
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic effects of CH3CO2H and Ca2+ on C–H bond activation by MnO4−†
Huatian
Shi‡
,
Lin
Cheng‡
,
Yi
Pan
 ,
Chi-Keung
Mak
*,
Kai-Chung
Lau
* and
Tai-Chu
Lau
*
,
Chi-Keung
Mak
*,
Kai-Chung
Lau
* and
Tai-Chu
Lau
*
Department of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon Tong, Hong Kong, China. E-mail: bhtclau@cityu.edu.hk; kaichung@cityu.edu.hk; chikmak6@cityu.edu.hk
First published on 13th September 2022
Abstract
The activation of metal-oxo species with Lewis acids is of current interest. In this work, the effects of a weak Brønsted acid such as CH3CO2H and a weak Lewis acid such as Ca2+ on C–H bond activation by KMnO4 have been investigated. Although MnO4− is rather non-basic (pKa of MnO3(OH) = −2.25), it can be activated by AcOH or Ca2+ to oxidize cyclohexane at room temperature to give cyclohexanone as the major product. A synergistic effect occurs when both AcOH and Ca2+ are present; the relative rates for the oxidation of cyclohexane by MnO4−/AcOH, MnO4−/Ca2+ and MnO4−/AcOH/Ca2+ are 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :73:198. DFT calculations show that in the active intermediate of MnO4−/AcOH/Ca2+, MnO4− is H-bonded to 3 AcOH molecules, while Ca2+ is bonded to 3 AcOH molecules as well as to an oxo ligand of MnO4−. Our results also suggest that these synergistic activating effects of a weak Brønsted acid and a weak Lewis acid should be applicable to a variety of metal-oxo species.
:73:198. DFT calculations show that in the active intermediate of MnO4−/AcOH/Ca2+, MnO4− is H-bonded to 3 AcOH molecules, while Ca2+ is bonded to 3 AcOH molecules as well as to an oxo ligand of MnO4−. Our results also suggest that these synergistic activating effects of a weak Brønsted acid and a weak Lewis acid should be applicable to a variety of metal-oxo species.
Introduction
The chemistry of metal-oxo species has been of great interest to chemists because of the key roles they play in various chemical and biological systems.1–4 In recent years, the use of Lewis acids to enhance the reactivity of metal-oxo species has attracted much attention,5,6 especially after the discovery of a Ca2+ in the active site of the oxygen-evolving center (OEC) of photosystem II, which contains a manganese calcium oxo cluster (Mn4CaO5).7,8Various metal-oxo species are readily activated by strong Lewis acids such as BF3, Sc(OTf)3 and other M3+ ions toward the oxidation of organic substrates.9–19 There are also a number of studies on the interaction of relatively weak Lewis acids such as Ca2+ and other group II ions with metal-oxo species, and as expected their activating effects are much smaller than those of Sc(OTf)3 or other M3+ ions.12,15,20–24
We recently reported the use of a weak Brønsted acid such as acetic acid in combination with a weak Lewis acid such as a group II ion to activate RuO4−.25 We found a remarkable cooperative activating effect of these two acids on RuO4− toward the oxidation of alkanes. The oxo ligands in RuO4− are basic and can be protonated by AcOH to generate [RuO3(OH)]OAc−, which readily abstracts H atoms from alkanes. In the presence of Ca2+, a more active intermediate is formed with [RuO3(OH)]OAc−, in which Ca(II) binds to both OAc− (which is H-bonded to OH) and an oxo ligand. In this work, we have carried out experimental and theoretical investigations on the use of AcOH and Ca2+ to activate MnO4− toward cyclohexane oxidation. MnO4− was chosen because of the relevance of Mn in the OEC of photosystem II. Remarkably, a similar synergistic activating effect of AcOH and Ca2+ is also observed for this oxoanion, although MnO4− is much less basic than RuO4− and is not protonated by AcOH. Our results suggest that the synergistic effects of a weak Brønsted and a weak Lewis acid should be applicable to a variety of metal-oxo species.
Results and discussion
Effects of AcOH on cyclohexane oxidation by KMnO4
KMnO4 does not react with cyclohexane at room temperature. However, upon addition of AcOH (3–9 M) to KMnO4 (0.011 M) in CH3CN in the presence of cyclohexane (1.0 M) at 23 °C, the purple color of the solution gradually changed to brown. Analysis of the solution by GC-MS and GC-FID revealed the formation of cyclohexanone (38.8%), together with a small amount of cyclohexanol (2.2%). The yields were calculated based on KMnO4 acting as a 3-electron oxidant (product is a Mn(IV) species, see below).Kinetics studies were carried out at constant [KMnO4] (0.011 M) and [cyclohexane] (1.0 M) by monitoring the growth of cyclohexanone using a GC-FID (gas chromatograph-flame ionization detector). The increase in [cyclohexanone] follows pseudo-first-order kinetics (Fig. 1a), and the pseudo-first-order rate constant kobs at [AcOH] = 3.0 M was found to be 4.3 × 10−6 s−1. kobs increases with increasing [AcOH] (3–9 M), and the plot of kobsversus [AcOH]2 is linear (Fig. 1b), suggesting that under these conditions the active oxidizing species involves MnO4− binding to two molecules of AcOH. The reaction can be represented by eqn (1) and (2).
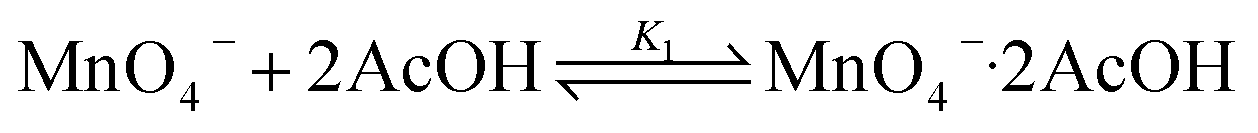 | (1) |
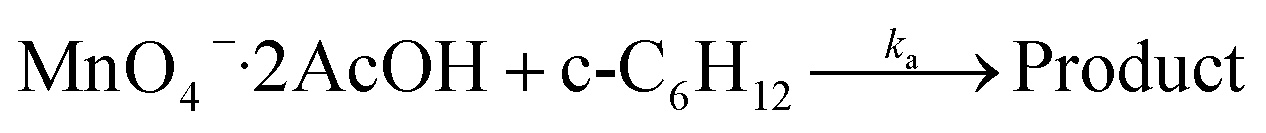 | (2) |
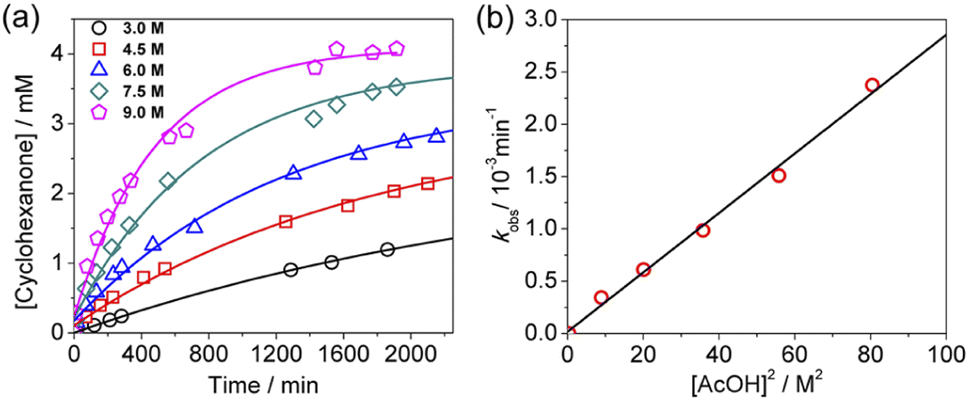 | ||
| Fig. 1 (a) Time course for the production of cyclohexanone at various [AcOH]. Conditions: KMnO4, 0.011 M; cyclohexane, 1.0 M; at 23 °C in CH3CN. (b) Linear plot of kobsvs. [AcOH]2. Slope = (2.84 ± 0.11) × 10−5, intercept = (2 ± 5) × 10−5, and r2 = 0.992. | ||
Under the condition that K1 ≪ 1, the rate-law is given by eqn (3).
| d[C6H10O]/dt = kobs[MnO4−] = K1ka[AcOH]2[c-C6H12][MnO4−] | (3) |
From the slope of Fig. 1b, K1ka = (4.73 ± 0.19) × 10−7 M−2 s−1 at 23.0 °C.
The kinetic isotope effect for cyclohexane oxidation determined by competitive oxidation of an equimolar mixture of c-C6H12 and c-C6D12 was found to be 7.5 ± 0.1, indicating C–H bond cleavage in the rate-limiting step.
Effects of Ca2+
The oxidation of cyclohexane by KMnO4 is also activated by Ca2+. Upon adding Ca(OTf)2 (5.5 × 10−3 to 4.4 × 10−2 M) to KMnO4 (0.011 M) in CH3CN containing cyclohexane (1.0 M), the purple color of the solution was gradually discharged together with the formation of a brown precipitate (Fig. 2). The UV/vis spectral changes indicate that there is no shift in the wavelengths of the peaks of MnO4−, just a gradual decrease in their absorbances (Fig. S1†). The same result was also observed in our earlier work on the effect of BF3 on KMnO4.18 The solution IR of KMnO4 and KMnO4 + Ca(OTf)2 in CH3CN was also performed (Fig. S2†). KMnO4 shows a M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 904 cm−1, but this peak was not shifted upon addition of Ca(OTf)2; presumably the binding of Ca2+ to MnO4− is relatively weak.
O stretch at 904 cm−1, but this peak was not shifted upon addition of Ca(OTf)2; presumably the binding of Ca2+ to MnO4− is relatively weak.
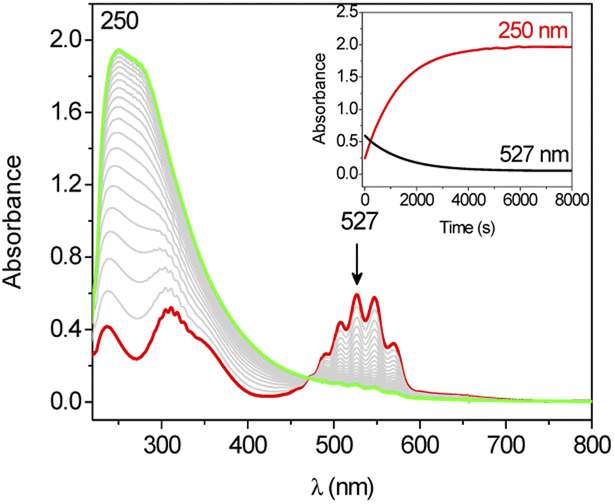 | ||
| Fig. 2 UV-vis spectral changes for the reaction of KMnO4 (2.35 × 10−3 M) with cyclohexane (1.0 M) in the presence of Ca(OTf)2 (2.35 × 10−3 M) in CH3CN at 25 °C (1 mm UV cell was used in the measurement). The inset shows the change of absorbance at 250 nm and 527 nm versus time. | ||
Analysis of the product solution by GC-FID and GC-MS revealed the formation of cyclohexanone (10.5%) and cyclohexanol (0.8%) in relatively low yields. The yields of products reached a maximum with just 1 equiv. of Ca2+.
Kinetics studies were also carried out at various [Ca(OTf)2] but at constant [KMnO4] (0.011 M) and [cyclohexane] (1.0 M). The increase in [cyclohexanone] follows pseudo-first-order kinetics (Fig. 3a), but saturation kinetics were observed on increasing [Ca(OTf)2] (Fig. 3b), and the plot of 1/kobsversus 1/[Ca(OTf)2] is linear. Kinetic studies were also conducted at various [cyclohexane] (Fig. 4a), while keeping both Ca(OTf)2 and KMnO4 at 0.011 M. The plot of kobsversus [cyclohexane] is linear with an intercept of (2.87 ± 3.3) × 10−4 s−1 (Fig. 4b), which should be due to the decomposition of KMnO4 in the absence of cyclohexane. An independent experiment showed that in the presence of 1 equiv. of Ca(OTf)2, the absorbance of KMnO4 (2.82 × 10−4 M) in CH3CN at around 527 nm gradually decreased with time with a rate constant of (9.5 ± 1.1) × 10−4 s−1 at 23 °C. Such a decomposition also accounts for the low yield of products. The kinetic results are consistent with a pre-equilibrium binding of Ca(II) to MnO4− to form an intermediate, which then oxidizes cyclohexane, as represented by eqn (4) and (5), and the rate law is shown in eqn (6).
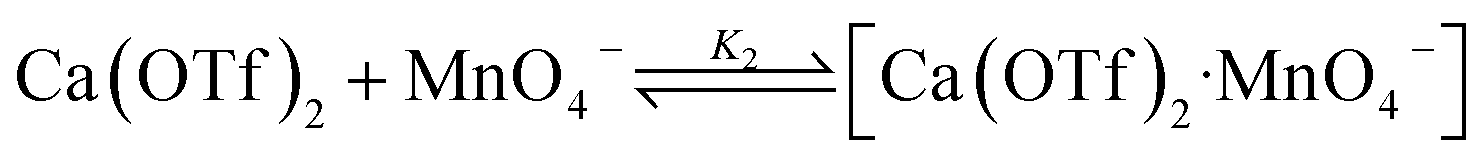 | (4) |
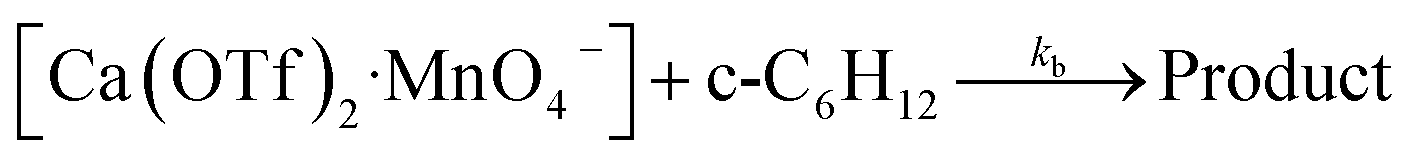 | (5) |
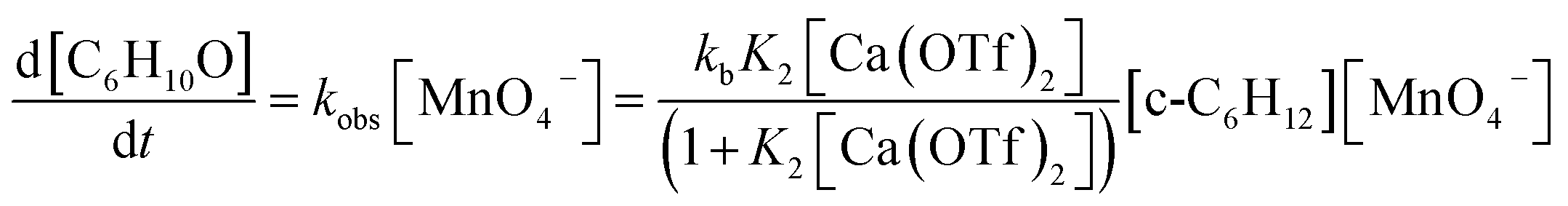 | (6) |
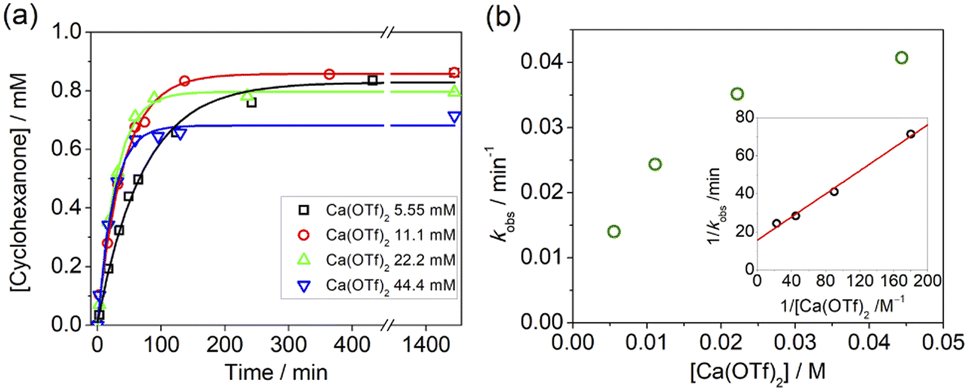 | ||
| Fig. 3 (a) Time course for the production of cyclohexanone at various [Ca(OTf)2]. Conditions: KMnO4, 0.011 M; cyclohexane, 1.0 M; at 23 °C in CH3CN. (b) Plot of kobsversus [Ca(OTf)2]. Inset: linear plot of 1/kobsvs. 1/[Ca(OTf)2]; slope = 0.303 ± 0.018, intercept = 15.8 ± 1.9, and r2 = 0.99. | ||
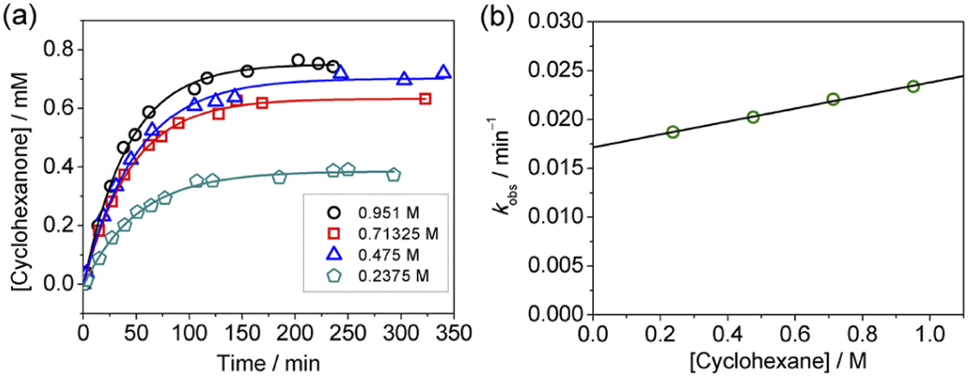 | ||
| Fig. 4 (a) Time course for the production of cyclohexanone at various [cyclohexane]. Conditions: KMnO4 (0.011 M), Ca(OTf)2 (0.011 M) and cyclohexane (0.238–0.951 M) in CH3CN at 23 °C. (b) Plot of kobsvs. [cyclohexane]. Slope = (6.64 ± 0.29) × 10−3, intercept = (1.72 ± 0.02) × 10−2, and r2 = 0.994. | ||
Synergistic effects of AcOH and Ca2+
The oxidation of cyclohexane in the presence of both AcOH and Ca2+ was investigated. As described above, the rate of oxidation of cyclohexane by KMnO4/AcOH is second-order in [AcOH]. However, in the presence of just 1 equiv. of Ca(OTf)2, the rate becomes first-order in [AcOH] (Fig. 5).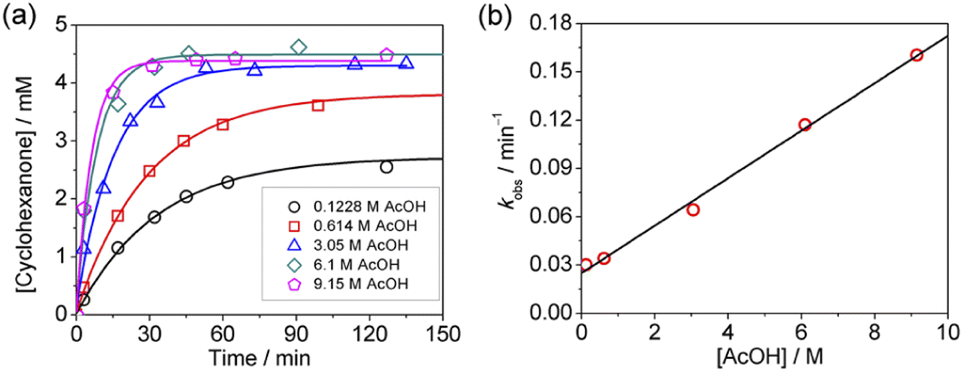 | ||
| Fig. 5 (a) Time course for cyclohexanone production by KMnO4 (0.011 M) in the presence of cyclohexane (1.0 M), 1 equiv. of Ca(OTf)2 (0.011 M) and various [AcOH] in CH3CN at 23.0 °C. (b) Plot of kobsvs. [AcOH]. Slope = (1.56 ± 0.06) × 10−2, y-intercept = 0.025 ± 0.003, and r2 = 0.995. | ||
On the other hand, at constant [AcOH] (2.79 M) and [cyclohexane] (1.0 M), saturation kinetics were observed on increasing [Ca(OTf)2], and the plot of 1/kobsversus 1/[Ca(OTf)2] is linear (Fig. 6).
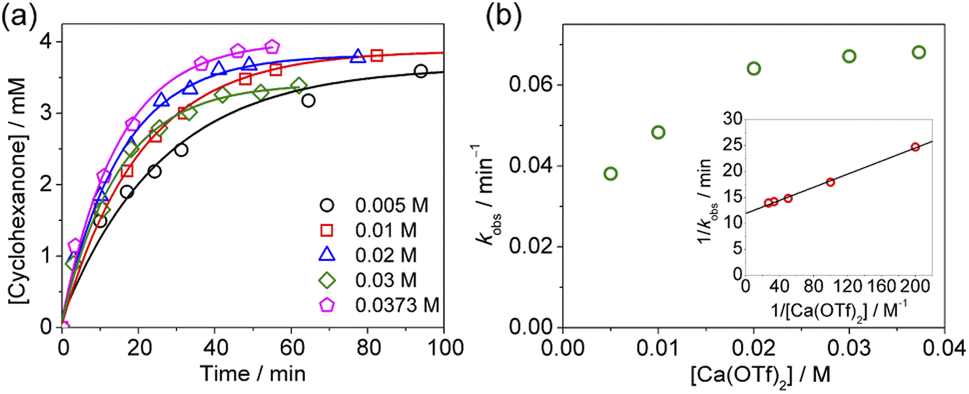 | ||
| Fig. 6 (a) Time course for cyclohexane production in CH3CN at various [Ca(OTf)2]. Conditions: cyclohexane, 1.0 M; KMnO4, 0.011 M; AcOH, 2.79 M. T = 23 °C. (b) Plot of kobsversus [Ca(OTf)2]. Inset: plot of 1/kobsversus 1/[Ca(OTf)2]; slope = (6.3 ± 0.2) × 10−2, y-intercept = 11.9 ± 0.2, and r2 = 0.995. | ||
These results are consistent with the reaction scheme shown in eqn (7)–(9).
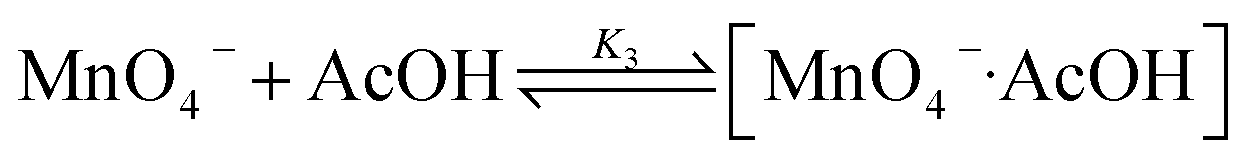 | (7) |
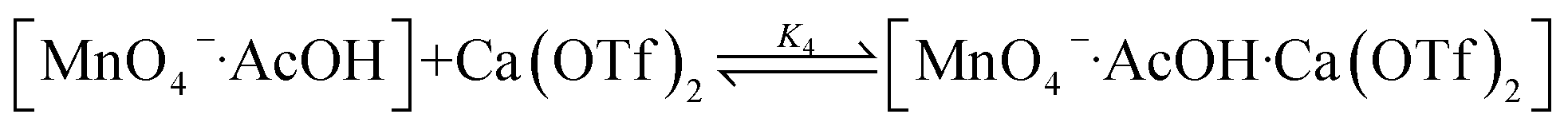 | (8) |
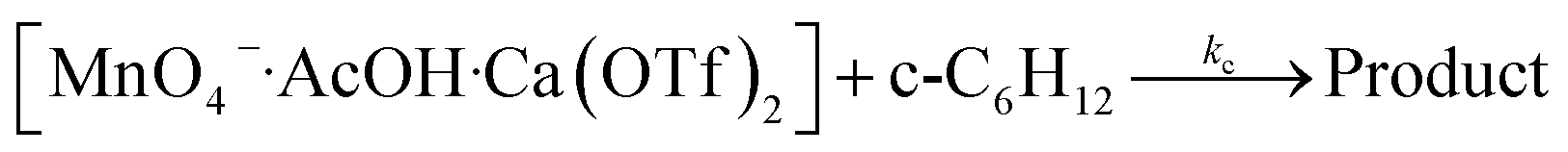 | (9) |
For K3 ≪ 1, the rate law is
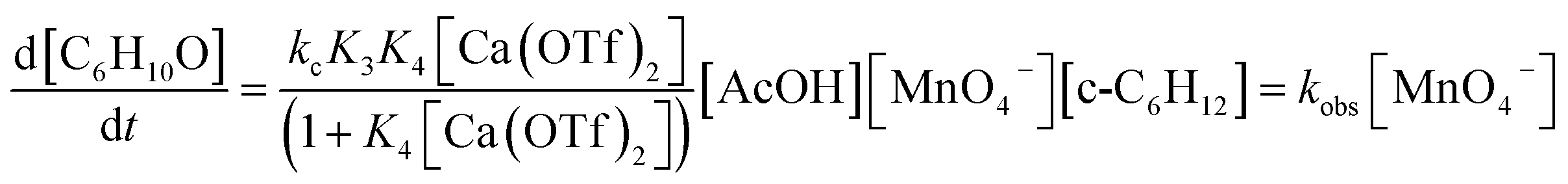 | (10) |
The kinetic isotope effect for cyclohexane oxidation by the KMnO4/AcOH/Ca(OTf)2 system was determined to be 5.7 ± 0.1 by competitive oxidation of an equimolar mixture of c-C6H12 and c-C6D12, indicating that C–H bond cleavage is the rate limiting step.
A comparison of the activating effects of AcOH, Ca(OTf)2 and AcOH + Ca(OTf)2 on cyclohexane oxidation by KMnO4 is shown in Table 1. The rate constant (kobs) for the oxidation of cyclohexane (1.0 M) by KMnO4 (0.011 M) in the presence of AcOH (3.0 M) at 23 °C was found to be 5.73 × 10−6 s−1. When AcOH was replaced by 1 equiv. of Ca(OTf)2 (0.011 M), the rate constant (4.21 × 10−4 s−1) is 73 times faster, but the yield is much lower (10.5%). On the other hand, in the presence of both Ca(OTf)2 and AcOH, the rate (1.14 × 10−3 s−1) increases by 198 fold relative to AcOH alone, and the product yield is also substantially increased to 57%, indicating synergistic effects of AcOH and Ca2+ in the activation of MnO4− toward C–H bond activation.
| 3.0 M AcOH (275 equiv.) | 1 equiv. Ca(OTf)2 (0.011 M) | 1 equiv. Ca(OTf)2 + 3.0 M of AcOH | |
|---|---|---|---|
| a Reaction conditions: KMnO4, 0.011 M, cyclohexane, 1.0 M, T = 23 °C. b KMnO4 alone does not oxidize cyclohexane for >48 h at 23 °C. c The final oxidation state of the Mn product after oxidation was determined by iodometric titration. d % Yield was calculated based on KMnO4 acting as a 3-electron oxidant. Cyclohexanol is the minor product in all cases. | |||
| k obs (relative rate) | 5.73 × 10−6 s−1 (1) | 4.21 × 10−4 s−1 (73) | 1.14 × 10−3 s−1 (198) |
| Oxidation state of Mn productc | 4.1 ± 0.2 | 4.1 ± 0.2 | 4.0 ± 0.2 |
| % Yield of cyclohexanoned | 38.8 | 10.5 | 57.3 |
| Cyclohexanone/cyclohexanol | 18 ± 1 | 14 ± 1 | 7 ± 1 |
The rate constant for the oxidation of 1.0 M toluene by (nBu4N)MnO4 was reported to be 4.2 × 10−7 s−1 at 25.0 °C.26 As a comparison, we also investigated the oxidation of 1.0 M toluene by KMnO4 in the presence of 1 equiv. Ca(OTf)2 and 3.0 M AcOH, and the rate constant was found to be 5.2 × 10−3 s−1 at 23 °C (Fig. S3†). Based on these data, the oxidation of toluene by KMnO4/Ca2+/AcOH is over 4 orders of magnitude faster than that of MnO4− alone.
Effects of other metal ions
The effects of other metal salts, including Ba(OTf)2, Mg(OTf)2, and Sc(OTf)3 on cyclohexane oxidation by KMnO4 in the presence of AcOH have also been investigated. As expected, both the rate and yield increase with increasing Lewis acidity of the metal ion (Fig. 7a and S4–S6†). The ratio of the rate constants obtained by GC for the 4 metal ions is found to be k(Ba2+):k(Ca2+):k(Mg2+):k(Sc3+) = 1:4.8:10.4:228; and the yields of cyclohexanone are 51%(Ba2+), 56%(Ca2+), 61%(Mg2+), and 75%(Sc3+). There is a fairly linear correlation of log(rate constant) with the pKa of the metal ion in water, Sc3+(pKa = 4.3), Mg2+(pKa = 11.2), Ca2+(pKa = 12.7), and Ba2+(pKa = 13.4),27 which is a measure of their Lewis acidity (Fig. 7b). Similar correlations of Lewis acid effects with their acidities have also been reported.15,23
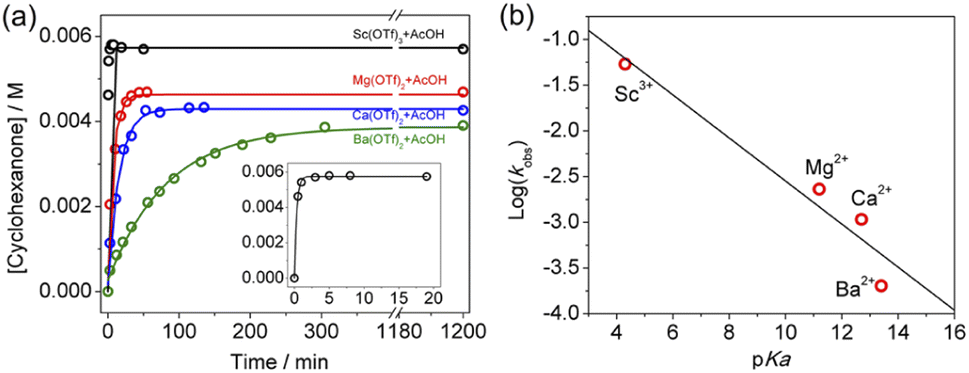 | ||
| Fig. 7 (a)Time course for the production of cyclohexanone by KMnO4 (0.01 M)/AcOH (2.78 M)/c-C6H12 (1.0 M) in the presence of 0.01 M of various metal salts. Pseudo-first-order fits give kobs: Sc(OTf)3 = 5.4 × 10−2 s−1, Mg(OTf)2 = 2.3 × 10−3 s−1, Ca(OTf)2 = 1.1 × 10−3 s−1, and Ba(OTf)2 = 2 × 10−4 s−1. The inset shows the time course for Sc(OTf)3. (b) Plot of log(kobs) vs. pKa of metal ions. Sc3+(pKa = 4.3), Mg2+(pKa = 11.2), Ca2+(pKa = 12.7), and Ba2+(pKa = 13.4). Slope = −0.24 ± 0.04, intercept = −0.2 ± 0.5, and r2 = 0.90. | ||
Nature of the manganese product
Addition of Ca(OTf)2 to KMnO4 in CH3CN gradually produced a brown solid and a colorless solution, irrespective of whether cyclohexane was present. When AcOH (3 M) was added to the resulting solution after cyclohexane oxidation, the brown solid dissolved but with only a small increase of 1–2% cyclohexanone. The oxidation state of Mn in the brown solution was determined to be 4.1 ± 0.2 by iodometric titration, which involves the addition of NBu4I to reduce the Mnn product to MnII and then determining the amount of I3− formed by UV-vis spectrophotometry.28Based on CHN elemental analysis, as well as K, Mn and Ca analysis by ICP-AES, it was found that the brown solids obtained from MnO4−/Ca(OTf)2 (1) and MnO4−/Ca(OTf)2/c-C6H12 (2) have similar compositions and are probably a MnIV–μ(O)–μ(Ca) polymer with an empirical formula close to “Ca3Mn7O14(H2O)10(OTf)6(CH3CN)2” (see the ESI†). The reason for the conversion of MnVII to MnIV in the absence of c-C6H12 is not clear, presumably due to oxidation of the solvent.
We have carried out X-ray photoelectron spectroscopy (XPS) of 1 and 2 (Fig. S7 and S8†). In the Mn3s XPS spectra (Fig. S8†), the peak splitting of ΔE = 4.8 eV indicates that the oxidation state of Mn is +4.29
We have also determined the magnetic susceptibility of the solid samples of 1 and 2 using a magnetic balance. The μeff for each Mn was found to be 3.96 and 4.05 μB for 1 and 2, respectively, consistent with that of high spin Mn(IV) with S = 3/2.
No brown precipitate was observed when cyclohexane oxidation was carried out in the presence of AcOH, irrespective of whether Ca(OTf)2 was added. The Mn oxidation state of the brown solution determined by iodometric titration is again 4.0 ± 0.2. Presumably the brown solution contains colloidal MnO2, as observed in alkane oxidation by BF3/MnO4−.18
Theoretical calculations
DFT calculations on the mechanisms of the oxidation of cyclohexane by MnO4−, MnO4−/AcOH, MnO4−/Ca(OTf)+ and MnO4−/AcOH/Ca(OTf)+ were performed. Solvent (CH3CN) effects have been included in the calculations.The potential energy surfaces (PESs) for all calculated reactions are summarized in Fig. S9.† In the c-C6H12 oxidation by MnO4− in CH3CN (Fig. 8), c-C6H12 and MnO4− first bind weakly together to form an intermediate [MnO4⋯C6H12]−, INT1(MnO4−). Hydrogen atom transfer (HAT) then occurs from c-C6H12 to MnO via a transition state, TS1(MnO4−), to give a second intermediate, INT2(MnO4−). The HAT step is rate-determining with a barrier height 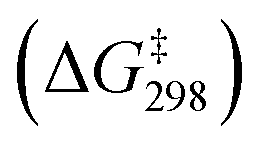 of 22.7 kcal mol−1. Such a high barrier suggests that MnO4− will hardly oxidize c-C6H12 at room temperature. The carbon atom in the cyclohexyl radical of INT2(MnO4−) bears a −1.01 electron spin, consistent with the spin distribution for a HAT. In the next step, the cyclohexyl radical binds to another MnO to generate an alkoxo intermediate [MnO2(OH)(OC6H11)]−via TS2(MnO4−). Subsequently, proton transfer from Mn–OH to the alkoxide in INT3(MnO4−) occurs via TS3(MnO4−) to generate the cyclohexanol product. The reaction mechanism is similar to that of c-C6H12 oxidation by RuO4− found in our previous study.25
of 22.7 kcal mol−1. Such a high barrier suggests that MnO4− will hardly oxidize c-C6H12 at room temperature. The carbon atom in the cyclohexyl radical of INT2(MnO4−) bears a −1.01 electron spin, consistent with the spin distribution for a HAT. In the next step, the cyclohexyl radical binds to another MnO to generate an alkoxo intermediate [MnO2(OH)(OC6H11)]−via TS2(MnO4−). Subsequently, proton transfer from Mn–OH to the alkoxide in INT3(MnO4−) occurs via TS3(MnO4−) to generate the cyclohexanol product. The reaction mechanism is similar to that of c-C6H12 oxidation by RuO4− found in our previous study.25
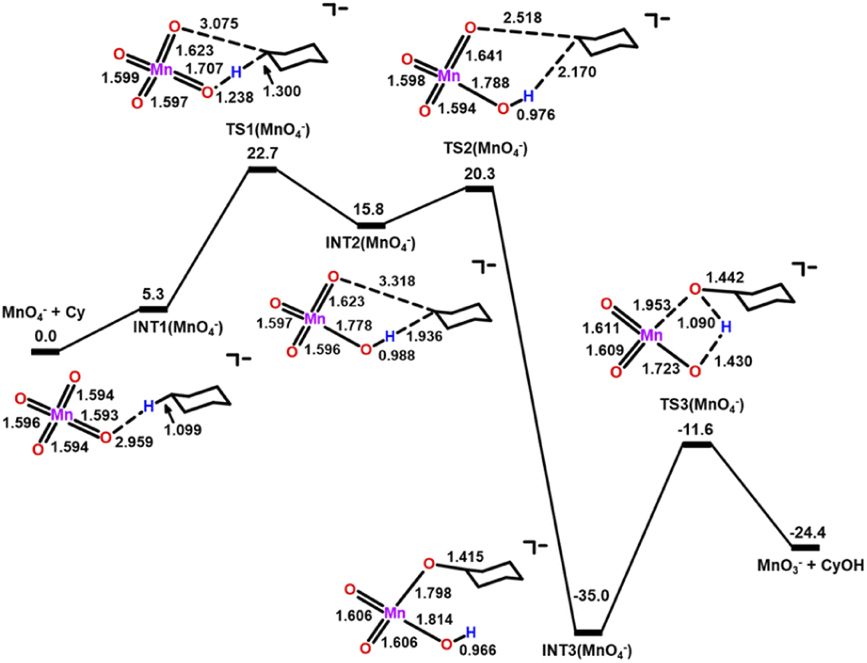 | ||
| Fig. 8 PES and structures for cyclohexane oxidation by [MnO4]− at the B3LYP-D3(BJ)/def2-SVPD level. The relative 298 K Gibbs free energies in acetonitrile are given in kcal mol−1. | ||
In the presence of an acetic acid molecule (Fig. S10†), AcOH and MnO4− first form INT1(AcOH), [MnO4(AcOH)⋯C6H12]−, through a hydrogen bond. The MnO that is H-bonded to AcOH is elongated to 1.611 Å, while the other three MnO bonds become more electrophilic and shorter with distances of 1.587–1.590 Å, compared with the bond length of 1.594 Å in MnO4−. In TS1(AcOH), HAT occurs from c-C6H12 to another free but shorter MnO bond (bond length = 1.588 Å). The 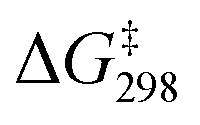 of TS1(AcOH) is 20.4 kcal mol−1, which is 2.3 kcal mol−1 lower than that of TS1(MnO4−). The subsequent steps are similar to those in c-C6H12 oxidation by MnO4−.
of TS1(AcOH) is 20.4 kcal mol−1, which is 2.3 kcal mol−1 lower than that of TS1(MnO4−). The subsequent steps are similar to those in c-C6H12 oxidation by MnO4−.
In the presence of three AcOH molecules, the AcOH molecules form hydrogen bonds with three MnO to give an intermediate, INT1(3AcOH) (Fig. 9). The bond lengths of the H-bonded MnO are slightly elongated from 1.591 to 1.601 Å. HAT then occurs from c-C6H12 to the free and more electrophilic MnO bond (bond length = 1.580 Å), with a 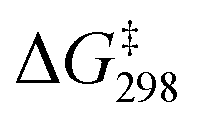 = 16.8 kcal mol−1. Compared with c-C6H12 oxidation by MnO4−, the
= 16.8 kcal mol−1. Compared with c-C6H12 oxidation by MnO4−, the  of HAT is further reduced by 5.9 kcal mol−1.
of HAT is further reduced by 5.9 kcal mol−1.
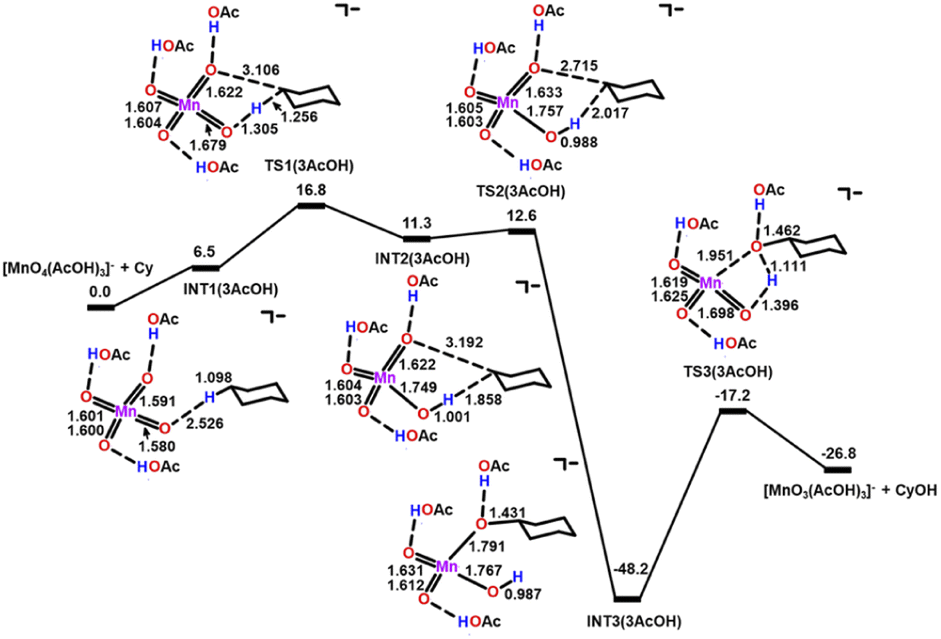 | ||
| Fig. 9 PES and structures for cyclohexane oxidation by [MnO4(AcOH)3]− at the B3LYP-D3(BJ)/def2-SVPD level. The relative 298 K Gibbs free energies in acetonitrile are given in kcal mol−1. | ||
In the presence of [Ca(OTf)]2 (Fig. 10), Ca(II) binds to two of the MnO bonds to form INT1(CaOTf), [MnO4(CaOTf)⋯C6H12]. The two MnO bonds that bind to Ca(II) in INT1(CaOTf) are longer (1.611 and 1.615 Å) than the unbound MnO bonds (1.576 Å) in INT1(MnO4−), as a result of the electron withdrawing effect of Ca(II). Subsequent HAT from c-C6H12 to an unbound MnO occurs with a  of 12.7 kcal mol−1via TS1(CaOTf), which is lower than that of MnO4− and MnO4−/3AcOH by 10.0 and 4.1 kcal mol−1, respectively; consistent with the experimental observation that the accelerating effect of Ca(II) is much higher than that of AcOH.
of 12.7 kcal mol−1via TS1(CaOTf), which is lower than that of MnO4− and MnO4−/3AcOH by 10.0 and 4.1 kcal mol−1, respectively; consistent with the experimental observation that the accelerating effect of Ca(II) is much higher than that of AcOH.
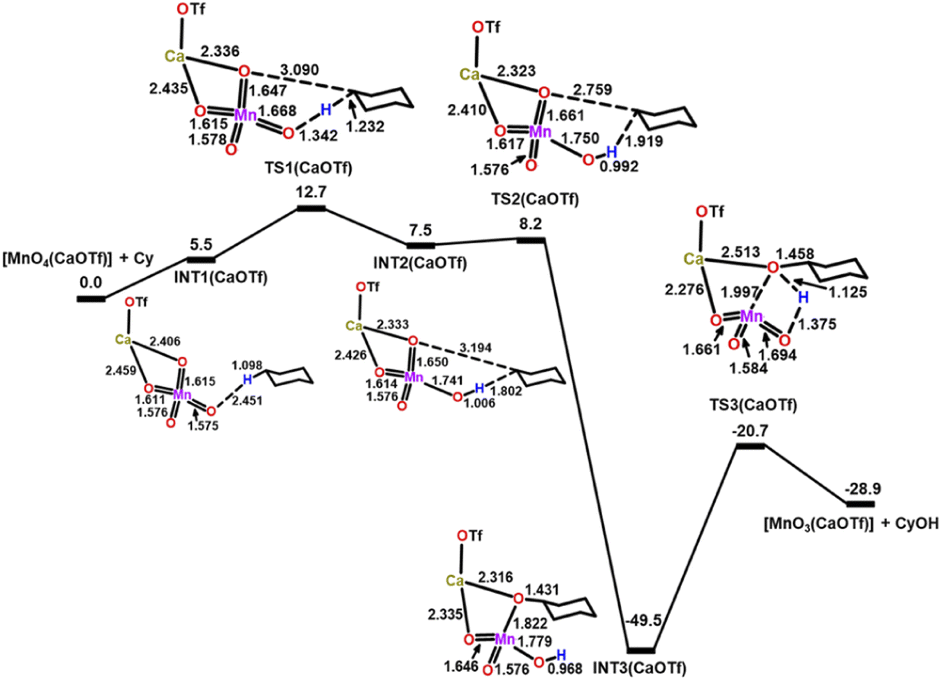 | ||
| Fig. 10 PES and structures for cyclohexane oxidation by [MnO4(CaOTf)] at the B3LYP-D3(BJ)/def2-SVPD level. The relative 298 K Gibbs free energies in acetonitrile are given in kcal mol−1. | ||
The activation barrier is further lowered when both [Ca(OTf)]+ and AcOH are present. In the presence of one molecule of [Ca(OTf)]+ and three molecules of AcOH, each AcOH is H-bonded to one MnO, while the [Ca(OTf)]+ is bonded to each AcOH and one MnO to form a relatively stable intermediate, INT1(CaOTf/3AcOH), as shown in Fig. 11. HAT then occurs from c-C6H12 to the free and shortest MnO bond with the lowest 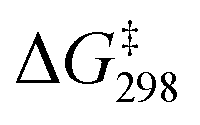 of 9.7 kcal mol−1 (
of 9.7 kcal mol−1 (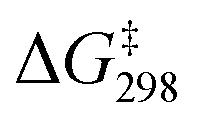 = 11.5 kcal mol−1 for one Ca(OTf)+ and one AcOH, Fig. S11†), in agreement with the observed synergistic activating effects of AcOH and Ca(II). It should be noted that in this case HAT results in the direct formation of the alkoxo intermediate without going through a cyclohexyl radical intermediate.
= 11.5 kcal mol−1 for one Ca(OTf)+ and one AcOH, Fig. S11†), in agreement with the observed synergistic activating effects of AcOH and Ca(II). It should be noted that in this case HAT results in the direct formation of the alkoxo intermediate without going through a cyclohexyl radical intermediate.
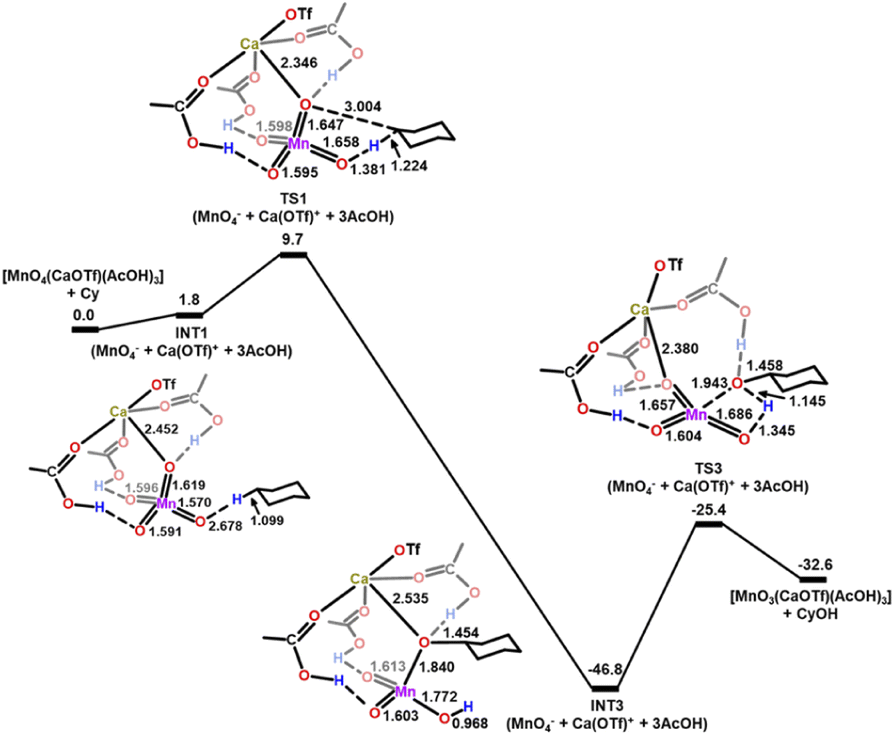 | ||
| Fig. 11 PES and structures for cyclohexane oxidation by [MnO4(CaOTf)(AcOH)3] at the B3LYP-D3(BJ)/def2-SVPD level. The relative 298 K Gibbs free energies in acetonitrile are given in kcal mol−1. | ||
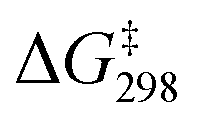 was also determined experimentally by performing kinetics studies at 20–50 °C. From the Arrhenius plot
was also determined experimentally by performing kinetics studies at 20–50 °C. From the Arrhenius plot 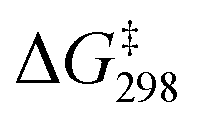 was found to be 10.4 ± 0.6 kcal mol−1, in agreement with the calculated value (Fig. S12†).
was found to be 10.4 ± 0.6 kcal mol−1, in agreement with the calculated value (Fig. S12†).
We have also calculated the TS barrier heights for hydroxylation via a rebound mechanism and they are higher than those of the TS forming the alkoxo intermediate by 2–3.4 kcal mol−1 (Table S1†).
Activating effects of Ca2+/AcOH on MnO4−vs. RuO4−
Although MnO4− and RuO4− are isostructural with similar E0 (0.56 V and 0.59 V, respectively), there is a great difference in the basicity of their oxo ligands; MnO4− is a weak base (pKa of HMnO4 is −2.25),30 whereas RuO4− is a much stronger base. Although the pKa of HRuO4 is not known, RuO4− is readily protonated by weak acids such as AcOH to give [RuO3(OH)(AcO)−], which readily undergoes HAT with c-C6H12 with a barrier that is lower than that of RuO4− by >10 kcal mol−1 (Table 2).25 In contrast, MnO4− is not protonated by AcOH. Nevertheless, AcOH can still exert a moderate activating effect on HAT by MnO4− through H-bonding with the oxo ligands, with the activation barrier lowered by 6 kcal mol−1 compared with MnO4− alone. On the other hand, Ca2+ has a higher activating effect on MnO4− than on RuO4− (lowering of the barrier is 10 vs. 8 kcal mol−1). Remarkably, for both complexes the presence of AcOH and Ca2+ together shows a similar synergistic activating effect, resulting in a lowering of the HAT barrier by 16 and 13 kcal mol−1 for RuO4− and MnO4−, respectively. DFT calculations show that Ca(II) forms a MO4−/AcOH/Ca(II) active intermediate by binding to AcO−/AcOH and an oxo ligand (Fig. 12). , kcal mol−1) for the oxidation of c-C6H12 by RuO4− and MnO4− under various conditions
, kcal mol−1) for the oxidation of c-C6H12 by RuO4− and MnO4− under various conditions
| RuO4− | MnO4− | |
|---|---|---|
| No additive | 26.8 | 22.7 |
| AcOH | 15.2 | 16.8 |
| Ca2+ | 18.5 | 12.7 |
| AcOH + Ca2+ | 10.8 | 9.7 |
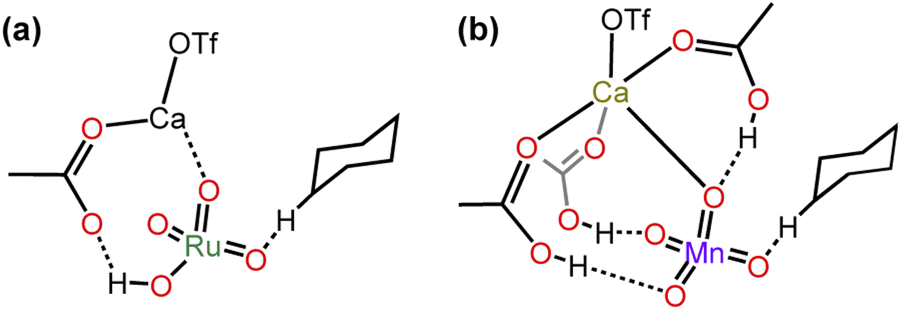 | ||
| Fig. 12 Structures of the intermediates (a) RuO4−/AcOH/Ca(OTf)2 and (b) MnO4−/AcOH/Ca(OTf)2. | ||
Conclusions
In conclusion, we have demonstrated synergistic activating effects of a weak Brønsted acid such as acetic acid and a weak Lewis acid such as Ca2+ on MnO4−. Although MnO4− is rather non-basic, it can readily be activated by AcOH + Ca2+ toward HAT of c-C6H12, resulting in a lowering of the barrier by 13 kcal mol−1. Remarkably, such a cooperative effect on MnO4− is similar to that on RuO4−, despite a great difference in the basicity of the two metal-oxo species, which should lead to vastly different affinities toward acids. Our studies suggest that such a synergistic activation is a general phenomenon which may occur in various other metal-oxo species.The combination of a weak Brønsted and a weak Lewis acid is a potentially useful strategy for the activation of a metal-oxo species toward oxidation of various organic compounds, particularly for substrates that may contain functional groups that are sensitive to strong acids. It is possible that this synergistic activating effect on metal-oxo species may also occur in biological systems, where only relatively mild Brønsted acids and Lewis acids (metal ions) are present.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
C. K. M. designed and carried out the experiments, analysed the data and wrote the manuscript. T. C. L. designed the experiments and wrote the manuscript. H. S. performed the experiments and helped in the analysis of the data and preparation of the manuscript. L. C., Y. P. and K. C. L. performed computational studies and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 26/09/2022, which contained errors in the position of the equilibrium constants in equations 1, 4, 7 and 8.Acknowledgements
This work was supported by the NSFC/RGC Joint Research Scheme (N_CityU111/20) and the Laboratory for Synthetic Chemistry and Chemical Biology Limited, LSCCB. The computational studies were carried out using the computational facilities at Burgundy at City University of Hong Kong.References
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2887 CrossRef CAS PubMed.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- X. P. Zhang, A. Chandra, Y. M. Lee, R. Cao, K. Ray and W. Nam, Chem. Soc. Rev., 2021, 50, 4804–4811 RSC.
- Y. Y. Liu and T. C. Lau, J. Am. Chem. Soc., 2019, 141, 3755–3766 CrossRef CAS PubMed.
- T. Devi, Y. M. Lee, W. Nam and S. Fukuzumi, Coord. Chem. Rev., 2020, 410, 213219 CrossRef CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–U65 CrossRef CAS.
- H. X. Du, P. K. Lo, Z. M. Hu, H. J. Liang, K. C. Lau, Y. N. Wang, W. W. Y. Lam and T. C. Lau, Chem. Commun., 2011, 47, 7143–7145 RSC.
- R. A. Baglia, C. M. Krest, T. Yang, P. Leeladee and D. P. Goldberg, Inorg. Chem., 2016, 55, 10800–10809 CrossRef CAS PubMed.
- J. Chen, Y. M. Lee, K. M. Davis, X. Wu, M. S. Seo, K. B. Cho, H. Yoon, Y. J. Park, S. Fukuzumi, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2013, 135, 6388–6391 CrossRef CAS PubMed.
- H. Yoon, Y. M. Lee, X. Wu, K. B. Cho, R. Sarangi, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 9186–9194 CrossRef CAS.
- Z. Q. Chen, L. Yang, C. Choe, Z. N. Lv and G. C. Yin, Chem. Commun., 2015, 51, 1874–1877 RSC.
- C. Choe, L. Yang, Z. A. Lv, W. L. Mo, Z. Q. Chen, G. X. Li and G. C. Yin, Dalton Trans., 2015, 44, 9182–9192 RSC.
- Y. Morimoto, H. Kotani, J. Park, Y. M. Lee, W. Nam and S. Fukuzumri, J. Am. Chem. Soc., 2011, 133, 403–405 CrossRef CAS PubMed.
- J. Park, Y. Morimoto, Y. M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 5236–5239 CrossRef CAS PubMed.
- S. M. Yiu, W. L. Man and T. C. Lau, J. Am. Chem. Soc., 2008, 130, 10821–10827 CrossRef CAS PubMed.
- W. W. Y. Lam, S.-M. Yiu, J. M. N. Lee, S. K. Y. Yau, H.-K. Kwong, T.-C. Lau, D. Liu and Z. Lin, J. Am. Chem. Soc., 2006, 128, 2851–2858 CrossRef CAS.
- T.-C. Lau, Z.-B. Wu, Z.-L. Bai and C.-K. Mak, J. Chem. Soc., Dalton Trans., 1995, 695–696 RSC.
- T. Devi, Y. M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2018, 140, 8372–8375 CrossRef CAS PubMed.
- T. Devi, Y. M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2020, 142, 365–372 CrossRef CAS PubMed.
- E. Y. Tsui and T. Agapie, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10084–10088 CrossRef CAS PubMed.
- E. Y. Tsui, R. Tran, J. Yano and T. Agapie, Nat. Chem., 2013, 5, 293–299 CrossRef CAS PubMed.
- M. M. Najafpour, T. Ehrenberg, M. Wiechen and P. Kurz, Angew. Chem., Int. Ed., 2010, 49, 2233–2237 CrossRef CAS PubMed.
- G. Chen, L. Ma, P. K. Lo, C. K. Mak, K. C. Lau and T. C. Lau, Chem. Sci., 2021, 12, 632–638 RSC.
- K. A. Gardner, L. L. Kuehnert and J. M. Mayer, Inorg. Chem., 1997, 36, 2069–2078 CrossRef CAS PubMed.
- S. J. Hawkes, J. Chem. Educ., 1996, 73, 516–517 CrossRef CAS.
- D. G. Lee and J. F. Perez-Benito, J. Org. Chem., 1988, 53, 5725–5728 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- N. Bailey, A. Carrington, K. A. K. Lott and M. C. R. Symons, J. Chem. Soc., 1960, 290–297 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03089f |
| ‡ These authors have equal contribution. |
| This journal is © The Royal Society of Chemistry 2022 |