
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of novel functional mini-receptors by combinatorial screening of split-WW domains†
Hermann
Neitz‡
 c,
Niels Benjamin
Paul‡
d,
Florian R.
Häge
ae,
Christina
Lindner
ae,
Roman
Graebner
ae,
Michael
Kovermann
*b and
Franziska
Thomas
*ae
c,
Niels Benjamin
Paul‡
d,
Florian R.
Häge
ae,
Christina
Lindner
ae,
Roman
Graebner
ae,
Michael
Kovermann
*b and
Franziska
Thomas
*ae
aInstitute of Organic Chemistry, Heidelberg University, Im Neuenheimer Feld 270, Heidelberg 69120, Germany. E-mail: franziska.thomas@oci.uni-heidelberg.de
bDepartment of Chemistry, University of Konstanz, Universitätsstraße 10, Konstanz 78457, Germany. E-mail: Michael.Kovermann@Uni-Konstanz.de
cInstitute of Organic Chemistry, University of Würzburg, Am Hubland, Würzburg 97074, Germany
dInstitute of Organic and Biomolecular Chemistry, University of Göttingen, Tammannstr. 2, Göttingen 37077, Germany
eCentre for Advanced Materials, Heidelberg University, Im Neuenheimer Feld 225, Heidelberg 69120, Germany
First published on 14th July 2022
Abstract
β-Sheet motifs such as the WW domain are increasingly being explored as building blocks for synthetic biological applications. Since the sequence-structure relationships of β-sheet motifs are generally complex compared to the well-studied α-helical coiled coil (CC), other approaches such as combinatorial screening should be included to vary the function of the peptide. In this study, we present a combinatorial approach to identify novel functional mini-proteins based on the WW-domain scaffold, which takes advantage of the successful reconstitution of the fragmented WW domain of hPin1 (hPin1WW) by CC association. Fragmentation of hPin1WW was performed in both loop 1 (CC-hPin1WW-L1) and loop 2 (CC-hPin1WW-L2), and the respective fragments were linked to the strands of an antiparallel heterodimeric CC. Structural analysis by CD and NMR spectroscopy revealed structural reconstitution of the WW-domain scaffold only in CC-hPin1WW-L1, but not in CC-hPin1WW-L2. Furthermore, by using 1H–15N HSQC NMR, fluorescence and CD spectroscopy, we demonstrated that binding properties of fragmented hPin1WW in CC-hPin1WW-L1 were fully restored by CC association. To demonstrate the power of this approach as a combinatorial screening platform, we synthesized a four-by-six library of N- and C-terminal hPin1WW-CC peptide fragments that was screened for a WW domain that preferentially binds to ATP over cAMP, phophocholine, or IP6. Using this screening platform, we identified one WW domain, which specifically binds ATP, and a phosphorylcholine-specific WW-based mini-receptor, both having binding dissociation constants in the lower micromolar range.
Introduction
Small, independently folding protein motifs are commonly used as scaffolds to mimic complex biological systems.1,2 Among the protein folding motifs available, the coiled coil (CC) has probably been most frequently used in such endeavours to date, as the sequence-structure relationship is well understood.3 Consequently, CCs have been applied in various synthetic biological designs including biomimetic receptors,4 catalysts,5 interaction domains,6 or vesicle fusion proteins.7 Despite the obvious advantages of CCs in the design of biomimetic systems, the disadvantages should not be overlooked and illustrate why further small protein folding motifs are increasingly being explored for applications in synthetic biology. For instance, the rigidity of CC assemblies might impede the effective design of biomimetic receptors and enzymes, since flexibility of the binding pocket is an important factor in ligand or substrate binding.2,8 Another obstacle is the frequent use of self-assembling CCs, very often homomers, which limits the engineerability and strand exchange is a significant problem.9Among others, WW domains are a good complement to the toolbox of small protein motifs used for synthetic biological applications. WW domains are stable antiparallel β-sheet peptides with a length of about 35 amino acids, which are protein–protein interaction modules and recognize proline-rich amino acid sequences.10 The name is derived from two characteristic and highly conserved tryptophan residues, which are crucial for structural stability and function.11,12 Calculations have shown that WW domains cover a very high sequence space, which makes this motif ideal for design and engineering purposes.13 This has been experimentally proven, as based on sequence alignments, artificial WW domains have been designed or redesigned to modulate folding properties.14 However, there are only a few examples showing successful engineering of WW domain function. In these cases, active sites located on the β-sheet surface of a natural protein were transferred more or less one-to-one to the β-sheet surface of a WW domain. For example, the WW domain of FBP11 was modified according to the natural model heat shock protein such that it binds specifically to single-stranded DNA,15 and a zinc-binding WW domain was obtained by transferring the His3-site of carbonic anhydrase II to the surface of the hPin1 WW domain and was further engineered to obtain a Zn (II) sensor.16
However, intuitive design of new functional WW domains becomes almost impossible if there is no natural model with similar structural features for the intended new function. Therefore, we considered a combinatorial approach that allows screening of multiple WW-domain variants. In the past, it has been shown that the function of fragmented proteins can be reconstituted if their fragments are forced into close proximity.17 For example, antiparallel CCs have been successfully used in the reconstitution of the green fluorescent protein or the enzyme luciferase.18 Recently, in-cell proteolysis-based signalling and logic circuits have been developed, in which split proteases are attached to orthogonal CC dimerization domains.19 Inspired by these reports and encouraged by a circularly permuted hyperstable WW domain,20 we identified a split-WW-domain approach as a potential screening platform for exploring engineered WW domains with novel functions, in which a fragmented WW domain is reconstituted by association of an antiparallel CC (ACCBCC, Fig. 1). This split approach would, if successful, facilitate the diversification of WW-domain sequences and thus the synthesis of WW-domain libraries. However, compared with previously reported systems, the WW domain is a small protein folding motif, and reconstitution of both structure and function in such a system requires thorough initial testing.
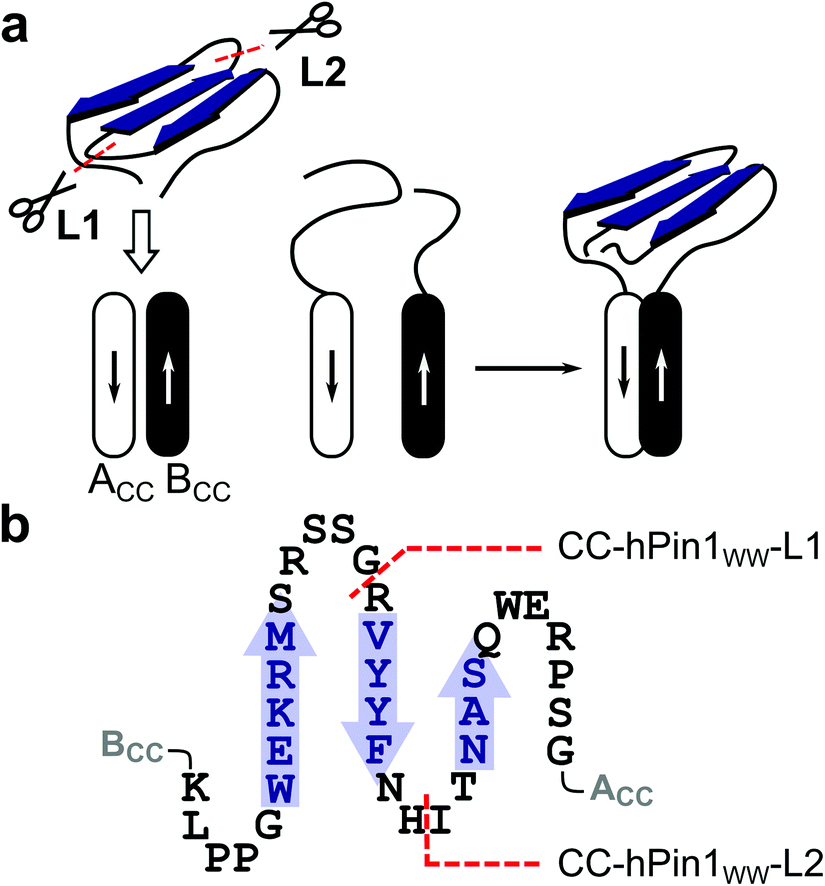 | ||
| Fig. 1 Reconstitution of a fragmented WW domain on an antiparallel heterodimeric CC (ACCBCC). (a) Schematic representation of the structural concept applied in this study (ACC – acidic coil strand; BCC – basic coil strand). (b) Two-dimensional representation of the folding motif of the WW domain of hPin1 (hPin1WW) and the two possible fragmentation sites defining CC-hPin1WW-L1 (fragmentation at the GR site in loop 1) and CC-hPin1WW-L2 (fragmentation at the HI site in loop 2) in a WW-domain-CC construct. The β-sheets comprising hPin1WW are highlighted in blue and represented as arrows. | ||
In this report, we present the successful reconstitution of a model WW domain, the WW domain of hPin1 (hPin1WW, hPin1(6-39)), by CC association. We have combined circular dichroism (CD) and nuclear magnetic resonance (NMR) spectroscopy to prove at first the proper structural reconstitution of hPin1WW. Then, NMR, fluorescence and CD spectroscopy was applied to investigate the functional binding of hPin1WW to the C-terminal domain peptide (CTD) of RNA polymerase II in a second step. As a result, our data show complete structural and functional reconstitution of fragmented hPin1WW in a CC-split-hPin1WW system. Based on this, we further developed the CC-split-hPin1WW concept into a combinatorial screening platform. As a proof-of-principle, we synthesized a four-by-six library of N- and C-terminal hPin1WW-CC peptide fragments that was successfully screened for WW domains possessing binding properties to organophosphates.
Results and discussion
Concept design and peptide synthesis
To demonstrate the concept of proximity-induced reconstitution of a fragmented WW domain based on CC association that could be used as a combinatorial screening platform, a suitable model WW domain had to be identified. It has been shown that a circularly permuted variant of hPin1WW reliably folds into a WW-domain scaffold.20 However, the amino acid sequence of this variant was significantly changed to achieve structural stability. For instance, β-caps were introduced at the permuted N- and C-termini and a tryptophan cage was required for structural stabilization. Since such severe structural changes contradict the design of a screening platform for functional WW domains and significantly limit the accessible sequence space, we relied on wild type hPin1WW as the target peptide in our studies. If a moderately stable WW domain such as hPin1WW does not fold in a proximity-induced manner after fragmentation, the envisaged concept would generally be unsuitable for the intended purpose.hPin1WW is a well characterized WW domain21–23 and has shown to be remarkably stable to engineering.20,24 Although the loops, more precisely loop1, play an important role in WW-domain folding,25,26 they are less conserved compared to the β-sheet regions (Fig. S1†). Hence, we split hPin1WW at the GR site (G20, R21) of loop 1 (CC-hPin1WW-L1) and the HI site (H27, I28) of loop 2 (CC-hPin1WW-L2), respectively (Fig. 1). The fragments were attached to the strands of an antiparallel heterodimeric CC, more precisely, to de novo designed Acid-Kg Base-Eg,27 which, in the following, are referred to as ACCBCC (Fig. 1). The conjugates of coil peptides and WW-domain fragments are referred to as BCC-hPin1WW-L1 or BCC-hPin1WW-L2 for the N-terminal WW-domain fragments linked to coil strand BCC, and hPin1WW-L1-ACC and hPin1WW-L2-ACC for the C-terminal WW-domain fragments linked to coil strand ACC (Table 1; note: The suffix L1/L2 identifies the fragmentation site – loop 1 or loop 2 – of the respective split-WW domains as well as their individual fragments). One-to-one mixtures of the related N- and C-terminal fragments then form CC-hPin1WW-L1 or CC-hPin1WW-L2.
| Peptide | Sequence |
|---|---|
| hPin1WW | H-KLPPGWEKRMSRSSGRVYYFNHITNASQWERPSG-OH |
| BCC-hPin1WW-L1 | BCC-KLPPGWEKRMSRSSG-OH |
| hPin1WW-L1-ACC | H-RVYYFNHITNASQWERPSG-ACC |
| BCC-hPin1WW-L2 | BCC-KLPPGWEKRMSRSSGRVYYFNH-OH |
| hPin1WW-L2-ACC | H-ITNASQWERPSG-ACC |
All peptides were synthesized using microwave-assisted Fmoc/tBu-based solid-phase peptide synthesis (SPPS). Interestingly, we observed an overall improved purity of the crude CC-hPin1WW peptides compared to crude hPin1WW (Fig. S2†), which we attribute to the known aggregation propensity of WW domains during the SPPS.28 As only fragments of hPin1WW were attached to the CC strands, this effect was less pronounced.
CD spectroscopic analysis of structural properties and thermodynamic stability of CC-hPin1WW variants
To study reconstitution of the fragmented CC-hPin1WW upon CC association, CD spectra and CD thermal denaturation profiles were recorded of both CC-hPin1WW constructs (CC-hPin1WW-L1 and CC-hPin1WW-L2), and hPin1WW and the antiparallel CC peptide ACCBCC were measured as references (Fig. 2). The CD spectrum of hPin1WW reveals the typical shape reported for WW domains showing a minimum at 197 nm, the characteristic shoulder or minimum at 206 nm and a maximum at 227 nm, which results from the disordered termini, the triple-stranded antiparallel β-sheet structure and the exciton coupling of the aromatic residues of the hydrophobic core, respectively.23 The antiparallel CC peptide was α-helical displayed by the strong minima at 208 and 222 nm. A CD spectrum of ACCBCC and hPin1WW in an one-to-one mixture was recorded as a reference spectrum (Fig. 2A, black dashed line). The CD spectra of CC-hPin1WW-L1 and CC-hPin1WW-L2 should have a similar profile in the case of a fully folded WW-domain region. Indeed, the CD spectrum of CC-hPin1WW-L1 (red) showed a good resemblance to this reference spectrum with an overall significantly reduced α-helical content compared to ACCBCC, whereas the CD spectrum of CC-hPin1WW-L2 (green) revealed a higher α-helical content (Fig. 2A). This observation alone led us to assume that the fragmented hPin1WW was more likely to be reconstituted in CC-hPin1WW-L1 than in CC-hPin1WW-L2. Deconvolution of the CD spectra using the CDSSTR algorithm on DichroWeb provided a more quantitative picture of the degree of folding of the fragmented WW-domain regions in CC-hPin1WW-L1 and CC-hPin1WW-L2.29 The fractions of secondary structural elements are listed in Table 2. As expected, ACCBCC revealed an α-helical content of almost 70% and hPin1WW a high β-strand content of 44% as well as 18% turn or 32% disordered regions. A folded WW-domain region in the CC-hPin1WW constructs should correspond to the fraction of secondary structure elements of the CD spectrum of the one-to-one mixture of hPin1WW and ACCBCC. The fraction of secondary structure in CC-hPin1WW-L1 very closely matches these values, which led us to conclude that WW-domain folding is indeed induced by CC association. In contrast, deconvolution of the CD spectrum of CC-hPin1WW-L2 revealed almost 60% α-helical content and only 15% β-sheet content indicating, if at all, only partial folding of the fragmented WW domain. | ||
| Fig. 2 CD spectroscopic data of CC-hPin1WW variants, hPin1WW and ACCBCC. (a) CD spectra of hPin1WW variants at 20 °C; (b) thermal denaturation profiles of hPin1WW, ACCBCC, CC-hPin1WW-L1, CC-hPin1WW-L2, and an one-to-one mixture of ACCBCC and hPin1WW to obtain a reference spectrum. Thermal denaturation profiles are depicted as fraction folded to enable comparison of the peptide species. Thermal denaturation profiles were fitted to a two-state folding model (see ESI†). Experimental conditions: PBS buffer, pH 7.4, 40 μM peptide concentration. | ||
| Peptide | Fraction secondary structurea | T m [°C] | |||
|---|---|---|---|---|---|
| α-Helix | β-Strand | Turn | Disordered | ||
| a Fraction of secondary structure was obtained from deconvolution of CD spectra on DichroWeb using the CDSSTR algorithm (data set 4).29 b T m values were calculated by least-square fitting of the thermal denaturation profiles assuming a two-state folding model for a monomer (hPin1WW) or a dimer (ACCBCC, CC-hPin1WW-L1, CC-hPin1WW-L2). | |||||
| ACCBCC (CC) | 0.69 | 0.11 | 0.10 | 0.12 | 58.5 ± 0.5 |
| hPin1WW | 0.04 | 0.44 | 0.18 | 0.32 | 57.5 ± 1.0 |
CC/hPin1WW (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
0.48 | 0.23 | 0.14 | 0.19 | — |
| CC-hPin1WW-L1 | 0.47 | 0.24 | 0.11 | 0.18 | 65.0 ± 0.5 |
| CC-hPin1WW-L2 | 0.58 | 0.15 | 0.07 | 0.20 | 64.0 ± 0.5 |
Thermal denaturation profiles of hPin1WW, ACCBCC and the two CC-hPin1WW variants were recorded as the thermal stability gives additional information about the degree of folding. Thermodynamic analysis of the obtained profiles revealed cooperative unfolding and melting temperatures, Tm, of Tm = 58.5 °C and Tm = 57.5 °C for ACCBCC and hPin1WW, respectively. The Tm of hPin1WW is in accordance to the literature,20,23 whereas the Tm of ACCBCC was increased by 18 °C from the reported literature value.27 This discrepancy can best be explained by the lack of N- and C-terminal glycine residues in the studied sequences of the original publication (see Table S1†). The Tm values of the CC-hPin1WW variants were increased by 7 and 6 °C compared to the reference peptides, respectively. This led us to assume that the interaction of the CC is stabilized against thermal denaturation. However, CC-hPin1WW-L2 was slightly less stable, which indicates a less productive interaction of the WW-domain fragments.
The fact that the WW domain in CC-hPin1WW-L2 was not properly folded led to the hypothesis that the apparent reconstitution of the WW-domain structure in CC-hPin1WW-L1 was not solely due to CC association. Therefore, we synthesized the WW-domain fragments hPin1WW(6-20) and hPin1WW(21-39) lacking the CC tag and recorded CD spectra of an one-to-one mixture subsequently at different temperatures (Fig. S3†). When mixed at room temperature, the WW-domain structure was not reconstituted; however, at 0 °C, the CD spectrum showed the typical signals of an at least partially folded WW domain. This indicates that the structural information of split hPin1WW was not completely lost, which may positively contribute to the successful reconstitution into CC-hPin1WW-L1. However, the recorded thermal denaturation profile indicates a melting temperature below 0 °C, confirming that additional structural stabilization, e.g. by a CC dimerization domain, is required to achieve a stable assembly.
NMR spectroscopic analysis of reconstitution of structure and function in CC-hPin1WW variants
NMR spectroscopic studies were performed to substantiate the conclusions on reconstitution of fragmented hPin1WW upon CC association in the CC-hPin1WW variants as obtained by CD spectroscopy so far. First, one-dimensional (1D) proton NMR spectra of hPin1WW, CC-hPin1WW-L1, CC-hPin1WW-L2, and, as control, of ACCBCC as well as an 1:1-mixture of hPin1WW(6-20) and hPin1WW(21-39) were recorded. All NMR spectra except the one-to-one mixture of hPin1WW(6-20) and hPin1WW(21-39) reveal predominantly folded peptide species. The spectrum of hPin1WW gives characteristic isolated proton resonance signals in the low field range between 9 and 11 ppm, which correspond to the indole NH protons of W11 and W34, as well as to the NH backbone protons of E12, F25 and Q33 (Fig. 3A–C, see Fig. S4† for assignment of NH backbone protons comprising hPin1WW). In this spectral range, no signals from neither folded ACCBCC nor the 1:1 mixture comprising hPin1WW(6-20) and hPin1WW(21-39), which show a pattern typical for non-structured peptide motifs, are resolved (Fig. 3A, grey and black traces, Fig. S5†), making these signals a strong indicator for successful structural reconstitution of the WW domain in the CC-hPin1WW variants. Indeed, the characteristic signals were clearly visible in the 1D proton spectrum of CC-hPin1WW-L1 with slightly altered chemical shifts of indole signals (Fig. 3B). CC-hPin1WW-L2, however, does not give this characteristic pattern of signals, which supports our findings obtained from the CD measurements that hPin1WW is not reconstituted in CC-hPin1WW-L2 upon CC association. The analysis of the NMR structure of hPin1WW published by Luh et al. revealed a conformationally defined loop 2 with a stabilizing hydrogen bond between the side chain at N26 and amide protons comprising the loop backbone (Fig. S6†).22 Therefore, we assume that loop 2 is crucial for the structural integrity of hPin1WW and, for this reason, not suitable to act as a fragmentation site. Consequently, CC-hPin1WW-L2 was not considered in further experimental work in the present study.
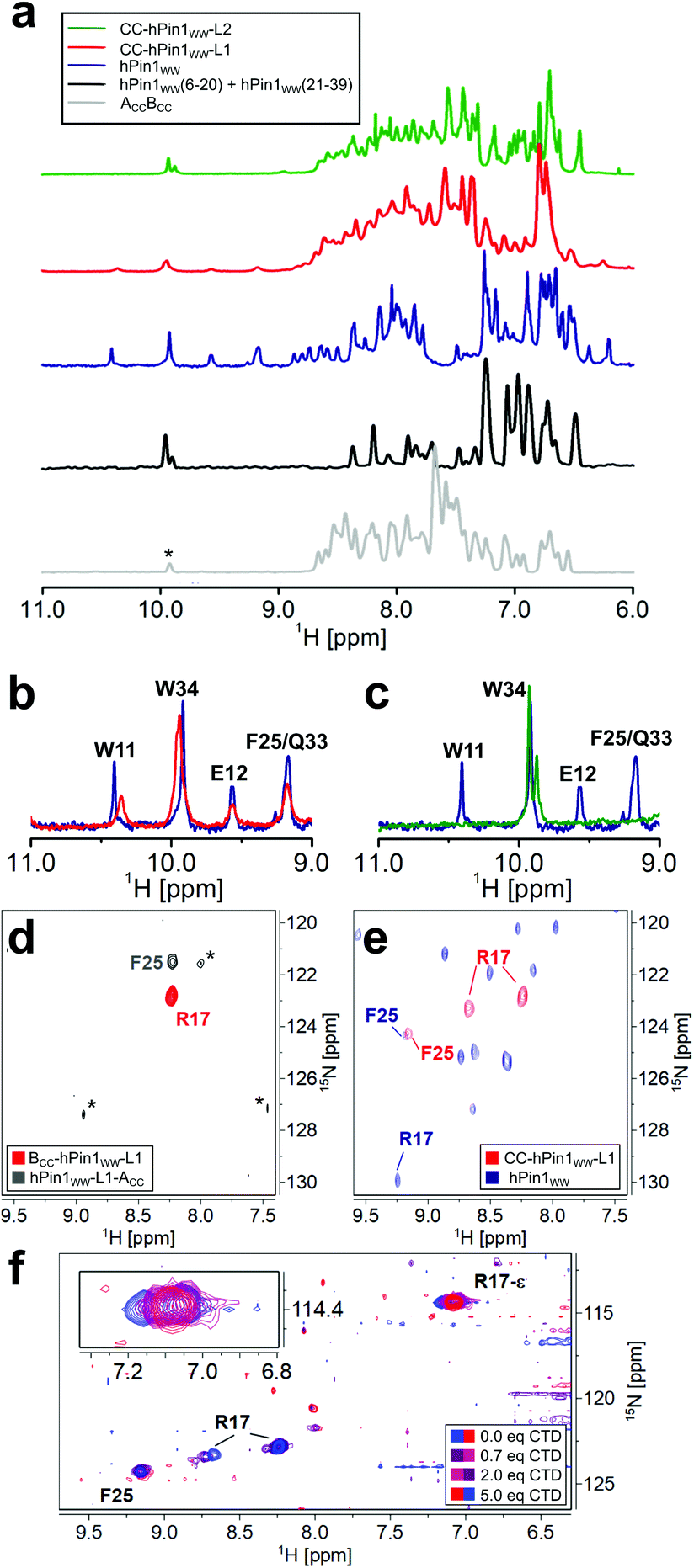 | ||
| Fig. 3 NMR spectroscopic analysis of CC-hPin1WW variants assessing reconstitution of structure and function of fragmented hPin1WW. (a) 1D proton NMR spectra of ACCBCC, a 1:1 mixture of hPin1WW(6-20) and hPin1WW(21-39), hPin1WW, CC-hPin1WW-L1, and CC-hPin1WW-L2. The isolated proton NMR signals between 9 and 11 ppm are characteristic for natively folded hPin1WW. (*: ACCBCC contains a tryptophan residue in ACC that is not present in the ACC-segment of hPin1WW-ACC). (b) Overlay of the low field proton NMR spectra of hPin1WW (blue) and CC-hPin1WW-L1 (red). The characteristic hPin1WW signals are also resolved in CC-hPin1WW-L1. (c) Overlay of the low field proton NMR spectra of hPin1WW (blue) and CC-hPin1WW-L2 (green). The characteristic hPin1WW signals are not resolved in CC-hPin1WW-L2. (d and e) Overlay of 2D 1H–15N HSQC NMR spectra of BCC-hPin1WW-L1-15N413C6-R17 and hPin1WW-L1-ACC-15N-F25 (d), and CC-hPin1WW-L1-15N413C6-R17, 15N-F25 and hPin1WW (e). Folding of CC-hPin1WW-L1 in (e) is apparent from the significant low field shift of F25 compared to (d) and the appearance of two cross-peaks for R17 indicating two backbone conformations. (*: refers to noise signals; low signal intensity of cross-peaks of F25 in apo state due to solvent exchange) (f) chemical shift perturbation of R17 and R17-ε in 1H–15N HSQC NMR spectra upon titration of CTD to CC-hPin1WW-L1 at T = 283 K. The signal of R17-ε is folded into the spectral range shown. | ||
Additionally, we have performed NMR diffusion experiments to determine the hydrodynamic radii (rh) of CC-hPin1WW-L1, hPin1WW and the monomers BCC-hPin1WW-L1 and hPin1WW-L1-ACC (Table 3, Fig. S7†). The hydrodynamic radius reports on a quantitative measure of the state of folding of the peptides investigated.30 The value determined for hPin1WW corresponds well with the theoretical value for a globularly folded protein structure comprising the same number of residues, while the experimental value for CC-hPin1WW-L1 slightly deviates from the theoretical value. We attribute this to the rod-shaped CC extension of the peptide. However, compared to CC-hPin1WW-L1 monomers, rh determined for CC-hPin1WW-L1 indicates a more globularly folded peptide, which becomes evident from the calculated average rh values of BCC-hPin1WW-L1 and hPin1WW-L1-ACC.
| Measured rh [nm] | Calculated rha [nm] | Meas./calc. | |
|---|---|---|---|
| a r h is calculated for a globularly folded protein comprising the same number of residues as the particular variant of hPin1WW which has been measured.30 | |||
| hPin1WW | 1.45 | 1.33 (35 aa) | 1.09 |
| CC-hPin1WW-L1 | 2.20 | 1.78 (94 aa) | 1.24 |
| BCC-hPin1WW-L1 | 2.15 | 1.43 (45 aa) | 1.50 |
| hPin1WW-L1-ACC | 1.78 | 1.48 (49 aa) | 1.20 |
| Calc. av. CC-hPin1WW monomers | 1.98 | 1.46 | 1.35 |
To obtain insights into the structural and functional characteristics of CC-hPin1WW-L1 at atomic resolution, we incorporated 15N-isotopically labelled amino acids, more specifically, 15N413C6-R17 and 15N–F25, since these are located (a) close to the fragmentation site and (b) to the ligand binding site. First, we studied the overall conformation of the fragmented WW domain by recording two-dimensional (2D) 1H–15N HSQC NMR spectra of the isotopically labelled monomers (Fig. 3D) and CC-hPin1WW-L1, respectively. The chemical shift values of the backbone amide protons of R17 (8.23 ppm) and F25 (8.23 ppm) found for the individual monomers are close to the reported random coil chemical shifts of 8.27 ppm and 8.23 ppm,31 thus indicating a disordered structure of the WW-domain fragments.
In CC-hPin1WW-L1 F25 is strongly low field shifted, indicating a β-sheet character, and also almost matches the chemical shift values found for F25 in hPin1WW (Fig. 3e). However, two conformations of the R17 backbone are evident, and the values of the 15N chemical shifts differ significantly from those of the reference peptide hPin1WW. Both phenomena are likely due to the fact that R17 is part of the unstructured and fragmented loop 1 and therefore can adopt different stable conformations compared to the non-fragmented parent peptide.
Since structural reconstitution of fragmented hPin1WW in CC-hPin1WW-L1 was proven independently by CD and NMR spectroscopy, we were particularly interested in whether this CC-induced reconstitution of structure also leads to reconstitution of the function. hPin1 is a peptidyl-prolyl-cis/trans isomerase that specifically isomerizes protein sequences with a phosphorylated S/TP motif recognized by hPin1WW.12,32 One of the target proteins is the RNA polymerase II, and the typical recognition motif is the heptapeptide YSPTpSPS (pS – phosphorylated serine residue), which occurs repeatedly in the C-terminal domain and is therefore called CTD peptide.33 We synthesized this peptide, titrated it to 15N-isotopically labelled CC-hPin1WW-L1 and monitored the changes in the chemical shift values of cross-peaks belonging to the backbone and side chain of R17 in 2D 1H–15N HSQC NMR spectra (Fig. 3f). By plotting the changes in chemical shift values against the stoichiometric ratio using the ligand and CC-hPin1WW-L1 concentrations, we were able to qualitatively estimate the binding dissociation constant (Kd) that was in the range of 50 μM (Fig. S8†). This is in good agreement with the Kd values previously reported for hPin1WW.34,35 Since R17 is directly involved in ligand binding in native hPin1WW, these results suggest successful reconstitution of binding capacity to the CTD peptide in CC-hPin1WW-L1.
hPin1WW and CC-hPin1WW-L1 show similar binding to CTD-peptide
Although the results obtained from the NMR titration experiment were promising, we aimed for an additional experiment to confirm the binding capacity between CTD and CC-hPin1WW-L1 and the value of the Kd. Furthermore, a direct comparison of the binding capacities to CTD peptide of hPin1WW and CC-hPin1WW-L1 improves the assessment of the extent of reconstitution regarding function of the fragmented WW-domain region in CC-hPin1WW-L1 significantly. Therefore, we measured the change in intrinsic tryptophan fluorescence of hPin1WW and CC-hPin1WW-L1 upon CTD peptide titration. It should be noted that direct titration of the ligand into the solution of the respective hPin1WW variant leads to false results, since tryptophan fluorescence in WW domains is highly sensitive to photobleaching.36 Therefore, we decided to perform this experiment in a microplate format, although signal scattering is sometimes an issue. An one-to-one dilution series of the ligand was prepared starting from 500 μM CTD concentration, each dilution equilibrated with 2 μM protein for 15 h at 4 °C, and measured in a microplate reader at room temperature to obtain the intrinsic tryptophan fluorescence at 350 nm. Both WW-domain peptides showed a very similar binding behaviour (Fig. 4), although the observed fluorescence enhancement was more pronounced for CC-hPin1WW-L1 than for hPin1WW. Surprisingly, the saturation binding curves indicated stronger binding for both peptides than initially indicated by NMR titration, and the non-linear least square fits (ESI Eqn. 14†) yielded Kd values of 2.2 ± 0.6 μM and 2.0 ± 0.7 μM for hPin1WW and CC-hPin1WW-L1, respectively. These values are by the order of one magnitude lower than those reported in the literature35 and estimated from NMR titration experiments (Fig. S8†). One possible explanation for this discrepancy could be a reduced effect of undesired ligand depletion, since the measurements were performed at peptide concentrations 30 times lower than the reported Kd.37 Additionally, we suspected conformational changes of the WW-domain backbone during ligand binding, which could result in cooperative changes of the fluorescence of both tryptophans, W11 in the hydrophobic core and W34 at the binding site, and thus to an apparent Kd value influenced by structural changes. At the extreme, such behaviour was observed in the Ess1WW domain during binding of similar CTD ligands. Ess1WW showed ligand-induced folding and provided a saturation-binding curve that indicated strong ligand binding.38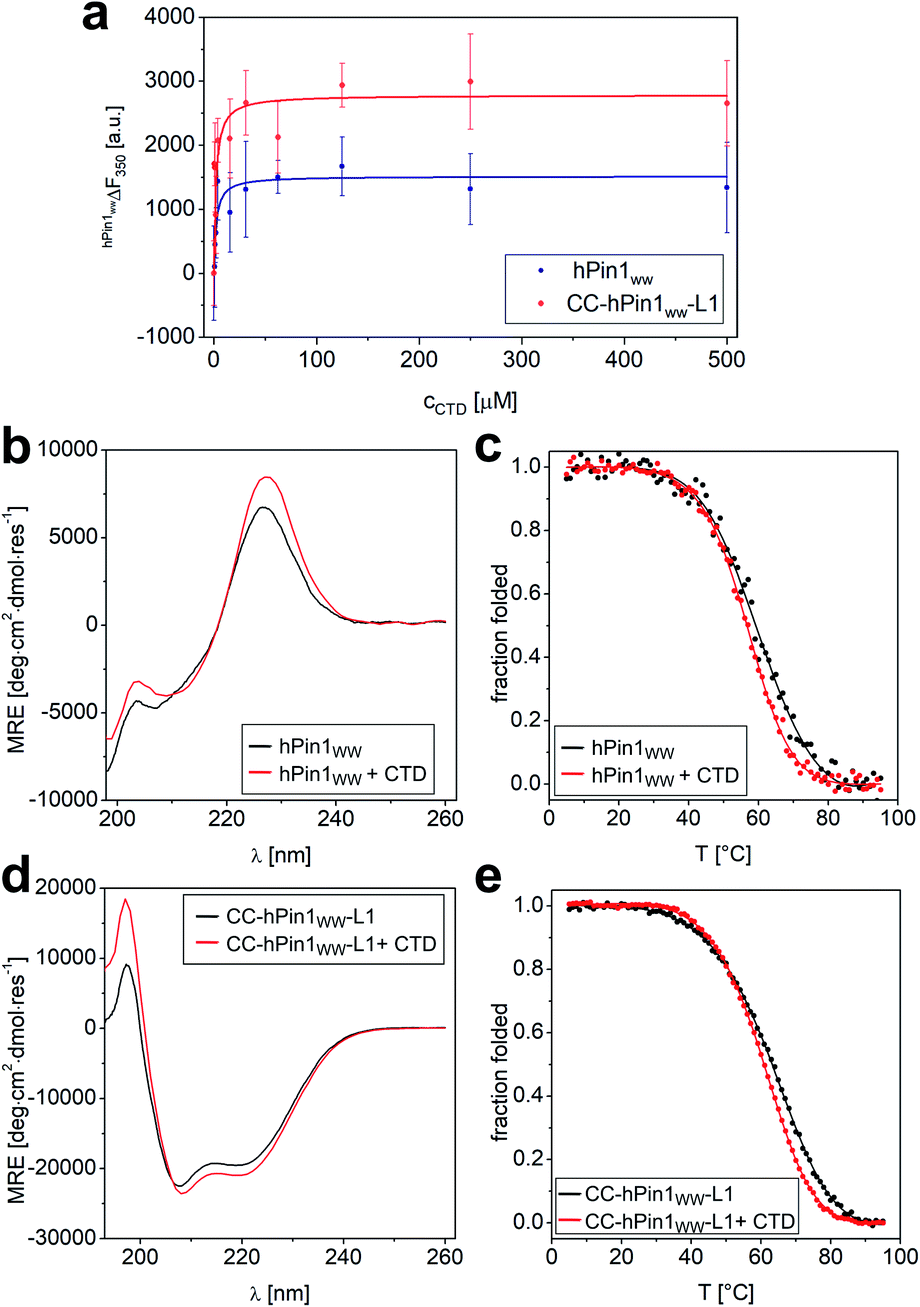 | ||
| Fig. 4 Fluorescence and CD spectroscopic analysis of CTD binding to hPin1WW and CC-hPin1WW-L1. (a) Intrinsic tryptophan fluorescence titrations to determine dissociation constants (Kd) of hPin1WW and CC-hPin1WW-L1. Saturation binding curves of hPin1WW (Kd = 2.2 ± 0.6 μM) and CC-hPin1WW-L1 (Kd = 2.0 ± 0.7 μM) obtained from tryptophan fluorescence enhancement upon CTD-peptide titration. Conditions: c (hPin1WW/CC-hPin1WW-L1) = 2 μM, PBS buffer, pH 7.4, room temperature; titration steps: 0–500 μM CTD peptide. All binding dissociation constants were obtained from non-linear least square fitting to the saturation bindings curves (see ESI Eqn. 14†). (b–e) Changes in CD spectra at 20 °C (b and d) and CD thermal denaturation profiles (c and e) of hPin1WW (b and c) and CC-hPin1WW-L1 (d and e) in the presence of equimolar amounts of CTD ligand. Thermodynamic data was obtained from non-linear least square fitting to thermal denaturation curves (Table 4). Experimental conditions: PBS buffer, pH 7.4, 40 μM peptide concentration. Thermal denaturation profiles were fitted using a two-state folding-unfolding model (see ESI†). | ||
Therefore, we also performed CD measurements of hPin1WW and CC-hPin1WW-L1 in the presence of equimolar amounts of ligand CTD to determine possible structural changes. Indeed, CD spectra showed increased signal intensities for both, hPin1WW and CC-hPin1WW-L1 (Fig. 4b and d). Furthermore, CD thermal denaturation revealed cooperative unfolding with significantly steeper transition profiles and slightly lower melting temperatures (Fig. 4d, e and Table 4). The thermodynamic analysis of the thermal denaturation curves focusing on ΔHm, ΔCp,m and ΔGDi at 20 °C using a two-state unfolding-folding model gave thermodynamic parameters that indicate a moderate structural stabilization of both hPin1WW and CC-hPin1WW-L1 upon ligand binding, respectively. Taken together, ligand titration monitored by NMR and fluorescence spectroscopy and CD measurements of hPinWW and CC-hPin1WW-L1 gave similar results for both peptides, which suggest that both the structure and function of the fragmented WW-domain region, are reconstituted successfully in CC-hPin1WW-L1 upon CC association.
| T m (TmK) [°C] | ΔHm [kJ mol−1] | ΔCp,m [J mol−1 K−1] | ΔGDiT=293K [kJ mol−1] | |
|---|---|---|---|---|
| a Thermodynamic parameters were obtained from non-linear least square fits using a two-state folding/unfolding model for a monomer.39Tm is the midpoint of thermal denaturation at fraction folded α = 0.5; ΔHm is the change in enthalpy at Tm; ΔCp,m is the change in heat capacity at Tm and ΔGT=293KDi is the change in free enthalpy at 20 °C. b Thermodynamic parameters were obtained from non-linear least square fits using a two-state folding/unfolding model for a dimer.39TmK is the concentration independent melting temperature at unfolding constant K = 1.40 | ||||
| hPin1WWa | 57.5 ± 0.9 | −120 ± 12 | −3300 ± 500 | −6.0 ± 1.4 |
| hPin1WW + CTDa | 56.0 ± 0.6 | −135 ± 6 | −2600 ± 1100 | −9.6 ± 0.8 |
| CC-hPin1WW-L1b | 65.0 ± 0.4 (124.5 ± 0.1) | −168 ± 2 | −190 ± 40 | −41.3 ± 0.2 |
| CC-hPin1WW-L1 + CTDb | 60.5 ± 0.1 (111.5 ± 0.7) | −187 ± 5 | −200 ± 120 | −42.3 ± 0.2 |
A library of split-WW domains
As mentioned in the introduction, the concept of CC mediated reconstitution of split-WW domains was developed here to be used as a combinatorial approach to design WW domains possessing new functions. Advantageously, sets of N-terminal and C-terminal fragments can be combined, significantly expanding the size of the library while minimizing synthesis effort. To illustrate this, a simple example is given: Four N-terminal fragments combined with four C-terminal fragments yield a relatively small library of sixteen split-WW domains at half the synthetic effort, since only eight peptides need to be synthesized rather than sixteen; ten N-terminal fragments combined with ten C-terminal fragments, on the other hand, would expand the library to 100 members, requiring the synthesis of only twenty peptides, a comparatively small synthetic effort.As a proof-of-concept, we chose to synthesize a small split-WW domain library derived from hPin1WW to be screened specifically for adenosine triphosphate (ATP) binding. Weak ATP binding with a binding dissociation constant of 0.6 mM was previously demonstrated with hPin1WW-M15C modified with a fluorescent stilbazole to which a Zn (II) dipicolylamine was attached as an additional phosphate binding motif.34 However, our goal was to find an ATP-binding WW domain that consists exclusively of natural amino acids and, in addition, binds other biologically relevant organophosphates such as cyclic adenosine monophosphate (cAMP), phosphorylcholine (PC), or inositol hexaphosphate (IP6, Fig. 5a) less well, i.e., that is ATP-specific. In addition to the original CC-hPin1WW-L1 fragments, in the context of this library now referred to as BCC-WW-1 and ACC-WW-5, eight other peptides – three N-terminal fragments and five C-terminal fragments that were derived from BCC-WW-1 and ACC-WW-5, respectively – were synthesized. This resulted in a total of four N-terminal fragments (BCC-WW-1 to BCC-WW-4) and six C-terminal fragments (BCC-WW-5 to BCC-WW-10), which after combination yielded a library of 24 split-WW domains (Fig. 5b). The most relevant interactions of ATP-binding proteins with the ligand are hydrogen bonds with the ribose moiety, aromatic interactions with the nucleobase, or electrostatic interactions of the negatively charged triphosphate with positively charged amino acid residues. Therefore, the variants of BCC-WW-1 and ACC-WW-5 contained mutations with positively charged lysine or arginine residues (BCC-WW-2, BCC-WW-3, ACC-WW-7), polar serine or glutamate residues (ACC-WW-6, ACC-WW-8, ACC-WW-10), and polar aromatic residues such as tyrosine and histidine (BCC-WW-4, ACC-WW-9, ACC-WW-10, Fig. 5b).
 | ||
| Fig. 5 Split-WW domain library design and screening for binding activity to organophosphates. (a) Organophosphate ligands for library screening: ATP (adenosine triphosphate), cAMP (cyclic adenosine monophosphate), PC (phosphorylcholine), IP6 (inositol hexaphosphate). (b) Sequences of the BCC-hPin1WW-L1 (BCC-WW-1 – BCC-WW-4) and hPin1WW-L1-ACC (ACC-WW-5 – ACC-WW-10) used in split-WW-domain library design. BCC-WW-1 corresponds to parent BCC-hPin1WW-L1 and ACC-WW-5 to hPin1WW-L1-ACC. Combination of the N- and C-terminal fragments results in a library of 24 split-WW domains. The amino acids highlighted in red mark the mutation sites. (c) Screening readout based on saturation binding monitored by intrinsic tryptophan fluorescence changes in split-WW domains upon ligand binding and quantified by calculation of the association constants (1/Kd). Note: For practical reasons, only elementary titrations were performed. Therefore, the association constants obtained in this screening serve only as a relative comparison of the binding of the four ligands to a respective split-WW domain variant (for experimental details see ESI†). | ||
Library screening was performed by measuring the changes in intrinsic tryptophan fluorescence upon ligand binding. Since the change in tryptophan fluorescence is highly dependent on the peptide, elementary ligand titrations were performed to provide the binding association constants (Fig. S9–S12†). In a screening format, this methodology represents an estimate, but provides a relative comparison of the binding of the different ligands to one peptide species in a qualitative manner. The results of the library screening are summarized in Fig. 5c. Many of the peptide variants did not exhibit preferential binding to any of the organophosphates tested or showed ambiguous results. Only two combinations – CC-WW-2-6 and CC-WW-2-10 – showed specific binding to ATP over the other organophosphates. Of these two, CC-WW-2-10 was investigated further, because ATP binding appeared to be more selective for this particular split-WW domain. In addition to CC-WW-2-10, we also decided to study CC-WW-1-8 and CC-WW-2-8, as these variants showed selective binding to PC and IP6.
WW-domain-derived peptides as mini-receptors for organophosphates
Based on the results of the library screening, WW domains WW-2-10, WW-1-8, and WW-2-8 were synthesized to further investigate their selective binding properties to ATP, PC, or IP6. Since these WW domains, unlike the CC-fusion variants, were not stabilized by CC interaction, CD spectroscopy was employed to characterize their structure and thermodynamic stability (Fig. 6a and b). The CD spectrum of WW-2-10 exhibits prominent features of a WW domain by showing the typical maximum at 227 nm resulting from the exciton coupling of the aromatic residues. However, compared to hPin1WW, the signal intensity is lower, mainly due to the exchange of W34 for a serine residue. In the CD spectrum of WW-1-8, the exciton coupling is even less pronounced, and WW-2-8 appears to be mainly a random coil in solution. To investigate the overall thermodynamic stability of these WW domains, thermal denaturation profiles were recorded by monitoring the change in CD signal at a wavelength of 227 nm, which gave a Tm of 36.5 °C for WW-2-10 and of 14 °C for WW-1-8 (Fig. 6a and b). WW-2-8 did not show a cooperative folding to unfolding transition, allowing us to conclude that it represents an unfolded peptide species at ambient temperature. However, WW-1-8 also exhibits lower thermodynamic stability, suggesting that mutations in the C-terminal fragment ACC-WW-8 significantly affect the structural integrity of the WW domains. WW-2-10, on the other hand, is a stably folded WW domain possessing a cooperative denaturation profile, although it shows lower thermodynamic stability compared with wild type hPin1WW. Taken together, these data clearly demonstrate that the split-WW-domain approach indeed allows screening for function and not primarily for thermodynamic stability.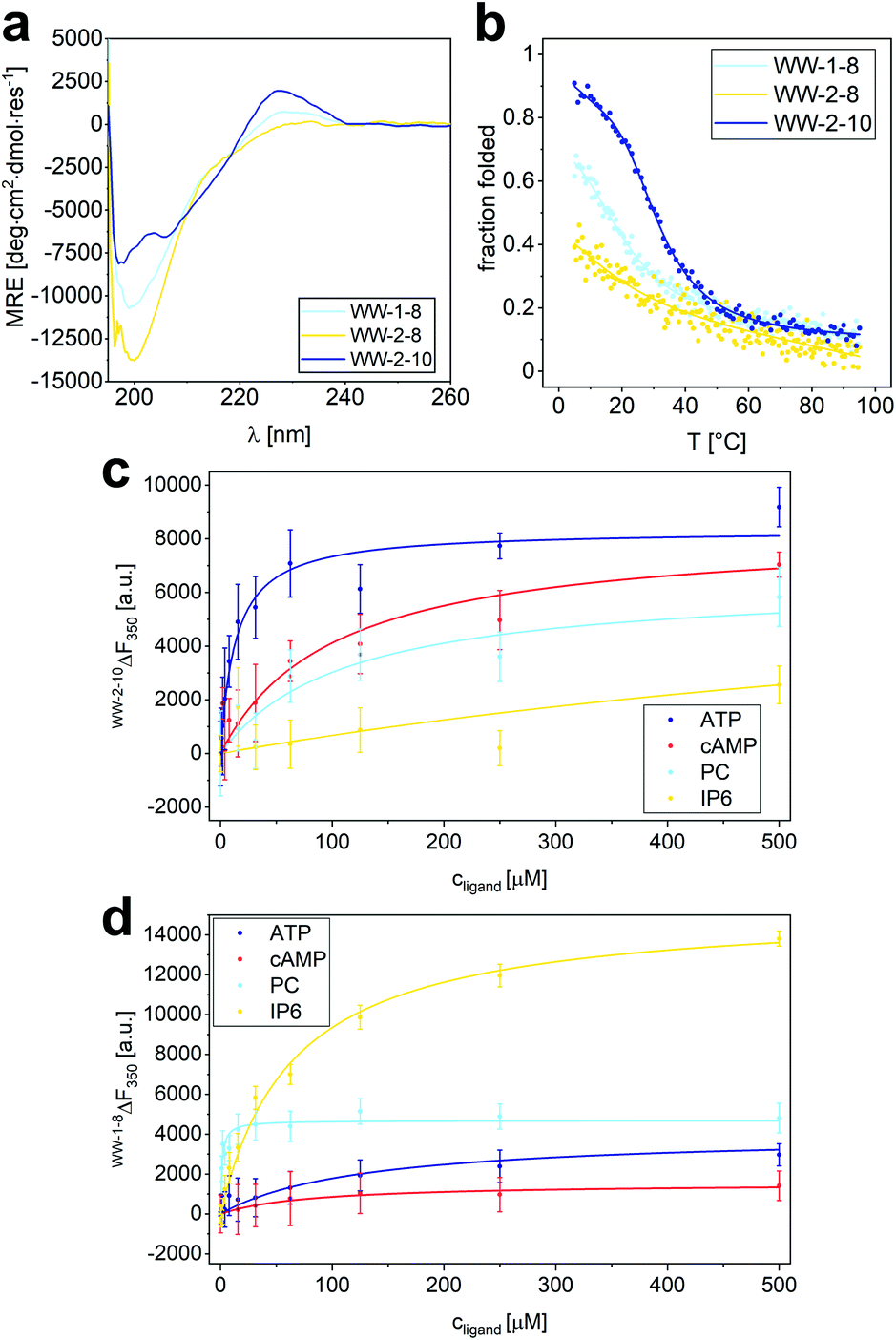 | ||
| Fig. 6 Characterization of structure and binding properties of WW-1-8, WW-2-8 and WW-2-10. (a) CD spectrum of WW-1-8, WW-2-8 and WW-2-10 at T = 20 °C; (b) thermal denaturation profile of WW-1-8, WW-2-8 and WW-2-10 depicted as fraction folded. The thermal denaturation profile was fitted to a two-state folding model giving a Tm value of 36.5 ± 0.5 °C (see ESI†). Experimental conditions: PBS buffer, pH 7.4, 40 μM peptide concentration. (c) Intrinsic tryptophan fluorescence titrations of ATP, cAMP, phosphorylcholine, and IP6 to WW-2-10 lead to the following Kd values: Kd (ATP) = 14 ± 3.4 μM, Kd (cAMP) = 102 ± 22 μM, Kd (PC) = 112 ± 51 μM, Kd (IP6) = 1.1 ± 0.3 mM. (d) Intrinsic tryptophan fluorescence titrations of ATP, cAMP, phosphorylcholine, and IP6 to WW-1-8 lead to the following Kd values: Kd (ATP) = 130 ± 40 μM, Kd (cAMP) = 81 ± 28 μM, Kd (PC) = 1.1 ± 0.3 μM, Kd (IP6) = 64 ± 6 μM. Experimental conditions: c (peptide) = 2 μM, PBS buffer, pH 7.4, room temperature; titration steps: 0–500 μM phosphate ligand. All Kd values were obtained from non-linear least square fitting using the saturation bindings curves (see ESI Eqn. 14†). | ||
After investigating the structure and thermodynamic stability, we analyzed the binding of organophosphates to WW-2-10 and WW-1-8 by measuring the changes in intrinsic tryptophan fluorescence. An one-to-one dilution series of each ligand was prepared starting from a concentration of 500 μM, each dilution was equilibrated with a concentration of 2 μM of the respective WW-domain variant at a temperature of 4 °C for a period of 15 h and measured in a microplate reader at room temperature to obtain the intrinsic tryptophan fluorescence at a wavelength of 350 nm.
When analyzing the binding properties of WW-2-10, tryptophan fluorescence quenching was observed upon ATP binding, while binding of the other ligands resulted in weak tryptophan fluorescence enhancement. By fitting the resulting titration curves to an one-site binding model, a Kd of 14 ± 4 μM was determined for ATP binding. In comparison, the Kd values of cAMP and PC were increased by an order of magnitude, and IP6 showed only weak binding possessing a Kd value in the mM range (Fig. 6c). This confirmed the results from the library screening, which indicated a specific binding activity of WW-2-10 to ATP. To our knowledge, only one small peptide folding motif has been reported so far that exhibits specific ATP-binding properties, namely a β-hairpin of 11 amino acids.43 ATP binding was achieved by electrostatic and aromatic interactions, but is one order of magnitude weaker than found for WW-2-10, which with its 34 amino acids can still be considered a peptide and, in this case, a mini-ATP receptor that binds comparatively strongly to ATP. By comparison, natural ATP-binding proteins bind their ligand in the high nM to mM range.44
To further investigate the relationship between sequence and activity, we also relied on computational methods. A structural model of WW-2-10 was constructed with the Rosetta Relax application41 based on an available NMR structure of hPin1WW (pdb code 1i6c), in which the corresponding amino acid residues were mutated. Subsequently, AutoDock Vina42 was used to perform molecular docking with the organophosphates considered in this study to predict their binding mode (Fig. S13†). Interestingly, WW-2-10 showed two binding pockets on the surface, a polar pocket (P1) and a positively charged pocket (P2). The key of this structural feature lies in the two mutations F25H and S32E. The two amino acid residues H and E form a hydrogen bond through which H25 moves out of the outer hydrophobic core, constituting a cavity (Fig. S13A and B†). While the long negatively charged phosphate tail of ATP binds to P2, where it is stabilized by electrostatic interactions and hydrogen bonds with the three arginine residues, its nucleoside moiety appears to fit perfectly into P1 and is stabilized mainly by hydrogen bonds. The other organophosphates that were investigated either do not fit into the cavities or are too small to occupy both pockets, which is for instance the case for phosphorylcholine (Fig. S13†).
Although WW-1-8 exhibits lower thermodynamic stability, we investigated its binding behaviour to the four different organophosphates (Fig. 6d). In all titration experiments, we observed an enhancement of tryptophan fluorescence, and Kd values were determined by fitting the resulting titration curves to a one-site-binding model. For titration of phosphorylcholine to WW-1-8, we determined a Kd value of 1.3 ± 0.3 μM, whereas titrations of ATP and cAMP yielded Kd values that were increased by approximately two orders of magnitude. Interestingly, titration of IP6 to WW-1-8 resulted in a strong tryptophan fluorescence enhancement. Although fitting the titration curve yielded a Kd value of only 65 μM, a factor of 1.5 increased compared with the Kd value of PC, we wondered whether this phenomenon was due to the lower thermodynamic stability of WW-1-8. Therefore, we also performed quantitative titrations of the phosphate ligands with the corresponding split variant CC-WW-1-8, which is thermodynamically stable due to the fused CC dimerization domain (Fig. S14†). Interestingly, CC-WW-1-8 showed a slightly different binding behaviour than the non-fusion WW domain WW-1-8. PC was similarly bound with a Kd value of 1.7 ± 0.4 μM, however, the Kd values for ATP and cAMP were reduced by one order of magnitude, and IP6 was only weakly bound without causing a strong enhancement of tryptophan fluorescence that was observed upon binding of IP6 to WW-1-8. Thus, we can conclude that the thermodynamic stability of the modified WW domain most likely has a significant effect on ligand binding, although an influence of the fragmentation in loop 1 in CC-WW-1-8 cannot be ruled out completely. However, we state that both WW-1-8 and CC-WW-1-8 show the highest binding affinity for PC, which is consistent with the library screening.
Conclusions
In this report, we have successfully introduced the concept of CC-mediated proximity-induced reconstitution of fragmented WW domains, which has high potential as a combinatorial approach for identifying WW domains with novel functions as demonstrated here. To develop this approach, we chose wild type hPin1WW as model peptide, which inherent structural and functional properties can be well investigated by applying a synergistic combination of CD, fluorescence and high-resolution NMR spectroscopy. Two fragmentation sites have been investigated: fragmentation in loop 1 (CC-hPin1WW-L1) at the GR site and fragmentation in loop 2 (CC-hPin1WW-L2) at the HI site. By using CD spectroscopy and 1D proton NMR experiments we could show that in CC-hPin1WW-L1 the fragmented WW domain is structurally reconstituted, although loop 1 is reported to be the folding nucleation site of hPin1WW.25,26 These results are in accordance to the findings of Kier et al. who studied a circularly permuted and hyperstable variant of hPin1WW.20 In contrast, the WW-domain region in CC-hPin1WW-L2 is not properly folded, which we attribute to the fact that loop 2 is conformationally well-defined and probably key to the overall stability of the WW-domain structure. A Φ-value analysis of hPin1WW by Gruebele et al. indicated that loop 1 and loop 2 play a similar role in the folding process of hPin1WW, anticipating a two-state folding mechanism and not, as previously reported,25 a sequential folding mechanism with loop 1 as initial nucleation site.45Reconstitution of function was investigated by titration of the CTD peptide of RNA polymerase II, a natural hPin1WW ligand, to solutions of CC-hPin1WW-L1 and hPin1WW and monitored by recording both two-dimensional heteronuclear 1H–15N HSQC NMR spectra of isotopically labelled CC-hPin1WW-L1 and intrinsic tryptophan fluorescence spectroscopy, respectively. Furthermore, structural changes upon ligand binding were investigated by CD spectroscopy and the acquisition of thermal denaturation profiles. These experiments show that not only the structure but also the binding properties of hPin1WW are fully reconstituted in CC-hPin1WW-L1, although loop 1 contains the phosphate binding site and is therefore directly involved in ligand binding. To put it in a nutshell, the split hPin1WW described here is the smallest protein/peptide to date that has been shown to reconstitute completely by CC association. This highlights the general robustness of WW-domain structure.
The WW domain has great potential as a scaffold for the development of biomimetic peptides and mini-proteins. The folding motif is extremely robust to modifications and mutations. However, unlike CCs, the sequence-structure relationships are less straightforward and therefore not as well studied. The split-WW domain approach presented here allows us to explore the sequence space of the WW domain in a combinatorial manner, facilitating the identification of novel functional peptides that exhibit WW-domain folding. We demonstrated the potential of this engineering strategy with a 24-member split-WW-domain library built from only ten peptides, from which we identified one specific ATP binder, namely WW-2-10, possessing a remarkable binding dissociation constant in the low micromolar range. This peptide competes very well with other engineered systems43 and natural ATP-binding proteins, the latter having Kd values from the high nanomolar to the millimolar regime.44 Sequence optimization of WW-2-10 could improve ATP binding even further. Only a single artificial ATP-binding protein of 80 amino acids, identified by in vitro selection of a messenger RNA displayed protein library with 6 × 1012 members, showed a Kd value of 0.1 μM.46 This is obviously very impressive, but the advantage of our approach is that the WW domain is comparatively small and therefore the peptides can be chemically synthesized in an automated manner, allowing library members with artificial amino acid residues. In addition to the ATP binder, we have also identified a phophorylcholine-selective WW domain, WW-1-8, which, although not thermodynamically very stable, binds to PC with a Kd value of about 1 μM and could be developed into a mini-receptor for PC or PC derivatives such as lysophosphatidylcholine, which is found, for example, as a so-called “Find-Me signal” on apoptotic cells.47 We are convinced that the results reported here are promising for future applications, such as the development of other mini-phosphate receptors or mini-phosphatases. Thus, we intend to establish this split-WW-domain approach as a platform for the development and identification of a variety of functional WW domains in future.
Data availability
All experimental data associated with this work are available in the ESI.†Author contributions
FT and MK designed the project. HN and NBP synthesised the CC-hPin1WW constructs and investigated the reconstitution of split-hPin1WW by coiled-coil association using CD spectroscopy. FH synthesised WW-1-8, WW-2-8 and WW-2-10 and performed CD spectroscopic analysis. CL synthesised the split-WW-domain library and performed the library screening. FH and RG performed the computational modelling and docking of WW-2-10. MK performed all NMR experiments and analysed the NMR data. FT investigated the binding of CC-hPin1WW-L1, hPin1WW, WW-1-8 and WW-2-10 to their respective ligands using fluorescence spectroscopy and CD spectroscopy. She also designed the split-WW-domain library and screening experiment. The data were analysed and discussed by all authors. FT and MK wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 414261058 and supported by the DFG under Germany's Excellence Strategy 2082/1 390761711. MK acknowledges financial support from the Young Scholar Fund and the permanent financial support provided by the University of Konstanz enabling to operate a state-of-the-art NMR core facility. FH thanks the Carl Zeiss Foundation for the financial support. We would also like to thank the Dr Sophie-Bernthsen-Stiftung for financial support in building our computational modeling infrastructure. We are grateful to Prof. Dr Michael Mastalerz (Heidelberg University) for providing access to a CD spectrometer. We thank the Centre for Advanced Materials (Heidelberg University) and Ulf Diederichsen (University of Göttingen) for general access to technical equipment.Notes and references
- (a) K. J. Grayson and J. L. R. Anderson, J. R. Soc., Interface, 2018, 15, 20180472 CrossRef PubMed; (b) L. R. Marshall, O. Zozulia, Z. Lengyel-Zhand and I. V. Korendovych, ACS Catal., 2019, 9, 9265–9275 CrossRef CAS PubMed.
- W. M. Dawson, G. G. Rhys and D. N. Woolfson, Curr. Opin. Chem. Biol., 2019, 52, 102–111 CrossRef CAS PubMed.
- D. N. Woolfson, Fibrous Proteins, 2017, 82, 35–61 CAS.
- (a) I. Nakase, S. Okumura, G. Tanaka, K. Osaki, M. Imanishi and S. Futaki, Angew. Chem., Int. Ed., 2012, 51, 7464–7467 CrossRef CAS PubMed; (b) F. Thomas, W. M. Dawson, E. J. M. Lang, A. J. Burton, G. J. Bartlett, G. G. Rhys, A. J. Mulholland and D. N. Woolfson, ACS Synth. Biol., 2018, 7, 1808–1816 CrossRef CAS PubMed.
- W. M. Rink and F. Thomas, Chem. – Eur. J., 2019, 25, 1665–1677 CrossRef CAS PubMed.
- (a) K. E. Thompson, C. J. Bashor, W. A. Lim and A. E. Keating, ACS Synth. Biol., 2012, 1, 118–129 CrossRef CAS PubMed; (b) A. J. Smith, F. Thomas, D. Shoemark, D. N. Woolfson and N. J. Savery, ACS Synth. Biol., 2019, 8, 1284–1293 CrossRef CAS PubMed; (c) C. L. Edgell, A. J. Smith, J. L. Beesley, N. J. Savery and D. N. Woolfson, ACS Synth. Biol., 2020, 9, 427–436 CrossRef CAS PubMed; (d) T. Lebar, D. Lainšček, E. Merljak, J. Aupič and R. Jerala, Nat. Chem. Biol., 2020, 16, 513–519 CrossRef CAS PubMed.
- (a) H. Robson Marsden, N. A. Elbers, P. H. H. Bomans, N. A. J. M. Sommerdijk and A. Kros, Angew. Chem. Int. Ed., 2009, 48, 2330–2333 CrossRef PubMed; (b) K. Meyenberg, A. S. Lygina, G. van den Bogaart, R. Jahn and U. Diederichsen, Chem. Commun., 2011, 47, 9405 RSC.
- (a) B. Ma and R. Nussinov, Curr. Opin. Chem. Biol., 2010, 14, 652–659 CrossRef CAS PubMed; (b) C. Micheletti, Phys. Life Rev., 2013, 10, 1–26 CrossRef PubMed.
- (a) K. M. Wilcoxen, L. J. Leman, D. A. Weinberger, Z.-Z. Huang and M. R. Ghadiri, J. Am. Chem. Soc., 2007, 129, 748–749 CrossRef CAS PubMed; (b) M. C. Groth, W. M. Rink, N. F. Meyer and F. Thomas, Chem. Sci., 2018, 9, 4308–4316 RSC.
- (a) Z. Salah, A. Alian and R. I. Aqeilan, Front. Biosci., 2012, 17, 331–348 CrossRef CAS PubMed; (b) J. L. Ilsley, M. Sudol and S. J. Winder, Cell. Signalling, 2002, 14, 183–189 CrossRef CAS PubMed.
- (a) M. J. Macias, M. Hyvönen, E. Baraldi, J. Schultz, M. Sudol, M. Saraste and H. Oschkinat, Nature, 1996, 382, 646–649 CrossRef CAS PubMed; (b) J. R. Pires, F. Taha-Nejad, F. Toepert, T. Ast, U. Hoffmüller, J. Schneider-Mergener, R. Kühne, M. J. Macias and H. Oschkinat, J. Mol. Biol., 2001, 314, 1147–1156 CrossRef CAS PubMed; (c) M. Meiyappan, G. Birrane and J. A. A. Ladias, J. Mol. Biol., 2007, 372, 970–980 CrossRef CAS PubMed.
- R. Ranganathan, K. P. Lu, T. Hunter and J. P. Noel, Cell, 1997, 89, 875–886 CrossRef CAS PubMed.
- P. Tian and R. B. Best, Biophys. J., 2017, 113, 1719–1730 CrossRef CAS PubMed.
- (a) M. J. Macias, V. Gervais, C. Civera and H. Oschkinat, Nat. Struct. Biol., 2000, 7, 375–379 CrossRef CAS PubMed; (b) M. Socolich, S. W. Lockless, W. P. Russ, H. Lee, K. H. Gardner and R. Ranganathan, Nature, 2005, 437, 512–518 CrossRef CAS PubMed; (c) W. P. Russ, D. M. Lowery, P. Mishra, M. B. Yaffe and R. Ranganathan, Nature, 2005, 437, 579–583 CrossRef CAS PubMed; (d) J. M. Anderson and N. H. Andersen, Angew. Chem., Int. Ed., 2017, 56, 7074–7077 CrossRef CAS PubMed.
- A. L. Stewart, J. H. Park and M. L. Waters, Biochemistry, 2011, 50, 2575–2584 CrossRef CAS PubMed.
- (a) T. L. Pham, M. Kovermann and F. Thomas, ACS Synth. Biol., 2022, 11, 254–264 CrossRef CAS PubMed; (b) M. Werner, J. Pampel, T. L. Pham and F. Thomas, Chem.–Eur. J., 2022, e202201339 Search PubMed.
- (a) S. S. Shekhawat and I. Ghosh, Curr. Opin. Chem. Biol., 2011, 15, 789–797 CrossRef CAS PubMed; (b) E. Michel, A. Plückthun and O. Zerbe, Angew. Chem., Int. Ed., 2018, 57, 4576–4579 CrossRef CAS PubMed.
- (a) I. Ghosh, A. D. Hamilton and L. Regan, J. Am. Chem. Soc., 2000, 122, 5658–5659 CrossRef CAS; (b) S. S. Shekhawat, J. R. Porter, A. Sriprasad and I. Ghosh, J. Am. Chem. Soc., 2009, 131, 15284–15290 CrossRef CAS PubMed; (c) T. Azad, A. Tashakor and S. Hosseinkhani, Anal. Bioanal. Chem., 2014, 406, 5541–5560 CrossRef CAS PubMed.
- T. Fink, J. Lonzarić, A. Praznik, T. Plaper, E. Merljak, K. Leben, N. Jerala, T. Lebar, Ž. Strmšek and F. Lapenta, et al. , Nat. Chem. Biol., 2019, 15, 115–122 CrossRef CAS PubMed.
- B. L. Kier, J. M. Anderson and N. H. Andersen, J. Am. Chem. Soc., 2014, 136, 741–749 CrossRef CAS PubMed.
- (a) R. Wintjens, J. M. Wieruszeski, H. Drobecq, P. Rousselot-Pailley, L. Buée, G. Lippens and I. Landrieu, J. Biol. Chem., 2001, 276, 25150–25156 CrossRef CAS PubMed; (b) J. A. Kowalski, K. Liu and J. W. Kelly, Biopolymers, 2002, 63, 111–121 CrossRef CAS PubMed; (c) C. Schelhorn, P. Martín-Malpartida, D. Sunõl and M. J. Macias, Sci. Rep., 2015, 5, 1–12 Search PubMed; (d) D. E. Mortenson, D. F. Kreitler, H. G. Yun, S. H. Gellman and K. T. Forest, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 2506–2512 CrossRef CAS PubMed.
- L. M. Luh, R. Hänsel, F. Löhr, D. K. Kirchner, K. Krauskopf, S. Pitzius, B. Schäfer, P. Tufar, I. Corbeski and P. Güntert, et al. , J. Am. Chem. Soc., 2013, 135, 13796–13803 CrossRef CAS PubMed.
- M. Jäger, M. Dendle and J. W. Kelly, Protein Sci., 2009, 18, 1806–1813 CrossRef PubMed.
- (a) W. Chen, S. Enck, J. L. Price, D. L. Powers, E. T. Powers, C. H. Wong, H. J. Dyson and J. W. Kelly, J. Am. Chem. Soc., 2013, 135, 9877–9884 CrossRef CAS PubMed; (b) D. E. Mortenson, D. F. Kreitler, N. C. Thomas, I. A. Guzei, S. H. Gellman and K. T. Forest, ChemBioChem, 2018, 19, 604–612 CrossRef CAS PubMed; (c) A. A. Fuller, D. Du, F. Liu, J. E. Davoren, G. Bhabha, G. Kroon, D. A. Case, H. J. Dyson, E. T. Powers and P. Wipf, et al. , Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 11067–11072 CrossRef CAS PubMed.
- D. E. Shaw, P. Maragakis, K. Lindorff-Larsen, S. Piana, R. O. Dror, M. P. Eastwood, J. A. Bank, J. M. Jumper, J. K. Salmon and Y. Shan, et al. , Science, 2010, 330, 341–346 CrossRef CAS PubMed.
- (a) M. Jäger, Y. Zhang, J. Bieschke, H. Nguyen, M. Dendle, M. E. Bowman, J. P. Noel, M. Gruebele and J. W. Kelly, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10648–10653 CrossRef PubMed; (b) F. Liu, D. Du, A. A. Fuller, J. E. Davoren, P. Wipf, J. W. Kelly and M. Gruebele, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2369–2374 CrossRef CAS PubMed.
- D. L. McClain, H. L. Woods and M. G. Oakley, J. Am. Chem. Soc., 2001, 123, 3151–3152 CrossRef CAS PubMed.
- (a) I. Coin, J. Pept. Sci., 2010, 16, 223–230 CrossRef CAS PubMed; (b) I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed.
- (a) L. Whitmore and B. A. Wallace, Nucleic Acids Res., 2004, 32, 668–673 CrossRef PubMed; (b) L. Whitmore and B. A. Wallace, Biopolymers, 2008, 89, 392–400 CrossRef CAS PubMed.
- D. K. Wilkins, S. B. Grimshaw, V. Receveur, C. M. Dobson, J. A. Jones and L. J. Smith, Biochemistry, 1999, 38, 16424–16431 CrossRef CAS PubMed.
- K. Wüthrich, NMR of Proteins and Nucleic Acids, Wiley, 1986 Search PubMed.
- K. P. Lu, G. Finn, T. H. Lee and L. K. Nicholson, Nat. Chem. Biol., 2007, 3, 619–629 CrossRef CAS PubMed.
- (a) Y. X. Xu, Y. Hirose, X. Z. Zhou, K. P. Lu and J. L. Manley, Genes Dev, 2003, 17, 2765–2776 CrossRef CAS PubMed; (b) J. P. Hsin and J. L. Manley, Genes Dev., 2012, 26, 2119–2137 CrossRef CAS PubMed.
- T. Anai, E. Nakata, Y. Koshi, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2007, 129, 6233–6239 CrossRef PubMed.
- M. A. Verdecia, M. E. Bowman, K. P. Lu, T. Hunter and J. P. Noel, Nat. Struct. Biol., 2000, 7, 639–643 CrossRef CAS PubMed.
- A. Yammine, J. Gao and A. H. Kwan, Bio-Protoc., 2019, 9, e3253 CAS.
- C. M. S. Carter, J. R. Leighton-Davies and S. J. Charlton, J. Biomol. Screening, 2007, 12, 255–266 CrossRef CAS PubMed.
- J. K. Myers, D. P. Morris, A. L. Greenleaf and T. G. Oas, Biochemistry, 2001, 40, 8479–8486 CrossRef CAS PubMed.
- N. J. Greenfield, Methods Enzymol., 2004, 383, 282–317 CAS.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2527–2535 CrossRef CAS PubMed.
- (a) F. Khatib, S. Cooper, M. D. Tyka, K. Xu, I. Makedon, Z. Popović, D. Baker and F. Players, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18949–18953 CrossRef CAS PubMed; (b) J. B. Maguire, H. K. Haddox, D. Strickland, S. F. Halabiya, B. Coventry, J. R. Griffin, S. V. S. R. K. Pulavarti, M. Cummins, D. F. Thieker, E. Klavins, T. Szyperski, F. DiMaio, D. Baker and B. Kuhlman, Proteins: Struct., Funct., Bioinf., 2021, 89, 436–449 CrossRef CAS PubMed.
- S. Forli, R. Huey, M. E. Pique, M. F. Sanner, D. S. Goodsell and A. J. Olson, Nat. Protoc., 2016, 11, 905–919 CrossRef CAS PubMed.
- S. M. Butterfield and M. L. Waters, J. Am. Chem. Soc., 2003, 125, 9580–9581 CrossRef CAS PubMed.
- D. J. Woodbury, E. C. Whitt and R. E. Coffman, Biophys. Rep., 2021, 1, 100012 CAS.
- K. Dave, M. Jäger, H. Nguyen, J. W. Kelly and M. Gruebele, J. Mol. Biol., 2016, 428, 1617–1636 CrossRef CAS PubMed.
- A. D. Keefe and J. W. Szostak, Nature, 2001, 410, 715–718 CrossRef CAS PubMed.
- T. O. J. Cockram, J. M. Dundee, A. S. Popescu and G. C. Brown, Front. Immunol., 2021, 12, 1–33 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The Supporting Information contains the complete description of the experimental procedures, additional tables and figures as well as the full characterization of the peptides investigated in this study. See https://doi.org/10.1039/d2sc01078j |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |