
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSub-1.5 nm-gapped heterodimeric plasmonic nanomolecules†
Xiaojun
Song‡
a,
Yueliang
Wang‡
b,
Yan
Hao
a,
Qingqing
Zhu
a,
Yanjuan
Li
a,
Lei
Song
a and
Zhaoxiang
Deng
 *a
*a
aCenter for Bioanalytical Chemistry, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: zhxdeng@ustc.edu.cn
bCollege of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing, Zhejiang 314001, China
First published on 1st April 2022
Abstract
Plasmonic molecules are discrete assemblies of similar/dissimilar nanomaterials (atomic equivalents) with efficient inter-unit coupling toward electromagnetic hybridization. Albeit fundamentally and technologically very important, these structures are rare due to the lack of a general way to manipulate the structure, composition, and coupling of the nanoassemblies. While DNA nanotechnology offers a precious chance to build such structures, the weak coupling of DNA-bonded materials and the very limited material building blocks are two obstacles. This work aims to remove the bottlenecking barriers on the road to dimeric (and possibly more complicated) plasmonic molecules. After solving key synthetic issues, DNA-guided, solvo-driven Ag ion soldering is utilized to build a whole set (10 combinations of 4 metals) of homo/heterodimeric plasmonic nanomolecules with prescribed compositions. Importantly, strong in-solution electric-dipole coupling mediated by a sub-1.5 nm interparticle dielectric gap is achieved for materials with strong (Au, Ag) or damped (Pt, Pd) plasmonic responses. The involvement of Pt/Pd materials is of great value for plasmon-mediated catalysis. The broken dimeric symmetry is desirable for Fano-like resonance and photonic nanodiode devices, as well as lightening-up of plasmon dark states. The generality and reliability of the method would allow excitonic, nonlinear-optical, and magnetic units to be involved toward correspondingly enhanced functions.
Introduction
Bottom-up nanofabrication takes advantage of synthetic material building blocks and molecular (or colloidal) assembly strategies to build well-defined nanoarchitectures. In a DNA-directed process, it is virtually possible to define the xyz coordinates of every structural unit toward specific functions.1,2 For example, DNA-assembled protein arrays can catalyze enzymatic reaction cascades or activate a bipartite protein complex.3–7 In this case, diffusion fields of closely adjacent reactions are effectively overlapped (“coupled”) to enable a “chemical crosstalk”. Besides biomaterials, chiroplasmonics is realizable via DNA-guided organization of achiral nanocrystals.8–12 Additionally, there is a strong will to obtain nearly touching nano-objects in a DNA-guided structure to achieve coupling-derived new functions. The strongly coupled nanoassembly behaves like a “nanomolecule” where a slight structural alteration dramatically modifies its behaviors.13–18 Localized surface plasmon resonance (LSPR) originating from light-induced collective electron oscillations in a metal nanostructure provides an ideal system to study subwavelength electromagnetic interactions.19–21 We previously revealed that silver ion or less-polar solvents can induce strong plasmon coupling in DNA-tethered gold nanoparticle (AuNP) homodimers.22–25 Differently, size- or composition-asymmetric heterodimers possess unique properties related to their broken parity, including excitable plasmonic dark states,26 unidirectional light propagation (photonic diode),27,28 and Fano resonance.29,30 The prevalence of gold homodimers in plasmonic molecules can be attributed to the easy synthesis and good stability of AuNPs. Superior to AuNPs, silver NPs (AgNPs) have less damped LSPR and thus much stronger and sharper plasmon resonance.31 However, their weak chemical and colloidal stabilities are often unwelcome.32,33 Heterodimers involving Pt/Pd NPs are very rare despite their superior catalytic activities.34 Besides, the strongly damped LSPRs of Pt/PdNPs often make their plasmonic coupling hard to distinguish.35–37Despite the great promise, another reason for the less popularity of heterodimers lies in their fabrication difficulties. Top down processes are unsuitable for structures with composition heterogeneity, not mentioning the inability to confidently make a sub-2 nm interunit gap. DNA nanotechnology is good at building geometrically and compositionally complicated nanoassemblies with some key issues to be solved. First, DNA-linked nanoparticle dimers exhibit negligible coupling in solution such that dried samples have to be considered due to their shrunk gaps (still beyond a strong coupling range of <2 nm).38 Second, the purity of as-prepared heterodimers is sometimes unsatisfactory, leading to mixed optical signals hard to deconvolute. Third, NPs with strongly damped LSPRs (e.g. Pt and Pd) are seldom considered.
The present work aims to remove the bottlenecking barriers on the road toward dimeric and possibly even more complicated plasmonic nanomolecules benefitting from the structural programmability of DNA-directed nanoassemblies. We explore a newly developed ethanol-assisted Ag ion soldering (eAIS) in conjunction with DNA-directed assembly to address the above challenges (Fig. 1). Solution-dispersible, strongly coupled heterodimers involving different binary combinations among Au, Ag, Pt, and Pd NPs (Fig. 2 and S1†) are to be constructed. Due to the good colloidal stability of as-prepared NPs (Fig. S2†), DNA decoration can be minimized to favor a narrow (<1.5 nm) interparticle gap toward a strongly bonding dipole plasmon mode (BDP, in-phase longitudinal dipole–dipole coupling) (Fig. S3†).39–41 Briefly, NPs conjugated with minimal complementary DNA strands are hybridized into a heterodimer under the help of preparative electrophoretic isolation. The loosely-tethered NPs by DNA in a dimer are made closely apposed via van der Waals (VDW) colloidal interactions achieved in ethanol due to neutralized double layer charges.42 Such a process can be reversed when the coupled dimers are brought back to water (Fig. S4†), which actually provides a way to re-verify coupling-induced LSPR hybridizations (Fig. S3†). The coupled dimers are fixed by Ag ion soldering (AIS) to maintain their coupling after being transferred back to water, where Ag+ serves as a ligand remover by forming Ag+–ligand complexes to enhance the VDW “gluing”. The fixed dimers are re-capped by fish sperm DNA (FSDNA) to achieve colloidal stability in water. While AIS is directly useable to make DNA-linked strongly-coupled AuNP dimers in water,25 it does not work well for materials with a poorer colloidal stability (Fig. S5†). The “round-trip” process of ethanol-driven AIS (eAIS) is thus preferred.
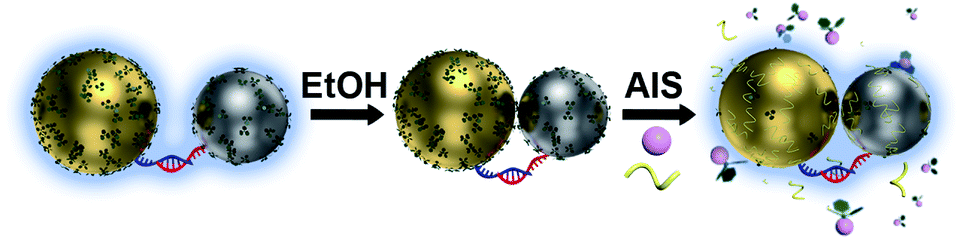 | ||
| Fig. 1 Ethanol-assisted Ag ion soldering (eAIS) of DNA-linked size/composition-asymmetric nanoparticle heterodimers based upon a whole set of combinations of Au, Ag, Pt, and Pd plasmonic building blocks. | ||
 | ||
| Fig. 2 TEM images of as-synthesized 23 (a) and 30 nm (b) AuNPs, 28 nm AgNPs (c), 31 nm g-AgNPs (d), 48 nm PtNPs (e), and 37 nm PdNPs (f) with uniform size distributions. Insets are optical extinction profiles of the NPs, reflecting their characteristic LSPRs. | ||
Results and discussion
Compared to AuNPs (Fig. 2a and b), AgNPs (Fig. 2c) are chemically more active (Fig. S6†) and vulnerable to aggregation. To stabilize AgNPs, surface passivation by a dense layer of thiolated polyethylene glycol (PEG) is often required.26 The PEG shell not only results in relatively large interparticle gaps of 4–5 nm (in dried state), but also prohibits further surface functionalization of the AgNPs. To circumvent these pitfalls, we synthesized AgNPs bearing very thin (ca. 2 nm) gold skins (Fig. S7†) (namely g-AgNPs).43,44 The electroless gold plating was optimized to guarantee a good colloidal stability without significantly altering the LSPRs of AgNPs (Fig. 2c, d and S8†). The somehow broadened (increased damping by gold) plasmon resonance of the gold-plated AgNPs (g-AgNPs) only slightly deviated (ca. 8 nm) from the LSPR peak of pristine AgNPs (centered at 397 nm) (Fig. S8†). Oxidative etching verified the enhanced chemical stability of g-AgNPs (Fig. S9†). Here a commonly adopted polyvinylpyrrolidone (PVP) capping ligand was replaced by FSDNA. We believe the rich negative charges on the phosphate backbone of surface-adsorbed FSDNA contributed to the impressive colloidal stability of g-AgNPs (Fig. S2c, S10 and S11†). PtNPs and PdNPs (spheroids of randomly associated or “fused” 3–5 nm Pt/Pd sub-particles, Fig. 2e and f) were prepared by a seeded nanosynthesis. Agarose gel electrophoresis (AGE) verified their colloidal stability (Fig. S2†).45,46The Au, Ag (with a protective gold shell), Pt, and Pd NPs had a good size monodispersity revealed by transmission electron microscopy (TEM) imaging and AGE assaying (Fig. S1 and S2†). The gold shells on AgNPs and the polycrystalline nature of Pt/Pd NPs were clearly resolved by TEM (Fig. S7†, 2e and f). Optical extinctions (Fig. 2a, b and d) of the AuNPs and g-AgNPs featured prominent LSPR peaks at 525 and 405 nm, respectively. In contrast, the LSPRs of as-synthesized PtNPs and PdNPs were broadened, attenuated, and overlapped with interband transitions in the ultraviolet (UV) region (Fig. 2e and f). Accordingly, only a tailing of their extinction toward the visible domain could be observed. All the NP materials were then grafted by a minimum of specific DNA strands (see experimental section in ESI†) via metal–thiol bonding. The LSPR profiles of the freely dispersed DNA-conjugates resembled those of the unconjugated counterparts (Fig. S12†), meaning DNA strands had marginal influence on the dielectric environments of the metal NPs (helpful for a theoretical LSPR consideration without the need of tentatively assigning a refractive index). Such an advantage contrasts surface-deposited NPs which can have a large LSPR shift due to highly refractive dry ligand layers and a planar supporting substrate (both complicate theoretical modeling and result in compromised reliability of calculations).
Upon DNA conjugation, the NPs bearing DNA complements were allowed to hybridize into clusters, among which dimeric products were isolated via AGE separation (Fig. 3 and S13–S18†). In addition to heterodimers, homodimers containing identical NPs were also prepared (Fig. S13–S18†). Under TEM, the characteristic NP sizes and morphologies as well as image contrasts helped distinguish different metal materials in an asymmetric dimer (Fig. 3). In addition, element mapping was also conducted to verify g-Ag31Au30 dimers (Fig. S19†). Note that capillary forces during sample drying caused the NPs in some dimers being closely associated,38 which did not reflect their states in a native solution (Fig. 3). This argument was supported by spectral data which did not show any coupling-related extinction features for DNA-linked dimers in water (Fig. 4). We should mention that the pinhole-free Au layers on the g-AgNPs were critical for their assembly with other NPs. Otherwise oxidative leaching of Ag+ would result in NP aggregates or hollow gold (more precisely, Au/Ag alloy) shells in the products (Fig. S6, S9 and S20†). All the DNA-tethered nanodimers could be pre-coupled in an ethanol solution. Fixation of the ethanol-coupled dimers was easily fulfilled in an AIS cocktail containing Ag ions and FSDNA (Fig. 4).
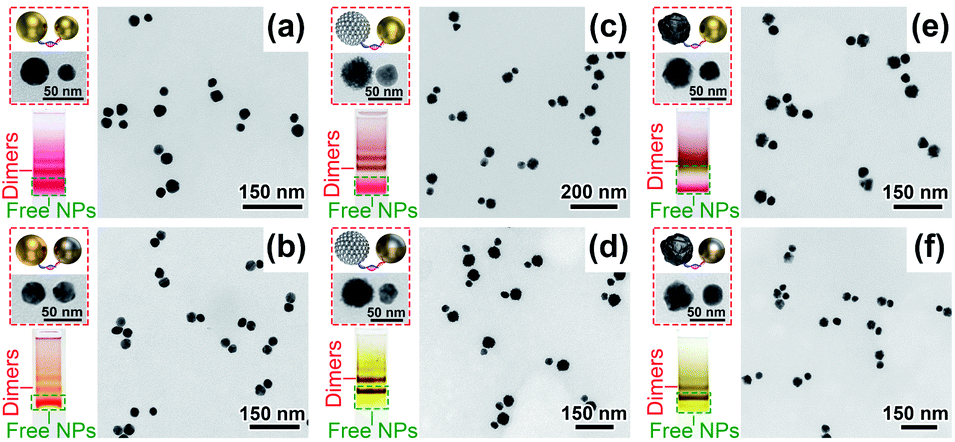 | ||
| Fig. 3 DNA-assembled heterodimeric plasmonic molecules with different NP compositions including (a) Au30Au23, (b) g-Ag31Au30, (c) Au30Pt48, (d) g-Ag31Pt48, (e) Au30Pd37, and (f) g-Ag31Pd37 (subscripts in the formulas denote NP diameters). Insets are schematic and magnified views of heterodimers and isolating agarose gels for the products. | ||
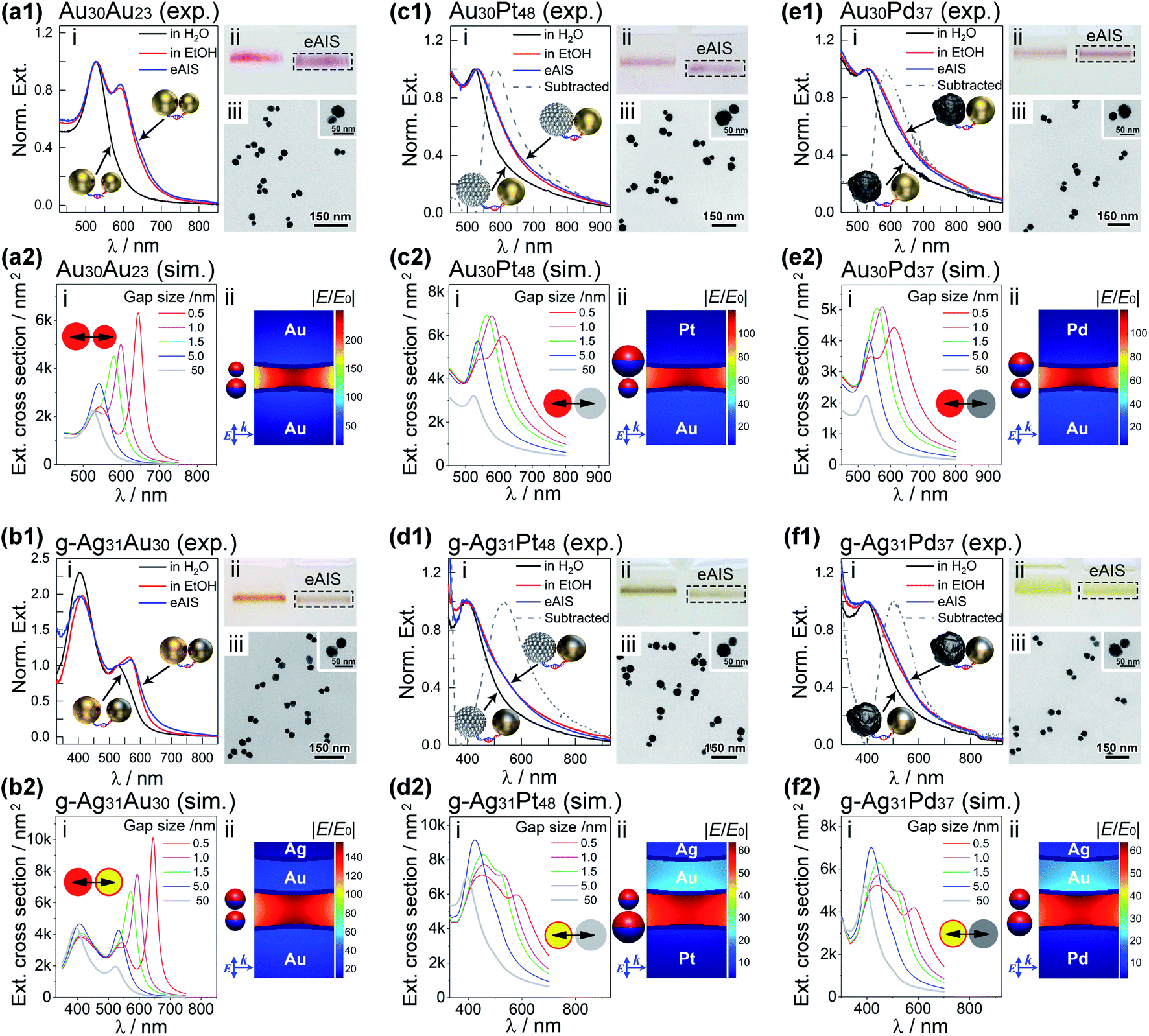 | ||
| Fig. 4 Ethanol-assisted Ag ion soldering (eAIS) of DNA-linked heterodimeric plasmonic molecules with different NP sizes and compositions including (a) Au30Au23, (b) g-Ag31Au30, (c) Au30Pt48, (d) g-Ag31Pt48, (e) Au30Pd37, and (f) g-Ag31Pd37. Panels (i)–(iii) in experimental data (marked as “exp.”) are optical extinction, gel electrophoresis, and TEM evidences, respectively, in support of strongly coupled dimers by eAIS. Dashed lines in (c1–f1) represent spectral differences between coupled and uncoupled dimers. Panels (i) and (ii) in simulated data (labelled as “sim.”) are extinction spectra corresponding to different gap sizes (0.5–50 nm), and electric field mappings for dimers with 1.0 nm (a2, c2 and e2) and 1.5 nm (b2, d2 and f2) gaps excited at peak extinctions, respectively. Dimers adjacent to nearfield mappings bear light-induced positive (red) and negative (blue) surface charges. Directions of light propagation (k) and excitation electric field (E) relative to the dimer orientation are also indicated along with the simulated data. | ||
The nanomolecule formed by two size-mismatched AuNPs (23 and 30 nm, respectively) was termed as an Au30Au23 dimer that exhibited excellent stability during AGE analyses (Fig. 4a1(ii)). The gel bands of the coupled and uncoupled Au30Au23 dimers exhibited sharply different colors (purple vs. red). Since no aggregation happened to the samples as judged from the gel data, this color transition truly reflected their different coupling states. TEM images in Fig. 4a1(iii) clearly supported such a situation. The eAIS-treated Au30Au23 dimers had very small interparticle gaps, in sharp contrast to their uncoupled precursors (Fig. 3a). The coupled Au30Au23 dimers exhibited a new LSPR peak at 595 nm, which was about 70 nm redshifted from the uncoupled dimers (same as unassembled AuNPs, Fig. S21†). Such a BDP resonance gave an interparticle gap of ca. 1.0 nm based on theoretical modelling (Fig. 4a1(i) and a2(i)).47 In addition, extinctions of 30 and 23 nm AuNP homodimers showed BDP peaks at 605 and 590 nm, respectively (Fig. S22†), after the eAIS coupling. It is noteworthy that the AIS process did not significantly alter the LSPRs of the ethanol-coupled dimers. However, in the absence of the AIS step, the BDP of ethanol-coupled dimers immediately disappeared upon being transferred back to water (Fig. S4†). This reversibility necessitated the use of AIS to permanently maintain the LSPR coupling in an aqueous solution. In addition, the completely reversed coupling also ruled out other possibilities (e.g. destabilization and aggregation) that might contribute to the emergent LSPR peaks.
An aqueous dispersion of composition-asymmetric g-Ag31Au30 heterodimers exhibited a prominent BDP peak located at 570 nm upon eAIS treatment (Fig. 4b1(i)). In contrast, symmetric Au30Au30 and g-Ag31g-Ag31 homodimers showed BDPs resonating at 600 nm and 525 nm, respectively (Fig. S23 and S24†). The relative energies of these BDPs could be rationalized by a plasmon hybridization theory (Fig. S3†), where the dipoles of two paired NPs and their spatial separations dictate the energy of a BDP. The nondegenerate LSPRs of Au and Ag NPs in a heterodimer result in a BDP mode leveled between two homodimers. A boundary element method (BEM) (see ESI†) was then employed to calculate the BDPs of these systems, giving a gap size close to 1.5 nm to replicate experimentally observed BDP wavelengths (Fig. 4b2(i)). The slightly larger (1.5 nm vs. 1.0 nm (for Au30Au23)) gap size estimated for g-Ag31Au30 dimers is in line with their relatively thick FSDNA capping layer. Theoretically, AuAg dimers can generate a Fano dip in the absorbance of gold through near-field coupling of the AgNP's discrete plasmon with the interband continuum of the AuNP in close nearby.29 In this regard, the tunable size of NPs and their strong coupling bring a chance to fine-tune the Fano resonance.30 Unfortunately, such a hidden Fano signal is often hard to observe due to spectral overlapping. However, our work may facilitate an experimental observation of this prediction based on nonlinear spectroscopy to selectively probe the gold component,29,48 or by finely tuning the size and coupling strength of the Au/Ag heterodimers in order to visualize the resulting more prominent Fano dip by regular spectroscopy.29,30
Apart from Au and Ag, Pt and Pd metals are rarely employed in nanoplasmonics due to their strongly damped and attenuated LSPRs. The broadened plasmon resonances of the Pt/Pd NPs in the UV region overlap with interband transition continuums, making them barely distinguishable. The sub-1.5 nm coupling achieved by eAIS for the Au/Au and Au/Ag heterodimers encouraged us to develop these “unpopular” structures toward visible plasmonic coupling. Benefitting from the very small interparticle gaps achieved by eAIS, the Au/Pt, g-Ag/Pt, Au/Pd, and g-Ag/Pd heterodimers consistently showed new LSPR features on the long wavelength edges of the AuNP and AgNP extinctions. Similar coupling-induced peak broadening to low-frequency sides was also observed for homodimers of identical Pt/Pd NPs (Fig. S23 and S24†). By subtracting the spectra of uncoupled structures from those of coupled ones, peak-shaped LSPR profiles were revealed (Fig. 4c1–f1). The above structures featuring strongly coupled plasmonic/catalytic metal nanounits are attractive as emerging materials in plasmon-mediated nanocatalysis. Theoretical results revealed broadened BDP profiles for dimers incorporating Pt/Pd materials (Fig. 4c2(i)–f2(i)), in sharp contrast to the intensive coupling peaks of Au/Au and Au/Ag dimers (Fig. 4a2(i) and b2(i)). It is therefore understandable why peak-shaped BDPs did not appear on the experimental spectra of Pt/PdNP-involved dimers, different from those of Au/AgNP-derived ones (Fig. 4a1(i)–f1(i)). In addition, calculated surface charge distributions at peak excitations revealed some quadrupole contributions (except Au30Au23) due to mixed dipolar and higher-order couplings (Fig. 4a2(ii)–f2(ii), Fig. S25 and S26†), which were avoidable by off-setting light excitations to the red-edges (at half-maxima) of the extinction peaks (Fig. S25†). In all cases, strong nearfield enhancements were observed based on simulated data (Fig. 4a2(ii)–f2(ii) and S26†). It is noteworthy that the in-gap electric fields for Au30Au23 heterodimers are only slightly asymmetric, without being apparently stronger on the larger particle side. In the case of heterodimers containing Pt/Pd particles, the electric fields are somehow pulled toward the Au/Ag surfaces.
We want to emphasize that the sub-1.5 nm gaps of the eAIS dimers are easily addressable by small molecules. For example, a p-aminothiophenol (p-ATP) molecule easily got adsorbed into the nanogaps of g-Ag31Au30 heterodimers. The strongly coupled dimers provided hotspots to achieve gap-enhanced Raman signals of p-ATP (Fig. S27†). Meanwhile, the dimers are ideal for hot carrier generation under laser irradiation. Accordingly, p,p′-dimercaptoazobenzene (DMAB) was observed on the Raman spectra as an oxidized form of p-ATP due to plasmon-medicated photocatalysis (Fig. S27†). Interestingly, g-Ag31Au30 behaved much better than an Au30Au30 counterpart, probably due to the better-functioning Ag@Au material. In addition, coating of NPs by polyethylene glycol (PEG) layers was possible, which provided a chance to adjust the gap size and the BDP resonance (Fig. S28†). While the present work was focused on spherical NPs, involvement of nonspherical ones is possible as no clear size and shape limits of NPs were seen at this stage. This is an interesting issue because shape-directed NP assembly/coupling may be exploited. Besides UV-vis extinctions of solution-borne dimers, we prepared well-deposited samples on a substrate for dark field optical imaging (Fig. S29–S32†) and single-particle spectroscopy (Fig. S33†). In this case, dimers formed by Au and Ag@Au NPs were chosen considering their strong and distinctive light scatterings. The dimers were encapsulated in thick silica shells to prevent interdimeric coupling signals in an aggregate. The excellent purity and uniformity of the samples helped our dark-field observation a lot. The imaging and spectral data showed a good coupling yield and polarization-dependent optical scattering. Heterogeneity did exist, which probably came from a variation of NP shape and size as well as coupling strength. A detailed investigation in various regards has been planned for our future research, benefiting from the easy-to-obtain high-quality heterostructures.
Conclusions
In summary, we have successfully constructed a whole set (10 combinations of 4 different metal colloids) of homo/heterodimeric plasmonic meta-molecules featuring strong electromagnetic interactions between light-excited electric dipoles. Nanoparticles of Au, Ag, Pt, and Pd metals are firstly incorporated into DNA-directed dimeric nanoparticle assemblies. Prominent dipolar plasmon coupling is confidently observed for materials with strong or damped plasmonic responses upon ethanol-assisted Ag ion soldering in solution, benefiting from a very narrow sub-1.5 nm interparticle gap. Our work represents a marked step on the way to DNA-programmable nanoplasmonics with quantitatively definable functions. The guaranteed purity of the synthetic plasmonic molecules would facilitate a fine characterization of them at single-particle levels toward nanoscale device development. For example, the broken symmetry of the nanodimers is suitable to generate Fano-like resonance and unidirectional light propagation (photonic diode), as well as lightening-up of dark state plasmons. This work significantly expands the library of building blocks for DNA-programmable strongly-coupled plasmonic nanomolecules. Subsequently, excitonic, nonlinear optical, and magnetic materials might be incorporated into the plasmonic molecules as novel functional substituents. The ability to build topologically and compositionally complicated nanomolecules would enable probing of structure–activity relationships in physical, chemical, and biological applications.Data availability
Available upon reasonable request to the corresponding author.Author contributions
X. S., Y. W. and Z. D. conceived the ideas and planned the research. X. S. and Y. H. conducted eAIS experiments. X. S. and Y. W. did dark field imaging and measured single particle spectra. X. S. and Q. Z. synthesized the nanoparticles. X. S., Y. L. and L. S. functionalized nanoparticles with DNA. X. S. assembled the dimers and measured the optical properties. Z. D. did theoretical simulations and supervised the project. X. S. and Z. D. analysed the data and wrote the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (Grants 2021YFA1200101 and 2018YFA0702001) and the National Natural Science Foundation of China (Grants 21972130, 21425521, and 21521001).Notes and references
- N. C. Seeman, Nature, 2003, 421, 427 CrossRef PubMed.
- M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 1260901 CrossRef PubMed.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516 CrossRef CAS.
- C. Wang, L. Yue and I. Willner, Nat. Catal., 2020, 3, 941 CrossRef CAS.
- L. Sun, Y. Gao, Y. Xu, J. Chao, H. Liu, L. Wang, D. Li and C. Fan, J. Am. Chem. Soc., 2017, 139, 17525 CrossRef CAS.
- Y. Chen, G. Ke, Y. Ma, Z. Zhu, M. Liu, Y. Liu, H. Yan and C. J. Yang, J. Am. Chem. Soc., 2018, 140, 8990 CrossRef CAS PubMed.
- H. Li, M. Wang, T. Shi, S. Yang, J. Zhang, H. H. Wang and Z. Nie, Angew. Chem., Int. Ed., 2018, 57, 10226 CrossRef CAS PubMed.
- C. Zhou, X. Duan and N. Liu, Acc. Chem. Res., 2017, 50, 2906 CrossRef CAS PubMed.
- N. Liu and T. Liedl, Chem. Rev., 2018, 118, 3032 CrossRef CAS PubMed.
- X. Lan and Q. Wang, Adv. Mater., 2016, 28, 10499 CrossRef CAS PubMed.
- X. Lan, T. Liu, Z. Wang, A. O. Govorov, H. Yan and Y. Liu, J. Am. Chem. Soc., 2018, 140, 11763 CrossRef CAS.
- X. Shen, C. Song, J. Wang, D. Shi, Z. Wang, N. Liu and B. Ding, J. Am. Chem. Soc., 2012, 134, 146 CrossRef CAS.
- A. M. Funston, C. Novo, T. J. Davis and P. Mulvaney, Nano Lett., 2009, 9, 1651 CrossRef CAS.
- Y. Li, Z. Liu, G. Yu, W. Jiang and C. Mao, J. Am. Chem. Soc., 2015, 137, 4320 CrossRef CAS PubMed.
- L. Song and Z. Deng, ChemNanoMat, 2017, 3, 698 CrossRef CAS.
- J. M. Nam, J. W. Oh, H. Lee and Y. D. Suh, Acc. Chem. Res., 2016, 49, 2746 CrossRef CAS PubMed.
- N. J. Halas, S. Lal, W.-S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913 CrossRef CAS.
- Q. Zhu, X. Song and Z. Deng, Acta Chim. Sin., 2020, 78, 675 CrossRef CAS.
- L. Zheng, Z. Liu, D. Liu, X. Wang, Y. Li, M. Jiang, F. Lin, H. Zhang, B. Shen, X. Zhu, Y. Gong and Z. Fang, Nat. Commun., 2021, 12, 291 CrossRef CAS PubMed.
- D. C. J. Neo, C. Yang, Y. Shi, Q. Y. S. Wu, J. Deng, Y. Xu, A. A. Bettiol, Y. Chan and E. J. Teo, ACS Photonics, 2019, 6, 93 CrossRef CAS.
- F. Yang, S. Ye, W. Dong, D. Zheng, Y. Xia, C. Yi, J. Tao, C. Sun, L. Zhang, L. Wang, Q. Chen, Y. Wang and Z. Nie, Adv. Mater., 2021, 33, 2100325 CrossRef CAS.
- Y. Li and Z. Deng, Acc. Chem. Res., 2019, 52, 3442 CrossRef CAS.
- M. Liu, L. Fang, Y. Li, M. Gong, A. Xu and Z. Deng, Chem. Sci., 2016, 7, 5435 RSC.
- Y. Wang, L. Fang, G. Chen, L. Song and Z. Deng, Small, 2018, 14, 1703303 CrossRef PubMed.
- H. Wang, Y. Li, M. Liu, M. Gong and Z. Deng, Small, 2015, 11, 2247 CrossRef CAS PubMed.
- S. Sheikholeslami, Y. W. Jun, P. K. Jain and A. P. Alivisatos, Nano Lett., 2010, 10, 2655 CrossRef CAS.
- T. Pakizeh and M. Käll, Nano Lett., 2009, 9, 2343 CrossRef CAS.
- L. V. Brown, H. Sobhani, J. B. Lassiter, P. Nordlander and N. J. Halas, ACS Nano, 2010, 4, 819 CrossRef CAS.
- G. Bachelier, I. Russier-Antoine, E. Benichou, C. Jonin, N. Del Fatti, F. Vallée and P.-F. Brevet, Phys. Rev. Lett., 2008, 101, 197401 CrossRef CAS PubMed.
- O. Peña-Rodríguez, U. Pal, M. Campoy-Quiles, L. Rodríguez-Fernández, M. Garriga and M. I. Alonso, J. Phys. Chem. C, 2011, 115, 6410 CrossRef.
- N. G. Bastús, F. Merkoçi, J. Piella and V. Puntes, Chem. Mater., 2014, 26, 2836 CrossRef.
- J. S. Lee, A. K. R. Lytton-Jean, S. J. Hurst and C. A. Mirkin, Nano Lett., 2007, 7, 2112 CrossRef CAS PubMed.
- Y. Zheng, Y. Li and Z. Deng, Chem. Commun., 2012, 48, 6160 RSC.
- L. Tian, C. Wang, H. Zhao, F. Sun, H. Dong, K. Feng, P. Wang, G. He and G. Li, J. Am. Chem. Soc., 2021, 143, 8631 CrossRef CAS PubMed.
- D. Manchon, J. Lermé, T. Zhang, A. Mosset, C. Jamois, C. Bonnet, J. M. Rye, A. Belarouci, M. Broyer, M. Pellarin and E. Cottancin, Nanoscale, 2015, 7, 1181 RSC.
- C. Langhammer, Z. Yuan, I. Zorić and B. Kasemo, Nano Lett., 2006, 6, 833 CrossRef CAS PubMed.
- S. De Marchi, S. Núñez-Sánchez, G. Bodelón, J. Pérez-Juste and I. Pastoriza-Santos, Nanoscale, 2020, 12, 23424 RSC.
- P. A. Kralchevsky and K. Nagayama, Langmuir, 1994, 10, 23 CrossRef CAS.
- E. Prodan, C. Radloff, N. J. Halas and P. Nordlander, Science, 2003, 302, 419 CrossRef CAS.
- P. Nordlander, C. Oubre, E. Prodan, K. Li and M. I. Stockman, Nano Lett., 2004, 4, 899 CrossRef CAS.
- T. Atay, J.-H. Song and A. V. Nurmikko, Nano Lett., 2004, 4, 1627–1631 CrossRef CAS.
- Y. Hao, L. Fang and Z. Deng, CCS Chem., 2020, 2, 1359 Search PubMed.
- H. Liu, T. Liu, L. Zhang, L. Han, C. Gao and Y. Yin, Adv. Funct. Mater., 2015, 25, 5435 CrossRef CAS.
- Z. Zhang, K. Bando, A. Taguchi, K. Mochizuki, K. Sato, H. Yasuda, K. Fujita and S. Kawata, ACS Appl. Mater. Interfaces, 2017, 9, 44027 CrossRef CAS PubMed.
- D. Zanchet, C. M. Micheel, W. J. Parak, D. Gerion and A. P. Alivisatos, Nano Lett., 2001, 1, 32 CrossRef CAS.
- H. Wang and Z. Deng, Chin. Chem. Lett., 2015, 26, 1435 CrossRef CAS.
- U. Hohenester and A. Trügler, Comput. Phys. Commun., 2012, 183, 370 CrossRef CAS.
- A. Lombardi, M. P. Grzelczak, E. Pertreux, A. Crut, P. Maioli, I. Pastoriza-Santos, L. M. Liz-Marzán, F. Vallée and N. Del Fatti, Nano Lett., 2016, 16, 6311 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sc01171a |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |