
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering lanmodulin's selectivity for actinides over lanthanides by controlling solvent coordination and second-sphere interactions†
Joseph A.
Mattocks
 a,
Joseph A.
Cotruvo
Jr
*a and
Gauthier J.-P.
Deblonde
*bc
a,
Joseph A.
Cotruvo
Jr
*a and
Gauthier J.-P.
Deblonde
*bc
aDepartment of Chemistry, The Pennsylvania State University, University Park, Pennsylvania 16802, USA. E-mail: juc96@psu.edu
bPhysical and Life Sciences Directorate, Lawrence Livermore National Laboratory, Livermore, California 94550, USA. E-mail: deblonde1@LLNL.gov
cGlenn T. Seaborg Institute, Lawrence Livermore National Laboratory, Livermore, California 94550, USA
First published on 26th April 2022
Abstract
Developing chelators that combine high affinity and selectivity for lanthanides and/or actinides is paramount for numerous industries, including rare earths mining, nuclear waste management, and cancer medicine. In particular, achieving selectivity between actinides and lanthanides is notoriously difficult. The protein lanmodulin (LanM) is one of Nature's most selective chelators for trivalent actinides and lanthanides. However, mechanistic understanding of LanM's affinity and selectivity for f-elements remains limited. In order to decipher, and possibly improve, the features of LanM's metal-binding sites that contribute to this actinide/lanthanide selectivity, we characterized five LanM variants, substituting the aspartate residue at the 9th position of each metal-binding site with asparagine, histidine, alanine, methionine, and selenomethionine. Spectroscopic measurements with lanthanides (Nd3+ and Eu3+) and actinides (243Am3+ and 248Cm3+) reveal that, contrary to the behavior of small chelator complexes, metal-coordinated water molecules enhance LanM's affinity for f-elements and pH-stability of its complexes. Furthermore, the results show that the native aspartate does not coordinate the metal directly but rather hydrogen bonds to coordinated solvent. By tuning this first-sphere/second-sphere interaction, the asparagine variant nearly doubles LanM's selectivity for actinides versus lanthanides. This study not only clarifies the essential role of coordinated solvent for LanM's physiological function and separation applications, but it also demonstrates that LanM's preference for actinides over lanthanides can be further improved. More broadly, it demonstrates how biomolecular scaffolds possess an expanded repertoire of tunable interactions compared to most small-molecule ligands – providing an avenue for high-performance LanM-based actinide/lanthanide separation methods and bio-engineered chelators optimized for specific medical isotopes.
Introduction
The design of chelators that are selective for particular metal ions is at the core of numerous applications, including anti-cancer drugs,1,2 medical imaging,3,4 hydrometallurgy,5–7 analytical chemistry,8–10 and nuclear spent fuel reprocessing.11 Some of the most studied metal chelators are those for the extraction and purification of rare earth elements,5–7 and for actinide/lanthanide partition in the frame of nuclear spent fuel reprocessing.12–17 However, f-element separations are among the most difficult due to the chemical similarities among trivalent actinide (An3+) and lanthanide (Ln3+) ions, both within and between the 4f and 5f series.18,19 For instance, separation of lanthanide fission products from heavy actinides (i.e., Am3+ and Cm3+) present in nuclear spent fuel is a critical challenge to overcome for closing the nuclear fuel cycle, but processes able to separate the 4f and 5f series are notoriously arduous to develop and implement11 – equivalent to the separation of individual lanthanides but with additional radiation-related constraints. Typical approaches to increase the selectivity of small-molecule20,21 and peptide22 ligands for actinides focus on alteration of the first coordination sphere, exploiting differential aqueous ligand complexation kinetics,23 or differences in the extractant speciation in the organic phase24 (in the case of liquid–liquid extraction). Here, we show how dissection of the supramolecular architecture of the metal-binding sites in lanmodulin – a natural protein highly selective for f-elements – allows engineering of higher An3+/Ln3+ selectivity by a new approach, tuning solvent and second-sphere interactions.With the accelerating interest in f-element chemistry over the past decades,5,25 highly effective and selective chelators for trivalent actinides and lanthanides, and for their separation, remain some of the most coveted molecules.26–28 The recent discovery that numerous bacteria selectively acquire and use lanthanide ions for alcohol oxidation and perhaps other biochemical processes29,30 has the potential to provide new, efficient ligands for extraction and separations of lanthanides and actinides.31,32 In this context, a small (12 kDa) natural protein that selectively complexes trivalent lanthanide ions, named lanmodulin (LanM) because its structure is modulated by lanthanide binding, was identified in 2018.33 LanM is found in a gene cluster encoding the first identified lanthanide uptake system.33–36 Multiple lines of evidence, from the details of LanM's discovery and its biophysical properties to its expression being strongly induced in vitro and under physiologically relevant conditions in the presence of lanthanides,34,37 point to an important role in uptake of the light lanthanides preferred by the bacteria, although it is non-essential for growth in the presence of soluble La3+.34,36 Natural proteins that bind trivalent lanthanides and actinides had been studied38–47 before LanM but these metalloproteins bind Ln3+ and An3+ ions relatively weakly (Kd in the millimolar to micromolar range) and non-selectively, as they are instead meant to bind other cations in the natural environment. By contrast, LanM is naturally optimized to complex Ln3+ ions and exhibits unusually strong affinity and selectivity for them. LanM has three high-affinity metal binding sites and forms Ln3LanM complexes with Kds in the low picomolar range.33,35,48 Among the lanthanide series, its affinity is highest for Pr3+ and Nd3+,33,35,48 although the La3+ complex is most resistant to physiological chelators such as citrate.7 In fact, LanM is the strongest known natural macrochelator for lanthanides, outcompeting previously studied proteins and even the synthetic peptide “lanthanide-binding tags” (LBTs)49–51 by multiple orders of magnitude.
Our team has recently demonstrated that LanM's unique properties can be leveraged for a number of applications for f-elements. LanM can selectively scavenge Ln3+ ions even in the presence of billons of equivalents of competing metal ions, and its complexes remain stable as low as pH ∼2.5 and at least up to 95 °C.33,35,48 It allows for selective recovery of rare earths from low-grade industrial feedstocks,48 or for detection of nanomolar concentrations of lanthanides in highly complex matrices,35,52 or even for lanthanide–lanthanide separation using immobilized LanM.7 In line with our results, Hussain et al.53 recently leveraged the thermal stability of LanM to recover lanthanides from steel slag leachates using a biomaterial based on LanM and an elastin-like polypeptide via heat-induced precipitation cycles. Furthermore, our team demonstrated54 that LanM is even more effective at binding trivalent actinides, capable of scavenging actinium (Ac3+) down to femtomolar concentrations while remaining selective against radium (Ra2+) or even >10+10 equivalents of endogenous cations (e.g., Ca2+, Mg2+, Cu2+, Zn2+, Mn2+). Radiochemistry experiments contrasted with circular dichroism measurements revealed54 that the actinium complex of LanM is more stable than its lanthanum counterpart with Kd values of 0.865 pM and 1.8 pM (at pH 7.0) for Ac3LanM and La3LanM, respectively. Using independent spectroscopic techniques, we determined55 the stability constants of the Am3+ and Cm3+ complexes of LanM and also found the actinide complexes to be more stable than their lanthanide analogues, with Kd values of 1.3 and 1.2 pM (at pH 5.0) for Am3LanM and Cm3LanM, compared to ∼10–20 pM for Pr3LanM, Nd3LanM, and Sm3LanM. It was also found that NpO2+ does not interact significantly with LanM, allowing its convenient separation from the LanM-bound Am3+ by simple filtration.55 More recently, Singer et al.56 performed lanthanide–lanthanide and lanthanide-actinide competition assays with LanM in the presence of super-stoichiometric amounts of f-elements, at pH 6.7 without pH buffer capacity. Although their conclusions56 appear largely in line with our prior studies,33,35,48,54,55 under those authors' conditions the formation of metal hydroxides as well as multi-metal species (e.g., LuOH2+, CmOH2+, NdCm2LanM, La2EuLanM, etc.) cannot be avoided and would have likely contributed to the spectroscopic properties and trends observed.
The reasons for LanM's combined high affinity and selectivity for trivalent f-elements are not fully understood. Moreover, all previously published studies on LanM have been done with unmodified coordination environments. It therefore remained unknown if proteins with enhanced properties relative to wild-type (WT) LanM could be obtained via select mutations. In this study, five LanM variants were engineered and their complexes with Am3+, Cm3+, and lanthanide ions (Nd3+, for its closest ionic radius to those actinides, as well as Eu3+, because of its intrinsic luminescence) were characterized. The study shows that the LanM framework still provides high-affinity metalloproteins able to challenge known f-element chelators, even for variants with altered metal-binding sites, thus suggesting a broader role for LanM homologs in nature. Additionally, results obtained with Am3+, Cm3+, and Ln3+, using a combination of solution thermodynamic and spectroscopy experiments, provide evidence that the presence of water molecules coordinating the metal ions within LanM's binding sites is a key factor driving the complexes' stability. The results strongly suggest that hydrogen bond interactions between second-sphere residues and first-sphere water molecules stabilize LanM's complexes and that disrupting these interactions yields weaker metal–LanM complexes even if the number of organic chelating moieties around the metal ion is increased. This mechanism departs from traditional small chelator–metal complexes where ligands with high denticity typically lead to stronger complexes.57 Finally, taking advantage of the ability to fine-tune these second-sphere interactions within the protein framework, we identify and test a variant with two times higher selectivity for actinides over lanthanides, compared to WT LanM. These results both provide fundamental understanding of metal selectivity in LanM and demonstrate the promise of further exploiting this remarkable protein for high-performance separation and chelation applications.
Results and discussion
Rational design of LanM variants for lanthanide and actinide binding
Three of LanM's four EF hands have high affinity for trivalent lanthanides and actinides (Fig. 1).33,35,48,52,54,55 The apo form of LanM is largely disordered33 and has not been structurally characterized. Structural data for a metal complex with LanM has been limited to a solution-state NMR structure58 of the protein's complex with Y3+, which confirmed EF hands 1, 2, and 3 as high-affinity binding sites.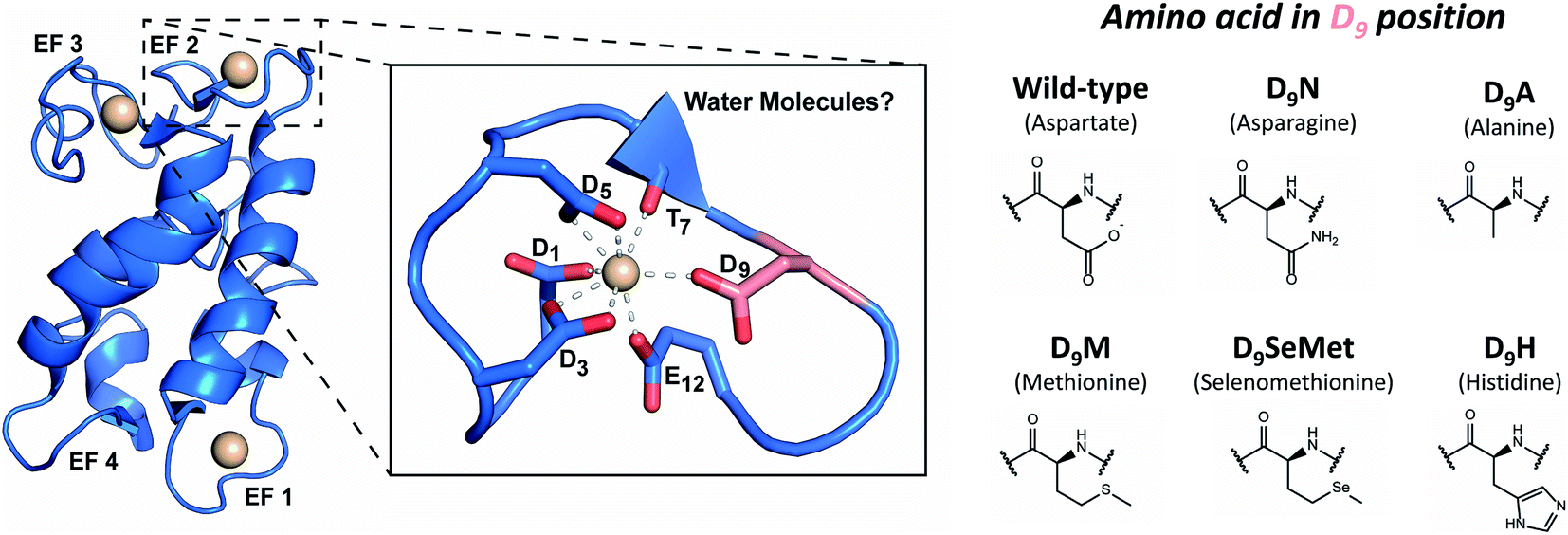 | ||
| Fig. 1 LanM's metal-binding sites and mutations performed in this study. Left: representation of the metal-bound WT LanM structure and the metal-binding site at EF2 based on the solution-state NMR structure of Y3LanM reported by Cook et al.58 (PDB code: 6MI5). The D9 residue is shown in salmon for clarity. Solvent was not explicitly modeled in the metal-binding sites but recent work has pointed to ∼2 coordinated solvent molecules on average per site in WT LanM.52,55 A detailed view of the metal binding site and notation of the metal-coordinating amino acids is provided. Right: amino acid substitutions at the D9 position of the EF hands 1, 2, and 3, studied in the present work. | ||
Recent studies have demonstrated that EF2 and EF3 bind Ln3+ ions cooperatively, with EF1 being slightly more labile.52 The fourth site (EF4) was not modeled due to chemical exchange of Y3+ on the NMR timescale,58 suggesting weaker than micromolar affinity, consistent with the biochemical studies.33,35,48,52 However, the NMR structure could not resolve details of the metal-binding sites (e.g., presence of metal-coordinated water molecules), but the lowest-energy structures suggested coordination of Y3+ through a backbone carbonyl (T7) and five conserved carboxylate sidechains (D1, D3, D5, D9, and E12) within the EF loops 1, 2 and 3.58 Because the residue at the 9th position of EF hands has been shown to play a key role in controlling affinity and kinetics in Ca2+-binding EF-hand proteins,59 we initially focused on this position in LanM. Although an aspartate is not an uncommon residue at this position in previously characterized EF-hand proteins,60M. extorquens LanM is unusual in that all four of its EF hands feature D9, and we have previously suggested that these residues may impact LanM's unprecedented selectivity pattern.33 Recent investigations into the coordination spheres of the WT Tb3LanM,52 Gd3LanM, and Cm3LanM55 complexes have demonstrated that, on average, ∼2H2O molecules are present in the first coordination sphere of these metals bound to LanM. This observation is similar to that in Tb3+ and Eu3+ complexes of calmodulin;38,61 the 9th position residue in Ca2+-bound EF hand proteins is often hydrogen bonded to a coordinated water.59 Together, these results raise the question whether the carboxylate sidechain of LanM's conserved D9 residue interacts directly with trivalent metals in the first coordination sphere or indirectly (in the second coordination sphere) via hydrogen bonding to coordinated solvent.
Considering the unique affinity and selectivity of LanM for lanthanides and actinides relative to other EF-hand proteins, we sought to both probe the importance of the conserved D9 position to f-element coordination in LanM and enhance the protein's selectivity for actinides over lanthanides, through strategic alteration of the metal coordination sphere at the 9th position in EF hands 1, 2, and 3. Therefore, we rationally altered the chelating, steric and/or electronic properties of this position: substituting D9 for a residue with comparable size but differing electronics (asparagine, N); or for sterically “bulky” residues (histidine, H; methionine, M; and selenomethionine, SeMet); or for a non-interacting residue (alanine, A). We postulated that in the case of direct metal coordination by D9 in WT LanM, variants with softer electron-donating side chains (i.e., M, N, H, SeMet) would have increased selectivity for actinides.26,62 The non-interacting alanine residue was chosen to help elucidate whether D9 participated in first- or second-sphere interactions.
Complexation of lanthanides and actinides by LanM variants
The five LanM variants were denoted 3D9N, 3D9A, 3D9M, 3D9SeMet, and 3D9H, as three EF hands were substituted at the D9 position. These variants were expressed and purified in good yield and first tested for complexation with lanthanides (see Methods in ESI†). Stoichiometric titrations utilizing direct excitation of Eu3+ luminescence, carried out at pH 5.0 (Fig. S1†), show that all five variants bind 3 equivalents of Eu3+ under these conditions, like the WT protein. Titration of LanM with solutions of lanthanides in equilibrium with ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) allows for the gradual formation of the lanthanide-LanM complexes, and the corresponding folding of the protein63,64 can be followed by circular dichroism (CD). Fig. 2 shows examples of CD titrations for WT LanM and the five variants, at pH 5, with Nd3+, with thermodynamic data summarized in Table 1. Within the lanthanide series, Nd3+ is the closest analogue for Am3+ in terms of ionic radius (1.161 Å for Nd3+vs. 1.157 Å for Am3+),18 motivating its selection for the comparative studies here. All variants undergo a significant conformational change in the presence of nanomolar to picomolar concentrations of free Nd3+, highlighting their strong affinity for trivalent lanthanides. For WT LanM, two consecutive conformational changes are observed, consistent with previous characterization.33,52 At near-neutral pH, the primary cooperative conformational change associated with EF2/3 occurs at slightly lower free lanthanide concentrations than the secondary change associated with EF1; at pH 5, however, EF1 is significantly more conformationally labile than EF2/3.33,52 Interestingly, only one conformational change was observed for each of the variants.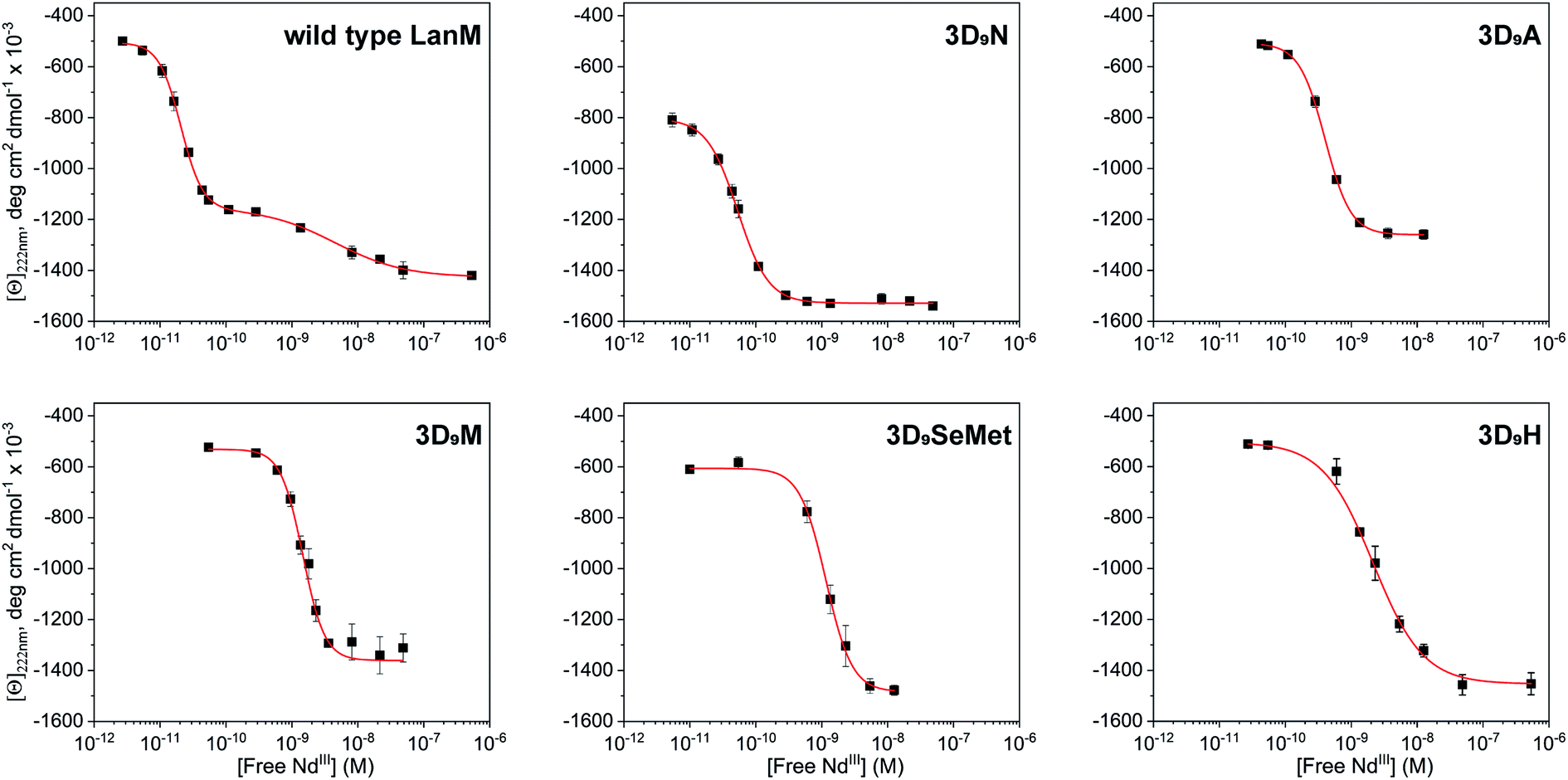 | ||
| Fig. 2 Characterization of WT LanM and five D9 variants using circular dichroism. CD data showing molar ellipticity values at 222 nm for WT LanM and LanM variants vs. calculated free Nd3+ based on Nd total concentration, pH, pKa's of EGTA, and Nd-EGTA stability constant. [Ln]total = 0–10 mM, [LanM] = 20 μM, [EGTA] = 10 mM. pH = 5.0. Buffer: 20 mM acetate, 100 mM KCl. The data were fitted to the Hill equation with 1 or 2 sets of sites, with apparent Kd values shown in Table 1 and complete parameters in Table S1.† | ||
| LanM variant | K d,average for Am3LanMa (in pM) | K d,app for Nd3LanMb (in pM) |
|---|---|---|
a From UV-vis spectrophotometric titrations.
b Apparent Kd from CD experiments. A conversion of Kd values into the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) βMLH scale (used for small-molecule chelators) is given Table S2. βMLH scale (used for small-molecule chelators) is given Table S2.
|
||
| WT LanM | 1.4 ± 0.5 | 21.3 ± 0.6 (EF2/3) |
| 4070 ± 1050 (EF1) | ||
| 3D9N | 3.4 ± 0.6 | 53.1 ± 1.9 |
| 3D9A | 9.7 ± 3.1 | 397 ± 6 |
| 3D9M | 17.1 ± 2.6 | 1460 ± 70 |
| 3D9SeMet | 16.4 ± 2.5 | 1160 ± 130 |
| 3D9H | 13.4 ± 2.1 | 2150 ± 380 |
Under the tested conditions, these conformational changes were triggered at free Nd3+ concentrations as low as ∼2.1 × 10−11, 5.3 × 10−11, 4.0 × 10−10, 1.5 × 10−9, 1.1 × 10−9, and 2.2 × 10−9 M, for WT LanM, 3D9N, 3D9A, 3D9M, 3D9SeMet, and 3D9H, respectively. The 3D9N variant is most like WT LanM in terms of sidechain size and electronics, exhibiting the most similar response to Nd3+ relative to the wild-type protein; interestingly, 3D9N also displays a greater degree of folding in the apoprotein than WT and the other variants, with a magnitude suggestive of pre-ordering of EF1 (Table S1†). The 3D9A variant's response to Nd3+ is only shifted by ∼20-fold relative to WT LanM (apparent Kd of 400 pM vs. 20 pM – Table 1), but the protein displays a conformational change consistent with ordering only of EF2/3.
The 3D9N and 3D9A data thus suggest that the 9th residue plays an especially important role in the structure of EF1. The variants containing amino acids with the bulkiest side chains (3D9M, 3D9H, and 3D9SeMet) exhibit similar, and the most adversely affected, response to Nd3+ (∼2 nM vs. 20 pM). All variants (and WT) showed positive cooperativity in conformational response, except for 3D9H (Table S1†).
These results lead to two important conclusions. First, the larger side chains chosen in this study fail to stabilize the interaction between the given variant and its coordinated Nd3+ ions, whether because of differences in main chain structure and communication between EF hands58,65 or because of suboptimal side chain steric effects.59 Second, complete loss of an interacting functionality at this position (3D9A variant) has only a modest effect on binding affinity, suggesting that direct interaction between the D9 side chain and coordinated Nd3+ may not be occurring in WT LanM (vide infra).
Analogous CD experiments cannot be performed with trivalent actinides (e.g., Ac3+, Am3+, Cm3+, Bk3+) due to radiation constraints and the scarcity of the research isotopes. The binding of the LanM variants to actinides was therefore evaluated via UV-visible spectrophotometry using 243Am3+ and EDTA as a reference ligand competitor. The Am3+ ion exhibits a narrow absorbance band (5f → 5f) at ∼500 nm that is observable at micromolar concentrations and is sensitive to the metal coordination environment.20,21,66 Measurement of the absorbance spectrum of Am3+ in the presence of the LanM variants at pH 5 confirmed that they all bind the actinide ion (Fig. S2 and Table S3†). The Am3+ complexes with the five variants (i.e., Am33D9N, Am33D9A, Am33D9M, Am33D9SeMet, and Am33D9H) have relatively similar absorbance features as the WT, with 3D9H being the most distinct, suggesting a slightly different coordination environment (vide infra). The Am3+ absorbance band shifts from 503.9 nm to 505–506 nm and a shoulder appears at 512–519 nm, which is direct evidence of the complexation of americium by the proteins. Using our previously established spectrophotometric titration method48,55 using EDTA as a suitable ligand competitor for LanM (Fig. 3), the formation constants of the five Am3+–LanM variant complexes were determined. While the sequential release of Am3+ from the three sites of LanM was not observed during these spectrophotometric experiments (indicative of similar affinities), the averaged dissociation constants (Kd,average, for metal–ligand dissociation) of Am3LanM can be calculated for comparison with the apparent dissociation constants (Kd,app, for metal-dependent conformational change) derived from the CD measurements with lanthanides. Table 1 summarizes these Kd values of WT LanM and its variants for americium and neodymium. The five variants tested exhibit slightly lower affinity for Am3+ than WT LanM but they nonetheless remain highly efficient actinide-binding proteins, with Kd values of 3–20 pM, at pH 5.0 (compared to 1.4 pM for WT Am3LanM under similar conditions). All variants exhibit higher stability constants for americium than for neodymium, which further evidences the superior affinity of LanM for trivalent actinides over lanthanides. The increased divergence between the Kd,average (Am3+) and Kd,app (Nd3+) values for all but 3D9N likely indicates less efficient coupling of metal binding and conformational response.48 Nevertheless, the overall stability trend observed among the variants at pH 5 is consistent for Am3+ and Nd3+ and is as follows: WT > 3D9N > 3D9A ≫ 3D9M ∼ 3D9SeMet ∼ 3D9H.
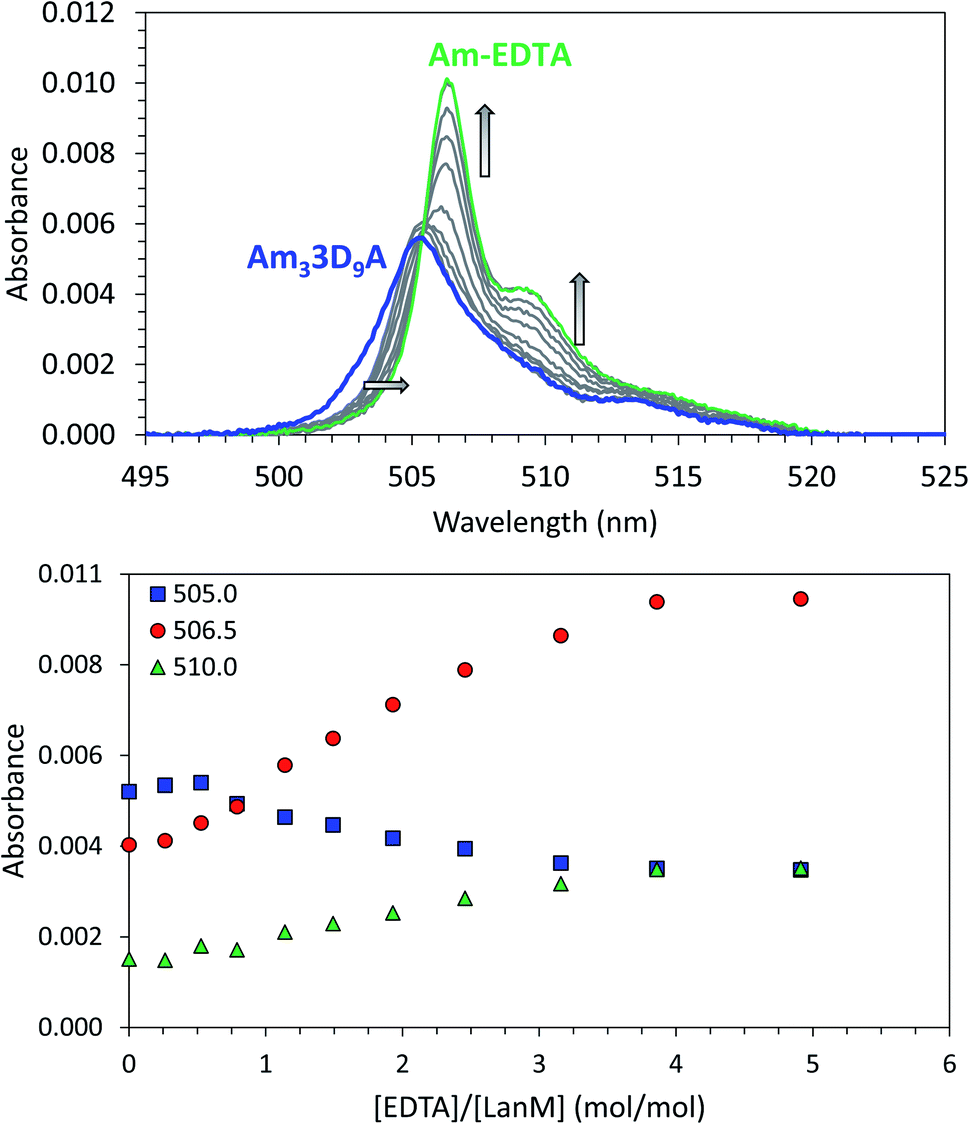 | ||
| Fig. 3 Complexation of americium(III) by LanM variants. Example of UV-vis spectrophotometric competition titration between 3D9A and EDTA for Am(III) binding. [Am] = 15 μM, [LanM] = 7.5 μM, [EDTA] = 0 to 37.5 μM. pH = 5.0, buffer: 25 mM acetate, 75 mM KCl. Top: full spectra. Bottom: absorbance variation at select wavelengths. See Fig. S3† for similar experiments with the LanM variants 3D9N, 3D9M, 3D9H, and 3D9SeMet. A similar titration of WT LanM for Am(III) has been reported elsewhere.55 | ||
Thermodynamic data on americium–protein species are scarce, and the five new stability constants for such complexes reported herein more than doubles the total number previously published. Besides these and the WT Am–LanM complex,55 the only other reported examples are siderocalin–siderophore–americium adducts45 with Kd values of 240–29000 pM, at pH 7.4, i.e., orders of magnitude less stable than for LanM and its variants. Other actinide(III)–protein complexes, notably with curium,38,39,41–43,47,67–69 also have Kds orders of magnitude weaker than LanM or its variants. The synthetic LBT peptides49 and their variants have been studied for lanthanide and also Am3+ complexation, but their Kds remained in the micromolar to nanomolar range (e.g., 45000 pM for Am3+–LBT and 230000–2700000 pM for nine LBT variants tested,22 at pH 7), highlighting the difficulty of designing high-affinity chelators for f-elements, and particularly for trivalent actinides.
To summarize these data so far, the thermodynamic comparisons between Am3+ and Nd3+ complexes of LanM and its variants allow us to draw several conclusions. First, as the ionic radii of Nd3+ and Am3+ are nearly identical,18 the higher stability of the Am–LanM complexes indicates that the metal ion size is not the only driver for LanM's binding affinity. In prior studies,7,33,35,48 LanM's affinity trend along the lanthanide series was found to not follow the lanthanide contraction (contrary to what is generally observed for small ligands1,12,70,71), which further suggests that effects other than size match and pure ionic interactions are at play in LanM's binding mechanism and preference for actinides. Second, the 3D9A and 3D9N variants retain strong affinity for actinides and lanthanides, akin to WT LanM. In particular, if the aspartate residue in WT LanM were a direct ligand to the metal ion, it would be surprising that high affinity is retained upon its substitution with alanine, the side chain of which does not have the ability to coordinate the metal ion (Fig. 1). This observation is the first evidence that the D9 residue does not interact directly with the metal ion in WT LanM and might instead interact with coordinated solvent; this interpretation will be further supported by additional experiments below. Third, substitution for a bulky residue in the D9 position interferes with LanM's folding, as exemplified with the D9H variant, strongly suggesting that the 9th position is critical for coupling of metal binding and the overall conformational change of the protein (Fig. 2). Finally, the substitution D → N is the best tolerated mutation at the 9th position. These results prompted further investigations into LanM complexes' selectivity and stability, as detailed below.
Acidity properties of lanthanide and actinide complexes with LanM variants
Whereas thermodynamic differences may not be enough to separate f-elements by directly using LanM at high pH, due to the presence of multiple binding sites and possible formation of multi-metal species (as previously observed at pH ≥ 5 for multi-lanthanide LanM complexes48), Dong et al. recently demonstrated7 that selective desorption of lanthanides from immobilized WT LanM can be obtained in the high-acidity range (pH < 3), closer to the apparent pKa of the metal-bound carboxylate groups of LanM's EF hands. The ability to bind lanthanides at low pH is another feature that is not observed in prior proteins studied for f-element complexation.7,48,52,54,55 We here posited that the variants constructed above may result in different pH values at which actinides and/or lanthanides are released, potentially enhancing selectivity compared to the wild-type protein. Europium and curium (Eu3+ and 248Cm3+) were used for these investigations as both are luminescent in the presence of LanM and can be quantitatively measured via fluorescence spectroscopy within the same concentration range. Fluorescence emission and excitation spectra were measured for Cm3+ and Eu3+ in the presence of LanM and variants from pH 1.0 to 8.5 (Fig. 4a–c and S4–S8†) using identical solution conditions for direct comparison. The emission/excitation spectra of the unbound Cm3+and Eu3+ readily differ from those of their LanM complexes (Fig. 4b, S9 and S10†), further confirming the ability of the five variants to complex actinides and lanthanides in solution.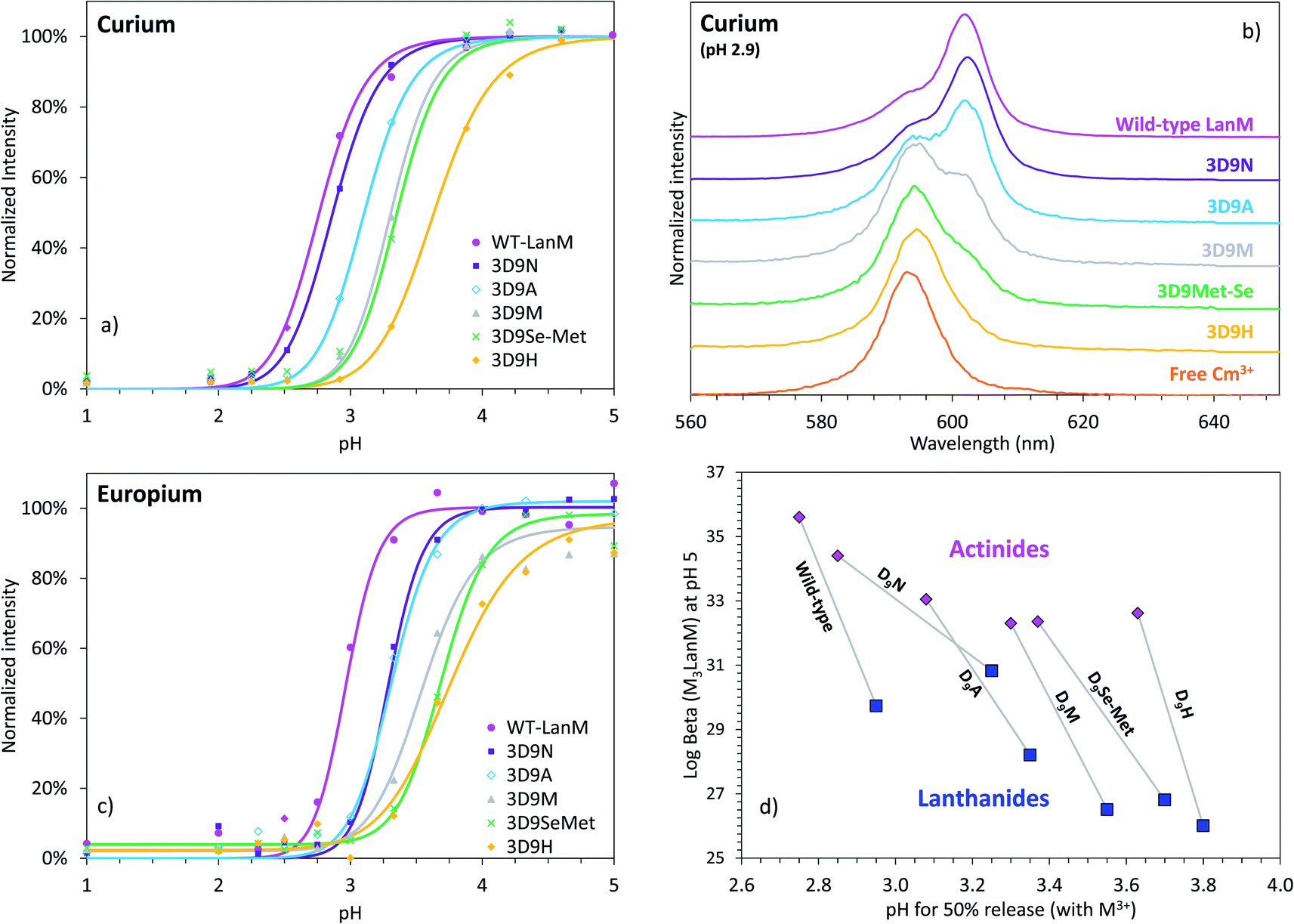 | ||
| Fig. 4 LanM variants binding to actinide and lanthanide ions and release at low pH. (a) Observed emission intensities for the fluorescence peak of the Cm3+–LanM complexes (602–603 nm) in the lower pH region (0.5 μM LanM, 1.0 μM Cm3+). Full titrations, up to pH ∼8.5, with emission spectra, excitation spectra, and lifetime measurements, as well as pH curve fitting are given in Fig. S4–S8.† (b) Fluorescence emission spectra for Cm3+ in the presence of LanM or its variants at pH 2.9, highlighting the different complexation behavior observed among the variants at low pH. A comparison of the Cm–LanM spectra at pH 7.0 and pH 2.9 is given in Fig. S9.† (c) Observed emission intensities for the fluorescence peak of the Eu3+–LanM complexes (615 nm) in the lower pH region (0.5 μM LanM, 1.0 μM Eu3+). Comparison of all variants at pH 3.3 and 7.0 is given in Fig. S10.† Full titrations, up to pH ∼8.5 are given in Fig. S11.† (d) pH50% values for actinide–LanM and lanthanide–LanM complexes plotted against the stability constant of Am3LanM and Nd3LanM (logβ31 at pH 5), for WT LanM and the five variants reported in this study, highlighting the distinct behavior of the 3D9N variant. Square symbols: lanthanides (Nd3+ and Eu3+). Diamond symbols: actinides (Am3+ and Cm3+). | ||
In line with our solution thermodynamic measurements on Am3+ and Nd3+ (vide supra), LanM binds Cm3+ at a lower pH than Eu3+. As shown in Fig. 4, 50% of Cm3+ is already bound to LanM at pH 2.75, while a pH of 2.95 is needed for Eu3+. All variants exhibit lower pH50% for Cm3+ relative to Eu3+, thus unequivocally demonstrating LanM's preference for the actinides over the lanthanides via a macroscopic observation, independent of thermodynamic measurements. The complexation order among the variants at low pH is the same for Cm3+ and Eu3+ (WT > 3D9N > 3D9A > 3D9M > 3D9SeMet > 3D9H) and is consistent with the stability constants determined for the M3LanM complexes at higher pH. The protein variants exhibiting the strongest metal affinity (i.e., lowest Kd value in Table 1) release their metal ion at lower pH. Interestingly, mutation of a single amino acid (out of 12) in LanM's EF loops can shift its pH50% value by one pH unit (from 2.8 for WT LanM to 3.8 for the 3D9H variant). This demonstrates that the release of actinides and lanthanides from LanM can be finely modulated for potential separation applications. Among the variants tested, 3D9N has a pH50% difference between actinides and lanthanides which is twice that of the WT, with pH50% values for Cm3+ and Eu3+ of 2.75 vs. 2.95 for WT (ΔpH50% = 0.20), compared to 2.85 vs. 3.25 for 3D9N (ΔpH50% = 0.40). The 3D9A, 3D9M, 3D9SeMet, and 3D9H variants have ΔpH50% of 0.27, 0.25, 0.33, and 0.17, respectively. Hence, the 3D9N variant offers the best combination of properties, with both high affinity for both actinides and lanthanides and improved selectivity between actinides and lanthanides at low pH.
The larger difference observed between actinides and lanthanides for the 3D9N variant, without compromising its efficiency at low pH, could be due to the conservation of the overall arrangement of the protein calix (the asparagine group being of similar size as aspartate) while introducing slightly softer donating atoms (neutral amide vs. negatively charged carboxylate functions). Introduction of nitrogen-donating groups in lieu of oxygen-donating functions has been shown to induce better selectivity for actinides over lanthanides in liquid–liquid extraction processes.62,72–75 However, because our thermodynamic data suggest that there is not a direct interaction between the D9/N9 residue and the metal ion, the pH50% results may instead point to a hydrogen bonding interaction between this residue and one of the coordinated solvent molecules being subtly tuned by the 3D9N substitution. Potential factors include donor–acceptor distance, potential for asparagine to act as hydrogen-bond donor (NH2) or acceptor (CO) depending on orientation of the sidechain, and Lewis acidity of the metal ion and its effects on hydrogen bonding. With regard to the latter point, we note that a decrease in Cm3+ fluorescence intensity was observed for all variants except 3D9N and WT in the pH range 6–10 (Fig. S4–S8†), concomitant with a slight shortening of the Cm3+ fluorescence lifetime (Tables S4 and S5†). While the fluorescence intensity decreases by up to 30% for the variants 3D9H, 3D9M, and 3D9SeMet, no free Cm3+ or Cm–hydroxide species were detected as the emission/excitation spectra and lifetimes clearly correspond to LanM-bound species. However, a LanM-bound Cm–hydroxide species could form via deprotonation of a coordinated water molecule. We propose that such deprotonation might be disfavored by hydrogen bonding between this solvent molecule and the 9th position residue (as suggested above and supported further below), but only when it is aspartate (WT) or asparagine (3D9N), of the substitutions investigated here. By contrast, no fluorescence decrease was observed for any variant in the case of Eu3+ for any variant (Fig. S11†), perhaps because it is predicted to be a weaker Lewis acid than Cm3+. These observations, which prompt further investigation, reinforce the complex metal-solvent-side chain interactions in the protein system.
Binding of LanM to f-elements at high pH
As detailed above, the 3D9H variant has the smallest pH50% difference between actinides and lanthanides and lowest stability constant among the variants tested. However, the mutated residue in this variant is a histidine, which is likely detrimental to metal binding at low pH since the pKa of unbound histidine is around 6.76 Hence, we also evaluated the behavior of the 3D9H variant at high pH where the protonation of the histidine group does not mask the binding potential of this variant. The binding of Cm3+ to 3D9H was evaluated at pH 8.0 and similar experiments were also performed with WT LanM and the 3D9N variant for comparison.In this high pH range, the small molecule siderophore desferrioaxmine B (DFOB) was used as ligand competitor since it is reputedly one of the strongest natural chelators for trivalent actinides at high pH.77,78Fig. 5 shows ligand–ligand competition titrations between LanM and DFOB for the binding of Cm3+. WT LanM strongly outcompetes DFOB in this pH range, with 50% of Cm3LanM still formed in the presence of ∼1800 equivalents of DFOB. These results are in excellent agreement with independent measurements55 at different pH values and slightly different Cm3+ concentrations. The results indicate that the 3D9N and 3D9H variants have lower affinity for Cm3+ than WT LanM. Both variants are nonetheless highly effective actinide chelators able to compete with DFOB. The 3D9N variant requires ∼250 equivalents of DFOB to release 50% of curium and 10 equivalents of DFOB in the case of 3D9H. These results are consistent with the behavior observed in the low-pH range and stability constants measured at pH 5.0 that indicate the complexes of the three protein types are within 3 orders of magnitude, and that 3D9H is weakest (vide supra). These results further confirm that the incorporation of a bulky residue within LanM's binding sites, even with a function amenable to potentially coordinate the f-element, is detrimental to complex stability.
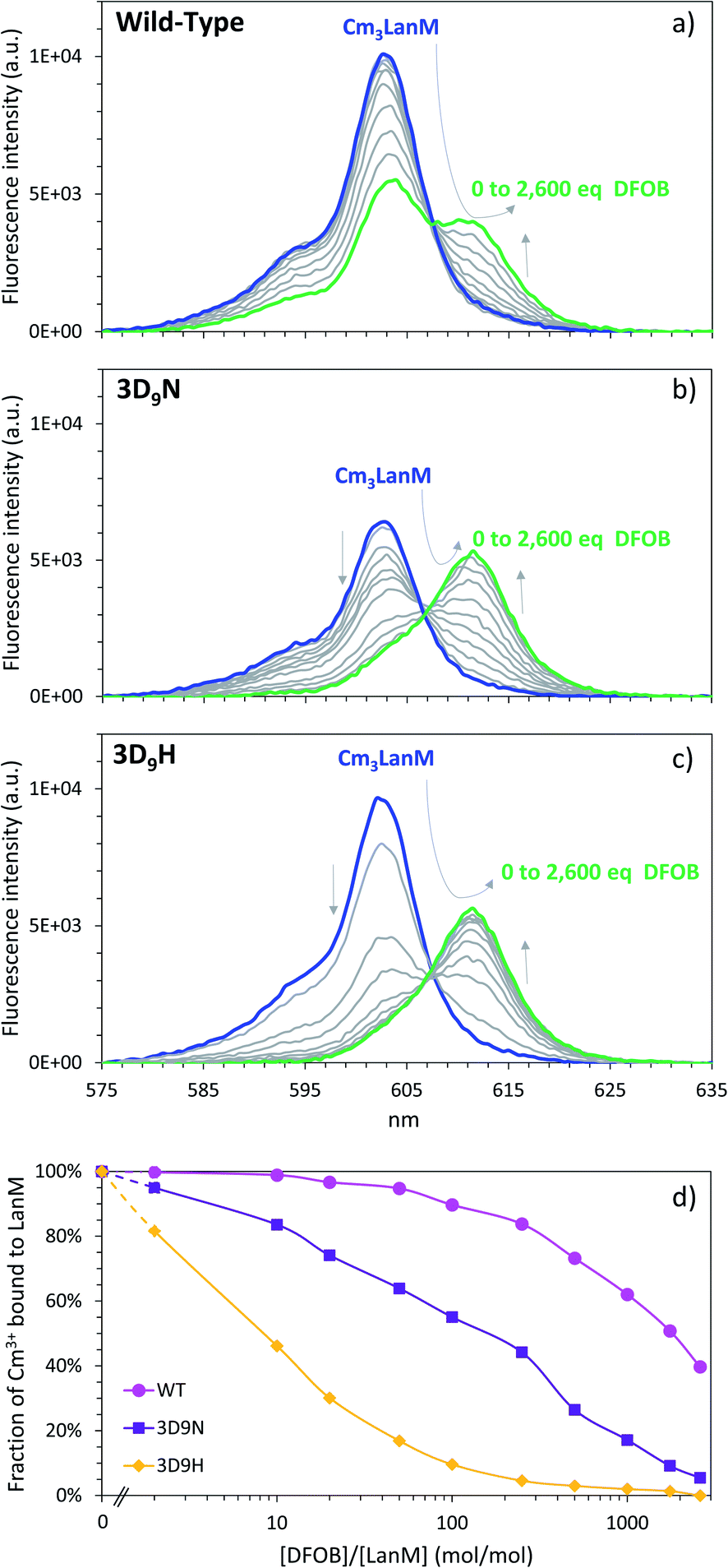 | ||
| Fig. 5 Affinity of LanM and its 3D9N and 3D9H variants for Cm(III) at high pH. Fluorescence spectroscopy titrations of Cm3LanM with DFOB. (a) Wild-type LanM. (b) 3D9N variant. (c) 3D9H variant. (d) Corresponding fraction of Cm(III) bound to LanM. [Cm] = 1.0 μM, [LanM] = 0.5 μM, [DFOB] = 0, 1, 5, 10, 25, 50, 125, 250, 500, 875, or 1300 μM. pH = 8.0. Buffer: 25 mM HEPES, 75 mM KCl. T = 22 °C. | ||
Fluorescence properties and role of coordinated water molecules
With the above data pointing to potential importance of coordinated solvent in LanM's metal selectivity, time-resolved fluorescence measurements were performed to determine the luminescence lifetimes of Cm3+ and Eu3+ in the presence of the LanM variants. Both metal ions exhibit longer lifetimes upon binding to the protein (∼200 μs for Cm3LanM and ∼420 μs for Eu3LanM – Table 2) relative to their free ions in aqueous solution (∼70 μs for free Cm3+ and ∼110 μs for free Eu3+).79 Except in the case of the 3D9N variant, the Cm3LanM and Eu3LanM complexes have longer lifetimes with the variants when compared to the wild-type protein. For both Cm3+ and Eu3+, the 3D9H variant has the longest lifetime (∼20% longer than in the case of WT LanM), while the 3D9N has the shortest (5% shorter than WT LanM). For aqueous complexes, a longer luminescence lifetime is typically associated with a decrease in the number of quenching water molecules around the emissive metal ion. Kimura et al.80 and Horrocks et al.61,81 developed empirical equations that allow for the calculation of the number of water molecules in the first coordination sphere of a metal ion based on its fluorescence lifetime. Based on these equations, Cm3+ and Eu3+ are surrounded by 2.3 and 2.0 water molecules, respectively, within the binding sites of WT LanM (Table 2). This is consistent with independent measurements recently performed52 with Tb3+ and LanM containing tryptophan residues as luminescence sensitizers. Based on the Kimura et al.80 and Horrocks et al.81 equations, the 3D9N variant complexes of Cm3+ and Eu3+ have comparable numbers of water molecules relative to their WT counterparts, whereas the 3D9A, 3D9M, 3D9SeMet, and 3D9H complexes have slightly fewer coordinated water molecules.| Variant | Curium(III) | Europium(III) | ||||
|---|---|---|---|---|---|---|
| In H2O (μs) | In D2O (μs) | n H2O | In H2O (μs) | In D2O (μs) | n H2O | |
| a Calculated using Kimura's equation.80 b Calculated using Horrocks' equation.81 c Previously reported in ref. 55. | ||||||
| WT LanM | 208 ± 2c | 936c | 2.3c | 400 ± 20 | 2560 ± 360 | 2.0 |
| 3D9N | 194 ± 4 | 853 ± 15 | 2.5 | 380 ± 10 | 2130 ± 110 | 2.0 |
| 3D9A | 219 ± 4 | 886 ± 16 | 2.1 | 410 ± 10 | 2490 ± 80 | 1.9 |
| 3D9SeMet | 220 ± 5 | 785 ± 18 | 2.1 | 410 ± 10 | 1260 ± 140 | 1.5 |
| 3D9M | 224 ± 5 | 815 ± 18 | 2.0 | 440 ± 10 | 2100 ± 250 | 1.6 |
| 3D9H | 245 ± 5 | 681 ± 14 | 1.8 | 480 ± 10 | 2330 ± 90 | 1.5 |
In the case of 3D9H, we observed sensitization of Cm3+via excitation of the histidine group at 270 nm and intramolecular energy transfer (Fig. S12†) whereas no luminescence is observed when exciting the WT Cm3LanM complex at this wavelength. This observation indicates that the histidine group coordinates Cm3+ in at least one EF hand in the 3D9H variant. The UV-vis absorbance spectrum of the Am3+ in the presence of 3D9H is also slightly different relative to the other variants (Fig. S2†), also suggesting participation of the histidine group in metal coordination.
Together, these results suggest that water molecules were displaced from the metal coordination sphere to accommodate the histidine side chain. Knowing that the 3D9H complexes are less stable than their wild-type analogues (vide supra), this suggests that the presence of water molecules within LanM's binding sites is more important for the stability of its complexes than the coordination of organic functionalities.
Since the equation from Horrocks et al.81 uses the lifetime values determined in H2O and D2O solutions, whereas the Kimura et al. equation79 only uses the lifetime measured in H2O, the direct comparison of Cm3+ and Eu3+ complexes and their calculated number of water molecules may be biased. Thus, the lifetimes of Cm3+–LanM complexes in deuterated solutions were also measured for a better comparison (Fig. 6, S13 and S14†). As H2O molecules are replaced by D2O ones, the non-radiative pathways are reduced, lengthening the fluorescence lifetime of the emissive f-element. Hence, the ratio between the lifetimes measured in D2O and H2O also informs us on the presence of water molecules in the vicinity of the metal and how they impact its coordination. Fig. 6 shows that the variants with highest lifetime ratios are also the most thermodynamically stable, thus confirming the key role of the water molecules in the actinide and lanthanide complexes of LanM.
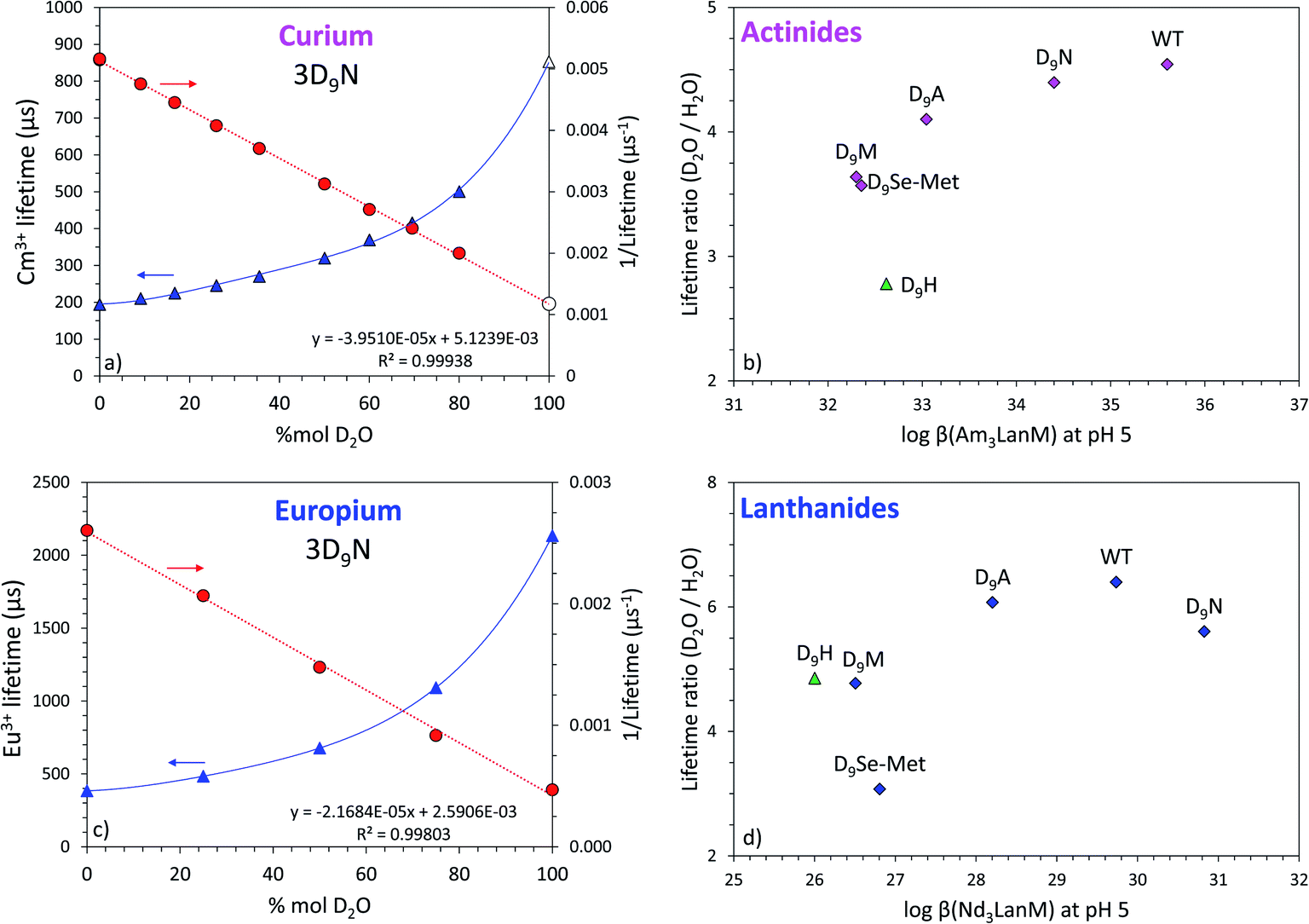 | ||
| Fig. 6 Luminescence lifetimes of Cm3+ and Eu3+ bound to LanM variants measured in D2O–H2O mixtures. (a) Fluorescence lifetime for Cm3LanM (3D9N variant) as a function of the content of D2O in solution. Triangles: lifetime values (left y-axis). Circles: inverse of the lifetime (right y-axis). The empty symbol for Cm3+ (100% D2O) was calculated based on the linear correlation as displayed on the graph. Initial pH in pure H2O = 7.0, buffer = 25 mM HEPES, 75 mM NaCl. Similar data for all the variants tested in this study (3D9M, 3D9A, 3D9H, 3D9SeMet) are given in Fig. S13–S15.† See Table S6† for numerical values. Corresponding values for WT LanM have been previously reported[55] and are also shown in Fig. S13† for comparison. (b) Correlation between the increase in the Cm3+ lifetime from pure H2O to pure D2O (lifetime ratio = τD2O/τH2O) for the different LanM variants as a function of the thermodynamic stability of their Am3+ complexes. The 3D9H variant is displayed separately (triangle) because of the potential protonation of the histidine group at pH 5. (c) Same as (a) but with Eu3+. (d) Same as (b) but with Eu3+ and Nd3+. | ||
This conclusion contrasts with what is generally observed with small molecule complexes of f-elements where ligands with higher denticity (i.e., fewer water molecules coordinated to the metal) yield stronger complexes,57,82–84 and it likely reflects the need to optimize both local (metal-site) and global (protein) structure to yield the highest-stability LanM complexes.48,85 In the case of Eu3+, where the equation from Horrocks et al.81 is well established, the results indicate that the metal ion is coordinated by 2 water molecules in the D9N and D9A variants, similar to WT LanM. In particular, the observation that substituting an Asp for an Ala residue, the side chain of which cannot coordinate the metal ion, does not increase the number of coordinated solvent molecules strongly supports our proposal that the Asp itself does not coordinate the metal ion directly in WT LanM. Because maintenance of hydrogen-bonding potential in 3D9N has only a minor effect on affinity, while the other substitutions are more disruptive, this residue still makes an important contribution to the metal site. Therefore the native D9 residue (as well as mutant N9) is most likely interacting with the coordinated solvent, as often observed in Ca2+-binding EF hands.59
Actinide–lanthanide separation using LanM
Based on the difference in pH50% values with Cm3+ and Eu3+ for WT LanM (Fig. 4), we hypothesized that, at low pH, LanM would be able to selectively complex the actinide ions while leaving the lanthanides unbound. Furthermore, because 3D9N exhibited a larger difference in pH50% it may perform even better than the WT protein. Fig. 7 displays the fluorescence emission spectra of Cm3LanM at pH 2.9 upon addition of Eu3+. An absence of selectivity would manifest as a 50% decrease in the fluorescence intensity after addition of 1 equivalent of Eu3+ relative to Cm3+ due to randomization of the species distribution (e.g., Eu3LanM, Eu2CmLanM, EuCm2LanM, Cm3LanM, Eu3+, Cm3+). However, the results clearly show that a large excess of Eu3+ is needed to bind Eu3+ to LanM at the expense of Cm3+. For instance, after addition of 10 equivalents of europium (enough metal ions to saturate the binding sites of LanM multiple times) ∼20% of curium is still bound to LanM. Similar results were obtained with Nd3+ (Fig. S16†). This definitively demonstrates the intrinsic selectivity of LanM for actinides over lanthanides. Moreover, similar experiments performed with the 3D9N variant show that it has better actinide/lanthanide selectivity than WT LanM, with ∼30% of curium bound to 3D9N after addition of 10 equivalents of europium. The calculated separation factors Cm/Eu for these experiments range from 2.6–5.2 for WT LanM, compared to 4.7–11.1 in the case of the 3D9N variant (Fig. 7). While these tests remain fundamental chemistry experiments and are not intended to represent the ultimate performance of a LanM-based process, they indicate that LanM could be used to selectively scavenge actinides from acidic mixtures containing lanthanides. One could imagine a system where the 3D9N variant is immobilized onto a solid support7 and could act as a passive device to preferentially extract and pre-concentrate actinides from acidic mixtures of actinides and lanthanide fission products. The results also demonstrate that select substitutions, including 3D9N, can improve the properties of the wild-type protein.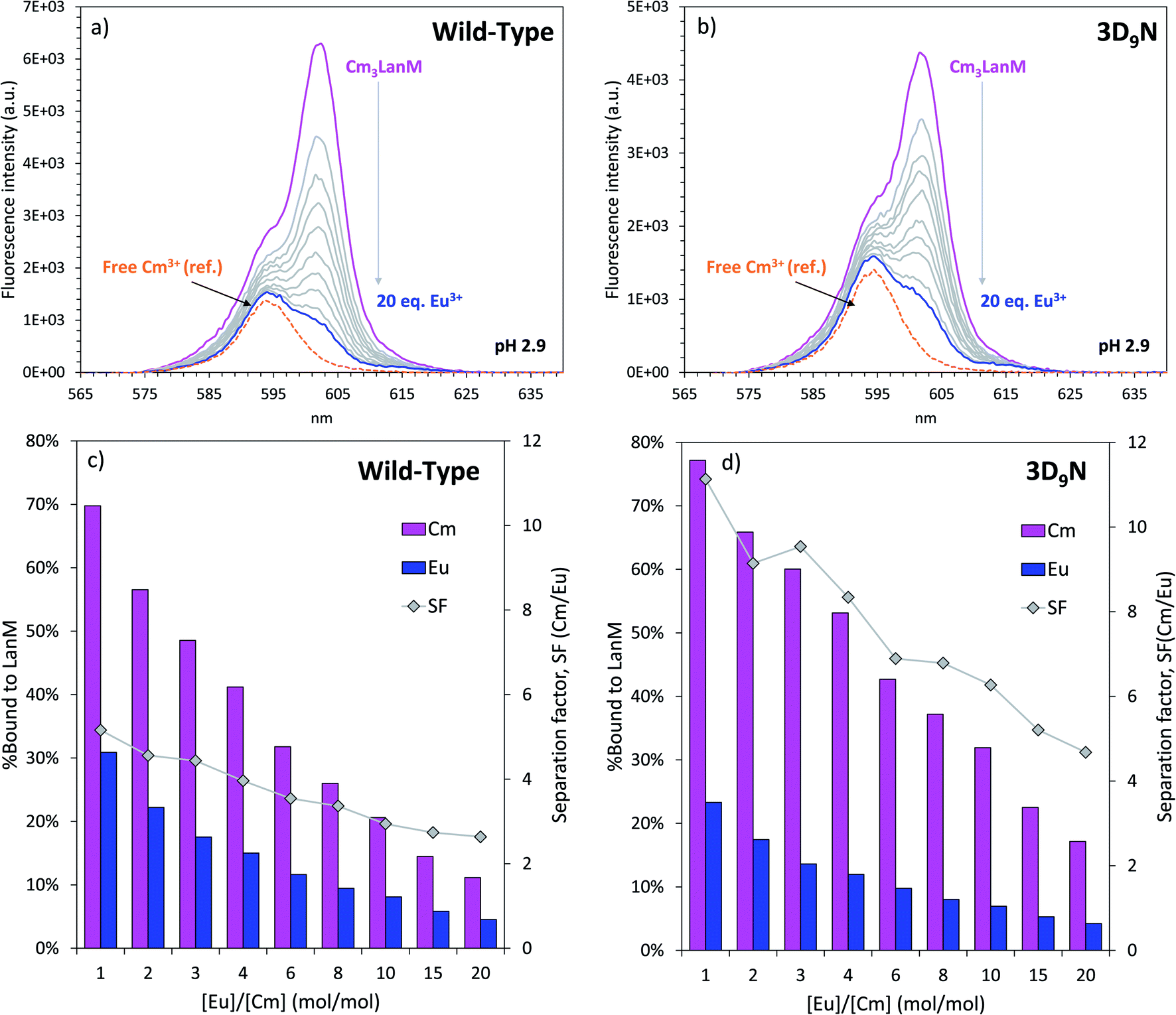 | ||
| Fig. 7 Observed selectivity of WT LanM and its variant 3D9N for the trivalent actinides over the trivalent lanthanides. Top panels: competition between Cm3+ and Eu3+, followed by fluorescence spectroscopy, in the presence of WT LanM (a) or its variant 3D9N (b). The dotted orange curve is the emission spectrum of Cm3+ in the absence of LanM and in the same buffer. The small shoulder appearing at 615 nm is due to Eu3+. Bottom panels: fraction of Cm3+ and Eu3+ bound to WT (c) and 3D9N (d) LanM (based on the fluorescence intensity) as a function of the ratio Eu/Cm, and corresponding separation factors Cm/Eu (SF). pH = 2.9. T = 22 °C. Buffer: 25 mM glycine, 75 mM NaCl. [Cm] = 1.0 μM, [LanM] = 0.5 μM, [Eu] = 0 to 20 μM. Similar results were obtained with Nd3+/Cm3+/LanM samples (see Fig. S16†). | ||
Conclusions
This work is the first to examine variants of LanM that have been strategically redesigned for altered metal-binding properties. Rational mutagenesis of the D9 position within each of LanM's high-affinity metal-binding loops reveals how the protein is optimized for first-sphere coordination of 2 solvent molecules (on average, at least for f-elements with similar ionic radius to Nd3+). It also demonstrates the crucial importance of second-sphere interactions with at least one of those waters impacting selectivity and affinity. Whereas other studies20,21,24,26 have focused on first-coordination-sphere effects to alter An3+/Ln3+ selectivity of ligands, here we show that LanM's selectivity can also be substantially enhanced by modulation of a single, second-sphere hydrogen bond.These results have important implications for both the physiological function and technological applications of LanM. All attempted mutations to the D9 position produced LanM variants that maintain the wild-type protein's overall function, demonstrating the robustness of the LanM architecture to natural variations. Importantly, however, the direct interaction between this residue and coordinated solvent is essential for optimal coupling of metal binding events to the protein's conformational change48,52,58,85 – which seems to be crucial for kinetic stability of the protein's metal complexes.7 Our work also shows that decreasing the number of these coordinated waters (e.g., 3D9H) does not increase complex stability. This surprising observation suggests that it is important for LanM to have open coordination sites to optimize not only f-element binding selectivity,52 but also associative metal transfer in the presence of a competitive ligand or a synergistic binding partner. Such a partner might be the outer membrane receptor in the lanM gene cluster, if the hypothesis that LanM is a secreted macromolecular lanthanophore/actinophore55 – to complement a separate small-molecule system recently described in a pre-print86 – is correct. Our observation also accounts for LanM's complexes with Ac3+ and La3+ being the most resistant to chelators that would be encountered in Nature like citrate,7 carbonate, and phosphate,54 despite having lower affinity55 than Nd3+, Am3+, and Cm3+ – an unusual property that can be exploited for separations.
Finally, our results solidify a new principle for fine-tunning f-element separations. Because the extent of coupling of metal binding and conformational change, and therefore complex stability, in LanM varies across the lanthanide series,48 it follows that second-sphere interactions also contribute to LanM's selectivity within the lanthanides.7,33,35,48 Specifically, the 3D9N variant may perform better than even the WT protein in REE–REE separations,7 improving separation factors while retaining high-affinity, high-selectivity, and low-pH binding. This observation motivates exploration of the numerous naturally occurring lanmodulins87 possessing potential hydrogen bond-donating residues in place of carboxylates in their EF hands, at the 9th position and elsewhere, for new selectivity trends. In turn, the diversity in these sequences could reflect distinct environmental niches and bioavailabilities of lanthanides/actinides for the bacteria expressing these lanmodulins. Such variants – whether naturally occurring or synthetically produced – or smaller units derived from them, may enable future applications in analysis of actinide samples, separation of actinides/lanthanides, and even variants tailored for specific medical radiometals.
Data availability
The data that support the findings of this study are available in the main manuscript and ESI.†Author contributions
J. A. M. and J. A. C. designed, and J. A. M. performed, experiments with lanthanides. G. J.-P. D. designed and performed experiments with radionuclides. All authors discussed the entirety of the results. All authors contributed to the writing and approved the manuscript as well as ESI.†Conflicts of interest
The authors declare the following competing financial interests: J. A. M, J. A. C., and G. J.-P. D. are listed as inventors on patent applications submitted by Lawrence Livermore National Laboratory and the Pennsylvania State University.Acknowledgements
This work was performed under the auspices of the U.S. Department of Energy (DOE) by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. Release number: LLNL-JRNL-831983. This work was funded by the Office of Defense Nuclear Nonproliferation Research and Development within the U.S. Department of Energy's National Nuclear Security Administration. J. A. M. and J. A. C. were supported by DOE grant DE-SC0021007. The authors thank Annie Kersting (LLNL) and Mavrik Zavarin (LLNL) for fruitful discussions.Notes and references
- A. Hu, S. N. MacMillan and J. J. Wilson, J. Am. Chem. Soc., 2020, 142, 13500–13506 CrossRef CAS PubMed.
- J. J. Wilson, M. Ferrier, V. Radchenko, J. R. Maassen, J. W. Engle, E. R. Batista, R. L. Martin, F. M. Nortier, M. E. Fassbender, K. D. John and E. R. Birnbaum, Nucl. Med. Biol., 2015, 42, 428–438 CrossRef CAS PubMed.
- E. Boros and A. B. Packard, Chem. Rev., 2019, 119, 870–901 CrossRef CAS PubMed.
- S. H. Ahn, B. A. Vaughn, W. A. Solis, M. L. Lupher, T. J. Hallam and E. Boros, Bioconjugate Chem., 2020, 31, 1177–1187 CrossRef CAS PubMed.
- T. Cheisson and E. J. Schelter, Science, 2019, 363, 489–493 CrossRef CAS PubMed.
- J. J. M. Nelson, T. Cheisson, H. J. Rugh, M. R. Gau, P. J. Carroll and E. J. Schelter, Commun. Chem., 2020, 3, 7 CrossRef CAS.
- Z. Dong, J. A. Mattocks, G. J.-P. Deblonde, D. Hu, Y. Jiao, J. A. Cotruvo Jr and D. M. Park, ACS Cent. Sci., 2021, 7, 1798–1808 CrossRef CAS PubMed.
- V. Radchenko, J. W. Engle, J. J. Wilson, J. R. Maassen, F. M. Nortier, W. A. Taylor, E. R. Birnbaum, L. A. Hudston, K. D. John and M. E. Fassbender, J. Chromatogr. A, 2015, 1380, 55–63 CrossRef CAS PubMed.
- B. Chen, M. He, H. Zhang, Z. Jiang and B. Hu, Phys. Sci. Rev., 2017, 2, 20160057 Search PubMed.
- A. G. Servis, T. Parsons-Davis, K. J. Moody and N. Gharibyan, Ind. Eng. Chem. Res., 2021, 60, 629–638 CrossRef CAS.
- P. Baron, S. M. Cornet, E. D. Collins, G. DeAngelis, G. Del Cul, Yu. Fedorov, J. P. Glatz, V. Ignatiev, T. Inoue, A. Khaperskaya, I. T. Kim, M. Kormilitsyn, T. Koyama, J. D. Law, H. S. Lee, K. Minato, Y. Morita, J. Uhlíř, D. Warin and R. J. Taylor, Prog. Nucl. Energy, 2019, 117, 103091 CrossRef CAS.
- K. L. Nash, Solvent Extr. Ion Exch., 2015, 33, 1–55 CrossRef CAS.
- G. A. Picayo, B. D. Etz, S. Vyas and M. P. Jensen, ACS Omega, 2020, 5, 8076–8089 CrossRef CAS PubMed.
- M. Carrott, A. Geist, X. Hères, S. Lange, R. Malmbeck, M. Miguirditchian, G. Modolo, A. Wilden and R. Taylor, Hydrometallurgy, 2015, 152, 139–148 CrossRef CAS.
- T. Lyseid Authen, A. Wilden, D. Schneider, F. Kreft, G. Modolo, M. R. StJ Foreman and C. Ekberg, Solvent Extr. Ion Exch., 2021, 40, 189–202 CrossRef.
- M. Miguirditchian, V. Vanel, C. Marie, V. Pacary, M.-C. Charbonnel, L. Berthon, X. Hérès, M. Montuir, C. Sorel, M.-J. Bollesteros, S. Costenoble, C. Rostaing, M. Masson and C. Poinssot, Solvent Extr. Ion Exch., 2020, 38, 365–387 CrossRef CAS.
- G. Modolo, A. Wilden, P. Kaufholz, D. Bosbach and A. Geist, Prog. Nucl. Energy, 2014, 72, 107–114 CrossRef CAS.
- D. Lundberg and I. Persson, Coord. Chem. Rev., 2016, 318, 131–134 CrossRef CAS.
- G. J.-P. Deblonde, M. Zavarin and A. B. Kersting, Coord. Chem. Rev., 2021, 446, 214130 CrossRef CAS.
- T. S. Grimes, C. R. Heathman, S. Jansone-Popova, V. S. Bryantsev, S. Goverapet Srinivasan, M. Nakase and P. R. Zalupski, Inorg. Chem., 2017, 56, 1722–1733 CrossRef CAS PubMed.
- T. S. Grimes, C. R. Heathman, S. Jansone-Popova, A. S. Ivanov, V. S. Bryantsev and P. R. Zalupski, Inorg. Chem., 2020, 59, 138–150 CrossRef CAS PubMed.
- S. Özçubukçu, K. Mandal, S. Wegner, M. P. Jensen and C. He, Inorg. Chem., 2011, 50, 7937–7939 CrossRef PubMed.
- J. A. Jackson, V. Linero, N. P. Bessen, K. L. Nash and J. C. Shafer, Sep. Purif. Technol., 2021, 274, 118919 CrossRef CAS.
- G. B. Hall, V. E. Holfeltz, E. L. Campbell, D. Boglaienko, G. J. Lumetta and T. G. Levitskaia, Inorg. Chem., 2020, 59, 4453–4467 CrossRef CAS PubMed.
- T. E. Albrecht-Schmitt, Inorg. Chem., 2019, 58, 1721–1723 CrossRef CAS PubMed.
- N. P. Bessen, J. A. Jackson, M. P. Jensen and J. C. Shafer, Coord. Chem. Rev., 2020, 421, 213446 CrossRef CAS.
- A. Geist, J.-M. Adnet, S. Bourg, C. Ekberg, H. Galán, P. Guilbaud, M. Miguirditchian, G. Modolo, C. Rhodes and R. Taylor, Sep. Sci. Technol., 2021, 56, 1866–1881 CrossRef CAS.
- S. Chapron, C. Marie, V. Pacary, M.-T. Duchesne, G. Arrachart, S. Pellet-Rostaing and M. Miguirditchian, Procedia Chem., 2016, 21, 133–139 CrossRef.
- T. Nakagawa, R. Mitsui, A. Tani, K. Sasa, S. Tashiro, T. Iwama, T. Hayakawa and K. Kawai, PLoS One, 2012, 7, e50480 CrossRef CAS PubMed.
- A. Pol, T. R. M. Barends, A. Dietl, A. F. Khadem, J. Eygensteyn, M. S. M. Jetten and H. J. M. O. den Camp, Environ. Microbiol., 2014, 16, 255–264 CrossRef CAS PubMed.
- J. A. Cotruvo Jr, ACS Cent. Sci., 2019, 5, 1496–1506 CrossRef PubMed.
- L. J. Daumann, Angew. Chem., Int. Ed., 2019, 58, 12795–12802 CrossRef CAS PubMed.
- J. A. Cotruvo Jr, E. R. Featherston, J. A. Mattocks, J. V. Ho and T. N. Laremore, J. Am. Chem. Soc., 2018, 140, 15056–15061 CrossRef PubMed.
- A. M. Ochsner, L. Hemmerle, T. Vonderach, R. Nüssli, M. Bortfeld-Miller, B. Hattendorf and J. A. Vorholt, Mol. Microbiol., 2019, 111, 1152–1166 CrossRef CAS PubMed.
- J. A. Mattocks, J. V. Ho and J. A. Cotruvo Jr, J. Am. Chem. Soc., 2019, 141, 2857–2861 CrossRef CAS PubMed.
- P. Roszczenko-Jasińska, H. N. Vu, G. A. Subuyuj, R. V. Crisostomo, J. Cai, N. F. Lien, E. J. Clippard, E. M. Ayala, R. T. Ngo, F. Yarza, J. P. Wingett, C. Raghuraman, C. A. Hoeber, N. C. Martinez-Gomez and E. Skovran, Sci. Rep., 2020, 10, 12663 CrossRef PubMed.
- S. Masuda, Y. Suzuki, Y. Fujitani, R. Mitsui, T. Nakagawa, M. Shintani and A. Tani, mSphere, 2018, 3, e00462-17 CrossRef PubMed.
- B. Drobot, M. Schmidt, Y. Mochizuki, T. Abe, K. Okuwaki, F. Brulfert, S. Falke, S. A. Samsonov, Y. Komeiji, C. Betzel, T. Stumpf, J. Raff and S. Tsushima, Phys. Chem. Chem. Phys., 2019, 21, 21213–21222 RSC.
- A. Barkleit, A. Heller, A. Ikeda-Ohno and G. Bernhard, Dalton Trans., 2016, 45, 8724–8733 RSC.
- M. P. Jensen, D. Gorman-Lewis, B. Aryal, T. Paunesku, S. Vogt, P. G. Rickert, S. Seifert, B. Lai, G. E. Woloschak and L. Soderholm, Nat. Chem. Biol., 2011, 7, 560–565 CrossRef CAS PubMed.
- G. J.-P. Deblonde, M. Sturzbecher-Hoehne, A. B. Mason and R. J. Abergel, Metallomics, 2013, 5, 619–626 CrossRef CAS PubMed.
- N. Bauer, D. R. Fröhlich and P. J. Panak, Dalton Trans., 2014, 43, 6689–6700 RSC.
- C. D. Auwer, I. Llorens, P. Moisy, C. Vidaud, F. Goudard, C. Barbot, P. L. Solari and H. Funke, Radiochim. Acta, 2005, 93, 699–703 CrossRef.
- M. Ali, A. Kumar, M. Kumar and B. N. Pandey, Biochimie, 2016, 123, 117–129 CrossRef CAS PubMed.
- B. E. Allred, P. B. Rupert, S. S. Gauny, D. D. An, C. Y. Ralston, M. Sturzbecher-Hoehne, R. K. Strong and R. J. Abergel, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10342–10347 CrossRef CAS PubMed.
- G. J.-P. Deblonde, M. Sturzbecher-Hoehne, P. B. Rupert, D. D. An, M.-C. Illy, C. Y. Ralston, J. Brabec, W. A. de Jong, R. K. Strong and R. J. Abergel, Nat. Chem., 2017, 9, 843–849 CrossRef CAS PubMed.
- H. Moll, F. Lehmann and J. Raff, Colloids Surf., B, 2020, 190, 110950 CrossRef CAS PubMed.
- G. J.-P. Deblonde, J. A. Mattocks, D. M. Park, D. W. Reed, J. A. Cotruvo Jr and Y. Jiao, Inorg. Chem., 2020, 59, 11855–11867 CrossRef CAS PubMed.
- M. Nitz, M. Sherawat, K. J. Franz, E. Peisach, K. N. Allen and B. Imperiali, Angew. Chem., Int. Ed., 2004, 43, 3682–3685 CrossRef CAS PubMed.
- L. J. Martin, B. R. Sculimbrene, M. Nitz and B. Imperiali, QSAR Comb. Sci., 2005, 24, 1149–1157 CrossRef CAS.
- L. J. Martin, M. J. Hähnke, M. Nitz, J. Wöhnert, N. R. Silvaggi, K. N. Allen, H. Schwalbe and B. Imperiali, J. Am. Chem. Soc., 2007, 129, 7106–7113 CrossRef CAS PubMed.
- E. R. Featherston, E. J. Issertell and J. A. Cotruvo Jr, J. Am. Chem. Soc., 2021, 143, 14287–14299 CrossRef CAS PubMed.
- Z. Hussain, S. Kim, J. Cho, G. Sim, Y. Park and I. Kwon, Adv. Funct. Mater., 2022, 2109158 CrossRef CAS.
- G. J.-P. Deblonde, J. A. Mattocks, Z. Dong, P. T. Wooddy, J. A. Cotruvo Jr and M. Zavarin, Sci. Adv., 2021, 7, eabk0273 CrossRef CAS PubMed.
- G. J.-P. Deblonde, J. A. Mattocks, H. Wang, E. M. Gale, A. B. Kersting, M. Zavarin and J. A. Cotruvo Jr, J. Am. Chem. Soc., 2021, 143, 15769–15783 CrossRef CAS PubMed.
- H. Singer, B. Drobot, C. Zeymer, R. Steudtner and L. J. Daumann, Chem. Sci., 2021, 12, 15581–15587 RSC.
- M. P. Kelley, N. P. Bessen, J. Su, M. Urban, S. I. Sinkov, G. J. Lumetta, E. R. Batista, P. Yang and J. C. Shafer, Chem. Commun., 2018, 54, 10578–10581 RSC.
- E. C. Cook, E. R. Featherston, S. A. Showalter and J. A. Cotruvo Jr, Biochemistry, 2019, 58, 120–125 CrossRef CAS PubMed.
- S. K. Drake and J. J. Falke, Biochemistry, 1996, 35, 1753–1760 CrossRef CAS PubMed.
- D. B. Halling, B. J. Liebeskind, A. W. Hall and R. W. Aldrich, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E1216–E1225 CrossRef CAS PubMed.
- J. Bruno, W. D. Horrocks and R. J. Zauhar, Biochemistry, 1992, 31, 7016–7026 CrossRef CAS PubMed.
- P. Kaufholz, G. Modolo, A. Wilden, F. Sadowski, D. Bosbach, C. Wagner, A. Geist, P. J. Panak, F. W. Lewis and L. M. Harwood, Solvent Extr. Ion Exch., 2016, 34, 126–140 CrossRef CAS.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- J. A. Mattocks, J. L. Tirsch and J. A. Cotruvo Jr, in Methods in Enzymology, ed. J. A. Cotruvo Jr, Academic Press, 2021, vol. 651, pp. 23–61 Search PubMed.
- S. C. Edington, D. B. Halling, S. M. Bennett, T. R. Middendorf, R. W. Aldrich and C. R. Baiz, Biochemistry, 2019, 58, 2730–2739 CrossRef CAS PubMed.
- J. M. Sperling, E. Warzecha, B. E. Klamm, A. N. Gaiser, C. J. Windorff, M. A. Whitefoot and T. E. Albrecht-Schönzart, Inorg. Chem., 2021, 60, 476–483 CrossRef CAS PubMed.
- N. Adam, M. Trumm, V. C. Smith, R. T. A. MacGillivray and P. J. Panak, Dalton Trans., 2018, 47, 14612–14620 RSC.
- M. Sturzbecher-Hoehne, C. Goujon, G. J.-P. Deblonde, A. B. Mason and R. J. Abergel, J. Am. Chem. Soc., 2013, 135, 2676–2683 CrossRef CAS PubMed.
- A. Barkleit, C. Wilke, A. Heller, T. Stumpf and A. Ikeda-Ohno, Dalton Trans., 2017, 46, 1593–1605 RSC.
- N. A. Thiele, D. J. Fiszbein, J. J. Woods and J. J. Wilson, Inorg. Chem., 2020, 59, 16522–16530 CrossRef CAS PubMed.
- G. J.-P. Deblonde, T. D. Lohrey and R. J. Abergel, Dalton Trans., 2019, 48, 8238–8247 RSC.
- A. V. Gelis, P. Kozak, A. T. Breshears, M. A. Brown, C. Launiere, E. L. Campbell, G. B. Hall, T. G. Levitskaia, V. E. Holfeltz and G. J. Lumetta, Sci. Rep., 2019, 9, 12842 CrossRef PubMed.
- N. Boubals, C. Wagner, T. Dumas, L. Chanèac, G. Manie, P. Kaufholz, C. Marie, P. J. Panak, G. Modolo, A. Geist and P. Guilbaud, Inorg. Chem., 2017, 56, 7861–7869 CrossRef CAS PubMed.
- C. Marie, P. Kaufholz, V. Vanel, M.-T. Duchesne, E. Russello, F. Faroldi, L. Baldini, A. Casnati, A. Wilden, G. Modolo and M. Miguirditchian, Solvent Extr. Ion Exch., 2019, 37, 313–327 CrossRef CAS.
- S. Jansone-Popova, A. S. Ivanov, V. S. Bryantsev, F. V. Sloop, R. Custelcean, I. Popovs, M. M. Dekarske and B. A. Moyer, Inorg. Chem., 2017, 56, 5911–5917 CrossRef CAS PubMed.
- R. J. Ouellette and J. D. Rawn, in Principles of Organic Chemistry, ed. R. J. Ouellette and J. D. Rawn, Elsevier, Boston, 2015, pp. 371–396 Search PubMed.
- H. Moll, M. Glorius and G. Bernhard, Bull. Chem. Soc. Jpn., 2008, 81, 857–862 CrossRef CAS.
- E. A. Christenson and J. Schijf, Geochim. Cosmochim. Acta, 2011, 75, 7047–7062 CrossRef CAS.
- T. Kimura, R. Nagaishi, Y. Kato and Z. Yoshida, Radiochim. Acta, 2001, 89, 125–130 CrossRef CAS.
- T. Kimura, G. R. Choppin, Y. Kato and Z. Yoshida, Radiochim. Acta, 1996, 72, 61–64 CrossRef CAS.
- D. Chaudhuri, W. DeW. Horrocks, J. C. Amburgey and D. J. Weber, Biochemistry, 1997, 36, 9674–9680 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- L. Li, J. Rousseau, M. de G. Jaraquemada-Peláez, X. Wang, A. Robertson, V. Radchenko, P. Schaffer, K.-S. Lin, F. Bénard and C. Orvig, Bioconjugate Chem., 2021, 32, 1348–1363 CrossRef CAS PubMed.
- A. E. V. Gorden, J. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207–4282 CrossRef CAS PubMed.
- E. R. Featherston and J. A. Cotruvo Jr, Biochim. Biophys. Acta, Mol. Cell Res., 2021, 1868, 118864 CrossRef CAS PubMed.
- A. M. Zytnick, N. M. Good, C. C. Barber, M. T. Phi, S. M. Gutenthaler, W. Zhang, L. J. Daumann and N. C. Martinez-Gomez, bioRxiv, 2022 DOI:10.1101/2022.01.19.476857 , pre-print.
- J. B. Glass, E. T. Cowan and K. H. Johannesson, FEMS Microbiol. Lett., 2020, 367, fnaa165 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Method sections, supplemental figures and tables including detailed circular dichroism, spectrophotometric, and fluorescence results, lifetime measurements, and actinide–lanthanide separation results. See https://doi.org/10.1039/d2sc01261h |
| This journal is © The Royal Society of Chemistry 2022 |