
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing cell membrane phase separation for inhibiting cancer metastasis with a stimuli-responsive DNA nanodevice†
Yingying
Su
a,
Xiaoqing
Chen
a,
Hui
Wang
a,
Lele
Sun
b,
Ying
Xu
c and
Di
Li
 *a
*a
aSchool of Chemistry and Molecular Engineering, East China Normal University, 200241, Shanghai, China. E-mail: dli@chem.ecnu.edu.cn
bSchool of Life Science, Shanghai University, Shanghai 200444, China
cDepartment of Pathophysiology, Key Laboratory of Cell Differentiation and Apoptosis of Ministry of Education, Shanghai Jiao-Tong University School of Medicine, Shanghai, 200025, China
First published on 2nd May 2022
Abstract
Phase separation in cell membranes promotes the assembly of transmembrane receptors to initiate signal transduction in response to environmental cues. Many cellular behaviors are manipulated by promoting membrane phase separation through binding to multivalent extracellular ligands. However, available extracellular molecule tools that enable manipulating the clustering of transmembrane receptors in a controllable manner are rare. In the present study, we report a DNA nanodevice that enhances membrane phase separation through the clustering of dynamic lipid rafts. This DNA nanodevice is anchored in the lipid raft region of the cell membrane and initiated by ATP. In a tumor microenvironment, this device could be activated to form a long DNA duplex on the cell membrane, which not only enhances membrane phase separation, but also blocks the interaction between the transmembrane surface adhesion receptor and extracellular matrix, leading to reduced migration. We demonstrate that the ATP-activated DNA nanodevice could inhibit cancer cell migration both in vitro and in vivo. The concept of using DNA to regulate membrane phase separation provides new possibilities for manipulating versatile cell functions through rational design of functional DNA structures.
Introduction
The cell membrane contains a tremendous complexity of lipids and proteins in heterogeneous phases.1–3 The phase separation in a cell membrane drives the formation of mesoscale structures, termed biomolecular condensates, for the spatiotemporal organization of several cellular processes.4 Recently, accumulated pieces of evidence have indicated that some families of transmembrane receptors undergo phase separation to open downstream signal pathways following exposure to extracellular stimuli.5,6 Thus, the clustering of transmembrane receptors is a common mechanism in cellular signaling, which functions to stabilize active conformations, amplify signals, or introduce switch like behaviors.2,7 However, in-depth understanding of interactions that regulate the membrane phase separation at the molecular level remains scarce.8,9 Consequently, it is still challenging to regulate signal transduction processes by harnessing spatiotemporal modulation of biomolecular condensates to develop therapeutic treatment options.Recently, some strategies, such as chemically induced dimerization (CID)10 and optogenetics,11 have been developed to artificially bring two transmembrane receptors into proximity to manipulate signal transduction. In addition to these strategies that rely on genetic engineering, nanomaterials are also introduced to direct the clustering of receptors.12 Particularly, DNA has been intensively used to induce the assembly of surface receptors.13 For example, aptamer-induced dimerization provides a spatially controllable redistribution of cell receptors, which has been proved to be an effective means to regulate various cellular functions, including adhesion, proliferation, migration, differentiation, and apoptosis.14–17 However, in addition to limited available aptamers for cell receptors, the affinity between up-to-date aptamers and receptors is relatively low.18,19 Moreover, the stability and robustness of dynamic DNA nanostructures at cell membranes are often affected by the internalization of DNA into the cell.20 As a result, the DNA-induced dimerization of cell receptors often leads to a comparatively unstable and weak regulation of signal transductions, and most of these studies were conducted in vitro instead of in vivo.21–25
In the present study, we developed a DNA nanodevice as an extracellular tool to regulate the cell membrane phase separation. We introduced an ATP-activated hybridization chain reaction (HCR) to gather Ganglioside GM1, a major component of lipid rafts, to enhance the membrane phase separation. In contrast to aptamer-induced dimerization of cell receptors, the HCR resulted in not only an enlargement of lipid rafts, but also a barrier between the cell membrane and extracellular matrix, leading to robust, long-lasting regulations of cellular behaviors. We demonstrated that this operation on the cell membrane could inhibit cancer cell migration in vitro and metastasis in vivo. The concept of using a stimuli-responsive DNA nanodevice to manipulate cell membrane phase separation provides new options for therapeutic treatments.
Results and discussion
The cell membrane contains some transient, phase-separated nanodomains with a high content of sphingolipids, sterols, and specific proteins, termed lipid rafts.26–29 Ganglioside GM1 is a common component of lipid rafts, and the high affinity of the cholera toxin B (CTxB) subunit towards ganglioside GM1 enables CTxB to be a marker of lipid rafts.30 The principle of ATP-initiated gathering of GM1 is outlined in Fig. 1. The lipid raft of living cells (human hepatoma cancer cell line, Hep G2) was first identified by incubating with CTxB. Then a single stranded DNA anchor containing three functional regions, i.e., CT916 (aptamer for CTxB), a spacer and the toehold (T), was designed to anchor to the lipid raft via the specific affinity of CT916 and CTxB (Fig. 1A).31 The HCR system contains three hairpin nucleic acid structures H1 and H2, and an initiator I. Noteworthily, the initiator I contains a sequence of ATP aptamers, and thus could be opened by ATP to initiate the consecutive HCR (Fig. 1B). Both H1 and H2 contain a sequence (T*) that is complementary with the toehold (T) region in the DNA anchor, thereby, H1 and H2 could be immobilized on the Hep G2 membrane through the hybridization between T and T*. In the presence of ATP, the complex of ATP with its aptamer region in I, opens the hairpin structure of I, and activates the consecutive HCR, giving rise to the formation of a long DNA duplex containing repeated anchor-H1 and anchor-H2 units on the cell membrane (Fig. 1C).32,33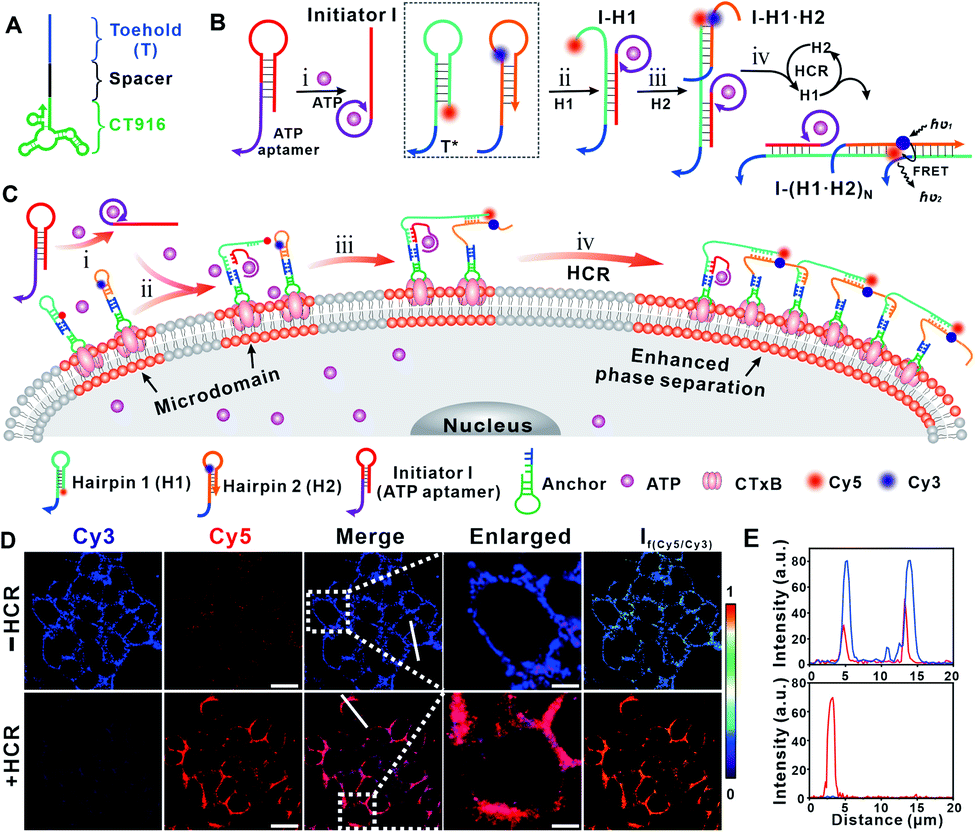 | ||
| Fig. 1 (A) Structure of the DNA anchor. (B) Scheme of the ATP-activated hybridization chain reaction. (C) Schematic illustration of the ATP-activated hybridization chain reaction on the cell membrane that enhances membrane phase separation. (D) Confocal fluorescence microscopy images of the cells before (top) and after (bottom) HCR treatment. “−HCR” indicates the cells after treatments with CTxB and the DNA anchor and “+HCR” indicates the cells after sequential treatments with CTxB, the DNA anchor and the ATP-activated HCR. The evenly distributed Cy3 fluorescence on the cell membrane was clustered into microdomains upon HCR treatment. The FRET ratio image was obtained from the fluorescence intensity collected in the Cy5/Cy3 channel. Scale bars = 10 μm. (E) Colocalization of Cy3 and Cy5 was analyzed by line profiling the fluorescence intensity along the line selected in confocal images in (D). | ||
We first applied Förster resonance energy transfer (FRET) to interrogate the ATP-activated HCR. H1 and H2 were functionalized with the fluorophores Cy5 and Cy3, respectively. The ATP-activated HCR was first tested in solution (Fig. S1†). In the presence of ATP, the fluorescence intensity of the donor Cy3 decreased while that of the acceptor Cy5 increased (Fig. S1A†), indicating the success of the HCR. The formation of the DNA duplex was confirmed by agarose gel electrophoresis (Fig. S1B†). The assembly of the DNA duplex at the cell membrane was interrogated by confocal microscopy. After incubating with Alexa-647 labeled CTxB and followed by the Alexa-488 labeled DNA anchor, the even distribution and colocalization of Alexa-647 and Alexa-488 fluorescence on the cell membrane verified the specificity of the CTxB aptamer and the efficient immobilization of the DNA anchor (Fig. S2†). In the absence of ATP (i.e., the −HCR condition), upon illuminating with a 561 nm laser (the excitation wavelength for Cy3), only the Cy3 fluorescence signal was observed. While in the presence of ATP (i.e., the +HCR condition), FRET-induced attenuated Cy3 fluorescence and concomitantly enhanced Cy5 fluorescence were observed (Fig. 1D). The enhanced FRET ratio indicated that H1 and H2 were brought into proximity, confirming the success of the ATP-activated HCR. In addition, we found that the Cy5 fluorescence could be maintained for at least 24 h on the cell membrane, suggesting the stability of the HCR product on the membrane without endocytosis (Fig. S3†).
Interestingly, we found that the ATP-activated HCR resulted in a redistribution of the Cy5 fluorescence on the cell membrane. After the HCR, the homogeneously distributed Cy5 fluorescence (upon illuminating at 647 nm, Fig. S4†) was gradually transformed into clusters with aggregates ranging from several hundred nanometers to micrometers (Fig. 1D, E and S18†). Since the binding between GM1 and CTxB is one of the strongest protein–carbohydrate interactions,30 we thus speculated that the ATP-activated HCR initiates the gathering of GM1 that stabilizes and enlarges the dynamic nanoscale lipid raft. To consolidate that the Cy5 lightened aggregates are associated with the lipid raft region, we performed control experiments by staining the HCR-treated cells with FITC-labeled CD59 and transferrin receptor (TfR) antibodies, respectively. CD59 is a glycosylphosphatidylinositol (GPI)-anchored membrane protein,26 while the transferrin receptor (TfR) is an extensively studied non-lipid component.28 A strong overlapping of CD59 with the Cy5 lightened aggregates was observed (Fig. S5†), while the overlapping of TfR and the Cy5 lightened aggregates was weak (Fig. S6†). Different distribution profiles of CD59 and TfR on the HCR-treated cell membrane verified that the Cy5 lightened microdomain was associated with an enlarged lipid raft region.
The enlarged raft domain may undergo enhanced phase separation, leading to fluidity and permeability changes in the cell membrane. We performed fluorescence recovery after the photobleaching (FRAP) experiment to assess the fluidity change (Fig. 2A).34 As shown in Fig. 2B, the recovery kinetics of Cy5 fluorescence were slowed down after HCR treatment, and the calculated FRAP ratio was 39%, much lower than that under the −HCR condition (62%). The calculated diffusion coefficient dropped from 0.11 μm2 s−1 (−HCR) to 0.09 μm2 s−1 (+HCR) (Fig. 2B, inset). The decreased diffusion coefficient of Cy5 indicated a weakened mobility of clustered GM1. The permeability change of the cell membrane was investigated by measuring the leakage of the cytoplasmic enzyme lactate dehydrogenase (LDH) into the culture medium, an indicative assay of cell membrane permeability.35 After treatment with CTxB, the percentage of released LDH increased from 11.5% to 15%. Upon activation by the HCR, the percentage of released LDH further increased to 32% (Fig. 2C). The increased permeability of the cell membrane confirmed that the HCR-enhanced membrane phase separation results in a disruption of the cell membrane. As a result, the viability of Hep G2 cells dropped to 47% after HCR treatment (Fig. 2D).
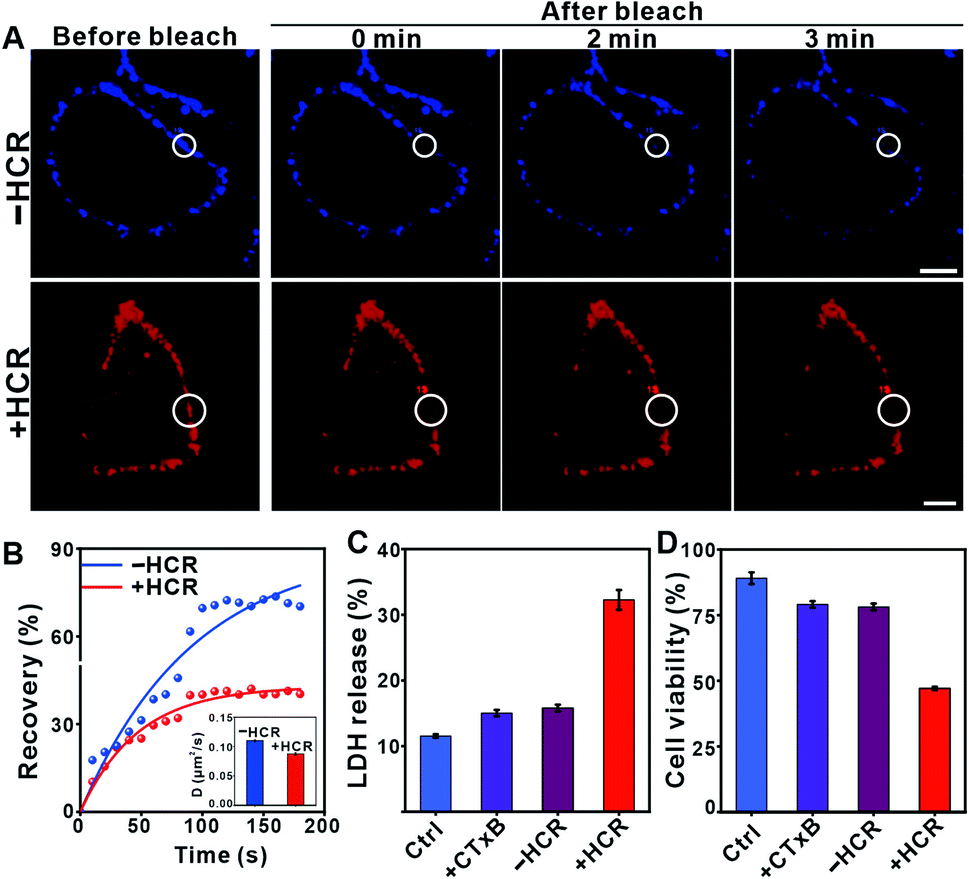 | ||
| Fig. 2 (A) Fluorescence recovery images of the cell membrane after photobleaching upon various treatments. “−HCR” indicates the cells after treatments with CTxB and the DNA anchor and “+HCR” indicates the cells after sequential treatments with CTxB, the DNA anchor and the ATP-activated HCR. The radius of the selected region is 4 μm for the top and 5 μm for the bottom image. Scale bar = 10 μm. (B) Plots of fluorescence intensity in the region of interest in (A) versus time after photobleaching. Inset: diffusion coefficients of Cy5 obtained from plots of normalized FRAP data. The mean and standard deviations were from at least three different samples with analysis of 20 bleach spots for each experiment. (C) LDH released from the Hep G2 cells collected after 1 h following different treatments. (D) The viability of Hep G2 cells upon different treatments. The data shown in (C) and (D) are presented as the mean ± SD of 5 parallel experiments. | ||
Interestingly, we found that the HCR-treated cells exhibited attenuated cell mobility. The attenuated cell mobility was examined by both the wound healing test and transwell chamber migration assay. In the presence of ATP, the cell population exhibited a slowed movement with a migration rate that decreased from ca. 56% to ca. 7.8% in 24 h (Fig. 3A). Moreover, we did not observe significant morphology changes in the HCR-treated cells (Fig. S7†), suggesting that the lowered cell viability did not lead to obvious cell death. The reduced cell motility and chemotaxis were also confirmed by transwell migration assay (Fig. S8†). As shown in Fig. 3B, 24 h later after HCR treatment, the migration rate of Hep G2 cells decreased from ca. 82% to ca. 16%. The comparison of cell mobility after different treatments is shown in Fig. 3C. The lowered migration of Hep G2 cells implies an inhibited epithelial–mesenchymal transition (EMT).36,37 We next performed western blotting analysis to study the expression of EMT markers, E-cadherin, N-cadherin, and vimentin (Fig. 3D). The HCR-treated Hep G2 cells exhibited increased expression of E-cadherin, while the expression of N-cadherin and vimentin decreased, confirming the inhibited EMT.
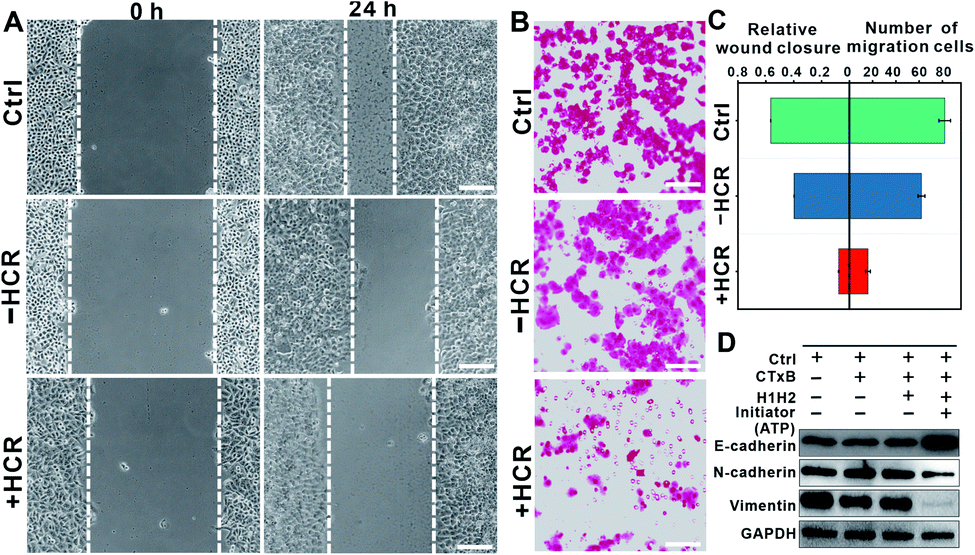 | ||
| Fig. 3 (A) In vitro scratch assay of Hep G2 cells after various treatments. “Control” indicates the cells without any treatment, “−HCR” indicates the cells after treatments with CTxB and the DNA anchor, and “+HCR” indicates the cells after sequential treatments with CTxB, and the DNA anchor and the ATP-activated HCR. The images were taken at 0 and 24 h after various treatments using an optical microscope. Scale bar = 100 μm. (B) Crystal-violet-stained images of transwell migration assay. Representative fluorescence images of crystal violet stained Hep G2 cells after various treatments as indicated. Scale bars = 100 μm. Characteristic pictures of migrated cells at the bottom of the filter after incubation for 24 h. Scale bar = 50 μm. (C) Quantitate analysis of the HCR-induced inhibition of cell mobility after various treatments. (left) The percentage of wound closure depicted as means ± SD, n = 6; (right) the number of migrated cells obtained from the inserted filter in the transwell. (D) Western blotting analysis of the expression of EMT representative proteins (E-cadherin, N-cadherin, and vimentin) in Hep G2 cells after various treatments. | ||
Since the lipid raft involves many membrane proteins that correlate with various cellular functions including cell adhesion, we thereby speculated that the HCR-enhanced membrane phase separation gives rise to a weakened formation of new focal adhesion that results in inhibited cell migration.2 CD44, a cell surface adhesion receptor that is rich in lipid rafts, is highly expressed in many tumor cells that regulates metastasis.38,39 Its interaction with appropriate extracellular matrix ligands promotes the migration and invasion processes involved in metastases.40,41 We hypothesized that the weakened cell adhesion is attributed to the HCR-enhanced phase separation that redistributes CD44 and blocks the interaction between CD44 and the extracellular matrix.
To confirm this hypothesis, we stained the HCR-treated cells with FITC-labeled CD44 antibody and studied the spatial distribution of CD44 and GM1. As shown in Fig. 4A, the colocalization of FITC and Cy5 indicated that the DNA-gathered GM1 region coincided with CD44, confirming the clustering of CD44 into the HCR-enlarged raft region. To further solidify the colocalization of CD44 and the enlarged lipid raft, we performed another control experiment to extract endogenous cholesterol from the cell membrane with MβCD to disrupt the lipid raft. Many studies have suggested that MβCD could remove cholesterol from rafts to induce significant changes in the physical properties of the plasma membrane by changing the distribution of raft associated proteins.26 As shown in Fig. 4B, the clustering of Cy5 suggested that the HCR product, a DNA duplex, was not influenced by the MβCD treatment. However, the FITC fluorescent aggregates were transferred back to an even distribution, further confirming the strong association of CD44 with the lipid raft region.
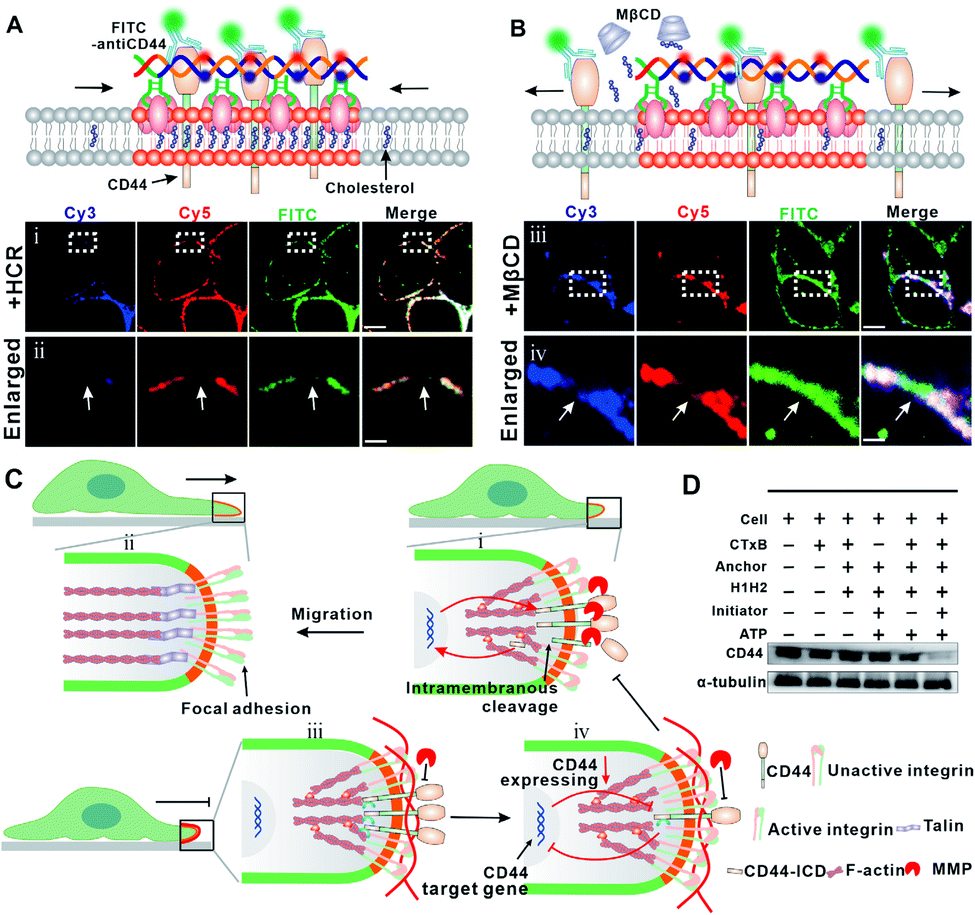 | ||
| Fig. 4 (A) Upper: scheme of the colocalization of CD44 and HCR products. Lower: confocal fluorescence microscopy images show the distribution of Cy5/Cy3 and FITC on the cell membrane after HCR treatment (i), scale bar = 5 μm and enlarged images of the selected regions (ii), scale bar = 0.5 μm. (B) Upper: scheme of the distribution of CD44 and HCR products on the cell membrane after the cells were treated by the HCR and then with MβCD. Lower: confocal fluorescence microscopy images show the distribution of Cy5, Cy3 and FITC on the cell membrane after HCR treatment (iii), scale bar = 5 μm and the enlarged images of the selected regions (iv), scale bar = 0.5 μm. (C) Schematic illustration of DNA nanodevice-enhanced cell membrane phase separation that clusters CD44 and blocks the interaction of MMPs with CD44, leading to an inhibition of the activation of F-actin and integrin. (D) Western blotting analysis of CD44 expression levels after different treatments as indicated. | ||
Previous studies suggested that CD44 undergoes sequential proteolytic cleavages by membrane-associated metalloproteases (MMPs) under physiological conditions (Fig. S9†), and this cleavage is responsible for dynamic regulation of the interaction between CD44 and the extracellular matrix during cell migration.39,40 Briefly, the intramembranous cleavage of CD44 induces a reorganization of F-actin and actin associated integrin to form focal adhesion, which plays a critical role in cell migration. Upon the intramembranous cleavage of CD44 to CD44-ICD, CD44-ICD translocates to the nucleus and promotes gene expression of CD44 (Fig. 4C, top). We assumed that the HCR product, a long DNA duplex, functions also as a barrier to block the cleavage of CD44 by MMPs (Fig. 4C, bottom). To confirm this, we performed another control experiment using a trigger DNA strand to initiate a dimerization of the DNA anchor (Fig. S10–11†). However, this DNA dimer only leads to a weak inhibition of cell migration with the migration rate dropping from ca. 67% to ca. 55% in the wound healing test (Fig. S12†). The different inhibition efficiencies of the HCR and dimer, indicated that the clustering of the lipid raft and formation of the HCR product contributed synergically to the inhibited cell migration. As a result of blocked cleavage, less CD44-ICD could be translocated to the nucleus, and gene expression of new CD44 was inhibited. To verify this assumption, we investigated the expression of CD44 through western blotting analysis. As shown in Fig. 4D, the expression of CD44 was downregulated after HCR treatments, further confirming the blocking of CD44 and MMPs by the HCR product.
In a tumor microenvironment (TME), extracellular ATP is highly elevated,23,24,42 and we thus expected that this ATP-activated DNA nanodevice could also function in TME to inhibit tumor metastasis. We chose melanoma, an aggressive and highly metastatic cancer,40,43,44 as a model to evaluate the in vivo therapeutic potential of the proposed DNA nanodevice. We first verified the feasibility of our DNA nanodevice on B16 cells in vitro and found a similar migration inhibition tendency (Fig. S13–17†). We then examined the in vivo therapeutic function of this DNA nanodevice on B16 cell-carrying immune-deficient mice. A melanoma xenograft model was created by injecting B16 cells into the flanks of immune-deficient mice. Mice displaying tumor volumes of around 100 mm3 were intratumorally injected with the DNA nanodevice (CTxB (5 μg mL−1), + DNA anchor (5 μM), initiator (1 μM), and H1 and H2, (both 5 μM)), while PBS, CTxB, and CTxB + DNA anchor + H1 and H2, were also intratumorally injected as controls (Fig. 5A). After 20 days, the tumor size increased to 2726.8 ± 110 mm3 for the PBS group, 2574.2 ± 141 mm3 for the −HCR group, and 1211.3 ± 381 mm3 for the +HCR group (Fig. 5B). According to photographs and tumor growth curves, the group with the injection of the DNA nanodevice could effectively inhibit the development of tumor growth (Fig. 5C and D). The body weights of the mice were monitored over the course of treatments every two days (Fig. 5E), and none of the mice showed notable changes, indicating that all the treatments had negligible toxic effects.
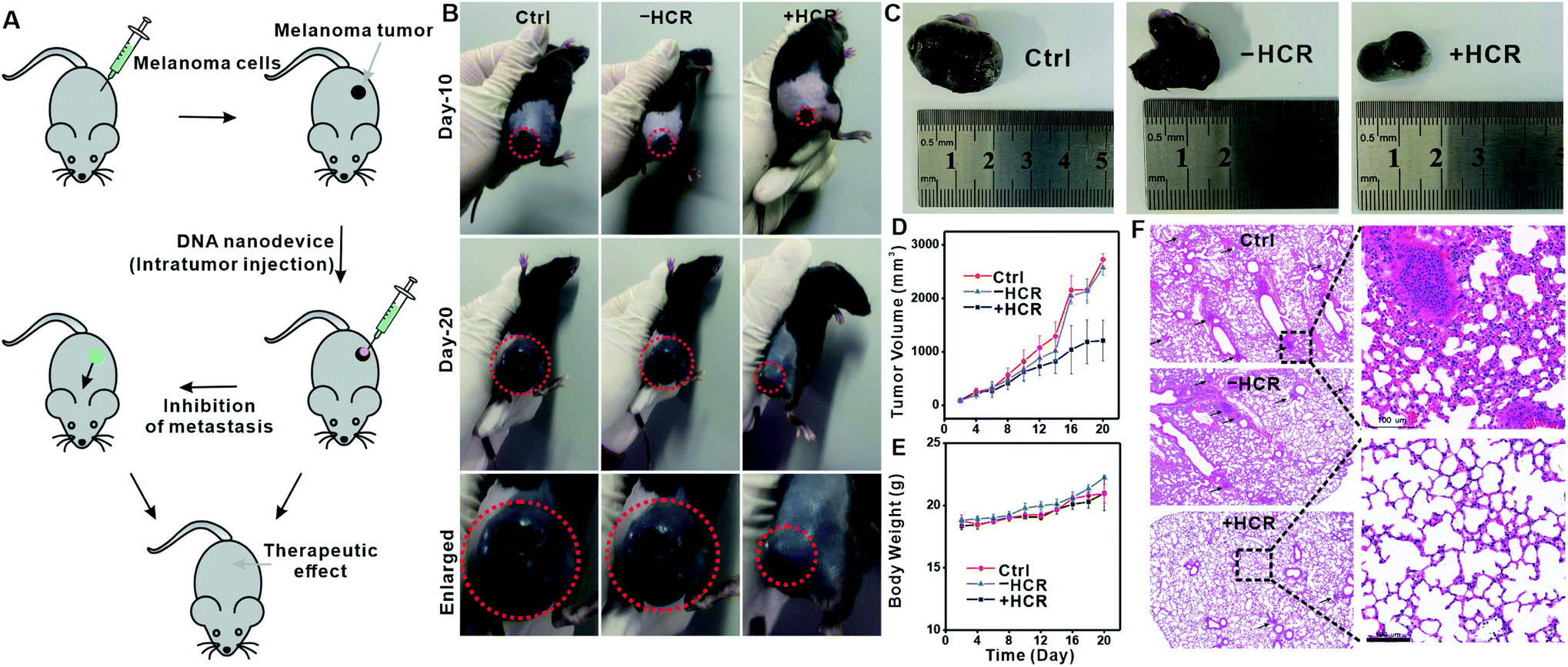 | ||
| Fig. 5 In vivo tumor therapy using a DNA nanodevice in a melanoma tumor model. (A) Schematic illustration of tumor formation and injection of the DNA nanodevice into the tumor. Mice were subcutaneously injected with 2 × 106 B16 cells. The anti-tumor effects and inhibition of metastasis were evaluated. PBS and “−HCR” were used as controls. (B) Representative image of mice after 10 days and 20 days of treatment. Enlarged photographic images of mice bearing B16 cells after treatments with PBS, “−HCR”, and “+HCR”. (C) Photographic images of postmortem tumors from B16-tumor-bearing mice after different treatments. (D) Tumor volume growth curves monitored at different time-points and the calculated tumor volume. The variation is represented by the standard deviation of three independent replicates in all graphs. (E) Body-weight measurements of B16-tumor-bearing mice per experimental group at different times after treatments. The variation is represented by the standard deviation of three independent replicates in all graphs. (F) Representative histopathology of the lungs from mice after different treatments. The lung slices were stained with hematoxylin and eosin for histological analysis. Scale bar = 100 μm. | ||
We next examined the metastasis of B16 cells to other organs upon DNA nanodevice injection. Fig. 5F shows the histological results of lung samples collected from the therapy and control groups. Melanin grains could be seen as small black spots that largely existed in lung samples from control groups, while in the DNA nanodevice treated group, melanin grains were barely seen. Collectively, these results were consistent with the in vitro results, which showed that the phase separation-regulated DNA nanodevice could effectively inhibit the migration of tumor cells.
Conclusions
In summary, we developed a DNA nanodevice to inhibit cell migration through enhancing phase separation in cell membranes. This DNA nanodevice is anchored at the raft domain of the cell membrane and triggered in the tumor microenvironment. The product of this nanodevice functions as a barrier for interactions between transmembrane receptors and the extracellular matrix, thus leading to the regulation of cellular functions. Our work provides a new possibility of controlling cell motility in vitro and in vivo through regulating membrane phase separation with extracellular DNA molecule tools. Importantly, the flexibility of DNA nanodevice and DNA computing provides versatile possibilities for therapeutic treatment options by regulating membrane phase separation, for example, logic-gate controllable regulation or light-induced regulation with photo-responsive nucleotides, thus shedding new light on therapy applications of lipid–lipid phase separation.Data availability
Most of the experimental data have been shown in the manuscript. Original data that support the findings of this study are available upon request from the corresponding author with an appropriate reason.Author contributions
D. Li conceived the idea and supervised, wrote and reviewed the manuscript. Y. Su carried out the experiments and drafted the manuscript. X. Chen, H. Wang, L. Sun and Y. Xu assisted in experiments and discussion.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21874046), Shanghai Municipal Commission for Science and Technology (19JC1411800), the Fundamental Research Funds for the Central Universities, and the original innovation project from 0 to 1 of the Basic Frontier Scientific Research Program of Chinese Academy of Sciences (ZDBS-LY-JSC010). We thank the East China Normal University Multifunctional Platform for Innovation (011) for supporting the mouse experiments.Notes and references
- P. L. Li, S. Banjade, H. C. Cheng, S. Kim, B. Chen, L. Guo, M. Llaguno, J. V. Hollingsworth, D. S. King, S. F. Banani, P. S. Russo, Q. X. Jiang, B. T. Nixon and M. K. Rosen, Nature, 2012, 483, 336–340 CrossRef CAS PubMed.
- I. de Curtis, Trends Cell Biol., 2021, 31, 145–148 CrossRef CAS PubMed.
- A. A. Hyman, C. A. Weber and F. Juelicher, Annu. Rev. Cell Dev. Biol., 2014, 30, 39–58 CrossRef CAS PubMed.
- Y. G. Zhao and H. Zhang, Dev. Cell, 2020, 55, 30–44 CrossRef CAS PubMed.
- Y. Wang, C. L. Zhang, W. Z. Yang, S. P. Shao, X. M. Xu, Y. J. Sun, P. L. Li, L. Liang and C. Y. Wu, Dev. Cell, 2021, 56, 1313–1325 CrossRef CAS PubMed.
- G. Y. Li, T. Sasaki, S. Asahina, M. C. Roy, T. Mochizuki, K. Koizumi and Y. Zhang, Chem, 2017, 2, 283–298 CAS.
- K. Simons and J. L. Sampaio, Cold Spring Harbor Perspect. Biol., 2011, 3, a004697 Search PubMed.
- D. Soloviov, Y. Q. Cai, D. Bolmatov, A. Suvorov, K. Zhernenkov, D. Zav'yalov, A. Bosak, H. Uchiyama and M. Zhernenkov, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4749–4757 CrossRef CAS PubMed.
- F. Yuan, H. Alimohamadi, B. Bakka, A. N. Trementozzi, K. J. Day, N. L. Fawzi, P. Rangamani and J. C. Stachowiak, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2017435118 CrossRef CAS PubMed.
- A. Fegan, B. White, J. C. T. Carlson and C. R. Wagner, Chem. Rev., 2010, 110, 3315–3336 CrossRef CAS PubMed.
- R. A. Hallett, S. P. Zimmerman, H. Yumerefendi, J. E. Bear and B. Kuhlman, ACS Synth. Biol., 2016, 5, 53–64 CrossRef CAS PubMed.
- K. X. Zhang, H. Gao, R. J. Deng and J. H. Li, Angew. Chem., Int. Ed., 2019, 58, 4790–4799 CrossRef CAS PubMed.
- J. Y. Li, L. P. Wang, J. M. Tian, Z. L. Zhou, J. Li and H. H. Yang, Chem. Soc. Rev., 2020, 49, 1545–1568 RSC.
- S. Chen, Z. F. Xu, W. Yang, X. H. Lin, J. Y. Li, J. Li and H. H. Yang, Angew. Chem., Int. Ed., 2019, 58, 18186–18190 CrossRef CAS PubMed.
- H. Li, J. Gao, L. Cao, X. Xie, J. H. Fan, H. D. Wang, H. H. Wang and Z. Nie, Angew. Chem., Int. Ed., 2021, 60, 26087–26095 CrossRef CAS PubMed.
- H. Li, M. Wang, T. H. Shi, S. H. Yang, J. H. Zhang, H. H. Wang and Z. Nie, Angew. Chem., Int. Ed., 2018, 57, 10226–10230 CrossRef CAS PubMed.
- J. H. Zhang, Z. Y. Qiu, J. H. Fan, F. He, W. Y. Kang, S. H. Yang, H. H. Wang, J. Huang and Z. Nie, Angew. Chem., Int. Ed., 2021, 60, 6733–6743 CrossRef CAS PubMed.
- X. Chang, C. Zhang, C. Lv, Y. Sun, M. Zhang, Y. Zhao, L. Yang, D. Han and W. Tan, J. Am. Chem. Soc., 2019, 141, 12738–12743 CrossRef CAS PubMed.
- L. P. Wang, H. Liang, J. Sun, Y. C. Liu, J. Y. Li, J. Y. Li, J. Li and H. H. Yang, J. Am. Chem. Soc., 2019, 141, 12673–12681 CrossRef CAS PubMed.
- S. P. Li, Q. Jiang, S. L. Liu, Y. L. Zhang, Y. H. Tian, C. Song, J. Wang, Y. G. Zou, G. J. Anderson, J. Y. Han, Y. Chang, Y. Liu, C. Zhang, L. Chen, G. B. Zhou, G. J. Nie, H. Yan, B. Q. Ding and Y. L. Zhao, Nat. Biotechnol., 2018, 36, 258–264 CrossRef CAS PubMed.
- T. Gao, T. S. Chen, C. Feng, X. He, C. L. Mu, J. Anzai and G. X. Li, Nat. Commun., 2019, 10, 2946 CrossRef PubMed.
- F. Li, W. T. Yu, J. J. Zhang, Y. H. Dong, X. H. Ding, X. H. Ruan, Z. Gu and D. Y. Yang, Nat. Commun., 2021, 12, 1138 CrossRef CAS PubMed.
- B. Liu, R. Ma, J. Zhao, Y. L. Zhao and L. L. Li, Sci. China: Chem., 2020, 63, 1490–1497 CrossRef CAS.
- J. Zhao, J. H. Gao, W. T. Xue, Z. H. Di, H. Xing, Y. Lu and L. L. Li, J. Am. Chem. Soc., 2018, 140, 578–581 CrossRef CAS PubMed.
- S. X. Jiang, Z. L. Ge, S. Mou, H. Yan and C. H. Fan, Chem, 2021, 7, 1156–1179 CAS.
- L. L. Sun, Y. Y. Su, J. G. Wang, F. Xia, Y. Xu and D. Li, Chem. Sci., 2020, 11, 1581–1586 RSC.
- Y. Bagheri, A. A. Ali, P. Keshri, J. Chambers, A. Gershenson and M. You, Angew. Chem., Int. Ed., 2022, 61, e202112033 CrossRef CAS PubMed.
- T. Harder, P. Scheiffele, P. Verkade and K. Simons, J. Cell Biol., 1998, 141, 929–942 CrossRef CAS PubMed.
- R. Ohtani, K. Kawano, M. Kinoshita, S. Yanaka, H. Watanabe, K. Hirai, S. Futaki, N. Matsumori, H. Uji-i, M. Ohba, K. Kato and S. Hayami, Angew. Chem., Int. Ed., 2020, 59, 17931–17937 CrossRef CAS PubMed.
- W. B. Turnbull, B. L. Precious and S. W. Homans, J. Am. Chem. Soc., 2004, 126, 1047–1054 CrossRef CAS PubMed.
- E. Frohnmeyer, N. Tuschel, T. Sitz, C. Hermann, G. T. Dahl, F. Schulz, A. J. Baeumner and M. Fischer, Analyst, 2019, 144, 1840–1849 RSC.
- J. Wei, X. Gong, Q. Wang, M. Pan, X. Q. Liu, J. Liu, F. Xia and F. Wang, Chem. Sci., 2018, 9, 52–61 RSC.
- F. Wang, B. Willner and I. Willner, Curr. Opin. Biotechnol., 2013, 24, 562–574 CrossRef CAS PubMed.
- N. O. Taylor, M. T. Wei, H. A. Stone and C. P. Brangwynne, Biophys. J., 2019, 117, 1285–1300 CrossRef CAS PubMed.
- R. C. Guo, X. H. Zhang, P. S. Fan, B. L. Song, Z. X. Li, Z. Y. Duan, Z. Y. Qiao and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 25128–25134 CrossRef CAS PubMed.
- S. A. Mani, W. Guo, M. J. Liao, E. N. Eaton, A. Ayyanan, A. Y. Zhou, M. Brooks, F. Reinhard, C. C. Zhang, M. Shipitsin, L. L. Campbell, K. Polyak, C. Brisken, J. Yang and R. A. Weinberg, Cell, 2008, 133, 704–715 CrossRef CAS PubMed.
- J. M. Lee, S. Dedhar, R. Kalluri and E. W. Thompson, J. Cell Biol., 2006, 172, 973–981 CrossRef CAS PubMed.
- J. Lesley, R. Hyman and P. W. Kincade, Adv. Immunol., 1993, 54, 271–335 CAS.
- R. Marhaba and M. Zoller, J. Mol. Histol., 2004, 35, 211–231 CrossRef CAS PubMed.
- Y. Guo, J. Ma, J. Wang, X. Che, J. Narula, M. Bigby, M. Wu and M.-S. Sy, Cancer Res., 1994, 54, 1561–1565 CAS.
- A. E. Faassen, J. A. Schrager, D. J. Klein, T. R. Oegema, J. R. Couchman and J. B. McCarthy, J. Cell Biol., 1992, 116, 521–531 CrossRef CAS PubMed.
- P. Pellegatti, L. Raffaghello, G. Bianchi, F. Piccardi, V. Pistoia and F. Di Virgilio, PLoS One, 2008, 3, e2599 CrossRef PubMed.
- L. Finn, S. N. Markovic and R. W. Joseph, BMC Med., 2012, 10, 23 CrossRef CAS PubMed.
- B. D. Curti and M. B. Faries, N. Engl. J. Med., 2021, 384, 2229–2240 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc00371f |
| This journal is © The Royal Society of Chemistry 2022 |