
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C2-ketonylation of carbohydrates via excited-state palladium-catalyzed 1,2-spin-center shift†
Gaoyuan
Zhao
a,
Upasana
Mukherjee
a,
Lin
Zhou
b,
Yue
Wu
b,
Wang
Yao
 a,
Jaclyn N.
Mauro
a,
Peng
Liu
*b and
Ming-Yu
Ngai
*a
a,
Jaclyn N.
Mauro
a,
Peng
Liu
*b and
Ming-Yu
Ngai
*a
aDepartment of Chemistry, Institute of Chemical Biology and Drug Discovery, The State University of New York at Stony Brook, Stony Brook, New York 11794, USA. E-mail: ming-yu.ngai@stonybrook.edu
bDepartment of Chemistry, Department of Chemical and Petroleum Engineering, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA. E-mail: pengliu@pitt.edu
First published on 11th May 2022
Abstract
C2-ketonyl-2-deoxysugars, sugars with the C2-hydroxyl group replaced by a ketone side chain, are important carbohydrate mimetics in glycobiology and drug discovery studies; however, their preparation remains a vital challenge in organic synthesis. Here we report the first direct strategy to synthesize this class of glycomimetics from readily available 1-bromosugars and silyl enol ethers via an excited-state palladium-catalyzed 1,2-spin-center shift (SCS) process. This step-economic reaction features broad substrate scope, has a high functional group tolerance, and can be used in late-stage functionalization of natural product- and drug-glycoconjugates. Preliminary experimental and computational mechanistic studies suggested a non-chain radical mechanism involving photoexcited palladium species, a 1,2-SCS process, and a radical Mizoroki–Heck reaction.
Introduction
Carbohydrates with substituents at C2 are ubiquitous in nature and feature prominently in bioactive agents and natural products, including many clinically significant antiviral, anticancer, and antibiotic drugs.1 For example, N-acetylglucosamine (GlcNAc), N-acetylmannosamine (ManNAc), and N-acetylgalactosamine (GalNAc) can be found in living organisms ranging from bacteria to vertebrates and are fundamental components of the cell wall, glycoproteins, and glycolipids (Fig. 1A).1a Consequently, their C2-carbon isosteres, C2-ketonylsugars such as 2-ketoGlc, 2-ketoMan, and 2-ketoGal, have been synthesized for the development of antibiotics2 and to study cell surface recognition, metabolic pathways, and the mechanism of polysaccharide formation and protein post-translational modifications.3 However, the preparation of C2-ketonylsugars is labor-intensive and time-consuming. It requires an 8-step procedure with less than 21% overall yield, multiple protection/deprotection protocols, and the use of toxic alkyl tin and OsO4 reagents (Fig. 1B).3a,b,4 This synthetic strategy is substrate-specific, and it is difficult to produce other such C2-ketonylsugars. Thus, the development of a concise, general method to access a wide array of C2-ketonylsugars from readily accessible starting materials can have a significant impact on glycobiology, medicinal chemistry, and drug discovery.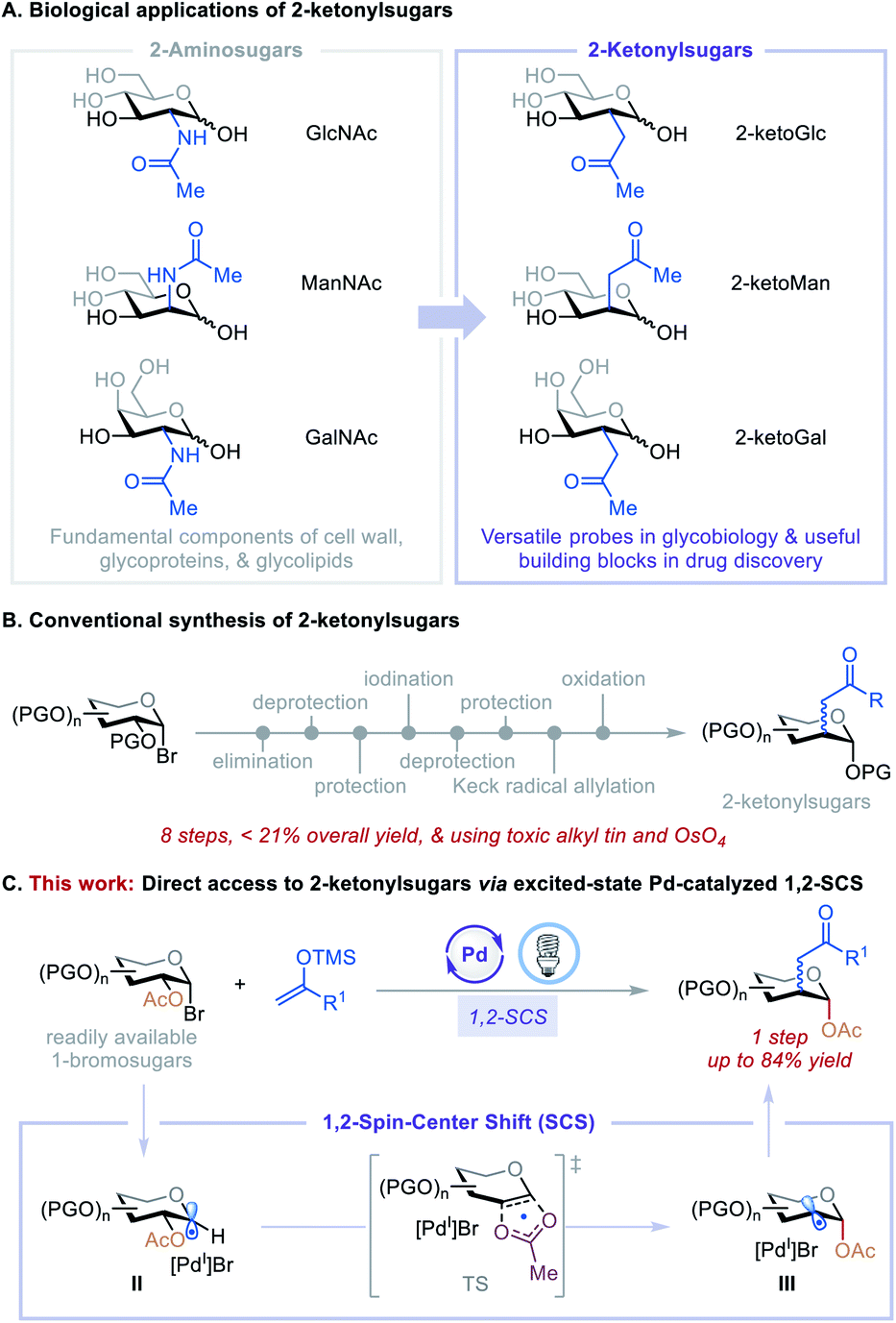 | ||
| Fig. 1 Applications and synthesis of C2-ketonylated carbohydrates. | ||
To develop such a transformation, we were drawn to a 1,2-spin-center shift (SCS) process that involves a 1,2-radical translocation and a group elimination/migration.5 This process has been observed and studied in various biological and chemical applications.6 For example, during DNA biosynthesis, ribonucleotide reductase mediates the formation of deoxyribonucleoside diphosphates via a 1,2-SCS process.6c In carbohydrate chemistry, a tin hydride-mediated 1,2-SCS of 1-bromosugars for the synthesis of 2-deoxysugars was developed by Giese et al.7 Inspired by these reports, we hypothesized that 1,2-SCS could serve as a reaction platform with which to develop a general, catalytic C2-functionalization of carbohydrates. The feasibility of this hypothesis was recently demonstrated by our preliminary studies using nickel catalysis8,9 and excited-state palladium catalysis.10,11 On the basis of these initial findings and the recent development of the excited-state Pd-catalyzed radical Mizoroki–Heck reactions,10i,j,12 we questioned whether we could merge these two reactivities to achieve a catalytic, one-step C2-ketonylation of 1-bromosugars using silyl enol ethers as coupling partners. The mechanism involves the generation of a 1-glycosyl radical (II) followed by a concerted β-C–O bond scission and acetoxyl migration leading to a transition state (TS), then forming a deoxypyranosan-2-yl radical (III) (Fig. 1C). A subsequent radical Mizoroki–Heck reaction and hydrolysis furnishes the desired C2-ketonylsugars. Realization of such a reaction would be novel and significant because it (i) greatly streamlines the synthesis of C2-ketonylsugars from an 8-step protocol to a single-step procedure, (ii) expands the reactivity profile of the excited-state Pd catalysis, and (iii) provides a new strategy for the preparation of useful glycomimetics to tackle fundamental questions in glycobiology and drug discovery.
Results and discussion
According to the postulated mechanism in Fig. 1C, we commenced our study by investigating the reaction of acetyl-protected 1-glucosyl bromide 1a with acetophenone trimethylsilyl enol ether 2a under photoexcited Pd-catalyzed conditions (Table 1). To our delighted, in the presence of 5.00 mol% Pd(PPh3)4, 6.00 mol% Xantphos, and 1.50 equiv. KOAc in benzene (0.015 M) at 90 °C under the irradiation of 36 W blue LEDs for 24 h, the desired C2-ketonylsugar 3a was obtained in 85% yield with 4.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 axial:equatorial (ax:eq) selectivity together with a small amount of the C1-ketonylsugar byproduct (entry 1). Pd(PPh3)4 was critical for this reaction since no reaction occurred in its absence, and only 10% of the desired product was obtained by replacing Pd(PPh3)4 with Pd(OAc)2 (entries 2 & 3). Removal of Xantphos or replacing it with BINAP decreased the reaction yield (entries 4 & 5). Other photosensitizers, such as Ir(ppy)3, Ru(bpy)3(PF6)2, and eosin Y free acid, were either inefficient or failed to catalyze the desired reaction (entries 6–8). Other bases, such as Cs2CO3, diminished the reaction efficiency (entry 9). The use of 1,4-dioxane as a solvent formed hydro-debromination side products, lowering the product yield (entry 10). Higher reaction concentrations or lower temperatures favored the formation of the C1-ketonylsugar side product, decreasing the yield of the desired product 3a (entries 11 & 12). Control experiments confirmed that both oxygen-free conditions and visible light were essential for product formation (entries 13 & 14).
:1 axial:equatorial (ax:eq) selectivity together with a small amount of the C1-ketonylsugar byproduct (entry 1). Pd(PPh3)4 was critical for this reaction since no reaction occurred in its absence, and only 10% of the desired product was obtained by replacing Pd(PPh3)4 with Pd(OAc)2 (entries 2 & 3). Removal of Xantphos or replacing it with BINAP decreased the reaction yield (entries 4 & 5). Other photosensitizers, such as Ir(ppy)3, Ru(bpy)3(PF6)2, and eosin Y free acid, were either inefficient or failed to catalyze the desired reaction (entries 6–8). Other bases, such as Cs2CO3, diminished the reaction efficiency (entry 9). The use of 1,4-dioxane as a solvent formed hydro-debromination side products, lowering the product yield (entry 10). Higher reaction concentrations or lower temperatures favored the formation of the C1-ketonylsugar side product, decreasing the yield of the desired product 3a (entries 11 & 12). Control experiments confirmed that both oxygen-free conditions and visible light were essential for product formation (entries 13 & 14).
|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) | ax/eq |
| a See ESI for Experimental details. Reaction yields and axial to equatorial (ax/eq) ratios were determined by 1H-NMR using CH2Br2 as an internal standard. Ac, acetyl; BINAP, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; LED, light-emitting diode; N.R., no reaction. | |||
| 1 | None | 85 | 4.5:1 |
| 2 | Without Pd(PPh3)4 | N.R. | — |
| 3 | Pd(OAc)2 instead of Pd(PPh3)4 | 10 | 3.2:1 |
| 4 | Without xantphos | 18 | 4.0:1 |
| 5 | BINAP instead of xantphos | 36 | 4.3:1 |
| 6 | Ir(ppy)3 as photocatalyst | 24 | 3.0:1 |
| 7 | Ru(bpy)3(PF6)2 as photocatalyst | N.R. | — |
| 8 | Eosin Y free acid as photocatalyst | N.R. | — |
| 9 | Cs2CO3 as base | 35 | 4.6:1 |
| 10 | Dioxane as solvent | 33 | 3.7:1 |
| 11 | 0.10 M | 25 | 5.0:1 |
| 12 | RT | 39 | 5.1:1 |
| 13 | Air | N.R. | — |
| 14 | Keep in dark | 0 | — |

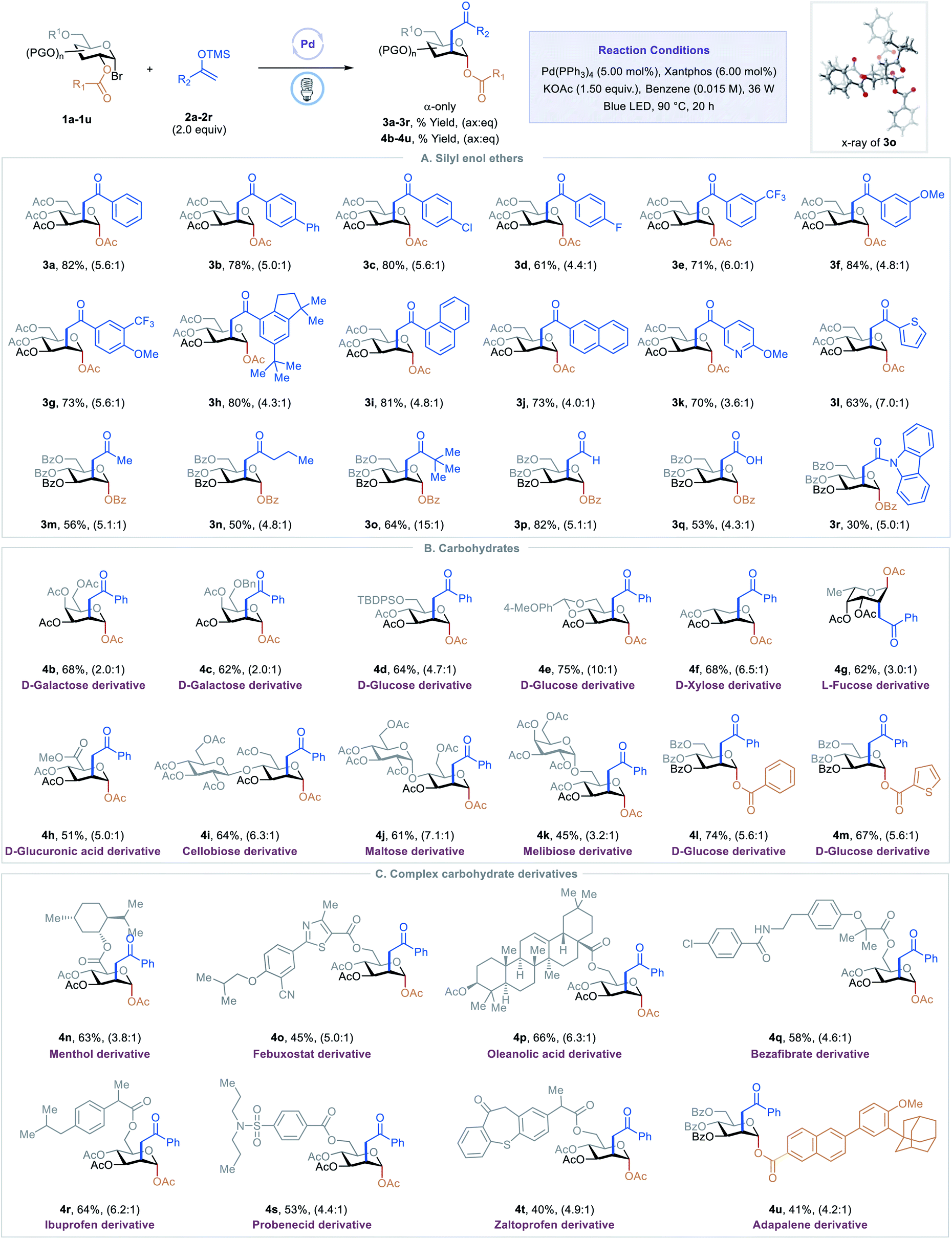
With optimized conditions in hand, we examined scope of the different silyl enol ethers in the reaction. As shown in Table 2, A, a diverse array of silyl enol ethers proved to be competent coupling partners for the C2-ketonylation protocol. Aryl silyl enol ethers with electron-neutral (2a), electron-withdrawing (2b–2e), or electron-donating (2f) substituents on the aryl ring reacted well, delivering the corresponding C2-ketonyl glucosides (3a–3f) in 61–84% yields and with 4.4:1 to 6.0:1 ax:eq selectivity. Aryl silyl enol ethers with multiple substituents (2g–2h) or extended conjugation (2i–2j) were compatible. The transformation is effective for compounds with medicinally relevant heterocyclic derivatives, such as pyridyl (2k) or thiophenyl (2l). Alkyl silyl enol ethers were also viable substrates, furnishing the desired products (2m–2o) with moderate to good yields. Silyl vinyl ether, a surrogate for acetaldehyde, couples with the C2-radical to give the C2-formylmethyl glucoside 3p in 81% yield and 5.1:1 ax:eq selectivity. When the silyl enol ether (2q) derived from tert-butyl acetate was employed as a substrate, the C2-carboxymethyl glucoside 3q was generated, presumably through the hydrolysis of the resulting silyl enol intermediate. Finally, the C2-acetamidated product 3r could also be obtained with this strategy. The absolute stereochemistry of the product was confirmed by a single-crystal analysis of 3o, as shown in Table 2.13
|
a See ESI for Experimental details. Isolated yield and ax:eq ratio are indicated below each entry.
|
|---|
|
Next, we evaluated the generality of this transformation with respect to 1-bromosugars (Table 2, B). A wide range of 1-bromosugars, including derivatives of D-galactose, D-glucose, D-xylose, and L-fucose (1b–1g), reacted with silyl enol ether 2a, affording the desired products (4b–4g) in 62–86% yields and with up to 10:1 ax:eq selectivity. It is noteworthy that peracetylated monosaccharide derivatives are particularly useful because they have been shown to passively diffuse through mammalian cell membranes and undergo subsequent deacetylation by cytosolic or ER esterases.14 Other protecting groups such as benzyl, tert-butyldiphenylsilyl, acetal, and benzoyl were well-tolerated as well. The D-glucuronic acid derivative (1h) was also a viable substrate, and disaccharide derivatives, such as cellobiose, maltose, and melibiose, proved to be compatible with the standard reaction conditions (4i–4k). In addition, C2-esters substituted with aryl or heteroaryl groups migrated smoothly, delivering the desired products (4l–4m) in good yields and with good selectivity.
Late-stage modification of complex molecules is often a key to identifying medicinal agents.15 To demonstrate the applicability of the excited-state Pd-catalyzed C2-ketonylation to late-stage syntheses, natural product- and drug-conjugated sugars were subjected to the standard reaction conditions (Table 2, C). For example, 1-bromosugar derivatives of L-menthol (decongestant and analgesic), febuxostat (an anti-hyperuricemic drug), oleanolic acid, bezafibrate (antilipemic agent), ibuprofen (non-steroidal anti-inflammatory drug, NSAID), probenecid (anti-gout), zaltoprofen (NSAID), and adapalene (antiacne agent) worked well under the standard conditions, affording the desired products 4n–4u in 40–66% yields and with up to 6.3:1 ax:eq selectivity.
The C2-ketonylsugar products are useful synthetic intermediates and can be converted into other novel glycomimetics (Table 3). For example, C2-ketonylated glycoside could be reduced to C2-hydroxyalkylated glycoside 5a and C2-alkylated glycoside 6a (Table 3, A and B). Cyclopropanated glycoside 7a, an important glycosylation donor,16 could be prepared from the C2-ketonylsugar (Table 3, C). Under Lewis acid activation conditions, 3a undergoes cyclization, and the resulting carbocation can be trapped with furan, affording perhydrofuro[2,3-b]pyran 8a in good yield (Table 3, D).17 C2-ketonylsugars can also serve as good glycosylation donors. For example, N/S/O-glycosylation of 3a proceeds smoothly, furnishing glycosyl azide 9a, thioglycoside 10a, trans-androsteronyl glycoside 12a, and disaccharide 14a in good yields and up to 20:1 α/β-selectivity (Table 3, E–H).
| a See ESI for Experimental details. Isolated yield and diastereomeric ratio are indicated below each entry. |
|---|

|
To better understand the mechanism of this excited-state Pd-catalyzed C2-ketonylation, we conducted a series of experimental and computational studies (Fig. 2). First, Stern–Volmer quenching studies showed that 1-glucosyl bromide 1a quenches the excited palladium species more efficiently than silyl enol ether 2a (Fig. 2A). The radical-trapping experiment using 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) as the scavenger significantly inhibited the reaction, and the radical clock reaction gave the desired product with a ring-opening (3s), implying the reaction proceeds through a radical pathway (Fig. 2B). However, radical chain propagation is unlikely as the quantum yield of the reaction was found to be 0.009 (Fig. S9 in the ESI†). When the 1,2-trans- and 1,2-cis 2-iodo-sugar (15a and 16a, respectively) were subjected to the reaction conditions, they both formed the desired product (3a) with similar yields and stereoselectivity as produced by the parent reaction (Fig. 2Cvs.Table 1, entry 1) and without the formation of C1-ketonylated side products. These results suggest that the reaction proceeds through a common deoxypyranosan-2-yl radical intermediate (III), and the reverse acetoxy migration is slower than the addition of radical III to silyl enol ethers. Furthermore, crossover experiments using substrates 1a and 1k afforded only the non-crossover products 3a and 4k, suggesting that the 1,2-SCS probably takes place through an in-cage or a concerted mechanism (Fig. 2D). DFT calculations showed that the addition of C2-radical III to the silyl enol ether via transition state TS1-ax to form IV-ax is more favorable than the formation of the equatorial isomer IV-eqviaTS1-eq (Fig. S11 in the ESI†).
 | ||
| Fig. 2 Mechanistic studies and proposed mechanism. See ESI† for Experimental details. | ||
Combining the insights gained from these experiments and published reports,10–12 a plausible mechanism is shown in Fig. 2E. The photoexcited species [Pd0]* abstracts a bromine atom from 1-bromosugar 1, forming a [PdI]Br complex and 1-glycosyl radical intermediate II. This radical intermediate then undergoes a 1,2-SCS pathway through a conformational change (IIa) followed by concerted [2,3]-acyloxy migration (IIb) under the standard reaction conditions, generating deoxypyranosan-2-yl radical III. Although the C2-radical is more reactive than the C1-radical, the formation of an anomeric C–O bond lowers the molecular energy of III and drives the migration.7b,9 C2-radical species III adds to silyl enol ether 2, furnishing intermediate IV. Pd-catalyzed β-hydride elimination or palladoradical H-atom abstraction liberates H[PdII]Br and silyl enol ether V, which upon hydrolysis affords the desired product 3. Meanwhile, base-assisted H–Br reductive elimination of H[PdII]Br regenerates the ground state [Pd0] catalyst, closing the catalytic cycle.18
Conclusions
In summary, we have developed a one-step synthesis of valuable C2-ketonylsugars from readily available 1-bromosugars and silyl enol ethers via a 1,2-SCS process catalyzed by excited-state palladium. The reaction features a broad substrate scope, tolerates a wide range of functional groups, and is amenable to late-stage modification of disaccharides, natural product- and drug-glycoconjugates. Preliminary experimental and computational mechanistic studies suggest a non-chain radical mechanism involving photoexcited Pd-complexes, a 1,2-SCS process, and a Mizoroki–Heck reaction. The catalytic 1,2-SCS process via a [2,3]-acyloxy migration could (i) offer a general catalytic strategy for the site-selective functionalization of carbohydrates to access a wide array of unexplored carbohydrate mimetics and (ii) guide the design and development of new transformations in organic synthesis.Data availability
All the data associated with this manuscript were provided in ESI.†Author contributions
G. Z., U. M., W. Y., and J. N. M performed the experiments, synthesized starting materials, developed substrate scope, and conducted detailed mechanistic studies. L. Z., Y. W., and P. L. designed and performed the DFT calculations. G. Z. and M.-Y. N. conceived the idea, designed the research, and wrote the manuscript. All the authors commented on the final draft of the manuscript and contributed to the analysis and interpretation of the data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this publication was supported by the National Institutes of Health (R35-GM119652 to M.-Y. N and R35-GM128779 to P. L.). DFT calculations were performed at the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the National Science Foundation grant number ACI1548562. The Shimadzu UPLC/MS used for portions of this work were purchased with funds from NIGMS equipment administrative supplement (R35-GM119652-04S1), Shimadzu Scientific Instruments grant, and Office of the Vice President for Research at Stony Brook University. We thank Dr Lev Zakharov (CAMCOR, University of Oregon) for the X-ray data of 3o.Notes and references
- (a) A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Essentials of Glycobiology, Cold Spring Harbor Press, Cold Spring Harbor, New York, 3rd edn, 2017 Search PubMed; (b) H. Ye, C. Xiao and L. Lu, Chin. J. Org. Chem., 2018, 38, 1897–1906 CrossRef CAS; (c) C. S. Bennett and M. C. Galan, Chem. Rev., 2018, 118, 7931–7985 CrossRef CAS PubMed.
- D. W. Gammon, R. Hunter, D. J. Steenkamp and T. T. Mudzunga, Bioorg. Med. Chem. Lett., 2003, 13, 2045–2049 CrossRef CAS PubMed.
- (a) H. C. Hang and C. R. Bertozzi, J. Am. Chem. Soc., 2001, 123, 1242–1243 CrossRef CAS PubMed; (b) N. Khidekel, S. Arndt, N. Lamarre-Vincent, A. Lippert, K. G. Poulin-Kerstien, B. Ramakrishnan, P. K. Qasba and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2003, 125, 16162–16163 CrossRef CAS PubMed; (c) N. Khidekel, S. B. Ficarro, E. C. Peters and L. C. Hsieh-Wilson, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 13132–13137 CrossRef CAS PubMed; (d) H.-C. Tai, N. Khidekel, S. B. Ficarro, E. C. Peters and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2004, 126, 10500–10501 CrossRef CAS PubMed; (e) N. Khidekel, S. B. Ficarro, P. M. Clark, M. C. Bryan, D. L. Swaney, J. E. Rexach, Y. E. Sun, J. J. Coon, E. C. Peters and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2007, 3, 339–348 CrossRef CAS PubMed; (f) J. E. Rexach, P. M. Clark and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2008, 4, 97–106 CrossRef CAS PubMed; (g) M. Pasek, B. Ramakrishnan, E. Boeggeman, M. Manzoni, T. J. Waybright and P. K. Qasba, Bioconjugate Chem., 2009, 20, 608–618 CrossRef CAS PubMed.
- L. Cai, W. Guan, W. Chen and P. G. Wang, J. Org. Chem., 2010, 75, 3492–3494 CrossRef CAS PubMed.
- P. Wessig and O. Muehling, Eur. J. Org. Chem., 2007, 2007, 2219–2232 CrossRef.
- For selected accounts and reviews on the spin-center shift process, see: (a) A. L. J. Beckwith, D. Crich, P. J. Duggan and Q. Yao, Chem. Rev., 1997, 97, 3273–3312 CrossRef CAS PubMed; (b) H. Zipse, Acc. Chem. Res., 1999, 32, 571–578 CrossRef CAS; (c) H. Eklund, U. Uhlin, M. Färnegårdh, D. T. Logan and P. Nordlund, Prog. Biophys. Mol. Biol., 2001, 77, 177–268 CrossRef CAS PubMed; (d) B. Halliwell and J. M. Gutteridge, Free radicals in biology and medicine, Oxford university press, USA, 2015 CrossRef; (e) C. E. Suh, H. M. Carder and A. E. Wendlandt, ACS Chem. Biol., 2021, 16, 1814–1828 CrossRef CAS PubMed; (f) C. Marino and A. V. Bordoni, Org. Biomol. Chem., 2022, 20, 934–962 RSC; (g) F.-L. Zhang, B. Li, K. Houk and Y.-F. Wang, JACS Au, 2022 DOI:10.1021/jacsau.2c00051 ; For selected examples, see:; (h) M. Newcomb, J. H. Horner, P. O. Whitted, D. Crich, X. Huang, Q. Yao and H. Zipse, J. Am. Chem. Soc., 1999, 121, 10685–10694 CrossRef CAS; (i) P. Wessig and O. Mühling, Angew. Chem., Int. Ed., 2001, 40, 1064–1065 CrossRef CAS; (j) J. H. Horner, L. Bagnol and M. Newcomb, J. Am. Chem. Soc., 2004, 126, 14979–14987 CrossRef CAS PubMed; (k) J. Jin and D. W. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS PubMed; (l) B. Bieszczad, L. A. Perego and P. Melchiorre, Angew. Chem., 2019, 131, 17034–17039 CrossRef; (m) V. Dimakos, D. Gorelik, H. Y. Su, G. E. Garrett, G. Hughes, H. Shibayama and M. S. Taylor, Chem. Sci., 2020, 11, 1531–1537 RSC; (n) H. Fuse, H. Nakao, Y. Saga, A. Fukatsu, M. Kondo, S. Masaoka, H. Mitsunuma and M. Kanai, Chem. Sci., 2020, 11, 12206–12211 RSC; (o) Y. Masuda, H. Tsuda and M. Murakami, Angew. Chem., Int. Ed., 2020, 59, 2755–2759 CrossRef CAS PubMed; (p) H. M. Carder, C. E. Suh and A. E. Wendlandt, J. Am. Chem. Soc., 2021, 143, 13798–13805 CrossRef CAS PubMed; (q) J. A. Turner, N. Rosano, D. J. Gorelik and M. S. Taylor, ACS Catal., 2021, 11, 11171–11179 CrossRef CAS; (r) Q. Zhu and D. G. Nocera, ACS Catal., 2021, 11, 14181–14187 CrossRef CAS.
- (a) B. Giese, K. S. Gröninger, T. Witzel, H. G. Korth and R. Sustmann, Angew. Chem., Int. Ed., 1987, 26, 233–234 CrossRef; (b) H. G. Korth, R. Sustmann, K. S. Groeninger, M. Leisung and B. Giese, J. Org. Chem., 1988, 53, 4364–4369 CrossRef CAS.
- For selected accounts and reviews on nickel catalysis: (a) M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525–1532 CrossRef CAS; (b) X. Hu, Chem. Sci., 2011, 2, 1867–1886 RSC; (c) J. Montgomery, in Organometallics in Synthesis, ed. B. H. Lipshutz, Wiley, Hoboken, NJ, 2013, pp. 319–428, DOI:10.1002/9781118651421.ch3; (d) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC; (e) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed; (f) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef CAS PubMed; (g) T. Iwasaki and N. Kambe, in Ni-and Fe-Based Cross-Coupling Reactions, ed. A. Correa, Springer International Publishing, Cham, 2017, pp. 1–36, DOI:10.1007/978-3-319-49784-6_1; (h) J. Twilton, P. Zhang, M. H. Shaw, R. W. Evans and D. W. MacMillan, Nat. Rev. Chem., 2017, 1, 1–19 CrossRef; (i) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed; (j) S. Ogoshi, Nickel Catalysis in Organic Synthesis: Methods and Reactions, John Wiley & Sons, 2020 CrossRef; (k) K. E. Poremba, S. E. Dibrell and S. E. Reisman, ACS Catal., 2020, 10, 8237–8246 CrossRef CAS PubMed; (l) J. Diccianni, Q. Lin and T. Diao, Acc. Chem. Res., 2020, 53, 906–919 CrossRef CAS PubMed.
- G. Zhao, W. Yao, I. Kevlishvili, J. N. Mauro, P. Liu and M.-Y. Ngai, J. Am. Chem. Soc., 2021, 143, 8590–8596 CrossRef CAS PubMed.
- For selected accounts and reviews on visible-light-induced excited-state Pd catalysis, see: (a) P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef CAS PubMed; (b) D. Kurandina, P. Chuentragool and V. Gevorgyan, Synthesis, 2019, 51, 985 CrossRef CAS PubMed; (c) R. Kancherla, K. Muralirajan, S. Arunachalam and M. Rueping, Trends Chem, 2019, 1, 510–523 CrossRef CAS; (d) W.-J. Zhou, G.-M. Cao, Z.-P. Zhang and D.-G. Yu, Chem. Lett., 2019, 48, 181–191 CrossRef CAS; (e) Z. Li, J. Jin and S. Huang, Chin. J. Org. Chem., 2020, 40, 563–574 CrossRef CAS; (f) W.-M. Cheng and R. Shang, ACS Catal., 2020, 10, 9170–9196 CrossRef CAS; (g) K. P. Shing Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef CAS PubMed ; For seminal work on visible-light-induced excited-state Pd catalysis, see:; (h) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340–6343 CrossRef CAS PubMed; (i) D. Kurandina, M. Parasram and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 14212–14216 CrossRef CAS PubMed; (j) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307–18312 CrossRef CAS PubMed; (k) W. J. Zhou, G. M. Cao, G. Shen, X. Y. Zhu, Y. Y. Gui, J. H. Ye, L. Sun, L. L. Liao, J. Li and D. G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15683–15687 CrossRef CAS PubMed; (l) L. Sun, J.-H. Ye, W.-J. Zhou, X. Zeng and D.-G. Yu, Org. Lett., 2018, 20, 3049–3052 CrossRef CAS PubMed.
- (a) G. Zhao, W. Yao, J. N. Mauro and M. Y. Ngai, J. Am. Chem. Soc., 2021, 143, 1728–1734 CrossRef CAS PubMed; (b) W. Yao, G. Zhao, Y. Wu, L. Zhou, U. Mukherjee, P. Liu and M.-Y. Ngai, J. Am. Chem. Soc., 2022, 144, 3353–3359 CrossRef CAS PubMed.
- For selected examples of excited-state Pd-catalyzed Mizoroki-Heck reactions, see: (a) G.-Z. Wang, R. Shang and Y. Fu, Org. Lett., 2018, 20, 888–891 CrossRef CAS PubMed; (b) D. Kurandina, M. Rivas, M. Radzhabov and V. Gevorgyan, Org. Lett., 2018, 20, 357–360 CrossRef CAS PubMed; (c) M. Koy, F. Sandfort, A. Tlahuext-Aca, L. Quach, C. G. Daniliuc and F. Glorius, Chem. - Eur. J., 2018, 24, 4552–4555 CrossRef CAS PubMed; (d) P. Chuentragool, D. Yadagiri, T. Morita, S. Sarkar, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 1794–1798 CrossRef CAS PubMed; (e) L. He, C. Jia, Y. Zhang and J. He, Macromol. Rapid Commun., 2020, 41, 1900640 CrossRef CAS PubMed; (f) L. Feng, L. Guo, C. Yang, J. Zhou and W. Xia, Org. Lett., 2020, 22, 3964–3968 CrossRef CAS PubMed; (g) M. Li, Y.-F. Qiu, C.-T. Wang, X.-S. Li, W.-X. Wei, Y.-Z. Wang, Q.-F. Bao, Y.-N. Ding, W.-Y. Shi and Y.-M. Liang, Org. Lett., 2020, 22, 6288–6293 CrossRef CAS PubMed; (h) R. Adamik, T. Földesi and Z. Novák, Org. Lett., 2020, 22, 8091–8095 CrossRef CAS PubMed; (i) H.-M. Huang, P. Bellotti, P. M. Pflueger, J. L. Schwarz, B. Heidrich and F. Glorius, J. Am. Chem. Soc., 2020, 142, 10173–10183 CrossRef CAS PubMed; (j) N. Kvasovs, V. Iziumchenko, V. Palchykov and V. Gevorgyan, ACS Catal., 2021, 11, 3749–3754 CrossRef CAS PubMed; (k) G. S. Lee, D. Kim and S. H. Hong, Nat. Commun., 2021, 12, 991 CrossRef CAS PubMed.
- CCDC 2142346 contains the supplementary crystallographic data for this paper.†.
- (a) A. K. Sarkar, T. A. Fritz, W. H. Taylor and J. D. Esko, Proc. Natl. Acad. Sci. U.S.A., 1995, 92, 3323–3327 CrossRef CAS PubMed; (b) C. L. Jacobs, K. J. Yarema, L. K. Mahal, D. A. Nauman, N. W. Charters and C. R. Bertozzi, Methods Enzymol., 2000, 327, 260–275 CAS.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- (a) J. Yin and T. Linker, Org. Biomol. Chem., 2012, 10, 2351–2362 RSC; (b) S. I. Awan and D. B. Werz, Bioorg. Med. Chem., 2012, 20, 1846–1856 CrossRef CAS PubMed; (c) R. Bielski and Z. J Witczak, Curr. Org. Chem., 2014, 18, 1897–1912 CrossRef CAS; (d) J. E. Harvey, R. J. Hewitt, P. W. Moore and K. K. Somarathne, Pure Appl. Chem., 2014, 86, 1377–1399 CAS; (e) Y. D. Vankar and T. Linker, Eur. J. Org. Chem., 2015, 2015, 7633–7642 CrossRef CAS.
- X. Ma, Q. Tang, J. Ke, J. Zhang, X. Yang, X. Shen and H. Shao, Org. Biomol. Chem., 2014, 12, 7381–7388 RSC.
- Another plausible mechanism involves bromine atom transfer from [PdI]Br to VI followed by H–Br elimination to form V and ground-state [Pd0]. For halogen atom transfer from [PdI]X to alkyl radicals, see (a) M. Ratushnyy, M. Parasram, Y. Wang and V. Gevorgyan, Angew. Chem., Int. Ed., 2018, 57, 2712–2715 CrossRef CAS PubMed; (b) D. Kim, G. S. Lee, D. Kim and S. H. Hong, Nat. Commun., 2020, 11, 1–13 CrossRef PubMed ; and ref. 12k..
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2142346. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01042a |
| This journal is © The Royal Society of Chemistry 2022 |