
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed arylative substitution of homoallylic alcohols†
Hai N.
Tran
 ,
Chau M.
Nguyen
,
Mason T.
Koeritz
,
Dustin D.
Youmans
and
Levi M.
Stanley
*
,
Chau M.
Nguyen
,
Mason T.
Koeritz
,
Dustin D.
Youmans
and
Levi M.
Stanley
*
Department of Chemistry, Iowa State University, Ames, IA 50011, USA. E-mail: lstanley@iastate.edu
First published on 12th August 2022
Abstract
Direct coupling of unactivated alcohols remains a challenge in synthetic chemistry. Current approaches to cross-coupling of alcohol-derived electrophiles often involve activated alcohols such as tosylates or carbonates. We report the direct arylative substitution of homoallylic alcohols catalyzed by a nickel-bisphosphine complex as a facile method to generate allylic arenes. These reactions proceed via formation of an allylic alcohol intermediate. Subsequent allylic substitution with arylboroxine nucleophiles enables the formation of a variety of allylic arenes. The presence of p-methoxyphenylboronic acid is crucial to activate the allylic alcohol to achieve high product yields.
Introduction
Alkanols are among the most common and accessible functional groups in organic synthesis and are attractive as potential C(sp3) coupling partners to form C(sp3)–C bonds. However, due to the relatively strong bond dissociation energy of the C–O bond and poor leaving group ability of the OH group,1 alcohols are rarely used directly as alkylating agents in cross-coupling chemistry.2 Although numerous alcohol derivatives (i.e. tosylates, carbonates, etc.) have been extensively explored in redox-neutral cross-coupling reactions,3,4 these derivatives typically require one or more preparation steps from the corresponding alcohol precursors. Therefore, processes that form C(sp3)–C bonds directly from unactivated alcohols have the potential to streamline synthetic routes.Transition metal-catalyzed hydroarylations of 1,3-dienes are privileged synthetic methods to generate allylic arenes and allylic heteroarenes.5–8 Traditionally, hydro(hetero)arylation of 1,3-dienes involves activation of an aryl C–H bond (Scheme 1a, top).5,6 Alternatively, hydro(hetero)arylations of 1,3-dienes have been developed by employing alcohols or silanes as external hydride sources (Scheme 1a, bottom).7,8 In the past decade, approaches to tandem transition metal-catalyzed hydro(hetero)arylation of 1,3-dienes generated in situ from homoallylic electrophiles have been reported.9–11 Sigman and co-workers developed Pd-catalyzed arylative substitutions of primary and secondary homoallylic tosylates with arylboronic acids (Scheme 1b).9 In 2020, Kawatsura and co-workers developed the arylative substitution of secondary homoallylic carbonates with arylboronic acids catalyzed by a nickel complex of a monophosphine ligand (Scheme 1c).10 These reactions proceed via in situ formation of a diene by a sequence of oxidative addition of the C–O bond to generate a metal-alkyl intermediate and subsequent β-hydride elimination. Alkene insertion to the metal-hydride bond, transmetallation with an arylboronic acid, and reductive elimination generate the allylic arene product. These formal hydroarylations of 1,3-dienes are attractive examples of coupling reactions involving secondary sp3 electrophiles. However, the opportunity exists to improve these reactions by eliminating the need for an activated homoallylic electrophile. To this end, we now report the development of nickel-catalyzed arylative substitution of unactivated secondary homoallylic alcohols with arylboroxine nucleophiles (Scheme 1d). These arylative substitution reactions occur via in situ formation of allylic alcohols through alkene isomerization, followed by allylic arylation12–14 to generate a variety of allylic arenes in short reaction times.
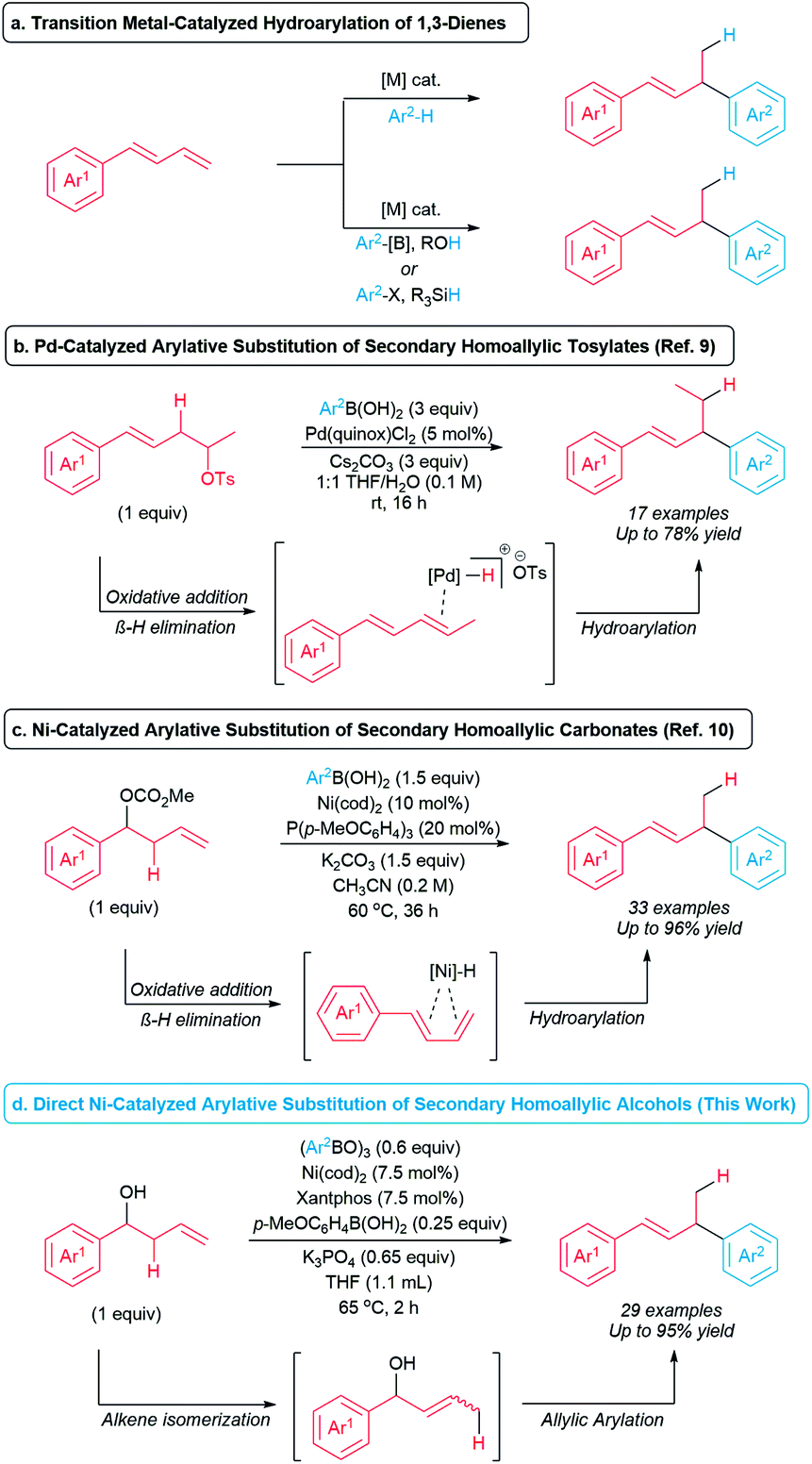 | ||
| Scheme 1 Transition metal-catalyzed formation of allylic arenes via hydroarylation and arylative substitutions. | ||
Results and discussion
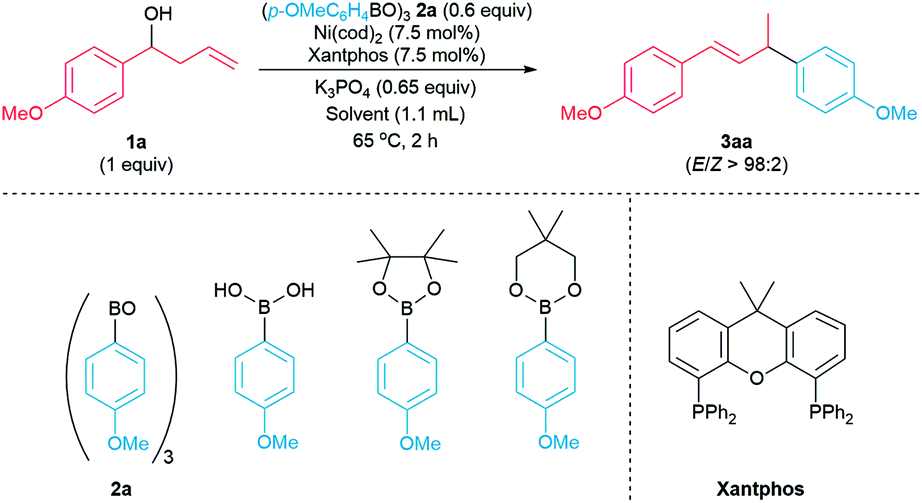
Initially, reactions of 1-(p-methoxyphenyl)but-3-en-1-ol 1a with common arylboron nucleophiles were evaluated to identify a suitable aryl coupling partner. Reactions did not occur when p-MeOC6H4B(OH)2, p-MeOC6H4Bpin, and p-MeOC6H4Bneop were used as the coupling partners (Table 1, entries 1–3). However, the reaction of 1 equiv. of p-methoxyphenylboroxine 2a (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of arylboroxine:arylboronic acid) with 1a formed the product 3aa in 95% yield (entry 4). The yield of the reaction remained high upon lowering the loading of 2a to 0.6 equiv. (entry 5). Interestingly, the reaction of 1a with arylboroxine 2a containing a higher ratio of boroxine to its corresponding boronic acid (arylboroxine:arylboronic acid = 16:1, entry 6) led to a drop in the yield of 3aa to 71%. We hypothesized that the catalytic amount of p-methoxyphenylboronic acid could play a role in activating the homoallylic alcohol 1a.15 Indeed, adding 0.25 equiv. of p-MeOC6H4B(OH)2 to the reaction of 0.6 equiv. of boroxine 2a with 1a led to the formation of the desired product 3aa in 98% yield (entry 7). The addition of p-methoxyphenylboronic acid allowed the amount of arylboroxine 2a to be lowered to 0.5, 0.4 or 0.33 equiv. without significant impact on the yield of 3aa (entries 8–10). Since electron-deficient arylboronic acids are expected to activate the alcohol more effectively,15a we attempted to use p-trifluoromethylphenylboronic acid and p-methoxycarbonyphenylboronic acid as additives in the model reaction to activate the homoallylic alcohol. However, due to high content of corresponding arylboroxines in these commercially available, electron-deficient arylboronic acids, we observed the formation of the allylic arene byproducts derived from these electron-deficient arylboroxines. In contrast, using p-methoxyphenylboronic acid as an additive, we did not observe the allylic arene byproduct derived from the p-methoxyphenylboroxine when evaluating the arylboroxine scope (see Scheme 4, below). Therefore, we chose p-methoxyphenylboronic acid as an additive in our arylative coupling reactions. In addition we re-evaluated the arylboronic ester nucleophiles with p-methoxyphenylboronic acid as an additive. Reactions did not occur when the arylboronic acid pinacol ester and arylboronic acid neopentylglycol ester were used as the coupling partner in the presence of 0.25 equiv. of p-MeOC6H4B(OH)2 (entries 11–12).16 We chose reaction conditions using 0.6 equiv. of 2a with the addition of 0.25 equiv. of p-MeOC6H4B(OH)2 to evaluate the scope of the arylative coupling reaction.
:1 mixture of arylboroxine:arylboronic acid) with 1a formed the product 3aa in 95% yield (entry 4). The yield of the reaction remained high upon lowering the loading of 2a to 0.6 equiv. (entry 5). Interestingly, the reaction of 1a with arylboroxine 2a containing a higher ratio of boroxine to its corresponding boronic acid (arylboroxine:arylboronic acid = 16:1, entry 6) led to a drop in the yield of 3aa to 71%. We hypothesized that the catalytic amount of p-methoxyphenylboronic acid could play a role in activating the homoallylic alcohol 1a.15 Indeed, adding 0.25 equiv. of p-MeOC6H4B(OH)2 to the reaction of 0.6 equiv. of boroxine 2a with 1a led to the formation of the desired product 3aa in 98% yield (entry 7). The addition of p-methoxyphenylboronic acid allowed the amount of arylboroxine 2a to be lowered to 0.5, 0.4 or 0.33 equiv. without significant impact on the yield of 3aa (entries 8–10). Since electron-deficient arylboronic acids are expected to activate the alcohol more effectively,15a we attempted to use p-trifluoromethylphenylboronic acid and p-methoxycarbonyphenylboronic acid as additives in the model reaction to activate the homoallylic alcohol. However, due to high content of corresponding arylboroxines in these commercially available, electron-deficient arylboronic acids, we observed the formation of the allylic arene byproducts derived from these electron-deficient arylboroxines. In contrast, using p-methoxyphenylboronic acid as an additive, we did not observe the allylic arene byproduct derived from the p-methoxyphenylboroxine when evaluating the arylboroxine scope (see Scheme 4, below). Therefore, we chose p-methoxyphenylboronic acid as an additive in our arylative coupling reactions. In addition we re-evaluated the arylboronic ester nucleophiles with p-methoxyphenylboronic acid as an additive. Reactions did not occur when the arylboronic acid pinacol ester and arylboronic acid neopentylglycol ester were used as the coupling partner in the presence of 0.25 equiv. of p-MeOC6H4B(OH)2 (entries 11–12).16 We chose reaction conditions using 0.6 equiv. of 2a with the addition of 0.25 equiv. of p-MeOC6H4B(OH)2 to evaluate the scope of the arylative coupling reaction.
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield 3aab (%) |
|
a Reaction conditions: 1a (0.25 mmol), 2a (0.15 mmol), Ni(cod)2 (0.0188 mmol), Xantphos (0.0188 mmol), K3PO4 (0.162 mmol), THF (1.1 mL) at 65 °C for 2 h under a N2 atmosphere. E/Z ratios were determined by 1H NMR spectroscopy of crude mixtures.
b Yields were determined by 1H NMR spectroscopy of the crude reaction mixture using CH2Br2 as an internal standard.
c
2a with arylboroxine:arylboronic acid ratio = 16:1.
d Isolated yield.
e 0.25 equiv. p-MeOC6H4B(OH)2 added.
|
||
| 1 | 2 equiv. p-MeOC6H4B(OH)2 instead of 2a | 0 |
| 2 | 2 equiv. p-MeOC6H4Bpin instead of 2a | 0 |
| 3 | 2 equiv. p-MeOC6H4Bneop instead of 2a | 0 |
| 4 | 1 equiv. 2a (arylboroxine:arylboronic acid = 12:1) |
95 |
| 5 | None (arylboroxine:arylboronic acid = 12:1) |
93 |
| 6 | None (arylboroxine:arylboronic acid = 16:1) |
71 |
| 7 | 0.25 equiv. p-MeOC 6 H 4 B(OH) 2 added | 98 (93) |
| 8c | 0.5 equiv. 2a, 0.25 equiv. p-MeOC6H4B(OH)2 added | 90 |
| 9c | 0.4 equiv. 2a, 0.25 equiv. p-MeOC6H4B(OH)2 added | 88 |
| 10c | 0.33 equiv. 2a, 0.25 equiv. p-MeOC6H4B(OH)2 added | 84 |
| 11e | 2 equiv. p-MeOC6H4Bneop instead of 2a | 0 |
| 12e | 2 equiv. p-OMeC6H4Bpin instead of 2a | 0 |
With reaction conditions identified that lead to high yields in our model reaction, we sought to establish the scope of the reaction by evaluating arylative substitutions of a variety of homoallylic alcohols (Scheme 2). An array of α-aryl-substituted homoallylic alcohols 1 react with p-methoxyphenylboroxine 2a to generate allylic arene products 3 in modest-to-high yields (33–95%). The reaction of chlorinated homoallylic alcohol (1d) furnished the product 3da in 33% yield with isomerization of 1d to a mixture of allylic alcohols accounting for the balance of the mass. It is likely that oxidative addition of the Ni catalyst to the Caryl–Cl bond is competitive with the desired arylate substitution. However, Suzuki–Miyaura coupling products are not observed. Notably, a homoallylic alcohol bearing an ester substituent at the aryl moiety (1f) was tolerated under the standard reaction conditions, furnishing the allyic arene 3fa in 82% yield. Homoallylic alcohols with polysubstituted α-aryl groups 1k–m and α-naphthyl homoallylic alcohol 1n react to form allylic arenes 3ka–3na in good-to-high yields (72–95%). Moreover, the reaction of α-thienyl homoallylic alcohol 1o generated the corresponding heteroarene-containing product 3oa in 48%. Additionally, the reaction of homoallylic alcohol 1p bearing a β-substituent furnished the corresponding allylic arene 3pa in 39% yield with 3:1 E/Z ratio. The reaction of α-alkyl-substituted homoallylic alcohol 1q with boroxine 2a formed the corresponding product 3qa in 63% yield with 2:1 rr. However, reactions of tertiary homoallylic alcohol 1r, homoallylic alcohols containing 1,2- and 1,1-disubstituted alkenes (1s and 1t), and bishomoallylic alcohol 1u did not occur under our reaction conditions.
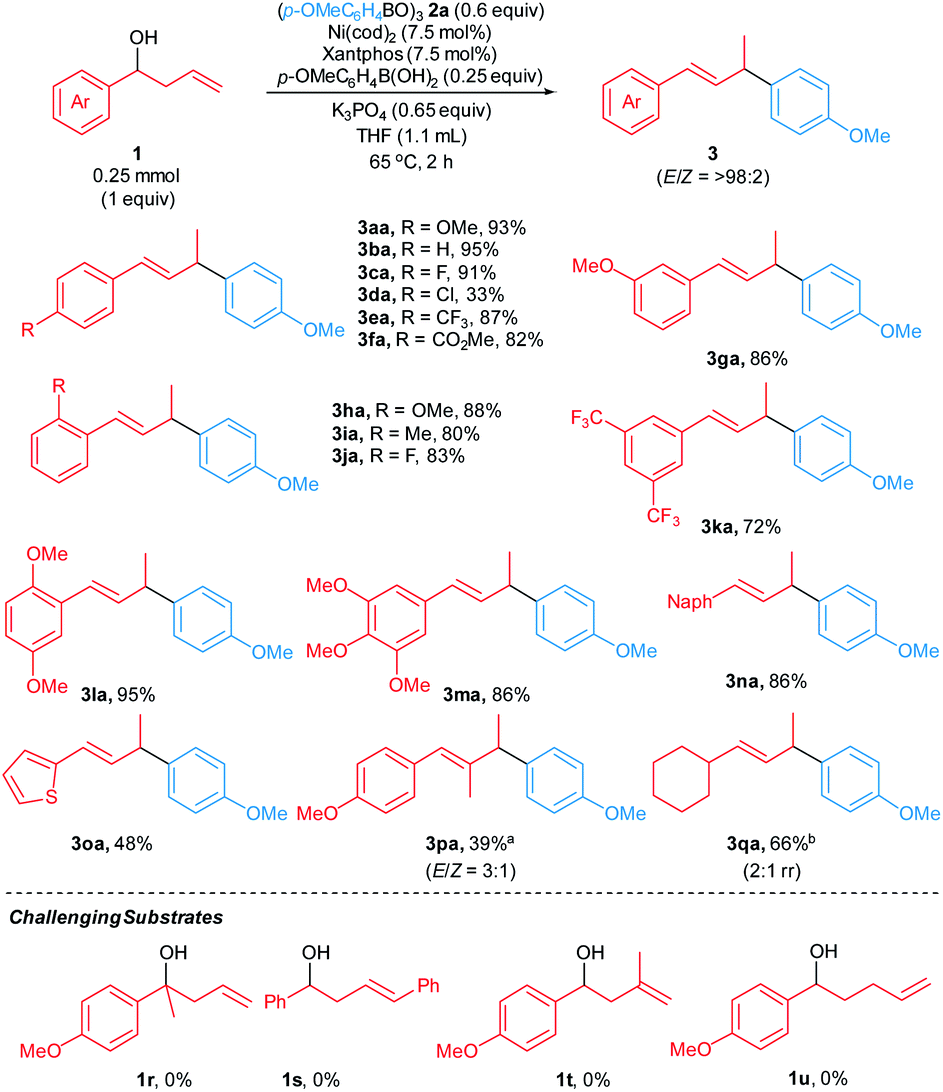 | ||
| Scheme 2 Homoallylic alcohol substrate scope. Reactions were carried out on a 0.25 mmol scale. 2a with arylboroxine:arylboronic acid = 16:1. E/Z ratios were determined by 1H NMR spectroscopy of crude mixtures. Yields of isolated products are shown. a Homoallylic alcohol 1p (syn:anti = 1:1). b Isolated yield of a mixture of 3,4- and 1,4-isomers. | ||
We attempted to extend the arylative substitution reaction to encompass α-styrenyl homoallylic alcohols (Scheme 3). Interestingly, the reaction of α-styrenyl homoallylic alcohol 1v with boroxine 2a did not generate the desired allylic arene 3va. Instead, product 4va was isolated in 53% yield. The observation of the aryl nucleophile replacing the OH at the α-position in product 4vain lieu of the γ-position like products 3aa–3qa suggests our arylative substitution reactions might proceed via a different mechanism compared to the previously reported formal hydroarylations of homoallylic alcohol derivatives (Scheme 1b and c).
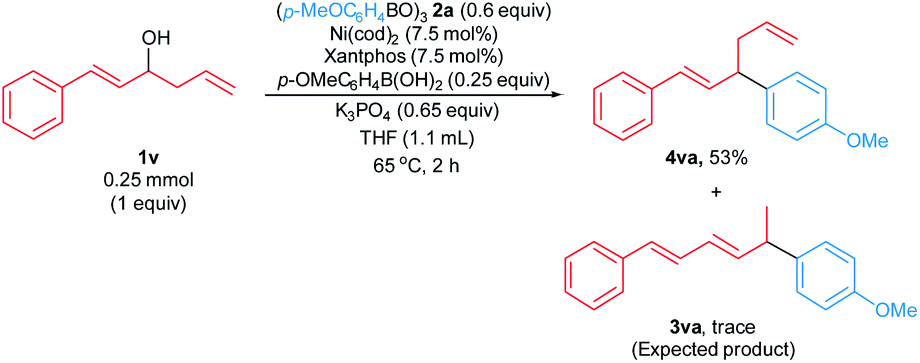 | ||
| Scheme 3 Arylative substitution of α-styrenyl homoallylic alcohol. Reaction was carried out on a 0.25 mmol scale. Yield of isolated product 4va is shown. | ||
 | ||
| Scheme 4 Arylboroxine substrate scope. Reactions were carried out on a 0.25 mmol scale. 2 with arylboroxine:arylboronic acid > 10:1. E/Z ratios were determined by 1H NMR spectroscopy of crude mixtures. Yields of isolated products are shown. | ||
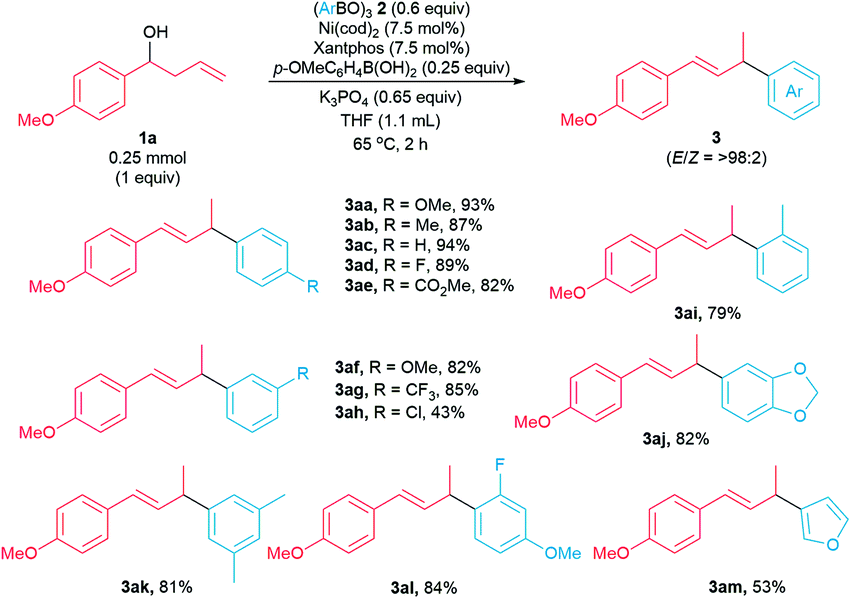
Next, we examined the scope of the arylative substitution reactions with respect to arylboroxine coupling partners (Scheme 4). Reactions of 1-(p-methoxyphenyl)but-3-en-1-ol 1a with a variety of substituted boroxines 2 generated allylic arenes 3 in modest-to-high yields (43–94%). The scope of these arylative substitution reactions encompasses a variety of para-, meta-, ortho-, and disubstituted arylboroxines. Notably, arylboroxines containing fluoride (2d), ester (2e), chloride (2h), and acetal (2j) substituents were tolerated under our reaction conditions. Moreover, furanyl-3-boroxine 2m reacts with homoallylic alcohol 1a to form the corresponding allylic heteroarene in 53% yield.
 | ||
| Scheme 5 Large-scale reaction and synthetic transformations. Yields of isolated products are shown. Reaction conditions for (a): 3aa (0.3 mmol), mCPBA (0.45 mmol), DCM (3 mL), rt, 24 h then sat. NaHCO3 (2 mL), rt, 5 min. Reaction conditions for (b): 3aa (0.3 mmol), K2S (0.9 mmol), DMSO (2 mL), 140 °C, 24 h under air. | ||
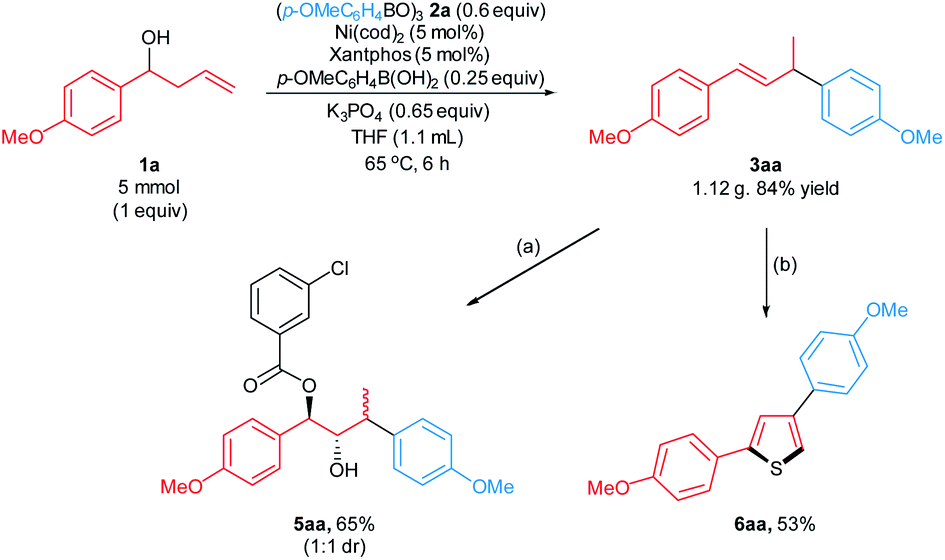
We examined the scalability of the arylative substitution in a 5 mmol scale reaction of homoallylic alcohol 1a (Scheme 5). This reaction formed the desired product 3aa in 84% yield when conducted in the presence of 5 mol% nickel precatalyst and 5 mol% Xantphos ligand. In addition, the synthetic utility was demonstrated by subjecting the allylic arene 3aa to alkene functionalization transformations. The reaction of 3aa with mCPBA, followed by workup with saturated aq. NaHCO3 furnished the hydroxyester 5aa in 65% yield as a 1:1 ratio of separable diastereomers. Transition metal-free sulfuration/annulation17 of 3aa formed thiophene 6aa in 53% yield.
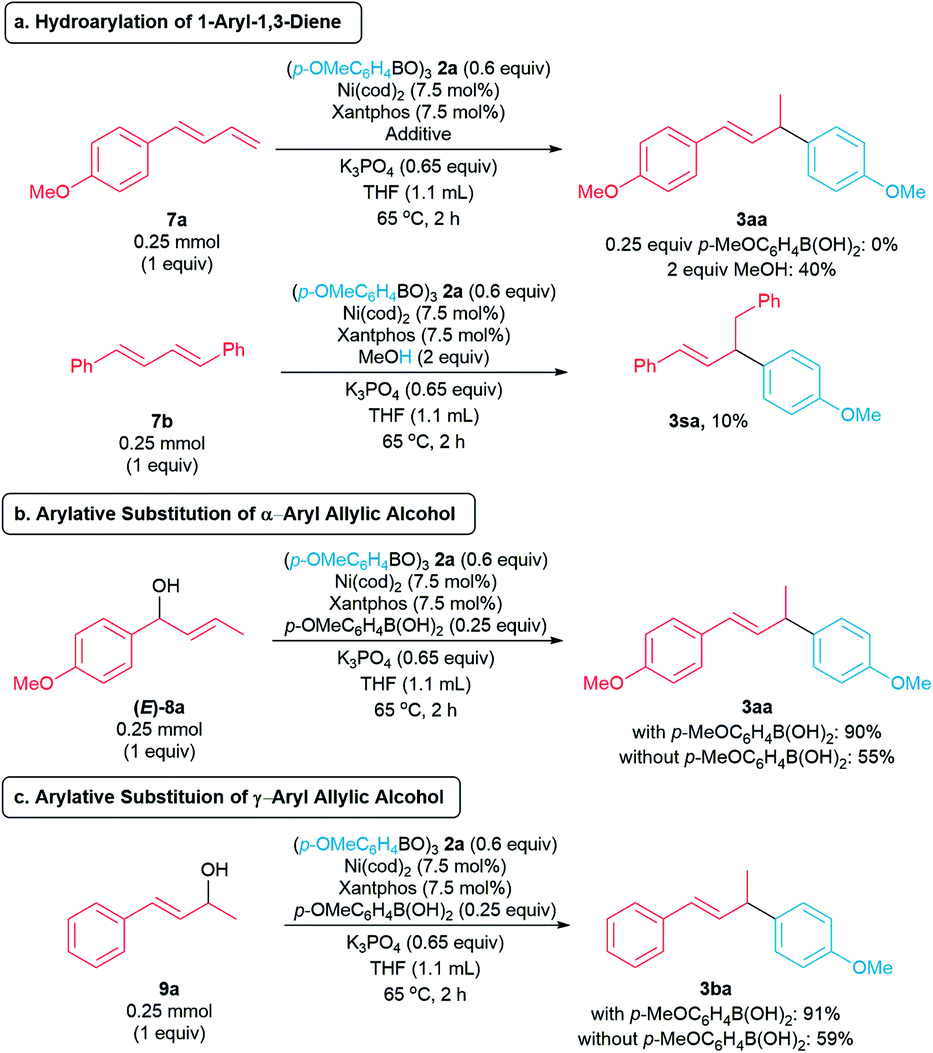
To gain insights into the reaction mechanism, we conducted a series of control experiments. In previous studies reported by the Sigman (Scheme 1b)9 and Kawatsura groups (Scheme 1c),10 a 1,3-diene is proposed to be generated in situ. To probe whether our arylative substitution proceeds via a similar mechanism, we subjected the diene 7a to the standard reaction conditions. Compound 7a was recovered in 97% yield, and the allylic arene 3aa was not detected (Scheme 6a, top). In the presence of MeOH as a hydride source,7b,8c,18 the hydroarylation of diene 7a formed the allylic arene 3aa in 40% yield. We also conducted the hydroarylation of 1,4-diphenyl-1,3-diene 7b, furnishing the allylic arene 3sa in 10% (Scheme 6a, bottom).7b In contrast, the reaction of homoallylic alcohol 1s with boroxine 2a did not generate 3sa (Scheme 2). These observations suggest the mechanism involving in situ generation of a 1,3-diene is not likely in our arylative substitution reactions.
 | ||
| Scheme 6 Control experiments. Yields were determined by 1H NMR spectroscopy of the crude reaction mixture using CH2Br2 as an internal standard. | ||
We then turned our focus to the potential for in situ formation of an allylic alcohol via isomerization of the homoallylic alcohol, followed by allylic arylation to form the allylic arene products 3. The reaction of allylic alcohol (E)-8a and boroxine 2a under the standard conditions produced allylic arene 3aa in 90% (with p-MeOC6H4B(OH)2) and 55% (without p-MeOC6H4B(OH)2) yields, suggesting the arylative substitution may occur via an allylic alcohol intermediate (Scheme 6b). In addition, allylic alcohol 9a reacts with boroxine 2a under standard reaction conditions to form allylic arene 3ba in 90% yield (Scheme 6c). These observations suggest the arylative substitutions of allylic alcohols (E)-8a and 9a proceed via π-allylnickel(II) intermediates.13,19 Furthermore, the reactions of these allylic alcohols produce the corresponding allylic arenes in higher yields in the presence of p-methoxyphenylboronic acid, suggesting p-methoxyphenylboronic acid is involved in activation of the allylic alcohols.15
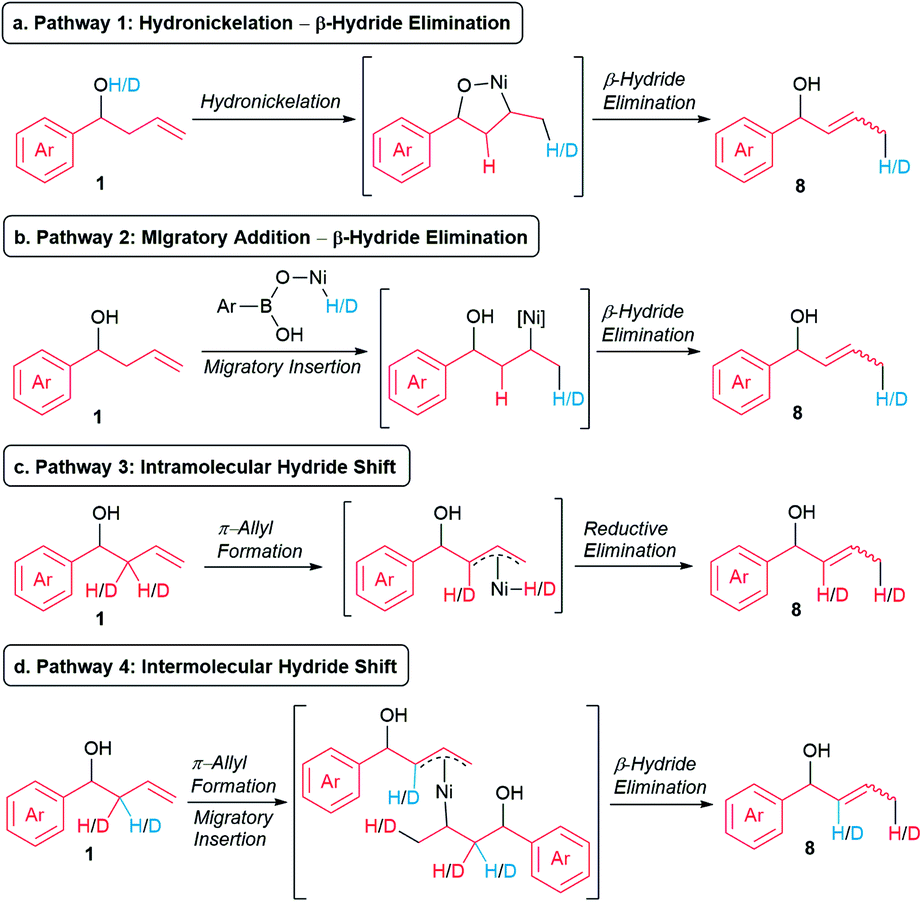
We continued our mechanistic studies by probing the in situ formation of the allylic alcohol. We envisioned four potential pathways to form the allylic alcohol. Pathway 1 involves the intramolecular hydronickelation of homoallylic alcohol 1 to form the oxanickelacycle, followed by β-hydride elimination (Scheme 7a).20 Pathway 2 consists of the oxidative addition of nickel catalyst to the O–H bond of p-MeOC6H4B(OH)2, subsequent migratory insertion of the alkene present in 1, and β-hydride elimination (Scheme 7b). Pathway 3 comprises an intramolecular hydride shift involving the formation of a π-allyl-Ni(II) to enable the migration of a hydride from the β position to the terminal position (Scheme 7c).21,22 Pathway 4 comprises an intermolecular hydride shift involving a sequential formation of a π-allyl-Ni(II), migratory insertion, and β-hydride elimination (Scheme 7d).22
 | ||
| Scheme 7 Possible pathways for the in situ formation of allylic alcohol. | ||
We conducted experiments with deuterium-labelled substrates to evaluate these pathways. The reaction of deuterated homoallylic alcohol 1a-OD with boroxine 2a produced allylic arene 3aa in 90% yield without any deuterium incorporation (Scheme 8a). Similarly, homoallylic alcohol 1a reacted with boroxine 2a in the presence of deuterated p-methoxyphenylboronic acid to form 3aa in 91% yield without any deuterium incorporation (Scheme 8b). These observations rule out the in situ formation of allylic alcohol via pathways 1 and 2. Notably, in the reaction of β-deuterated homoallylic alcohol 1b-D2, we observed deuterium incorporation (88% D) to the terminal position of product 3ba-D2 (Scheme 8c), suggesting that the pathways 3 and 4 are feasible in our arylative substitution reaction. To further distinguish these pathways, we performed the cross-over experiment using β-deuterated homoallylic alcohol 1b-D2 and nondeuterated homoallylic alcohol 1m (Scheme 8d). We observed deuterium incorporation into products arising from both homoallylic alcohol substrates. Higher amounts of deuterium were incorporated into the terminal position (31% D) of product 3ma-D2 derived from the nondeuterated homoallylic alcohol 1m compared to the product 3ba-D2 derived from the deuterated homoallylic alcohol 1b-D2 (12% D). This result indicates that the in situ formation of allylic alcohol can proceed through the intermolecular hydride shift mechanism (pathway 4, Scheme 7d), but we cannot rule out formation of the allylic alcohol via the intramolecular hydride shift mechanism (pathway 3, Scheme 7c) as a competing reaction pathway.
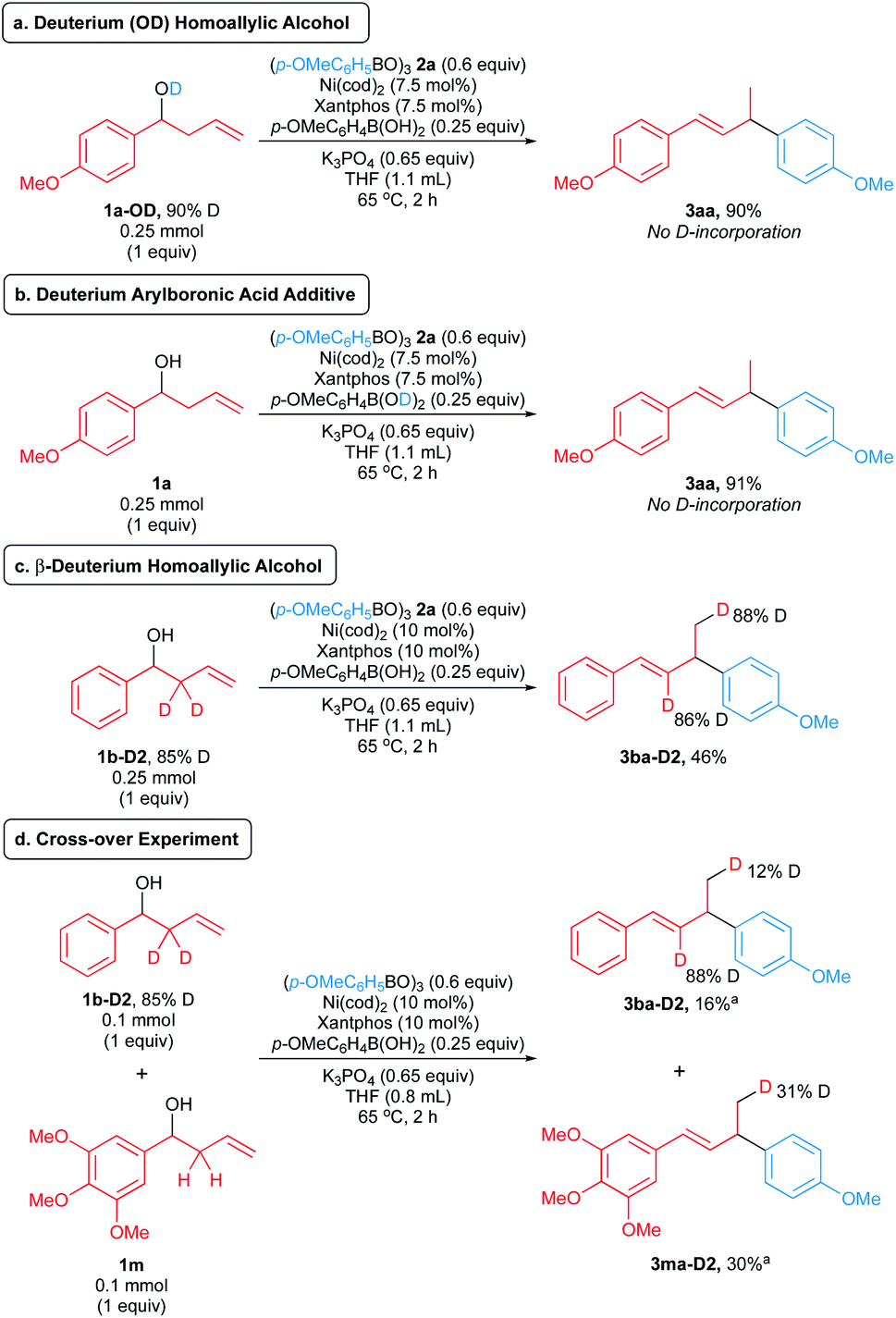 | ||
| Scheme 8 Deuterium-labelling experiments. Yields and deuterium ratios were determined by 1H NMR spectroscopy of the crude reaction mixture. a Isolated yields. | ||
We also subjected homoallylic alcohol 1a to the standard reaction conditions in the absence of arylboroxine nucleophile, p-methoxyphenylboronic acid additive, and K3PO4 (Scheme 9). The reaction formed the allylic alcohol 8a in 35% yield with 1.7:1 dr. This experiment, in combination with the allylic arylation of 8a (Scheme 6b) and the reactions of deuterium-labelled homoallylic alcohol 1b-D2 (Schemes 8c and d), is consistent with in situ formation of the allylic alcohol intermediate by pathways 3 and/or 4 (Schemes 7c and d). In addition, the 1,3-diene was not observed in the isomerization reaction of homoallylic alcohol (Scheme 9), which further suggests the mechanism involving the in situ generation of 1,3-diene is not active in our arylative substitutions of homoallylic alcohols.
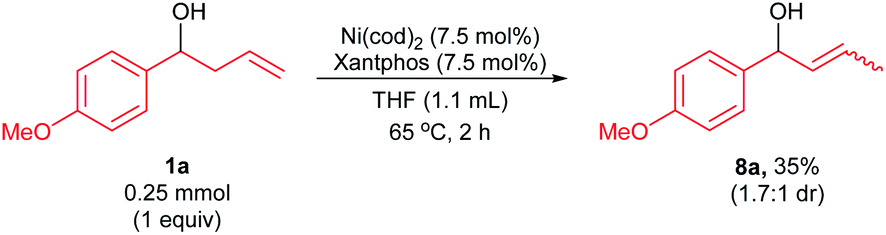 | ||
| Scheme 9 Isomerization of homoallylic alcohol 1a. Yield was determined by 1H NMR spectroscopy of the crude reaction mixture using CH2Br2 as an internal standard. | ||
Based on these control experiments, we propose the tandem catalytic cycles in Scheme 10. In cycle I, the isomerization of the homoallylic alcohol is proposed to occur via formation of a π-allyl-Ni(II) B from LNi(0) catalyst and homoallylic alcohol 1.21,22 Intermediate B undergoes migratory insertion to another homoallylic alcohol 1 to form intermediate C, followed by β-hydride elimination to generate allylic alcohol 8. Intermediate B can also undergo reductive elimination to form allylic alcohol 8. Next, the allylic alcohol 8 generated in cycle I is activated in situ (intermediate D) by coordination of the oxygen atom to the boron atom of p-methoxyphenylboronic acid.15 Intermediate D undergoes oxidative addition with Ni(0) catalyst in cycle II to produce π-allylnickel(II) intermediate F. Subsequent transmetallation with arylboroxine 2 facilitated by K3PO423 and reductive elimination forms the desired product 3 and regenerates the Ni(0) catalyst.12a In a recent study, Fang and coworkers proposed a related isomerization/substitution mechanism for nickel-catalyzed hydrocyanation of alkenyl alcohols.24
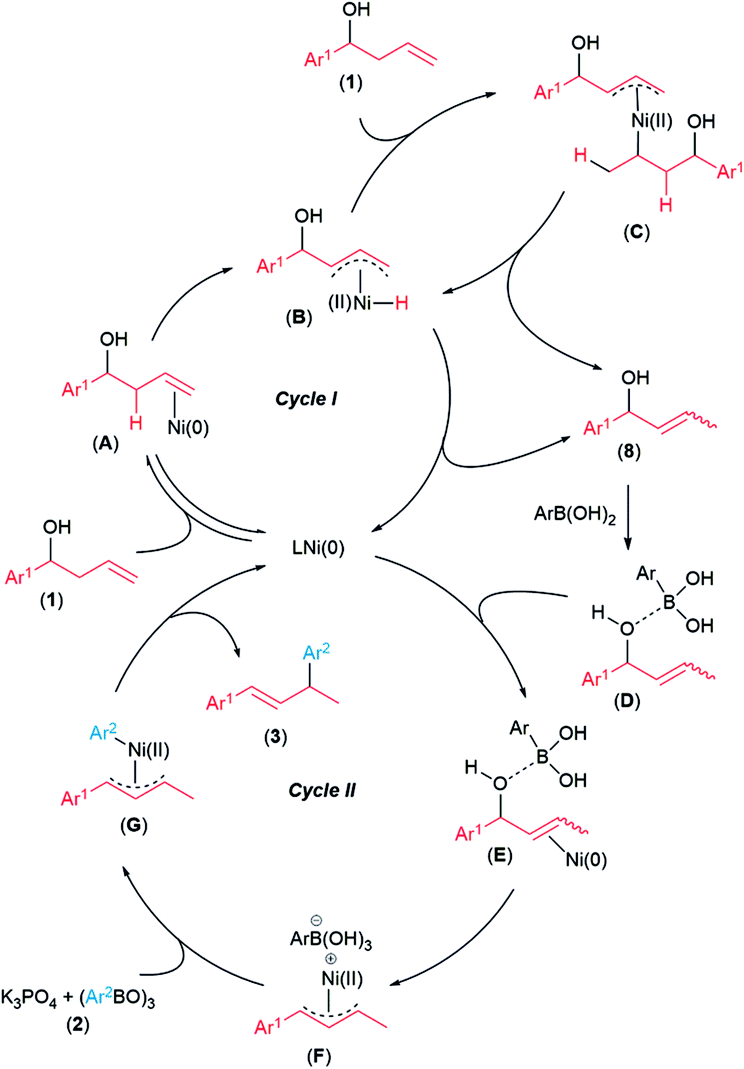 | ||
| Scheme 10 Proposed mechanism. | ||
Conclusions
We have developed an arylative substitution of homoallylic alcohols catalyzed by a nickel complex prepared in situ from Ni(cod)2 and Xantphos ligand to generate allylic arenes under mild reaction conditions. These reactions are fast and proceed via a tandem isomerization/allylic arylation mechanism. The use of p-methoxyphenylboronic acid to activate the in situ generated allylic alcohol leads to the formation of products in higher yields. These arylative substitutions demonstrate the utility of unactivated alcohols in transition metal-catalyzed cross-coupling chemistry, and the requirement of preactivated alcohols is not necessary. Additional studies on the use of unactivated alcohols in nickel-catalyzed cross-coupling reactions are ongoing.Data availability
Experimental data associated with this article are provided in the ESI.†Author contributions
H. N. T. and L. M. S. conceived the project. H. N. T. optimized the reactions conditions, performed the experimental mechanistic studies, and completed the synthetic applications. H. N. T. and C. M. N. synthesized starting materials and evaluated the scope. H. N. T. and L. M. S. wrote the manuscript with input from C. M. N. M. T. K. and D. D. Y. performed the deuterium-labelling experiments during the revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Foundation (CHE-1955529).Notes and references
- Handbook of Bond Dissociation Energies in Organic Compounds, ed. Y.-R. Luo, CRC Press, Boca Raton, FL, 2002 Search PubMed.
- (a) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS PubMed; (b) D.-H. Lee, K.-H. Kwon and C. S. Yi, J. Am. Chem. Soc., 2012, 134, 7325–7328 CrossRef CAS PubMed; (c) D.-H. Lee, K.-H. Kwon and C. S. Yi, Science, 2011, 333, 1613–1616 CrossRef CAS.
- For nickel-catalyzed stereospecific cross-coupling reactions using alcohol derivatives, see: (a) J. Xu, O. P. Bercher and M. P. Watson, J. Am. Chem. Soc., 2021, 143, 8608–8613 CrossRef CAS PubMed; (b) J. Xu, O. P. Bercher, M. R. Talley and M. P. Watson, ACS Catal., 2021, 11, 1604–1612 CrossRef CAS PubMed; (c) E. J. Tollefson, L. E. Hanna and E. R. Jarvo, Acc. Chem. Res., 2015, 48, 2344–2353 CrossRef CAS PubMed.
- For selected racemic transition metal-catalyzed cross-coupling reactions using alcohol derivatives, see: (a) M. Ohsumi, A. Ito and N. Nishiwaki, RSC Adv., 2018, 8, 35056–35061 RSC; (b) M. Ohsumi and N. Nishiwak, ACS Omega, 2017, 2, 7767–7771 CrossRef CAS PubMed; (c) X.-X. Wang, M.-J. Luo and J.-M. Lu, Org. Biomol. Chem., 2015, 13, 11438–11444 RSC; (d) X.-X. Wang, B.-B. Xu, W.-T. Song, K.-X. Suna and J.-M. Lu, Org. Biomol. Chem., 2015, 13, 4925–4930 RSC; (e) Q. Chen, X.-H. Fan, L.-P. Zhang and L.-M. Yang, RSC Adv., 2015, 5, 15338–15340 RSC; (f) R. Kuwano and M. Yokogi, Chem. Commun., 2005, 5899–5901 RSC; (g) M. McLaughlin, Org. Lett., 2005, 7, 4875–4878 CrossRef CAS PubMed; (h) R. Kuwano and M. Yokogi, Org. Lett., 2005, 7, 945–947 CrossRef CAS.
- (a) M. Nagamoto, H. Yorimitsu and T. Nishimura, Org. Lett., 2018, 20, 828–831 CrossRef CAS PubMed; (b) W.-C. Lee, W.-C. Shih, T.-H. Wang, Y. Liu, G. P. A. Yap and T.-G. Ong, Tetrahedron, 2015, 71, 4460–4464 CrossRef CAS; (c) Y. Nakao, N. Kashihara, K. S. Kanyiva and T. Hiyama, J. Am. Chem. Soc., 2008, 130, 16170–16171 CrossRef CAS PubMed.
- For related rhodium-catalyzed hydroarylation of diene electron-rich hetero- and homoarenes, see: (a) L. Gu, L. M. Wolf, A. Zielinski, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2017, 139, 4948–4953 CrossRef CAS PubMed; (b) C. C. Roberts, D. M. Matías, M. J. Goldfogel and S. J. Meek, J. Am. Chem. Soc., 2015, 137, 6488–6491 CrossRef CAS PubMed.
- (a) L. Liao and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 10209–10211 CrossRef CAS; (b) L.-J. Xiao, L. Cheng, W.-M. Feng, M.-L. Li, J.-H. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2018, 57, 461–464 CrossRef CAS; (c) C. Wang, Y. Guo, X. Wang, Z. Wang and K. Ding, Chem.–Eur. J., 2021, 27, 15903–15907 CrossRef CAS PubMed.
- For transition metal-catalyzed asymmetric hydro(hetero)arylation of 1,3-diene, see: (a) J. S. Marcum, T. R. Taylor and S. J. Meek, Angew. Chem., Int. Ed., 2020, 59, 14070–14075 CrossRef CAS PubMed; (b) X.-Y. Lv, C. Fan, L.-J. Xiao, J.-H. Xie and Q.-L. Zhou, CCS Chem., 2019, 1, 328–334 CrossRef CAS; (c) Y.-G. Chen, B. Shuai, X.-T. Xu, Y.-Q. Li, Q.-L. Yang, H. Qiu, K. Zhang, P. Fang and T.-S. Mei, J. Am. Chem. Soc., 2019, 141, 3395–3399 CrossRef CAS PubMed; (d) J. S. Marcum, C. C. Roberts, R. S. Manan, T. N. Cervarich and S. J. Meek, J. Am. Chem. Soc., 2017, 139, 15580–15583 CrossRef CAS PubMed; (e) S. M. Podhajsky, Y. Iwai, A. Cook-Sneathen and M. S. Sigman, Tetrahedron, 2011, 67, 4435–4441 CrossRef CAS PubMed.
- (a) B. J. Stokes, A. J. Bischoff and M. S. Sigman, Chem. Sci., 2014, 5, 2336–2339 RSC; (b) B. J. Stokes, S. M. Opra and M. S. Sigman, J. Am. Chem. Soc., 2012, 134, 11408–11411 CrossRef CAS PubMed.
- T. Hamaguchi, Y. Takahashi, H. Tsuji and M. Kawatsura, Org. Lett., 2020, 22, 1124–1129 CrossRef CAS PubMed.
- For a related arylative substitution of homoallylic alcohol derivatives to form linear products, see: K. Muto, T. Kumagai, F. Kakiuchi and T. Kochi, Angew. Chem., Int. Ed., 2021, 60, 24500–24504 CrossRef CAS.
- For reviews of transition metal-catalyzed allylic substitution of allylic alcohols, see: (a) D. Ghorai, À. Cristòfol and A. W. Kleij, Eur. J. Inorg. Chem., 2022, e202100820 CAS; (b) J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539–2555 CrossRef CAS PubMed; (c) N. A. Butt and W. Zhang, Chem. Soc. Rev., 2015, 44, 7929–7967 RSC; (d) B. Sundararaju, M. Achard and C. Bruneau, Chem. Soc. Rev., 2012, 41, 4467–4483 RSC.
- For selected direct nickel-catalyzed allylic arylation of allylic alcohols, see: (a) H. Yua and Z.-X. Wang, Org. Biomol. Chem., 2021, 19, 9723–9731 RSC; (b) Y.-B. Wang, B.-Y. Liu, Q. Bu, B. Dai and N. Liu, Adv. Synth. Catal., 2020, 362, 2930–2940 CrossRef CAS; (c) S. H. Nazari, J. E. Bourdeau, M. R. Talley, G. A. Valdivia-Berroeta, S. J. Smith and D. J. Michaelis, ACS Catal., 2018, 8, 86–89 CrossRef CAS; (d) X.-G. Jia, P. Guo, J. Duan and X.-Z. Shu, Chem. Sci., 2018, 9, 640–645 RSC.
- For selected nickel-catalyzed allylic arylations of allylic alcohol derivatives, see: (a) X. Li, Y. Li, Z. Zhang, X. Shi, R. Liu, Z. Wang, X. Li and D. Shi, Org. Lett., 2021, 23, 6612–6616 CrossRef CAS PubMed; (b) J.-L. Tao, B. Yang and Z.-X. Wang, J. Org. Chem., 2015, 80, 12627–12634 CrossRef CAS PubMed; (c) L. L. Anka-Lufford, M. R. Prinsell and D. J. Weix, J. Org. Chem., 2012, 77, 9989–10000 CrossRef CAS PubMed; (d) S. Wang, Q. Qian and H. Gong, Org. Lett., 2012, 14, 3352–3355 CrossRef CAS PubMed; (e) Y. Dai, F. Wu, Z. Zang, H. You and H. Gong, Chem.–Eur. J., 2012, 18, 808–812 CrossRef CAS PubMed.
- For in situ activation of allylic alcohols using arylboronic acids, see: (a) D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC; (b) H.-B. Wu, X.-T. Maa and S.-K. Tian, Chem. Commun., 2014, 50, 219–221 RSC; (c) J. Ye, J. Zhao, J. Xu, Y. Mao and Y. J. Zhang, Chem. Commun., 2013, 49, 9761–9763 RSC; (d) H. Tsukamoto, T. Uchiyama, T. Suzuki and Y. Kondo, Org. Biomol. Chem., 2008, 6, 3005–3013 RSC.
- The superiority of arylboroxines over other arylboron sources has been observed previously, see: R. Li, F. Liu and G. Dong, Chem, 2019, 5, 929–939 CAS.
- L. Chen, H. Min, W. Zeng, X. Zhu, Y. Liang, G. Deng and Y. Yang, Org. Lett., 2018, 20, 7392–7395 CrossRef CAS PubMed.
- For examples of MeOH as a hydride source, see: (a) H. N. Tran and L. M. Stanley, Org. Lett., 2022, 24, 395–399 CrossRef CAS; (b) H. N. Tran, R. W. Burgett and L. M. Stanley, J. Org. Chem., 2021, 86, 3836–3849 CrossRef CAS PubMed; (c) Y. He, C. Liu, L. Yu and S. Zhu, Angew. Chem., Int. Ed., 2020, 59, 9186–9191 CrossRef CAS PubMed; (d) J.-F. Li, Z.-Z. Wei, Y.-Q. Wang and M. Ye, Green Chem., 2017, 19, 4498–4502 RSC.
- For examples of formations π-allyl-nickel(II) intermediates from allylic alcohols, see: (a) Y. Gan, H. Hu and Y. Liu, Org. Lett., 2020, 22, 4418–4423 CrossRef CAS PubMed; (b) Y. Bernhard, B. Thomson, V. Ferey and M. Sauthier, Angew. Chem., Int. Ed., 2017, 56, 7460–7464 CrossRef CAS; (c) M. S. Azizi, Y. Edder, A. Karim and M. Sauthier, Eur. J. Org. Chem., 2016, 3796–3803 CrossRef CAS; (d) Y. Kita, H. Sakaguchi, Y. Hoshimoto, D. Nakauchi, Y. Nakahara, J.-F. Carpentier, S. Ogoshi and K. Mashima, Chem.–Eur. J., 2015, 21, 14571–14578 CrossRef CAS PubMed; (e) H. Bricouta, E. Monflier, J.-F. Carpentier and A. Mortreux, Eur. J. Inorg. Chem., 1998, 1739–1744 CrossRef.
- For examples of β-hydride elimination of metallacylces, see: (a) D. Jin, T. J. Schmeier, P. G. Williard, N. Hazari and W. H. Bernskoetter, Organometallics, 2013, 32, 2152–2159 CrossRef CAS; (b) X. Huang, J. Zhu and Z. Lin, Organometallics, 2004, 23, 4154–4159 CrossRef CAS.
- (a) K. Farshadfar, A. Chipman, M. Hosseini, B. F. Yates and A. Ariafard, Organometallics, 2019, 38, 2953–2962 CrossRef CAS; (b) J. Tan, Z. Zhang and Z. Wang, Org. Biomol. Chem., 2008, 6, 1344–1348 RSC.
- H. Iwamoto, T. Tsuruta and S. Ogoshi, ACS Catal., 2021, 11, 6741–6749 CrossRef CAS.
- For selected examples on the use of K3PO4 to facilitate the transmetallation of arylboroxines, see: (a) J. Zhu, L. Huang, W. Dong, N. Li, X. Yu, W.-P. Deng and W. Tang, Angew. Chem., Int. Ed., 2019, 58, 16119–16123 CrossRef CAS PubMed; (b) F. Pan, H. Wang, P.-X. Shen, J. Zhao and Z.-J. Shi, Chem. Sci., 2013, 4, 1573–1577 RSC.
- Y. Ding, J. Long, F. Sun and X. Fang, Org. Lett., 2021, 23, 6073–6078 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization data, and NMR spectra for all new compounds. See https://doi.org/10.1039/d2sc01716d |
| This journal is © The Royal Society of Chemistry 2022 |