aggregation in the crystal molecular packing to cover a wide spectral region of 300–610 nm. These properties endowed the luminescence of the materials with excitation-wavelength dependence and they can be utilized for the large-scale visual detection of variable light sources.
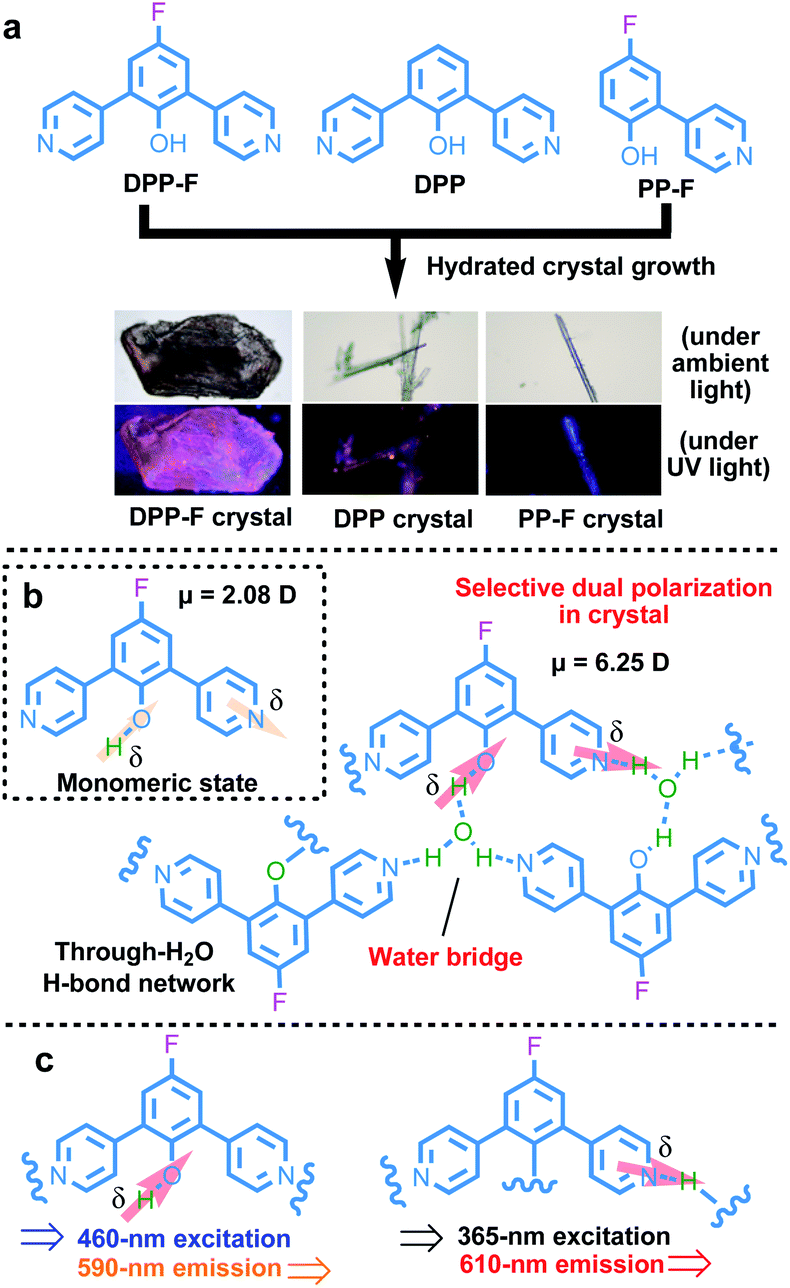 | ||
| Fig. 1 (a) The chemical structures of the D–A molecules and photographs of their hydrated crystals. (b) An illustration of the water-based molecular-bridge-stabilized H-bond network that can induce a strong selective dual polarization effect involving the phenolic hydroxyl group and the pyridinyl group, causing strengthened charge-separation orientation, taking the DPP-F crystal as an example. The direction and extent of electron flow upon charge separation are highlighted by arrows. (c) A summary of the excitation and emission characteristics of two differently polarized species. | ||
Results and discussion
Deprotonation in solution
Firstly, the deprotonation behavior of these D–A molecules, which can reflect their selective charge-separation abilities, was monitored in different solvents. Focusing on DPP-F, we find a dual absorption phenomenon (a band at 300–340 nm and another at 390–480 nm) in DMF and DMSO, compared to a single band (300–350 nm) in other benign solvents (Fig. 2a). Correspondingly, DPP-F could emit a strong band in DMSO and DMF with a nanosecond-scale (4.72 ns) fluorescent lifetime (Fig. S1†), compared to the other solvents, which showed barely any emission (Fig. 2b). As the phenol groups of ortho-pyridinil generally show absorption in a relatively short wavelength region without photoluminescence,8 these results indicated that DMF and DMSO played a prominent role in pushing the dynamic equilibrium towards deprotonation, and the formation of phenolate species was the root of the luminescent properties. It seems that highly polar solvents like DMF and DMSO could stabilize the transition state to promote deprotonation, owing to different solvent effects.10 This stabilization of the transition state probably involves H bonds,11 since ACN, which has large polarity, did not cause equivalent deprotonation due to its weak H-bond forming abilities. The other two compounds, DPP and PP-F, behaved similarly in DMF, showing relatively remarkable dual absorption and emission (Fig. S1†) featuring a similar type of deprotonation. However, the absorption and emission intensities of DPP and PP-F were weaker than those of DPP-F due to differences in chemical structure.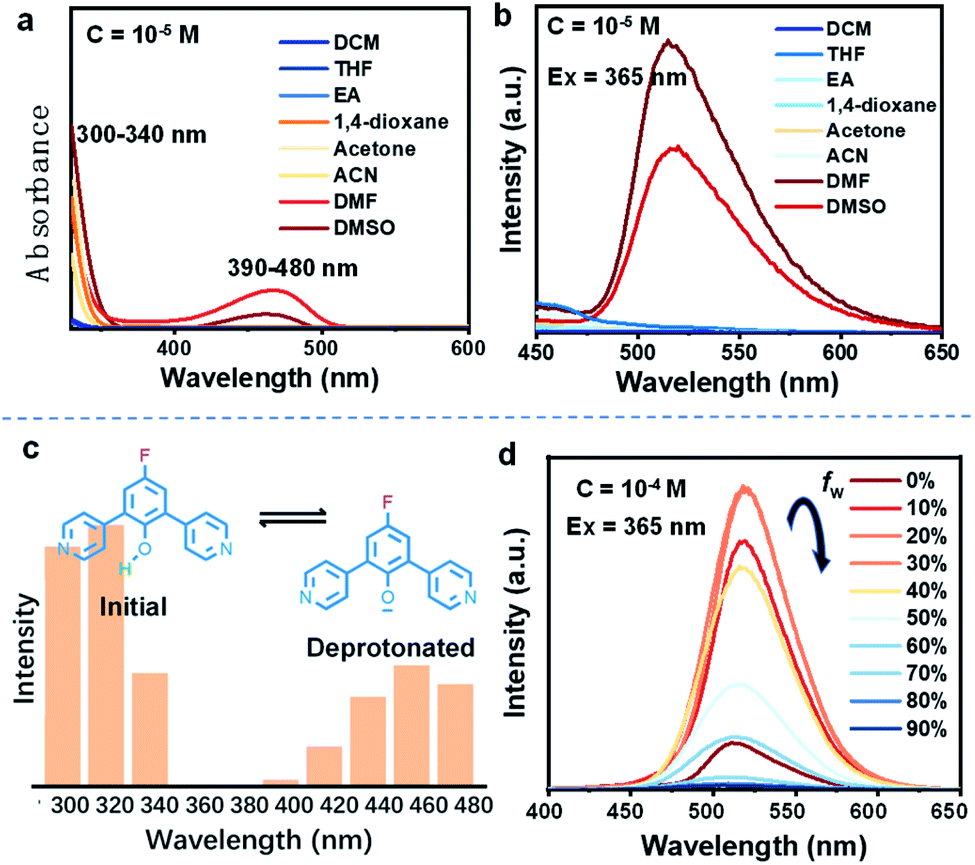 | ||
| Fig. 2 (a) Absorption and (b) emission spectra (λex = 365 nm) of DPP-F in different benign solvents. (c) The emission intensity distribution (515 nm) of DPP-F in DMF at different excitation wavelengths showing two distinct excitation events from the initial state and deprotonated state, respectively. (d) The emission spectra (λex = 365 nm) of DPP-F in DMSO with different water fractions (fw represents the water volume fraction). | ||
In addition, we altered the excitation wavelength to reveal its relationship with the emission intensity in DMF, and we observed a unique dual-excitation phenomenon (Fig. 2c). The first excitation was attributed to the initial state of the molecule, while the second originated from the deprotonated state, similar to its absorption characteristics; this could be further proved based on the calculated electronic transition data (see the neutral form (312 nm) and the anionic form (460 nm) in Fig. S2†). Here, molecular aggregation did not play a major role in regulating the photophysical properties of these molecules in solution, while DPP-F only presented a linear variation of the emission intensity as the concentration gradient increased rather than a wavelength shift (Fig. S3†). When water was introduced into DMSO solution of DPP-F, the emission intensity was continuously raised as the water fraction increased up to 30% (Fig. 2d). This increase was because the introduction of water further enhanced the polarity so as to promote the emission of DPP-F. A further increase in the water fraction offset the dynamic equilibrium and was not conducive to the deprotonation of DPP-F, which resulted in a drop in emission intensity (Fig. 2d).
Selective dual polarization in the hydrated crystals
When turning to the solid powder form of DPP-F, broad absorption characteristics could be seen, with a main band generally located below 400 nm and a broad band in the visible region from 400 to 650 nm (Fig. S4a†). DPP showed similar broad absorption, while PP-F showed weak absorption between 525 nm and 700 nm (Fig. S4b and c†). This originated from the existence of molecular dimers maintained by intermolecular H-bonding between the proton donor of one molecule and the acceptor of another in the solid state.8 Such intermolecular H-bonds in the bulk generated relatively weak polarization to produce new electronic transitions (a charge-transfer state) from polarized species. However, the photoluminescence of these bulk states is still weak.To strengthen the selective polarization effects, the hydrated crystal state was a focus in our system because of two reasons: (1) water, a solvent with high polarity, can be readily utilized through the immobilization of a rigid crystal cell via selective intermolecular H-bonds;12 and (2) rigid and regular molecular packing in the crystal state13 may be helpful for possible oriented dipole-moment regulation. Based on this kind of intrinsic structural design, we attempted to introduce water-based bridges into these systems to generate an ordered and alternating intermolecular H-bond mode to regulate charge separation within the rigid crystal conformation. Via a hydrated crystal growth strategy, we obtained a hydrated single crystal of DPP-F and the control compounds DPP and PP-F (Fig. S39–S41†) with significantly different apparent and luminescent colors (see the images captured under a microscope shown in Fig. 1a).
Two-stage weight-loss during thermal gravimetric analysis (TGA) and the analysis of single-crystal data signified the existence of crystalline water in the DPP-F crystal; the first-stage weight-loss corresponds to the amount of hydration. Similar weight-loss trends could be observed for DPP and PP-F as well (Fig. S5 and S39–S41†). The X-ray diffraction (XRD) results from DPP-F revealed the structure change upon the loss of crystalline water through dehydration, turning the crystals to powder form (see the XRD patterns in Fig. S6† and the simulated powder XRD patterns of reference compounds in Fig. S7†). Direct evidence allowing the characterization of the hydrated crystals could be obtained via single-crystal X-ray analysis. There were two conformations plus two water molecules coexisting in an asymmetric unit of a DPP-F crystal, while one conformation and one water molecule were present in the DPP crystal and PP-F crystal units (see Fig. 3a and S39–S41†). Through crystallization, intermolecular water-based bridges were formed, which firmly and alternately connect the phenolic hydroxyl group of one molecule and the pyridinyl group of another via intermolecular H-bonds in these crystals (see the packing mode in Fig. S8†). Through single-crystal analysis, we could find that in these stable H-bonded networks the O–H distance for phenolic hydroxyl groups in all compounds was <1 Å (within the typical covalent-bond range). Meanwhile, the N⋯H distance between the pyridine group and a hydrogen atom in an adjacent water molecule was maintained at >1.8 Å (within the typical non-covalent bond range). Different from the case in solution, these results indicated that hydrated crystallization could not cause full deprotonation or protonation. It is reasonable to speculate that this phenomenon involves molecular polarization.
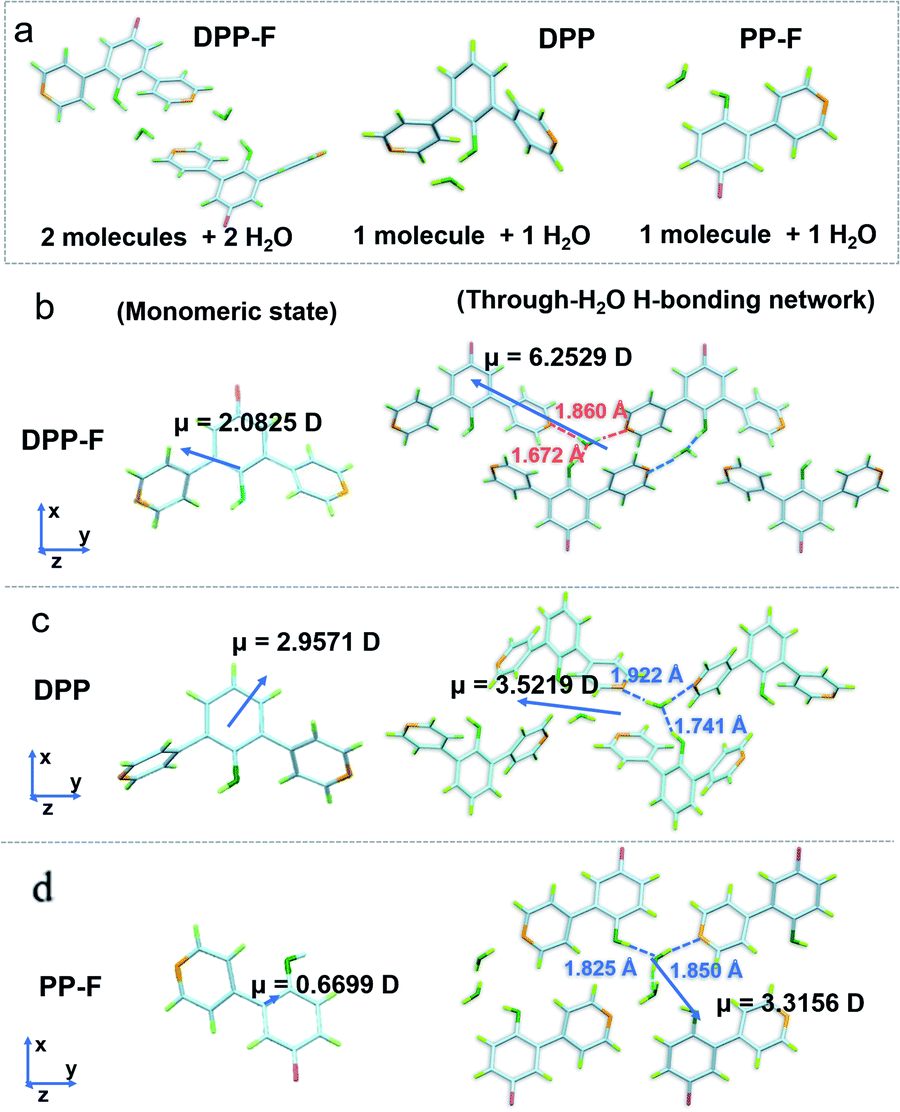 | ||
| Fig. 3 (a) The X-ray crystallographic structures of DPP-F, DPP, and PP-F in asymmetric units. The calculated dipole moments of (b) DPP-F, (c) DPP, and (d) PP-F in monomeric mode and through-H2O H-bonding network mode. The through-H2O H-bonding distances are also highlighted for each compound. | ||
On this basis, unique selective dual polarization was achieved in the crystal structures through double H-bond networks in the form of O⋯H or N⋯H, thus, strong charge-separation orientation could be established via water-based bridges in these systems. This strengthened charge-separation between molecules produced partially negatively charged and partially positively charged species existing simultaneously, which could be observed based on the electrostatic potential surfaces of the three crystals (as illustrated in Fig. 1, S9, and Table S1†). As a result, the total dipole moment in the hydrated crystals became considerably larger than in the corresponding monomeric state, due to the directional character and abundance of H bonds in the crystal structure. DFT simulations reveal that the dipole moments of the three monomers are 2.0825 D for DPP-F, 2.9571 D for DPP, and 0.6699 D for PP-F (Fig. 3b–d). However, with H-bond-network-regulated attraction, the dipole moments of these system increased to 6.2529 D, 3.5219 D, and 3.3156 D, respectively (Fig. 3). These results suggested that remarkable charge-separation orientation upon selective dual polarization inside the system could significantly enhance the dipole–dipole interactions. Furthermore, when water was removed from the crystal models, significant decreases in the dipole moments were observed (values of 1.4968 D for DPP-F, 0.0010 D for DPP, and 0.0974 D for PP-F, as shown in Fig. S10†). These results indicate that in the absence of water, the dipole moments of the molecules could be offset by each other, showing that the H-bond network created via water-based molecular bridges played a significant role in the dual polarization.
Multi-excitation and multi-emission characteristics
Based on the unique dual polarization, a red emission band (λmax = 612 nm) and an orange emission band (λmax = 590 nm), excited at 365 nm and 460 nm, respectively, could be observed from the DPP-F crystal. Two emission centers appeared for the DPP and PP-F crystals as well, located at 610 nm and 630 nm (DPP) and at 433 nm and 448 nm (PP-F) (see the three-dimensional photoluminescence spectral maps in Fig. 4a). The excitation abilities of these multiple emission events were also reflected through the presence of broad absorption bands, in spite of the scattering caused by the crystal structure (Fig. 4b). Upon irradiation, phenolate and pyridinium species were produced by an excited-state intermolecular proton transfer process assisted by water-based molecular bridges (probably owing to the favorable lengths of these intermolecular H-bonds, see Fig. S11† taking a DPP-F crystal as an example). This process led to the distinct dual-emission bands (Fig. 4a), and both of the emission bands exhibited short-lifetime fluorescence characteristics as well (Fig. S12†). To verify these species, fully protonated (reacted with HCl) and fully deprotonated (reacted with NaOH) species of DPP-F were prepared as reference materials (Fig. S13†), and their fluorescence-signal wavelengths were identical to the dual-emission characteristics of the DPP-F crystal (Fig. S14†). These results signified the formation of phenolate and pyridinium upon excitation, further supporting the emission theory. Then, absorption could be designated according to the relationship between excitation and emission based on the mapping shown in Fig. 4a, where the main absorption band located at ∼350 nm could be assigned to a local state and other polarized states (i.e., charge-transfer states) were responsible for absorption at 365 nm and 460 nm in the case of the DPP-F crystal (Fig. 4b). The excited-state intermolecular proton-transfer process can be explored based on the secondary excited-state absorption signal14 growing on the picosecond scale (see the transient absorption measurements in Fig. S15†) and the results of quantum-chemical calculations (Fig. S16†). The optimization of the excited-state geometry of DPP-F in the hydrated crystal state led to barrierless proton-transfer between two neighboring molecules mediated by a water-based bridge with the formation of phenolate and pyridinium species, and this could spontaneously return to the initial state.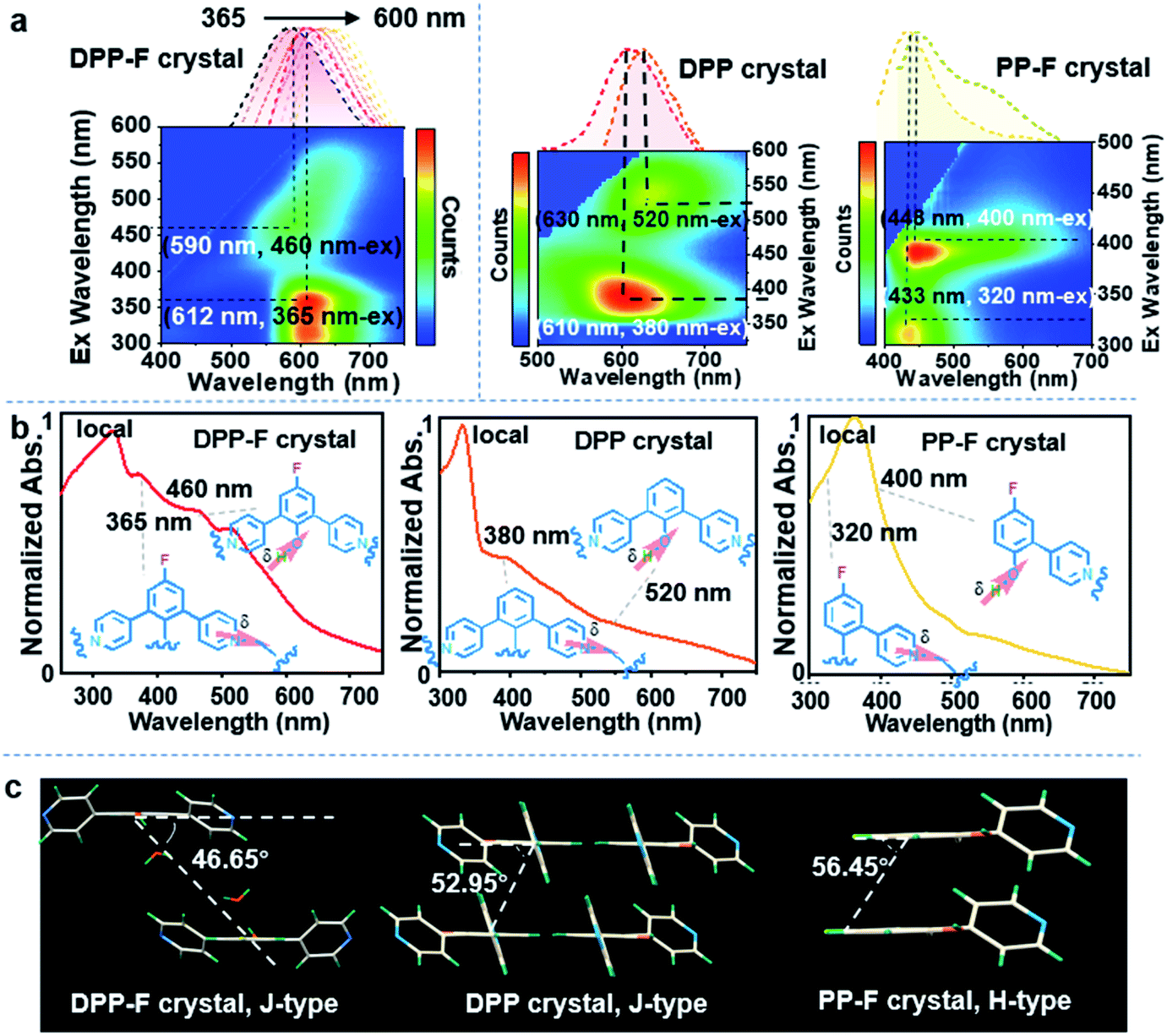 | ||
| Fig. 4 (a) Excitation–emission mapping of the three crystals at room temperature. The upper inset shows normalized emission spectra at different excitation wavelengths. (b) The absorption spectra of the three crystals at room temperature. (c) The molecular packing of the three crystals. The angle between the molecular plane and the arrangement direction of the molecular stacking mode is highlighted. | ||
It could be found that the dual excitation and dual emission of DPP-F and DPP crystals were red shifted overall relative to the monomeric states (referenced from the results in solution). In contrast, the dual excitation and dual emission of the PP-F crystal were blue shifted overall. After further analysis of the single-crystal structures, we found that a synergic factor involving F atoms and pyridinyl groups in these molecules played a non-negligible role in the establishment of different molecular stacking modes in these hydrated crystals. Specifically, the angles between the molecular plane and the arrangement direction of the molecular stacking mode in DPP-F and DPP crystals were 46.65° and 52.95°, respectively (Fig. 4c), and J-aggregation can be ascribed as bringing about the red shifting of absorption and emission. However, in the PP-F crystal, this angle was 56.45° (Fig. 4c), which could be explained as being above the cross-stacking mode (54.7°);15 this could lead to spectral blue-shifting due to the effects of H-aggregation.
Excitation-wavelength-dependent photoluminescence
Of these three crystals, the DPP-F crystal exhibited the largest dipole moment and the strongest absorption, covering a large UV-Vis region. Fig. 3b–d compares the H-bond distances of DPP-F, DPP, and PP-F in crystal form. The results revealed that DPP-F had the shortest intermolecular double hydrogen bond length (3.532 Å). DPP and PP-F formed double H-bonds with lengths of 3.663 Å and 3.675 Å, respectively. The short distance promoted charge transfer more efficiently. Based on the strong absorption and emission of the DPP-F crystal, covering a large UV-Vis region, relatively remarkable excitation-wavelength-dependent photoluminescence was observed from the DPP-F crystal (see Fig. 4a and S17†), and this could be rationally explained based on variations in the ratio of the two emission intensities excited at different excitation wavelengths, leading to distinct photoluminescent color conversion (Fig. 5). Exploring the practical usage of this phenomenon, we demonstrated the visual detection of a light source using this crystal material. Upon a change in excitation from 365 to 600 nm, the luminescent color of the DPP-F crystal switched from pink to orange and eventually to red (Fig. 5, see the CIE-1931 chromaticity diagram with white dots signifying these luminescent color coordinates). Although in the engineering field it is common to detect excitation light wavelength with a physical device, this showcase of naked-eye detection offers a simple solution for wavelength identification with a portable chemical material, relying on the material luminescent color having excitation-wavelength-dependent characteristics covering a large UV-Vis spectral region.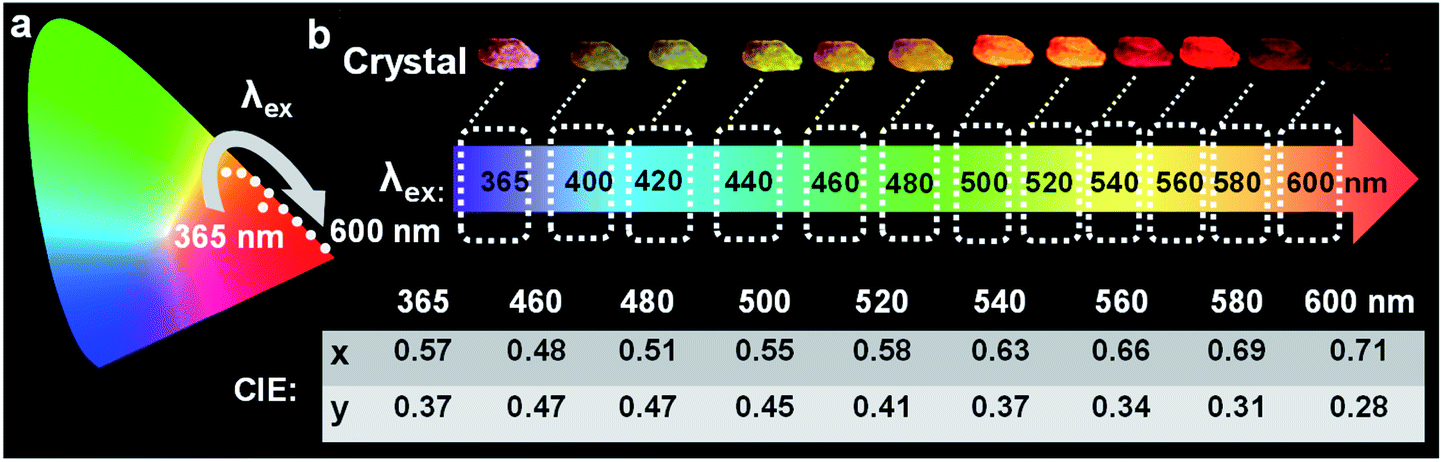 | ||
| Fig. 5 (a) The CIE-1931 chromaticity diagram with dots signifying the luminescent color coordinates of the DPP-F crystal in the corresponding states upon changing the excitation wavelength. (b) Corresponding DPP-F crystal photographs taken upon excitation at different wavelengths showing the ability of the crystal to visually detect specific wavelengths of light covering a large UV-Vis spectral region due to the excitation-dependent emission wavelength. | ||
Conclusions
We designed and fabricated a series of D–A molecules where the phenolic hydroxyl group acts as a proton donor and the pyridinyl group acts as an acceptor. Under the effects of water-based molecular bridges, the crystallization of these molecules with water promoted selective dual polarization, resulting in strong and oriented charge separation through this method due to the simultaneous deprotonation and protonation tendency of the D–A molecules. This is an internal chemical-based approach for obtaining large molecular dipole–dipole interactions based on ordered and alternating H-bond networks in these crystal platforms. Such dual polarization endowed the material with multi-excitation and multi-emission characteristics, with excitation-wavelength dependence. Harnessing these stable properties, we showcased a visual light detector that can work over a large UV-Vis spectral range. These results can be valuable for inspiring more advanced and sophisticated approaches for fabricating solid-state materials with distinct optoelectronic properties.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
L. Z. and Y. X. conceived this project and designed the experiments. Y. X., Z. L., and D. Y. performed the synthetic work. Y. X. carried out the spectral experiments. S. S. contributed to part of the crystal structural study. Y. X., G. V. B., and H. A. performed the quantum-chemical calculations. Y. X. prepared the manuscript, and the other authors helped revise the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by 2019 NSFC (21975046), and partially by the National Key Research and Development Program of China (2017YFA0207700). G. V. B. acknowledges financial support from the Swedish Research Council (starting grant no. 2020-04600). The quantum-chemical calculations were performed with computational resources provided by the Swedish National Infrastructure for Computing (SNIC 2020-3-29) at the High-Performance Computing Center North (HPC2N), partially funded by the Swedish Research Council through Grant Agreement No. 2018-05973. We thank Mr M. Lv and Prof. J. Chen (ECNU) for assistance with the TA study, and Ms X. Jiang and Prof. J. Li (Tsinghua) for assistance with the electrostatic potential surface calculations.Notes and references
- (a) H.-T. Feng, J. Zeng, P.-A. Yin, X.-D. Wang, Q. Peng, Z. Zhao, J. W. Y. Lam and B. Z. Tang, Nat. Commun., 2020, 11, 2617 CrossRef CAS PubMed; (b) J. Zhang, A. Li, H. Zou, J. Peng, J. Guo, W. Wu, H. Zhang, J. Zhang, X. Gu, W. Xu, S. Xu, S. H. Liu, A. Qin, J. W. Y. Lam and B. Z. Tang, Mater. Horiz., 2020, 7, 135–142 RSC; (c) J. Wang, S. Liu, K. Chang, Q. Liao, S. Li, H. Han, Q. Li and Z. Li, J. Mater. Chem. C, 2020, 8, 14453–14461 RSC; (d) Y. Wang, B. Chen, W. Wu, X. Li, W. Zhu, H. Tian and Y. Xie, Angew. Chem., Int. Ed., 2014, 53, 10779–10783 CrossRef CAS PubMed; (e) X. Li, G. Baryshnikov, C. Deng, X. Bao, B. Wu, Y. Zhou, H. Ågren and L. Zhu, Nat. Commun., 2019, 10, 731 CrossRef CAS PubMed; (f) Y. Zhou, Y. Zhuang, X. Li, H. Ågren, L. Yu, J. Ding and L. Zhu, Chem.–Eur. J., 2017, 23, 7642–7647 CrossRef CAS PubMed; (g) M. Luo, X. Li, L. Ding, G. Baryshnikov, S. Shen, M. Zhu, L. Zhou, M. Zhang, J. Lu, H. Ågren, X.-d. Wang and L. Zhu, Angew. Chem., Int. Ed., 2020, 59, 17018–17025 CrossRef CAS PubMed; (h) Y. Yang, J. Ouyang, L. Ma, R. J. H. Tseng and C. W. Chu, Adv. Funct. Mater., 2006, 16, 1001–1014 CrossRef CAS; (i) S. Miao, H. Li, Q. Xu, Y. Li, S. Ji, N. Li, L. Wang, J. Zheng and J. Lu, Adv. Mater., 2012, 24, 6210–6215 CrossRef CAS PubMed; (j) W. Li, L. Ye, S. Li, H. Yao, H. Ade and J. Hou, Adv. Mater., 2018, 30, 1707170 CrossRef PubMed; (k) J. Yang, J. Jing and Y. Zhu, Adv. Mater., 2021, 33, 2101026 CrossRef CAS PubMed; (l) M. Sarma and K.-T. Wong, ACS Appl. Mater. Interfaces, 2018, 10, 19279–19304 CrossRef CAS PubMed; (m) T. S. Metzger, C. G. Chandaluri, R. Tel-Vered, R. Shenhar and I. Willner, Adv. Funct. Mater., 2016, 26, 7148–7155 CrossRef CAS; (n) R. K. Konidena, J. Lim and J. Y. Lee, Chem. Eng. J., 2021, 416, 129097 CrossRef CAS.
- (a) S. Qi, S. Kim, V.-N. Nguyen, Y. Kim, G. Niu, G. Kim, S.-J. Kim, S. Park and J. Yoon, ACS Appl. Mater. Interfaces, 2020, 12, 51293–51301 CrossRef CAS PubMed; (b) X.-D. Xu, J. Zhang, X. Yu, L.-J. Chen, D.-X. Wang, T. Yi, F. Li and H.-B. Yang, Chem.–Eur. J., 2012, 18, 16000–16013 CrossRef CAS PubMed; (c) J.-X. Wang, L.-Y. Niu, P.-Z. Chen, Y.-Z. Chen, Q.-Z. Yang and R. Boulatov, Chem. Commun., 2019, 55, 7017–7020 RSC; (d) X. Li, S. Shen, C. Zhang, M. Liu, J. Lu and L. Zhu, Sci. China: Chem., 2021, 64, 534–546 CrossRef CAS; (e) F. Chen, H. Huang, L. Guo, Y. Zhang and T. Ma, Angew. Chem., Int. Ed., 2019, 58, 10061–10073 CrossRef CAS PubMed; (f) M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed; (g) R. S. Nobuyasu, Z. Ren, G. C. Griffiths, A. S. Batsanov, P. Data, S. Yan, A. P. Monkman, M. R. Bryce and F. B. Dias, Adv. Opt. Mater., 2016, 4, 597–607 CrossRef CAS.
- (a) L. Zhu, X. Li, Q. Zhang, X. Ma, M. Li, H. Zhang, Z. Luo, H. Ågren and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 5175–5182 CrossRef CAS PubMed; (b) X. Li, G. Baryshnikov, L. Ding, X. Bao, X. Li, J. Lu, M. Liu, S. Shen, M. Luo, M. Zhang, H. Ågren, X. Wang and L. Zhu, Angew. Chem., Int. Ed., 2020, 59, 7548–7554 CrossRef CAS PubMed; (c) K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS; (d) D.-d. Zhang, K. Suzuki, X.-Z. Song, Y. Wada, S. Kubo, L. Duan and H. Kaji, ACS Appl. Mater. Interfaces, 2019, 11, 7192–7198 CrossRef CAS PubMed; (e) Y. Geng, A. D'Aleo, K. Inada, L.-S. Cui, J. U. Kim, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2017, 56, 16536–16540 CrossRef CAS PubMed; (f) F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- I. V. Muralikrishna and V. Manickam, Environ. Manage., 2017, 463–494 Search PubMed.
- K. S. Goh, Y. Chen, J. Y. Chong, T. H. Bae and R. Wang, J. Membr. Sci., 2021, 621, 119008 CrossRef CAS.
- (a) P.-Y. Gu, J. Zhang, G. Long, Z. Wang and Q. Zhang, J. Mater. Chem. C, 2016, 4, 3809–3814 RSC; (b) J. Yeom, B. Yeom, H. Chan, K. W. Smith, S. Dominguez-Medina, J. H. Bahng, G. Zhao, W.-S. Chang, S.-J. Chang, A. Chuvilin, D. Melnikau, A. L. Rogach, P. Zhang, S. Link, P. Král and N. A. Kotov, Nat. Mater., 2015, 14, 66–72 CrossRef CAS PubMed; (c) H. Bin, L. Gao, Z.-G. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang, M. Xiao and Y. Li, Nat. Commun., 2016, 7, 13651 CrossRef CAS PubMed; (d) S. Li, L. Ye, W. Zhao, X. Liu, J. Zhu, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1704051 CrossRef PubMed; (e) J. Huang, B. Peng, W. Wang, H. Ji, L. Li, K. Xi, W. Lai, X. Zhang and X. Jia, Adv. Funct. Mater., 2016, 26, 1646–1655 CrossRef CAS.
- (a) C.-T. Poon, D. Wu and V. W.-W. Yam, Angew. Chem., Int. Ed., 2016, 55, 3647–3651 CrossRef CAS PubMed; (b) J. Lee, A. J. Kalin, C. Wang, J. T. Early, M. Al-Hashimi and L. Fang, Polym. Chem., 2018, 9, 1603–1609 RSC.
- (a) Z. Li, Y. Wang, G. Baryshnikov, S. Shen, M. Zhang, Q. Zou, H. Ågren and L. Zhu, Nat. Commun., 2021, 12, 908 CrossRef CAS PubMed; (b) A. Li, Z. Li, M. Zhang, B. Wu, Y. Xing and L. Zhu, Adv. Opt. Mater., 2022, 10, 2102146 CrossRef CAS.
- (a) S. Dai, F. Zhao, Q. Zhang, T.-K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336–1343 CrossRef CAS PubMed; (b) Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed; (c) H. Wu, W. Chi, G. Baryshnikov, B. Wu, Y. Gong, D. Zheng, X. Li, Y. Zhao, X. Liu, H. Ågren and L. Zhu, Angew. Chem., Int. Ed., 2019, 58, 4328–4333 CrossRef CAS PubMed.
- M. H. Abraham, J. Chem. Soc., Dalton Trans., 1969, 1307–1308 CAS.
- S. Arseniyadis, A. Valleix, A. Wagner and C. Mioskowski, Angew. Chem., Int. Ed., 2004, 43, 3314–3317 CrossRef PubMed.
- (a) F. Zhou, P. Gu, Z. Luo, H. K. Bisoyi, Y. Ji, Y. Li, Q. Xu, Q. Li and J. Lu, Nat. Commun., 2021, 12, 2339 CrossRef CAS PubMed; (b) J. Othong, J. Boonmak, F. Kielar and S. Youngme, ACS Appl. Mater. Interfaces, 2020, 12, 41776–41784 CrossRef CAS PubMed.
- (a) S. Shen, G. Baryshnikov, B. Yue, B. Wu, X. Li, M. Zhang, H. Ågren and L. Zhu, J. Mater. Chem. C, 2021, 9, 11707–11714 RSC; (b) T. Seki, N. Hoshino, Y. Suzuki and S. Hayashi, CrystEngComm, 2021, 23, 5686–5696 RSC.
- (a) H. Lin, X. Chang, D. Yan, W.-H. Fang and G. Cui, Chem. Sci., 2017, 8, 2086–2090 RSC; (b) Y.-C. Wei, Z. Zhang, Y.-A. Chen, C.-H. Wu, Z.-Y. Liu, S.-Y. Ho, J.-C. Liu, J.-A. Lin and P.-T. Chou, Commun. Chem., 2019, 2, 10 CrossRef; (c) H. Sun, S.-S. Sun, F.-F. Han, Z.-H. Ni, R. Zhang and M.-D. Li, J. Mater. Chem. C, 2019, 7, 7053–7060 RSC; (d) H. Sun, S.-S. Sun, F.-F. Han, Y. Zhao, M.-D. Li, B.-X. Miao, J. Nie, R. Zhang and Z.-H. Ni, Chem. Eng. J., 2021, 405, 127000 CrossRef CAS.
- (a) J. Seo, S. Kim, S. H. Gihm, C. R. Park and S. Y. Park, J. Mater. Chem., 2007, 17, 5052–5057 RSC; (b) F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed; (c) J. Gierschner and S. Y. Park, J. Mater. Chem. C, 2013, 1, 5818–5832 RSC; (d) E. G. McRae and M. Kasha, J. Chem. Phys., 1958, 28, 721–722 CrossRef CAS; (e) C. Zheng, C. Zhong, C. J. Collison and F. C. Spano, J. Phys. Chem. C, 2019, 123, 3203–3215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2090475–2090477. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00908k |
| This journal is © The Royal Society of Chemistry 2022 |