
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic kinetic resolution of transient hemiketals: a strategy for the desymmetrisation of prochiral oxetanols†
Alexander
Sandvoß
 *ab,
Henning
Maag
b,
Constantin G.
Daniliuc
a,
Dieter
Schollmeyer
b and
Johannes M.
Wahl
*b
*ab,
Henning
Maag
b,
Constantin G.
Daniliuc
a,
Dieter
Schollmeyer
b and
Johannes M.
Wahl
*b
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität, Corrensstraße 36, 48149 Münster, Germany
bDepartment Chemie, Johannes Gutenberg-Universität, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: wahl@uni-mainz.de
First published on 4th May 2022
Abstract
Identification of an electron poor trifluoroacetophenone allows the formation of uniquely stable hemiketals from prochiral oxetanols. When exposed to a cobalt(II) catalyst, efficient ring-opening to densely functionalized dioxolanes is observed. Mechanistic studies suggest an unprecedented redox process between the cobalt(II) catalyst and the hemiketal that initiates the oxetane-opening. Based on this observation, a dynamic kinetic resolution of the transient hemiketals is explored that uses a Katsuki-type ligand for stereoinduction (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 96:4 er) and allows a variety of 1,3-dioxolanes to be accessed (20 examples up to 98% yield).
:1 dr and 96:4 er) and allows a variety of 1,3-dioxolanes to be accessed (20 examples up to 98% yield).
Introduction
The stereoselective synthesis of polyols has been a cornerstone of organic chemistry due to the prevalence of this structural motif in biologically important molecules such as polyketides or carbohydrates (Scheme 1, top). As a result, continuous effort is devoted to the development of new methods that establish C–O bonds with high levels of stereocontrol. One possibility to accomplish such an endeavor invokes the application of a transient hemiacetal as an intramolecular nucleophile and was introduced by Evans and coworkers in 1993.1 Since this early example, many diastereoselective2 and enantioselective3 variants have been developed on the basis of Brønsted acid, as well as Lewis acid, catalysis (Scheme 1, middle). Recent extensions regarding the alkene-electrophile4 as well as applications in the synthesis of natural products5 have renewed the interest in hemiacetals as a platform for reaction development. However, nucleophilic attack of a transient hemiacetal to other groups than activated alkenes are rare,6 even though this would allow a much broader range of polyolic targets to be accessed.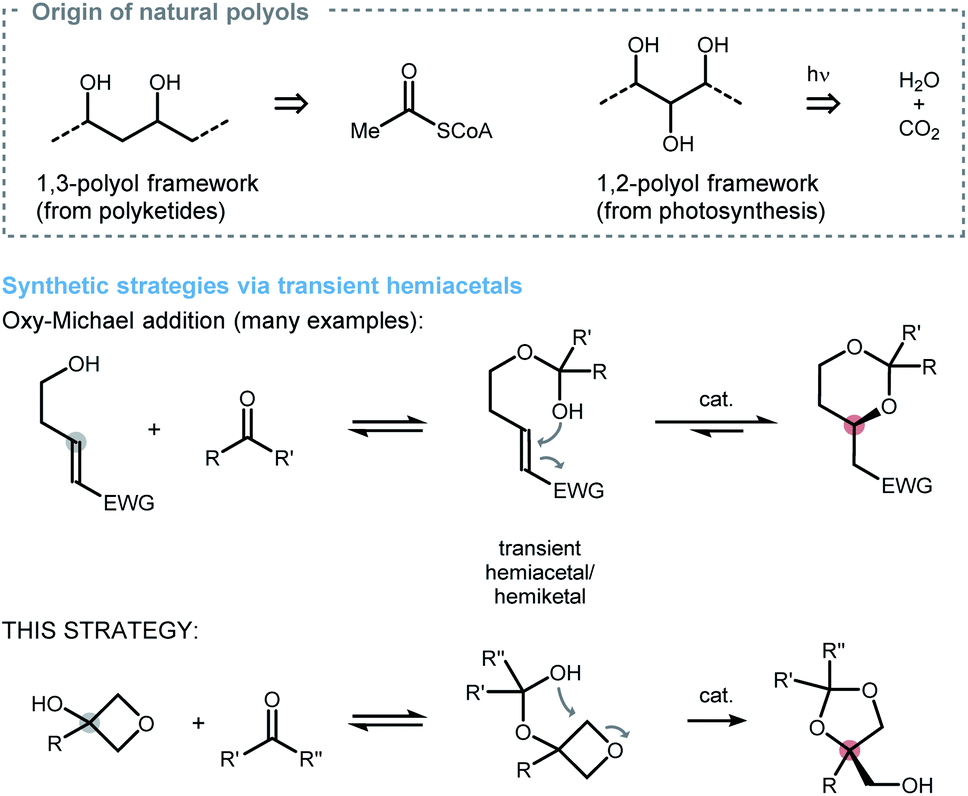 | ||
| Scheme 1 Overview of asymmetric strategies using hemiacetals to forge new C–O bonds. CoA = coenzyme A; EWG = electron withdrawing group. | ||
To address this limitation, we hypothesized that when adequately activated a transient hemiacetal would be able to undergo ring-opening at a strained heterocyclic core. Based on our laboratory's interest in desymmetrisation of prochiral four-membered rings,7 we considered 3-oxetanols as promising candidates to test this hypothesis (Scheme 1, bottom). Here, the preformed hemiacetal attacks the oxetane ring to form a dioxolane, which is energetically favourable due to the ∼20 kcal mol−1 of strain release, and opens the possibility to forge two new stereogenic centres along the way. According to literature precedent, a number of catalysts qualify to activate oxetanes towards desymmetrisation.8 Among them, CoIII·salen9 as well as chiral phosphoric acids10 are worth mentioning because they proved viable for the synthesis of various oxygen-heterocycles in the past. However, no synthetic strategy towards the formation of 1,3-dioxolanes was reported so far, which we now wish to report on along with our efforts identifying a suitable catalyst system for a highly selective desymmetrisation process.11
Results and discussion
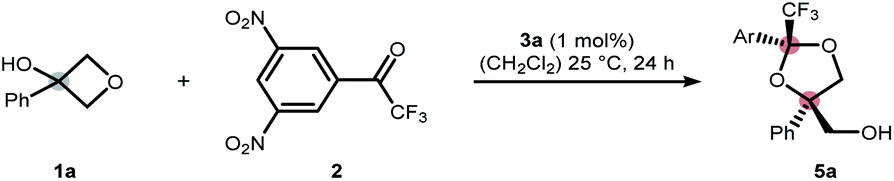
We commenced our study by exploring the ring-opening of 3-phenyloxetanol (1a) with simple aldehydes in the presence of a range of Brønsted acids as well as Lewis acids (see ESI† for further details). However, these attempts were met with limited success, presumably due to the low stability of the transient hemiacetal. We addressed this problem by installing electron withdrawing groups at the carbonyl core to increase the stability of the hemiacetal moiety.12 After exploring a range of electron withdrawing groups, we identified 3,5-dinitrotrifluoroacetophone 2 as a suitable candidate for the envisioned transformation.13 In the presence of 1 mol% of CoII·salen 3a, oxetanol 1a underwent cyclization to dioxolane 5a in 95% yield and in a diastereomeric ratio (dr) of 76:24 (Table 1, entry 1). As expected, no ring-opening was observed without the catalyst (Table 1, entry 2). Interestingly, when the parent trifluoroacetophenone was used instead of 2, no conversion to the respective dioxolane was detected (entry 3). The reaction was also found to be highly water-sensitive, which lends credibility to a transient hemiketal being a crucial reaction intermediate (Table 1, entry 4).
|
|
||||
|---|---|---|---|---|
| Entry | Changes from std. conditions | Yield 5ab | drc | erd |
| a Reactions were run on a 0.1 mmol scale using 1 equivalent of ketone 2 in 0.5 mL of dry solvent (0.2 M). b Yield of 5a (Ar = 3,5-(NO2)2-C6H3) determined by 19F NMR using trifluorotoluene as an internal standard. c Determined by 19F NMR from the crude reaction mixture. d Enantiomeric ratio of the major diastereomer was determined by HPLC analysis using a chiral stationary phase. e In this case, the expected product Ar = Ph was not detected. n.d. = not determined; Bz = benzoyl; OTf = trifluoromethanesulfonate. | ||||
| 1 | — | 95% | 76:24 |
— |
| 2 | No catalyst added | — | — | — |
| 3 | BzCF3 instead of 2 | —e | — | — |
| 4 | 1 equiv. H2O added | 7% | n.d. | — |
| 5 | Catalyst 3b instead of 3a | 92% | 79:21 |
60:40 |
| 6 | CoIII-catalyst 3c instead of 3a | <5% | n.d. | n.d. |
| 7 | Catalyst 3d instead of 3a | 48% | 78:22 |
60:40 |
| 8 | Katsuki-type cat 4a instead of 3a | 90% | 90:10 |
75:25 |
| 9 | Katsuki-type cat 4b instead of 3a | 86% | 96:4 |
86:14 |
|
||||
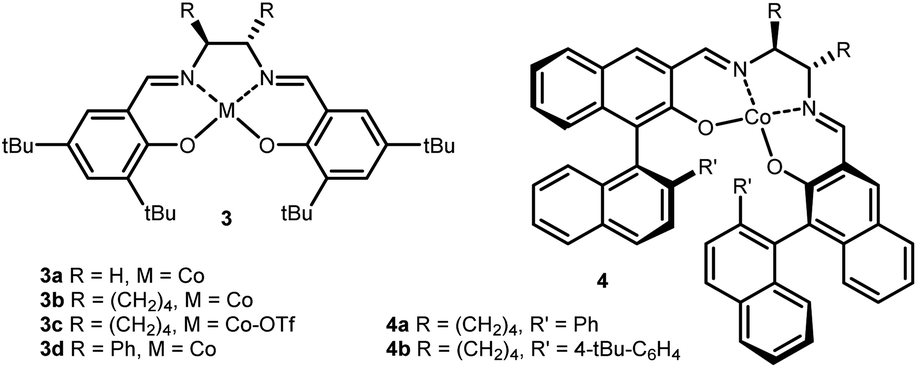
To develop an asymmetric variant, commercially available complex 3b was tested in the reaction sequence. To our delight, product 5a was obtained in a small but detectable enantiomeric ratio (er) of 60:40 (Table 1, entry 5). It was surprising that the oxidized form (TfO)CoIII·salen 3c, a privileged catalyst for the ring-opening of strained cyclic ethers, resulted in only traces of product (Table 1, entry 6).9 When replacing the cyclohexyl backbone with the sterically more demanding phenyl groups, no notable improvement in enantioselectivity was observed, but the overall yield dropped to 48% (Table 1, entry 7). Following that, we considered a distal group for stereoinduction and were attracted by Katsuki-type ligands 4.14 With ligand 4a an increase in selectivity to 90:10 dr and 75:25 er was detected (Table 1, entry 8). Finally, the overall performance of the ligand was further improved by the installation of pendant tert-butyl groups (complex 4b) providing dioxolane 5a in good yield and selectivity (Table 1, entry 9).
While some of the results from the optimization were in accordance with what we had envisioned, some were not and prompted us to conduct further mechanistic investigations. In particular, the origin of the unique activity of dinitrotrifluoroacetophenone 2 compared to trifluoroacetophenone remained unclear. First, we studied hemiketal formation using a range of para-substituted trifluoroacetophenones 6 (Scheme 2a). To quantify the electronic effect on hemiketal stability, a Hammett study was performed. A linear correlation between log(K) and the Hammett constant σ was found (Scheme 2a, entries 1–5).15 The sensitivity constant ρ was determined to be 1.8 highlighting the crucial role of the aryl-substitution on hemiketal stability. As a consequence, ketone 2 pushes the equilibrium far to the hemiketal side. Validation for the proposed structures was provided by X-ray diffraction of compound 8e, a stable hemiketal deriving from 4-nitroacetophenone 6e and benzyl alcohol (Scheme 2b). Next, we studied hemiketal formation of 2 with different oxetanols (Scheme 2c). Sterically accessible 3-oxetanol (1b) reversibly formed the expected hemiketal rac-9b, which could be monitored by NMR spectroscopy. Tertiary oxetanols such as 1a on the other hand, did not form detectible amounts of the respective hemiketal rac-9a. However, the formation of oxy-Michael product rac-10c from oxetanol 1c still points towards the transient formation of tertiary hemiketal rac-9c, albeit in low concentration. While these results show the steric and electronic contributions to hemiketal formation, they do not account for the relative reactivity towards ring-opening. To investigate the opening without having a pre-equilibrium step present, we designed substrate rac-11 (Scheme 2d). Surprisingly, rac-11 underwent no cyclization to oxolane rac-12 under the optimized conditions. While this may be explained by the reduced electrophilicity of rac-11 due to its lack of an oxygen atom at the 3-position, an insufficient activation of the oxygen nucleophile by the catalyst seems also reasonable. We hypothesized that an in situ aerobic CoII·salen oxidation initiated only by acidic hemiketals takes place increasing their inherent nucleophilicity.16 In accordance with literature reports, this oxidation process typically requires Brønsted acids with a pKa below 10.17 As this coincides well with the expected pKa for electron deficient hemiketals deriving from 2, we decided to study such an aerobic oxidation based on easily accessible hydrate 13 as a surrogate (Scheme 2e). When catalyst 3b was treated with 13 under ambient conditions, cobalt oxidation was observable by the naked eye through a color change from orange to green. UV-vis analysis showed good consistency with related complexes of the general type (X)CoIII·salen (see ESI† for further information).18 By contrast, less acidic trifluoroethanol showed only limited potential in aerobic oxidation of CoII. This not only explains the unsuccessful ring-opening of rac-11 but also underpins the inimitable role of ketone 2 to modulate both the stability and acidity of its respective hemiketals.
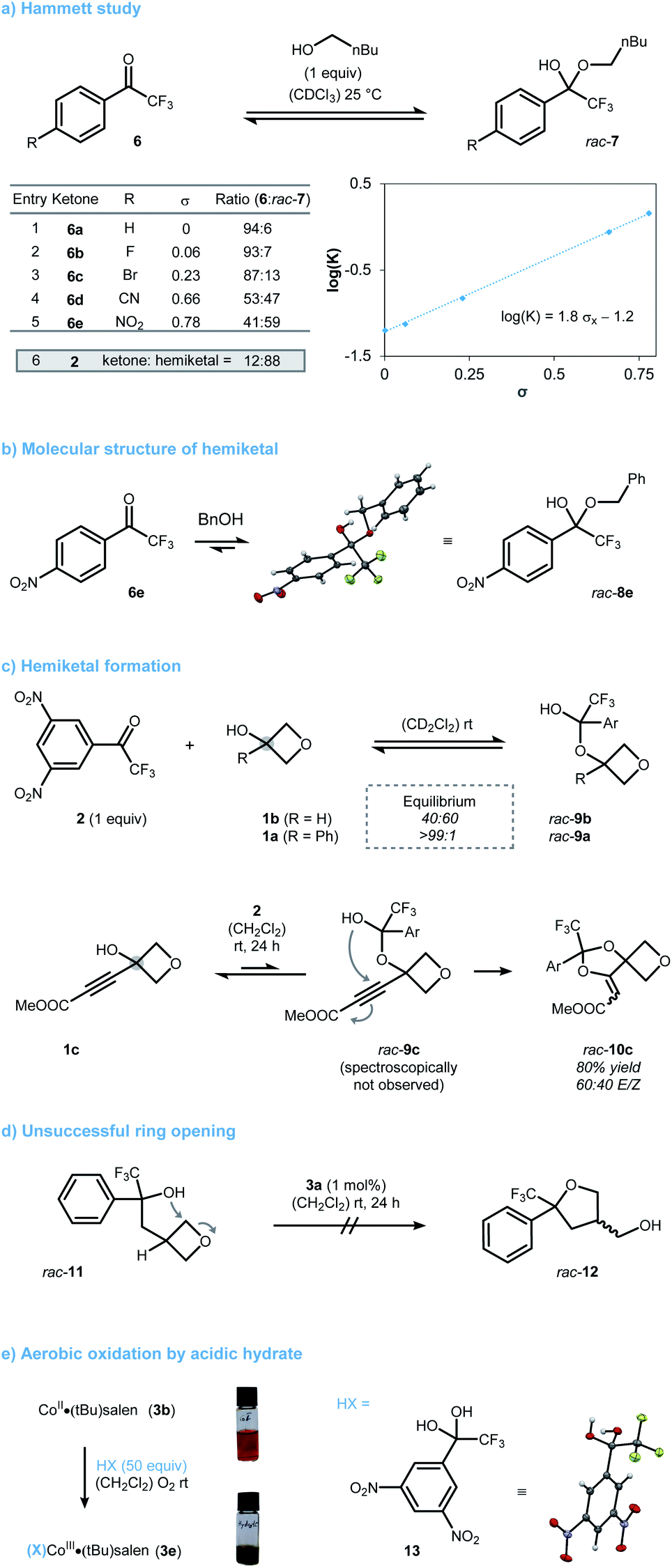 | ||
| Scheme 2 (a)–(e) Pieces of experimental data highlighting the unique character of ketone 2 for the mechanism. Ar = 3,5-(NO2)2-C6H3. CCDC 2130260 (rac-8e), CCDC 2130259 (13).† | ||
Based on the collected data, a mechanistic picture for the desymmetrization of oxetanols is proposed in Scheme 3.19 Reversible formation of the hemiketals 9a and ent-9a is facilitated by the electron deficient nature of ketone 2 and can occur without a catalyst as suggested by the findings from Scheme 2a. However, an influence of the catalyst on the underlying equilibrium can neither be excluded nor confirmed at this point. The enhanced acidity of the hemiketals triggers aerobic oxidation to furnish CoIII intermediates 14a and epi-14a, respectively. Cobalt-hemiketalate 14a exhibits an enhanced nucleophilicity triggering efficient ring-opening to furnish intermediate 15a. Catalyst turn-over can be explained by proton transfer from hemiketal 9a to 15a regenerating hemiketalate 14a and releasing the dioxolane product 5a. The overall high yield and selectivity can be rationalized by a dynamic kinetic resolution (DKR) of the racemic hemiketals.20 This implies that hemiketal ent-9a reacts much slower than its enantiomer 9a while the rate of racemization remains relatively fast. About a dual role of the catalyst can only be speculated based on our current data, but it seems reasonable that the catalyst also acts as an inter- or intramolecular Lewis acid further facilitating oxetane-opening.
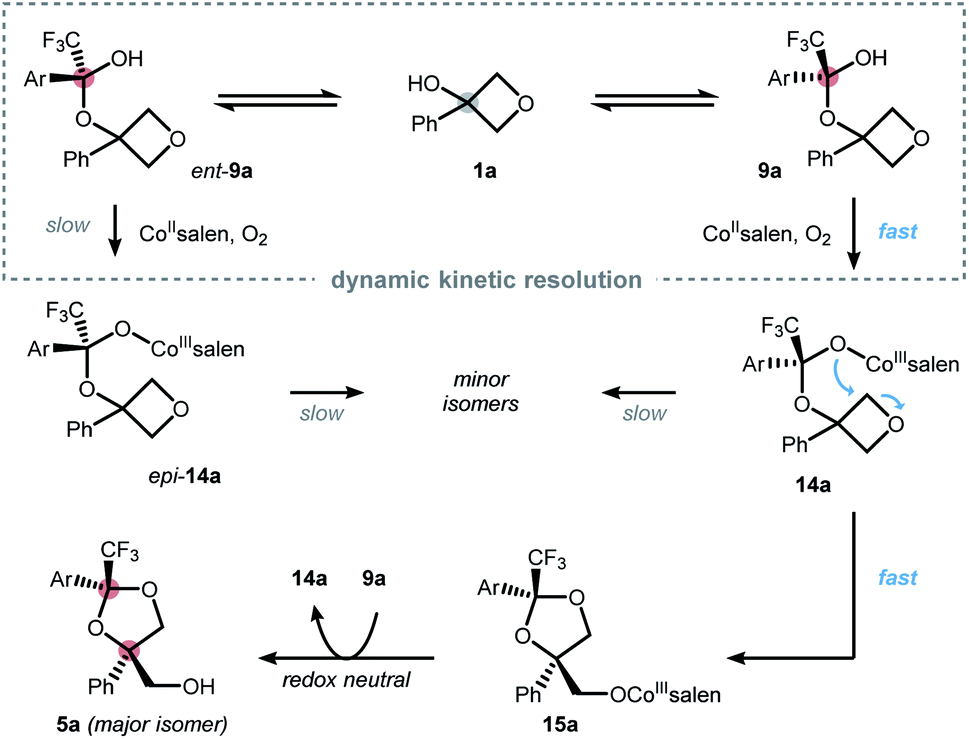 | ||
| Scheme 3 Mechanistic picture of the desymmetrization via a dynamic kinetic resolution of a transient hemiketal in best accordance with the findings from Scheme 2. Ar = 3,5-(NO2)2-C6H3. | ||
With a reasonable mechanistic picture in hand, we started exploring the scope of the oxetane desymmetrization (Scheme 4). Steric substitution at the ortho, meta, and para position of the phenyl core was well tolerated furnishing tolyl-dioxolanes 5d, 5e, and 5f. Electronic perturbations resulted in an overall improved enantioselectivity of up to 96:4 er for the corresponding electron-deficient dioxolanes 5g–5k. Moreover, gram-scale reaction was feasible with no loss in selectivity as indicated by bromophenyl dioxolane 5j. The very same dioxolane was also suitable for growing single crystals to determine the absolute configuration. Extended π-systems such as naphthyl and diphenylyl as well as heterocycles such as thiophene revealed good efficiency in the cyclization event (5l–5n). In addition, alkenyl as well as alkynyl substitution was tolerated (5o–5s). Replacing the aryl group with an n-butyl group resulted in a substantial decrease in enantioselectivity (5t). Furthermore, sterically encumbered aryl 1u as well as unsubstituted oxetanol (1b) successfully underwent ring-opening to the corresponding alcohols 5u and 5b, albeit in significantly lower diastereo- and enantioselectivity. These results suggest that some sort of π-interaction between the catalyst and substrate is crucial for a successful DKR.
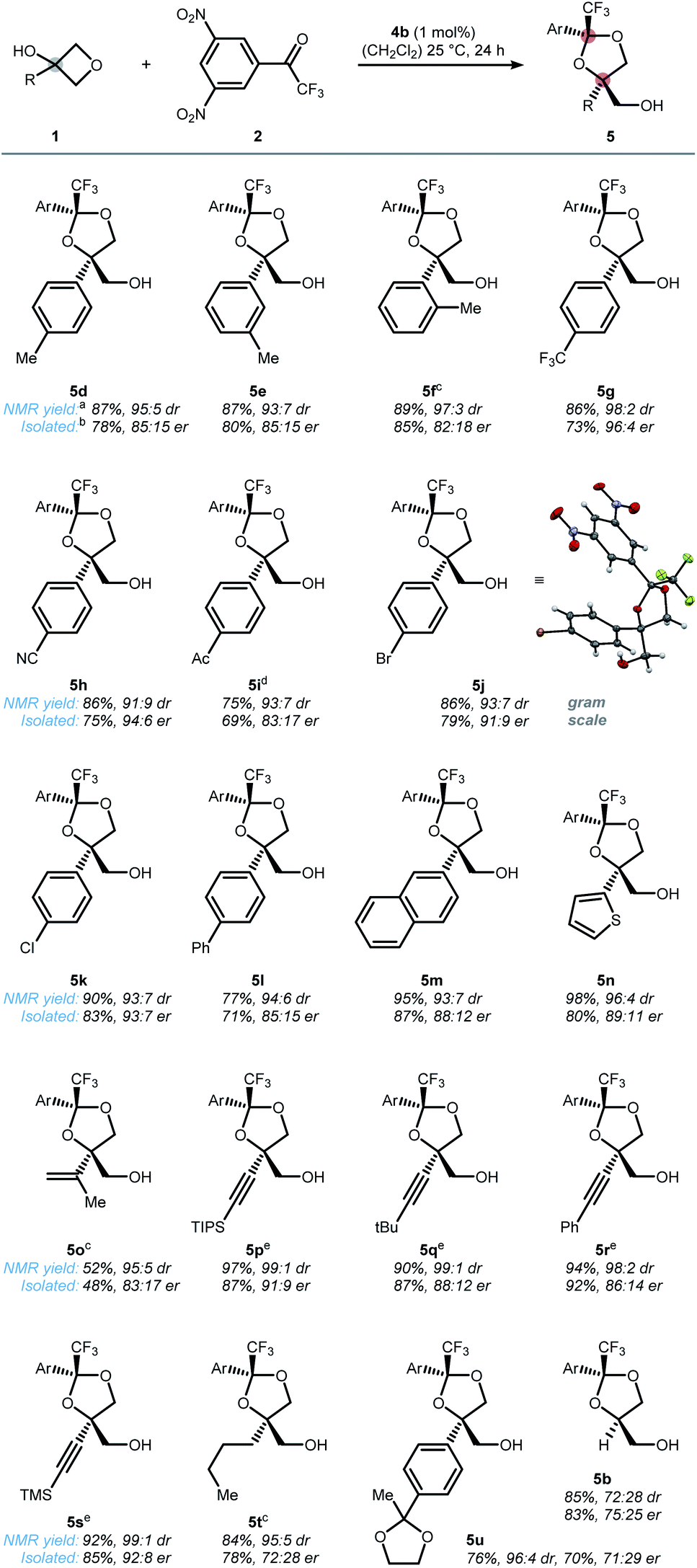 | ||
| Scheme 4 Scope of the desymmetrization of prochiral oxetanols 1via a DKR of the respective transient hemiketals. [a]Determined by 19F NMR using trifluorotoluene as an internal standard. [b]Isolated yield and enantiomeric ratio correspond to the major isomer only. [c]Reactions were run at 40 °C instead of 25 °C. [d]Fluorobenzene was used instead of trifluorotoluene as the internal standard. [e]Reactions run with catalyst 4a instead of 4b. Ar = 3,5-(NO2)2-C6H3. CCDC 2141905 (5j).† | ||
In contrast to the quite general scope of the oxetanol precursor, alterations at the ketone scaffold were less fruitful (Scheme 5). Changing from two to one nitro-groups (ketone 6e) significantly reduced the reaction rate and only 15% yield of dioxolane 16a were obtained after 24 h. On the other hand, replacing the CF3 by a CF2Cl group (ketone 17) allowed the synthesis of dioxolane 18a in reasonable yield and moderate selectivity. More drastic changes to the scaffold of ketone 2 completely shut down the reaction (for details see the ESI†). Altogether, these results underline the unique character of ketone 2, which was further elaborated upon in Scheme 6. First, a stereodivergent synthesis of dioxolanes 5r and 19r from phenylacetylated oxetanol 1r was achieved by switching the metal catalyst (Scheme 6a).6b,c Ring-opening at other strained rings such as epoxides was also feasible. This was showcased by the reaction of glycidol (rac-20) to dioxolane 5b using 1 mol% of cobalt catalyst 4b (Scheme 5b).21 It should be noted that for this epoxide-opening no signs of kinetic resolution were observed, presumably because the reaction is too fast. Nonetheless, no reaction was observed in the absence of catalyst 4b. Unlike 1,2-diol protecting groups such as acetonide or benzylidene, attempts to cleave the ketal group deriving from trifluoroacetophenone 2 were unsuccessful (see ESI† for a table of all tested conditions). While this currently limits the applicability of the presented method, there were some interesting observations made during the attempted cleavage. The complementary nature of electron deficient ketals allows a chemoselective deprotection indicated by the conversion of dioxolane 5u to ketone 5i upon treatment with hydrochloric acid (Scheme 6c). The unexpected stability towards acids might qualify trifluoromethyl-dioxolanes as a rare acetal motif suitable for (oral) drug discovery, an important property as elucidated by Yu and Meanwell in a recent review.22 Further studies to successfully cleave the trifluoroketal-motif and access the free triol motif are currently underway in our laboratory.
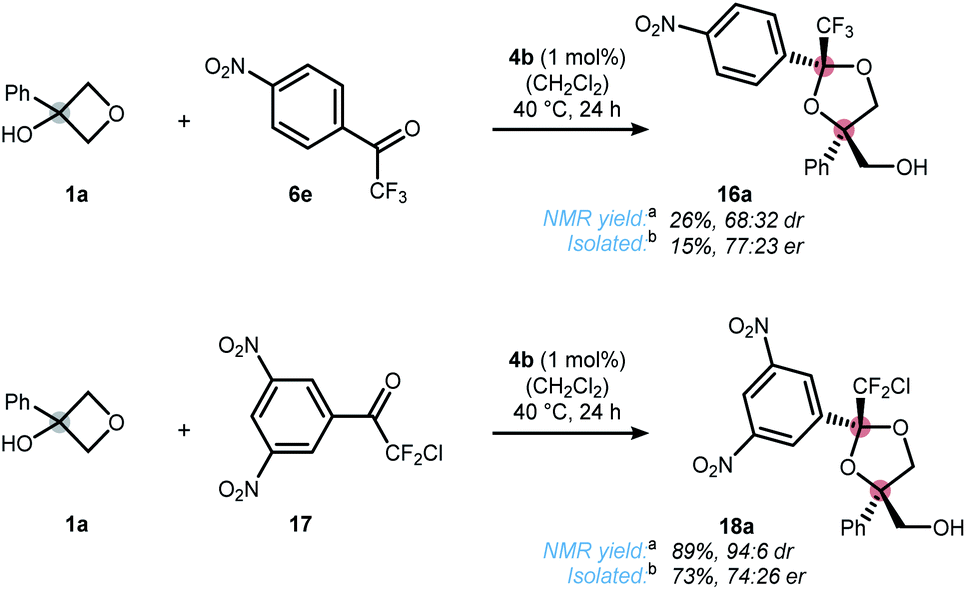 | ||
| Scheme 5 Ketone scope. [a]Determined by 19F NMR using trifluorotoluene as an internal standard. [b]Isolated yield and enantiomeric ratio correspond to the major isomer only. | ||
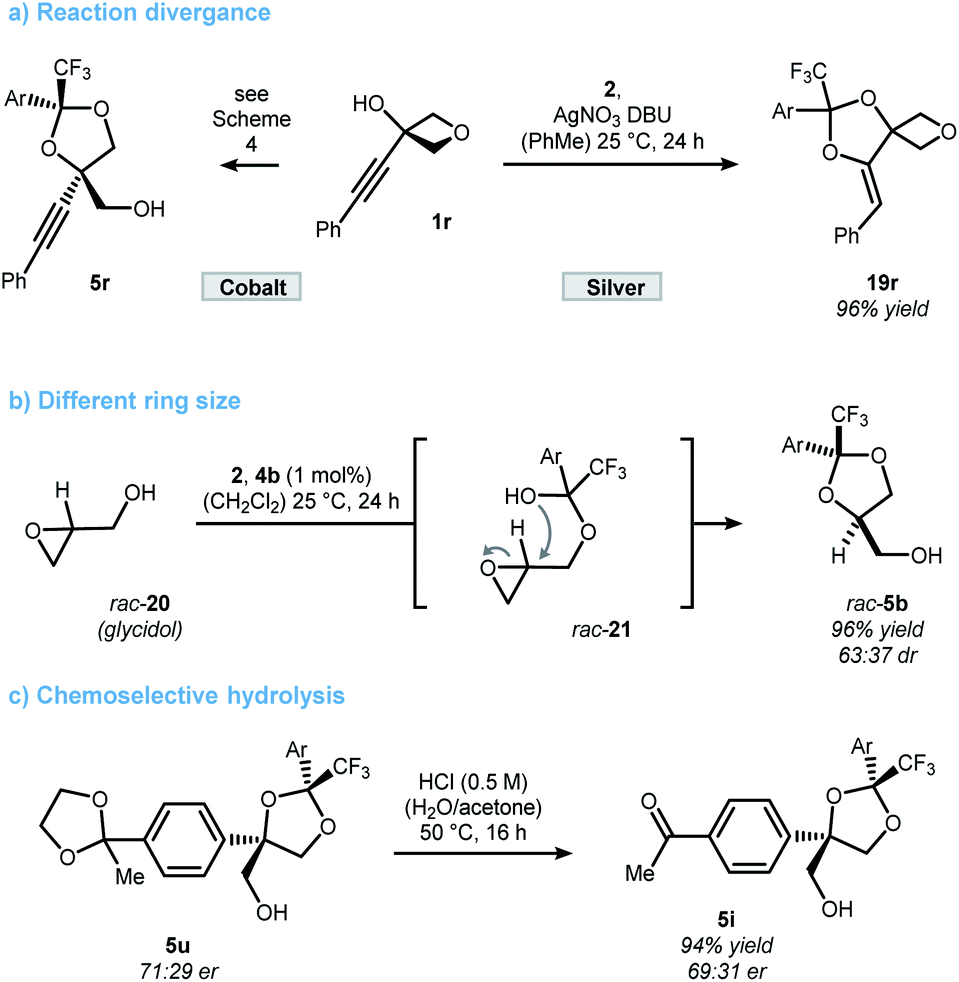 | ||
| Scheme 6 Reactions using hemiketal formation as a platform for reaction development (a and b). Chemical stability of ketals deriving from ketone 2 (c). Ar = 3,5-(NO2)2-C6H3. | ||
Conclusions
In this study, electron deficient trifluoroacetophenone 2 was identified to form remarkably stable hemiketals with unique properties. Based on this observation, a novel ring-opening of prochiral oxetanols was pursued, which relies on a dynamic kinetic resolution of transiently formed hemiketals and an in situ oxidation of the CoII catalyst. By providing access to a range of complex dioxolanes with good control of selectivity, this study not only widens the reach of hemiketals but also serves as a platform for future reaction development.Data availability
Crystallographic data for rac-8e, and 5j has been deposited at the CCDC and is available under 2130260, 2130259, and 2141905 respectively. Experimental data including detailed procedures, characterisation of new compounds as well as NMR and HPLC spectra is accessible in the ESI.†Author contributions
A. S. and H. M. conducted and analysed all experiments. C. G. D. and D. S. performed the X-ray analysis. A. S. and J. M. W. conceptualized the experiments and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous financial support from the Westfälische Wilhelms-Universität Münster, the Johannes Gutenberg-Universität Mainz, the deutsche Studienstiftung (fellowship to A. S.), and the Fonds der Chemischen Industrie (Liebig fellowship to J. M. W.) is acknowledged.References
- D. A. Evans and J. A. Gauchet-Prunet, J. Org. Chem., 1993, 58, 2446 CrossRef CAS.
- (a) P. A. Evans, A. Grisin and M. J. Lawler, J. Am. Chem. Soc., 2012, 134, 2856 CrossRef CAS PubMed; (b) A. Grisin, S. Oliver, M. D. Ganton, J. Bacsa and P. A. Evans, Chem. Commun., 2015, 51, 15681 RSC; (c) K. Murata, K. Sakamoto and H. Fuwa, Org. Lett., 2019, 21, 3730 CrossRef CAS PubMed; (d) H. Watanabe, K. Machida, D. Itoh, H. Nagatsuka and T. Kitahara, Chirality, 2001, 13, 379 CrossRef CAS PubMed.
- (a) D. M. Rubush, M. A. Morges, B. J. Rose, D. H. Thamm and T. Rovis, J. Am. Chem. Soc., 2012, 134, 13554 CrossRef CAS PubMed; (b) A. Matsumoto, K. Asano and S. Matsubara, Chem. Commun., 2015, 51, 11693 RSC; (c) K. Asano and S. Matsubara, Org. Lett., 2012, 14, 1620 CrossRef CAS PubMed.
- (a) A. Khan, R. Zheng, Y. Kan, J. Ye, J. Xing and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 6439 CrossRef CAS PubMed; (b) R. Zhang, W. Guo, M. Duan, K. N. Houk and J. Sun, Angew. Chem., Int. Ed., 2019, 58, 18055 CrossRef CAS PubMed; (c) L. Wang and D. Menche, Angew. Chem., Int. Ed., 2012, 51, 9425 CrossRef CAS PubMed; (d) K. Miura, T. Takahashi, H. Nishikori and A. Hosomi, Chem. Lett., 2001, 30, 958 CrossRef; (e) J. A. Goodwin, C. F. Ballesteros and A. Aponick, Org. Lett., 2015, 17, 5574 CrossRef CAS PubMed; (f) S. D. Dreher, K. R. Hornberger, S. T. Sarraf and J. L. Leighton, Org. Lett., 2000, 2, 3197 CrossRef CAS PubMed; (g) A. T. Herrmann, T. Saito, C. E. Stivala, J. Tom and A. Zakarian, J. Am. Chem. Soc., 2010, 132, 5962 CrossRef CAS PubMed.
- (a) P. A. Evans, M.-H. Huang, M. J. Lawler and S. Maroto, Nat. Chem., 2012, 4, 680 CrossRef CAS PubMed; (b) D. A. Evans, P. J. Coleman and L. C. Dias, Angew. Chem., Int. Ed. Engl., 1997, 36, 2738 CrossRef CAS; (c) N. Yoneda, Y. Fukata, K. Asano and S. Matsubara, Angew. Chem., Int. Ed., 2015, 54, 15497 CrossRef CAS PubMed; (d) A. Matsumoto, K. Asano and S. Matsubara, Asian J. Org. Chem., 2019, 8, 814 CrossRef CAS.
- (a) M. A. Williams, M. J. Miller and N. P. Rath, J. Org. Chem., 1991, 56, 1293 CrossRef CAS; (b) J. Wang, F. Li, H. Xie, M. Xu, X. Zhao, L. Liu and W.-X. Zhao, Appl. Organomet. Chem., 2017, 31, e3545 CrossRef; (c) X.-B. Ding, D. P. Furkert and M. A. Brimble, Angew. Chem., Int. Ed., 2019, 58, 11830 CrossRef CAS PubMed.
- J. Sietmann, M. Ong, C. Mück-Lichtenfeld, C. G. Daniliuc and J. M. Wahl, Angew. Chem., Int. Ed., 2021, 60, 9719 CrossRef CAS PubMed.
- (a) A. Sandvoß and J. M. Wahl, Chem.–Eur. J., 2021, 27, 5871 CrossRef PubMed; (b) Z. Wang, Z. Chen and J. Sun, Org. Biomol. Chem., 2014, 12, 6028 RSC.
- R. N. Loy and E. N. Jacobsen, J. Am. Chem. Soc., 2009, 131, 2786 CrossRef CAS PubMed.
- (a) W. Yang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 1868 CrossRef CAS PubMed; (b) X. Zou, G. Sun, H. Huang, J. Wang, W. Yang and J. Sun, Org. Lett., 2020, 22, 249 CrossRef CAS PubMed; (c) V. A. Bhosale, M. Nigríni, M. Dračínský, I. Císařová and J. Veselý, Org. Lett., 2021, 23, 9376 CrossRef CAS PubMed.
- (a) Z. X. Giustra and K. L. Tan, Chem. Commun., 2013, 49, 4370 RSC; (b) Y. Zhao, A. W. Mitra, A. H. Hoveyda and M. L. Snapper, Angew. Chem., Int. Ed., 2007, 46, 8471 CrossRef CAS PubMed.
- (a) S. Maeda, A. Sudo and T. Endo, Tetrahedron Lett., 2016, 57, 1061 CrossRef CAS; (b) J. F. Miller and A. Spaltenstein, Tetrahedron Lett., 1996, 37, 2521 CrossRef CAS; (c) R. Stewart and J. D. van Dyke, Can. J. Chem., 1970, 48, 3961 CrossRef CAS.
- (a) R. Stewart and D. G. Lee, Can. J. Chem., 1964, 42, 439 CrossRef CAS; (b) A. Ohno, H. Yamamoto and S. Oka, J. Am. Chem. Soc., 1981, 103, 2041 CrossRef CAS.
- R. Irie, T. Uchida and K. Matsumoto, Chem. Lett., 2015, 44, 1268 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- A. Nishinaga, T. Kondo and T. Matsuura, Chem. Lett., 1985, 14, 905 CrossRef.
- (a) R. Blaauw, I. E. Kingma, J. H. Laan, J. L. van der Baan, S. Balt, M. W. G. de Bolster, G. W. Klumpp, W. J. J. Smeets and A. L. Spek, J. Chem. Soc., Perkin Trans. 1, 2000, 1199 RSC; (b) J. M. Ready and E. N. Jacobsen, J. Am. Chem. Soc., 1999, 121, 6086 CrossRef CAS.
- (a) S. Kemper, P. Hrobárik, M. Kaupp and N. E. Schlörer, J. Am. Chem. Soc., 2009, 131, 4172 CrossRef CAS PubMed; (b) A. Kochem, H. Kanso, B. Baptiste, H. Arora, C. Philouze, O. Jarjayes, H. Vezin, D. Luneau, M. Orio and F. Thomas, Inorg. Chem., 2012, 51, 10557 CrossRef CAS PubMed; (c) T. Kurahashi and H. Fujii, Inorg. Chem., 2013, 52, 3908 CrossRef CAS PubMed; (d) M. Hatazawa, K. Nakabayashi, S. Ohkoshi and K. Nozaki, Chem.–Eur. J., 2016, 22, 13677 CrossRef CAS PubMed.
- (a) L. P. C. Nielsen, C. P. Stevenson, D. G. Blackmond and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 1360 CrossRef CAS PubMed; (b) D. D. Ford, L. P. C. Nielsen, S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 15595 CrossRef CAS PubMed.
- (a) B. Ghosh, R. Balhara, G. Jindal and S. Mukherjee, Angew. Chem., Int. Ed., 2021, 60, 9086 CrossRef CAS PubMed; (b) S. Müller, M. J. Webber and B. List, J. Am. Chem. Soc., 2011, 133, 18534 CrossRef PubMed; (c) K. Murata, K. Sakamoto and H. Fuwa, Org. Lett., 2019, 21, 3730 CrossRef CAS PubMed.
- (a) S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307 CrossRef CAS PubMed; (b) M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936 CrossRef CAS PubMed.
- Y.-J. Wu and N. A. Meanwell, J. Med. Chem., 2021, 64, 9786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2130260, 2130259 and 2141905. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01547a |
| This journal is © The Royal Society of Chemistry 2022 |