
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of oxaboranes via nickel-catalyzed dearylative cyclocondensation†
Mason T.
Koeritz‡
,
Haley K.
Banovetz‡
,
Sean A.
Prell
and
Levi M.
Stanley
 *
*
Department of Chemistry, Iowa State University, Ames, Iowa 50011, USA. E-mail: lstanley@iastate.edu
First published on 20th May 2022
Abstract
We report Ni-catalyzed dearylative cyclocondensation of aldehydes, alkynes, and triphenylborane. The reaction is initiated by oxidative cyclization of the aldehyde and alkyne coupling partners to generate an oxanickelacyclopentene which reacts with triphenylborane to form oxaboranes. This formal dearylative cyclocondensation reaction generates oxaboranes in moderate-to-high yields (47–99%) with high regioselectivities under mild reaction conditions. This approach represents a direct and modular synthesis of oxaboranes which are difficult to access using current methods. These oxaboranes are readily transformed into valuable building blocks for organic synthesis and an additional class of boron heterocycles. Selective homocoupling forms oxaboroles, oxidation generates aldol products, and reduction and arylation form substituted allylic alcohols.
Introduction
Nickel-catalyzed oxidative cyclization reactions of two π-components enable facile generation of nickelacycles with formation of a new C–C bond.1–6 These reactions encompass a broad array of suitable π-components that provide access to a range of substituted nickelacycles. Among these π-components, nickel-catalyzed oxidative cyclizations of aldehydes and alkynes are established methods to generate an array of oxanickelacyclopentenes.2 These oxanickelacyclopentenes are versatile intermediates that can be transformed into a variety of synthetically valuable building blocks. Early studies in this area focus on reductive and alkylative coupling reactions to form substituted allylic alcohols (Scheme 1a, path A).2a,b Subsequently, Montgomery and coworkers discovered that oxanickelacyclopentenes react with diethylsilane to form stable oxasilacyclopentenes via dehydrogenative cyclocondensation reactions (Scheme 1a, path B).7 Ogoshi and coworkers reported related dealkylative cyclocondensation reactions of oxanickelacyclopentenes with trimethylaluminum to form oxaaluminacyclopentenes (Scheme 1a, path C).8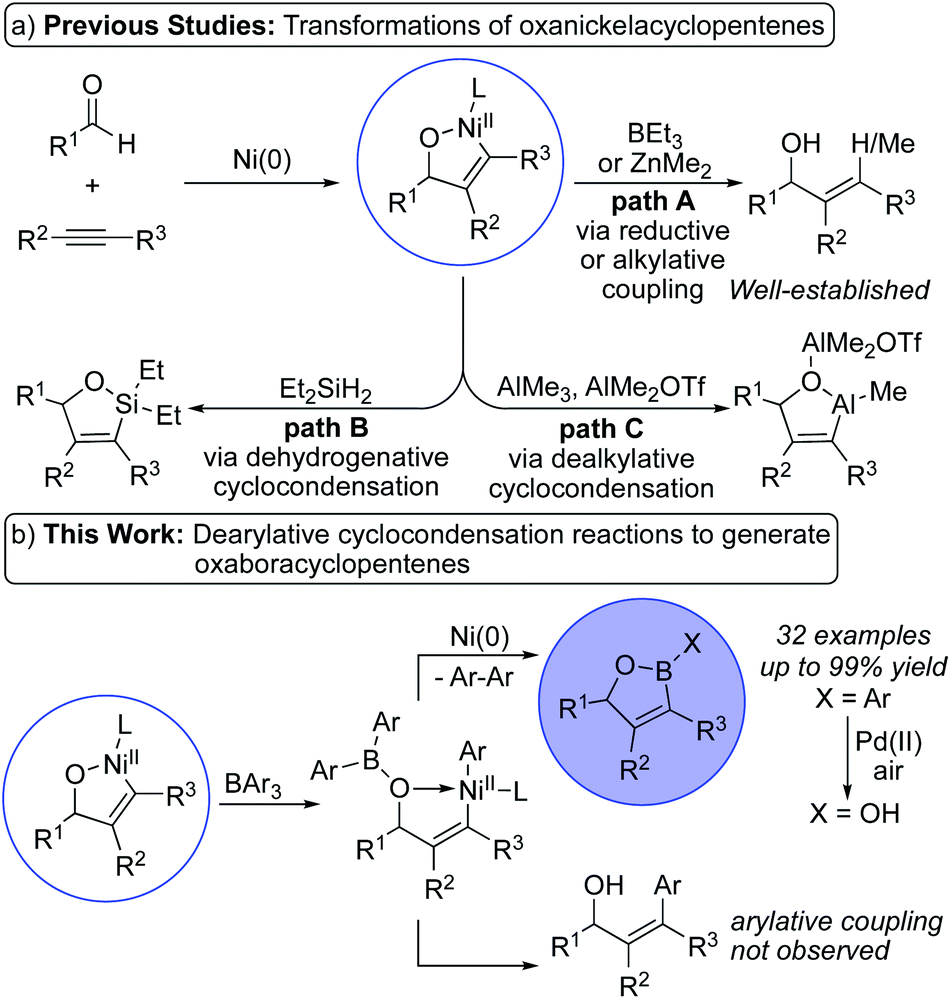 | ||
| Scheme 1 Reactivity of oxanickelacycles. | ||
Oxaboranes9 (X = Ar), oxaboroles10 (X = OH), and related boron heterocycles11 have garnered increased interest in recent years due to their biological activities,12 optical properties,13 and utility as synthetic building blocks. The ability to leverage the reactivity of oxaboracyclopentenes would enable rapid and tailorable syntheses of these boron heterocycles. However, dehydrogenative cyclocondensation reactions analogous to the synthesis of oxasilacyclopentenes would be challenging with dihydridoborane or trihydridoborane reagents due to competing hydroboration of the alkyne component.14 Analogous reactions with a trialkylborane lead to reductive coupling instead of dealkylative cyclocondensation.2b
We envisioned dearylative cyclocondensation reactions of aldehydes, alkynes, and a triarylborane as direct, catalytic syntheses of oxaboracyclopentenes (Scheme 1b, top). The use of a triarylborane reagent eliminates the potential for reductive coupling to form allylic alcohols as shown in Scheme 1a. This approach also eliminates the need for complex starting materials,10g strongly basic reaction conditions,10d and multi-step synthetic procedures.10e,f The development of dearylative cyclocondensation reactions to form oxaboracyclopentenes would provide a straightforward and modular entry into these boron heterocycles that addresses the limitations of current synthetic methods. Key to the development of these dearylative cyclocondensation reactions is the identification of a nickel catalyst that mitigates the potential for arylative coupling to form substituted allylic alcohols (Scheme 1b, bottom).3c Herein, we report the first nickel-catalyzed couplings of aldehydes, alkynes, and triphenylborane to form oxaboracyclopentenes in high yields with excellent regio- and chemoselectivities.
Results and discussion
In our initial studies, we found that the reaction of benzaldehyde 1a, 1-phenyl-1-propyne 2a, and triphenylborane in the presence of a catalyst generated from Ni(cod)2 and PBu3 produced oxaborane 3a in 70% yield and a 13.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 regioisomeric ratio (eqn (1)). Additionally, we observed the formation of equal amounts of biphenyl in the reaction with no observable arylative coupling product. This result is consistent with a reaction that proceeds via the dearylative cyclocondensation pathway shown in Scheme 1b.
:1 regioisomeric ratio (eqn (1)). Additionally, we observed the formation of equal amounts of biphenyl in the reaction with no observable arylative coupling product. This result is consistent with a reaction that proceeds via the dearylative cyclocondensation pathway shown in Scheme 1b.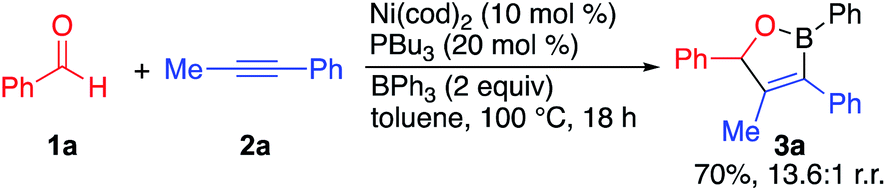 | (1) |
 | (2) |
We next chose to leverage design of experiment (DoE) to increase the yield of the reaction and lower the catalyst loading.15 We first evaluated categorical factors (ligand and solvent) in the reaction (Table S1†) and found that a catalyst generated from Ni(cod)2 and PBu3 produced oxaborane product 3a in quantitative yields when THF was used as the solvent. However, we recognized the potential to use DoE to further optimize the reaction by lowering the loadings of catalyst and the organoboron reagent (eqn (2)). We evaluated the level of significance of four continuous factors (catalyst loading, reaction concentration, temperature, and loading of the organoboron reagent, Scheme S1 and Table S2†) in 14 reactions using a linear model DoE. The results of this DoE showed that temperature and catalyst loading did not have a significant impact on the yield of the reaction, but the yield of the reaction is positively correlated with increased loadings of triphenylborane. We then performed a quadratic model DoE consisting of 15 reactions to further optimize our dearylative cyclocondensation reaction (Scheme S2 and Table S3†). This set of reactions showed that the loading of catalyst could be reduced if the loading of triphenylborane is increased (Fig. 1a). In addition, higher reaction temperatures generally lead to lower yields of the oxaborane product across a range of catalyst loadings (Fig. 1b). Based on the response surfaces in Fig. 1, we chose 5 mol% catalyst and 1.5 equivalents BPh3 at 50 °C as reaction conditions to evaluate the scope of the dearylative cyclocondensation reaction.
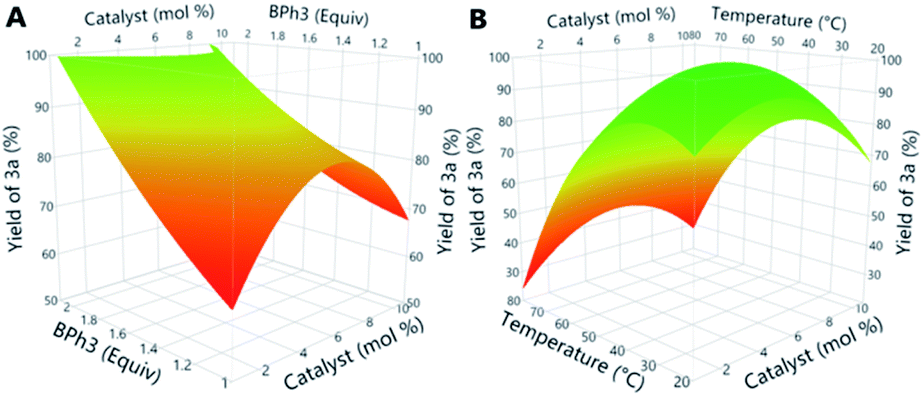 | ||
| Fig. 1 Response surface analysis of quadratic design of experiment. (A) Yield of 3aversus catalyst loading and equivalents of triphenylborane at 50 °C. (B) Yield of 3aversus catalyst loading and temperature with 1.5 equivalents of triphenylborane. | ||
We initially evaluated the scope of the dearylative cyclocondensation reaction with respect to the alkyne coupling partner (Scheme 2). The reaction of benzaldehyde with 1-aryl-1-hexyne derivatives 2b–2i bearing electron-neutral, electron-donating, and electron-withdrawing para-substituents generated oxaborane products 3b–3i in 73–98% yield with up to 11.4:1 regioisomeric ratios. A phenolic hydroxyl group was also tolerated in the reaction, with the reaction of alkyne 2i producing oxaborane 3i in 85% yield and 9.1:1 r.r. The reactions of alkynes containing meta-substituted aryl groups lead to oxaborane products 3j and 3k in 89% and 72% yield, respectively. The reaction of 1-(2-methylphenyl)-1-hexyne 2l formed oxaborane 3l in 80% yield and >20:1 regioselectivity as a 1.1:1 ratio of diastereomers when the reaction temperature was increased to 70 °C. The reaction of 2-(hex-1-yn-1-yl)naphthalene 2m generated oxaborane 3m in 99% yield and 11.1:1 r.r. A heteroaromatic ring was also tolerated in the reaction with benzofuran-derived alkyne 2n leading to oxaborane 3n in 90% yield. The reaction of enyne 2o formed oxaborane 3o in 40% isolated yield with 3.1:1 regioselectivity. The lower regioselectivity is presumably due to competing coordinating effects of the alkene.16 The reaction of benzaldehyde with diphenylacetylene produced oxaborane product 3p in 84% isolated yield. Aliphatic alkynes are also tolerated in the reaction, with reactions of 3-hexyne and 4-octyne leading to oxaboranes 3q and 3r in 80% and 94% yield, respectively. Limitations of the dearylative cyclocondensation reactions include: (1) aromatic aldehydes bearing strongly electron-withdrawing substituents, such as nitro groups; (2) alkynes containing bulky substituents, such as trimethylsilyl groups, and (3) terminal alkynes due to competing cyclotrimerization processes.2b,d,3b,17
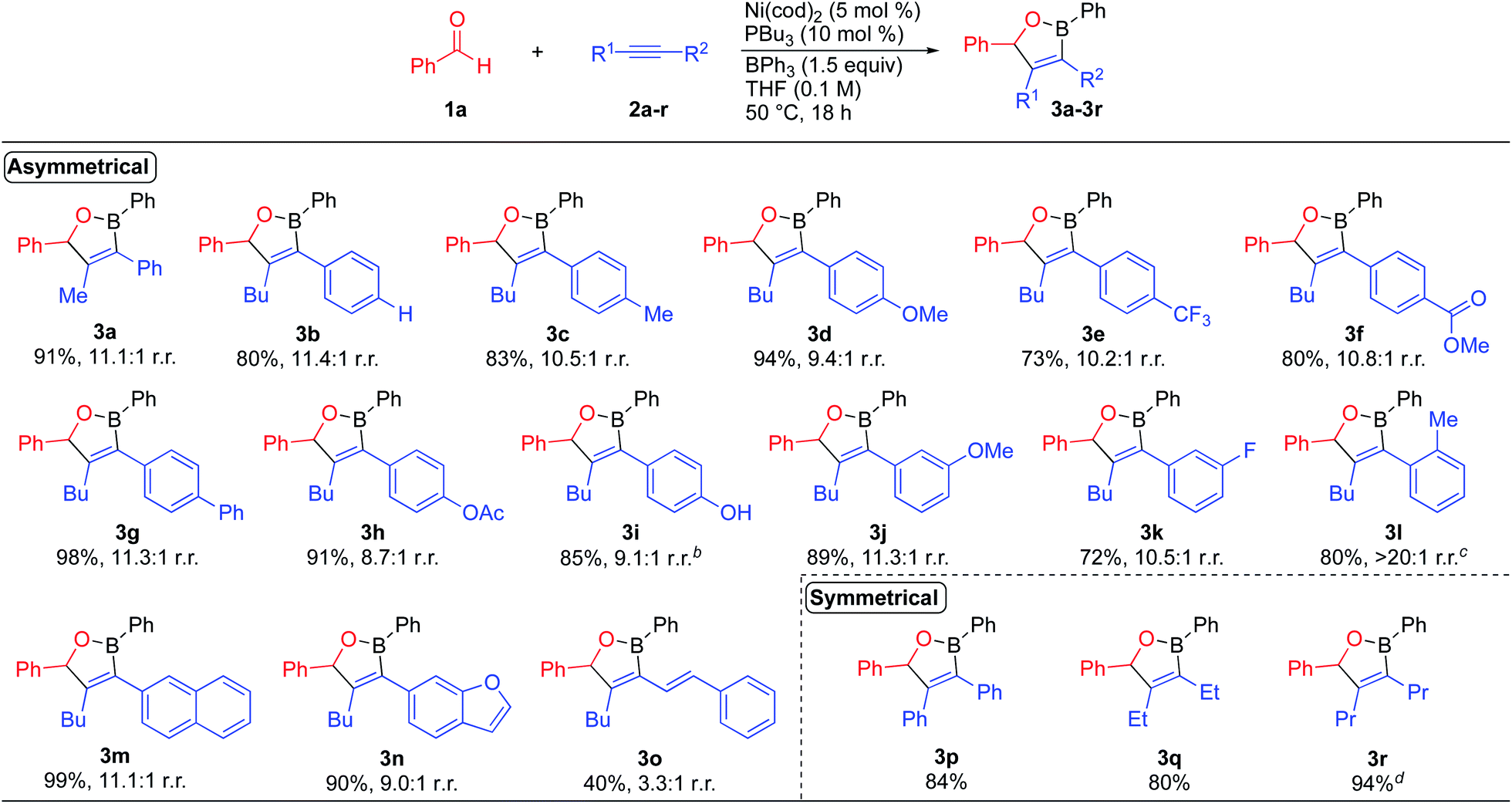 | ||
| Scheme 2 Alkyne substrate scope for the nickel-catalyzed dearylative cyclocondensations. aReaction conditions: 1a (0.200 mmol), 2 (0.220 mmol), BPh3 (0.300 mmol), Ni(cod)2 (0.010 mmol), PBu3 (0.020 mmol), THF (2.0 mL, 0.10 M), 50 °C, 18 h. bReaction run with 10 mol% Ni(cod)2, 20 mol% PBu3, 1.5 equiv. alkyne, and 2 equiv. BPh3. cReaction run at 70 °C. dReaction run with 2.5 mol% catalyst. | ||
We next turned our attention to establishing the scope of aldehyde coupling partners (Scheme 3). While investigating the scope of aldehydes with 4-octyne as the alkyne coupling partner, we found that only 2.5 mol% of the catalyst was required to form oxaboranes derived from aromatic aldehydes containing electron-donating and electron-withdrawing substituents. These reactions lead to the formation of oxaboranes 3s–3ab in good to excellent yields. In general, reactions of benzaldehyde derivatives bearing electron-donating substituents occur at lower temperatures and form oxaboranes in higher yields, while reactions of benzaldehyde derivatives bearing electron-withdrawing substituents require higher temperatures to generate the corresponding oxaboranes, such as 3w, 3x, and 3aa, in good yields. 2-Naphthaldehyde was also well tolerated, and this reaction formed oxaborane 3ac in 70% yield. The dearylative cyclocondensation reaction also tolerates heteroaromatic aldehydes, such as furfural and 2-thiophenecarboxaldehyde, generating oxaboranes 3ad and 3ae in 63% and 47% yield, respectively. Pivaldehyde also proved to be a viable substrate, and this reaction generated oxaborane 3af in 47% yield. However, aliphatic aldehydes containing alpha-hydrogens, such as cyclohexanecarboxaldehyde and 3-phenylpropanal, proved to be challenging substrates due to competing aldehyde oligomerization.2a,18 Reactions of these aldehydes typically formed the corresponding oxaborane products in <15% yield.
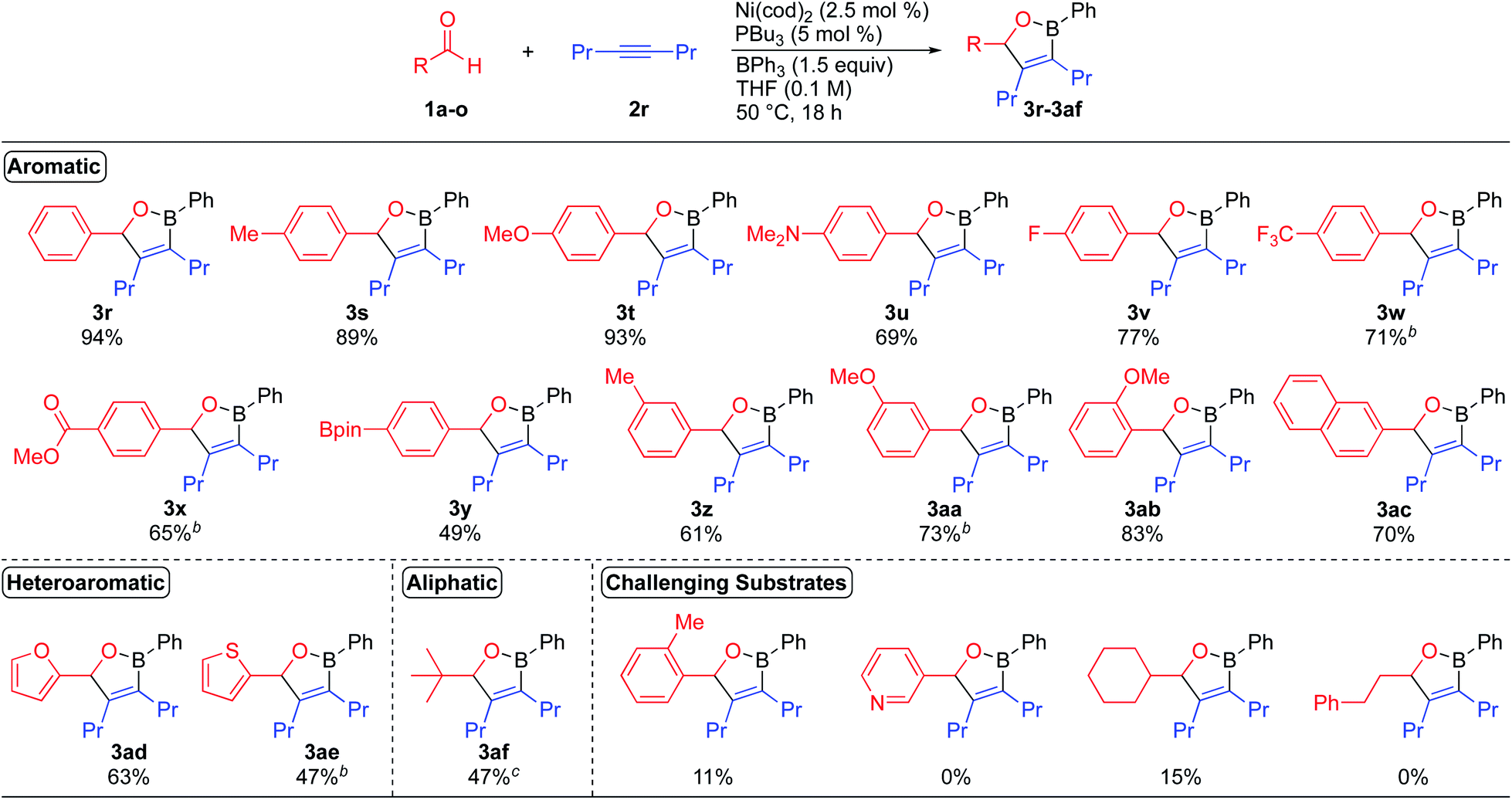 | ||
| Scheme 3 Aldehyde substrate scope for the nickel-catalyzed dearylative cyclocondensations. aReaction conditions: 1 (0.200 mmol), 2r (0.220 mmol), BPh3 (0.300 mmol), Ni(cod)2 (0.005 mmol), PBu3 (0.010 mmol), THF (2.0 mL, 0.10 M), 50 °C, 18 h. bReaction run at 70 °C. cReaction run with 10 mol% catalyst. | ||
The dearylative cyclocondensation reaction is currently limited to triphenylborane as the organoboron component. Reactions of other triarylboranes led to significantly lower yields of the corresponding oxaborane products. For example, the reaction of benzaldehyde, 1-phenyl-1-propyne, and tris(4-methoxyphenyl)borane formed the corresponding oxaborane in <20% yield.
The utility of the dearylative cyclocondensation reaction to generate oxaboranes is shown in Scheme 4. The gram-scale reaction of benzaldehyde (4.0 mmol) and 1-phenyl-1-propyne (4.4 mmol) occurs to form oxaborane 3a in 97% yield with an 11.8:1 regioisomeric ratio (Scheme 4a). We then conducted a series of transformations of oxaborane 3a to demonstrate oxaboranes as versatile synthetic intermediates (Scheme 4b).10e Oxidation of oxaborane 3a with sodium perborate produced β-hydroxy ketone 4 in 93% yield and 1.5:1 d.r.19 The sequence of dearylative cyclocondensation and oxidation provides an alternative pathway to cross-aldol addition reactions. Protodeboronation of oxaborane 3a leads to allylic alcohol 5 in 81% yield.20
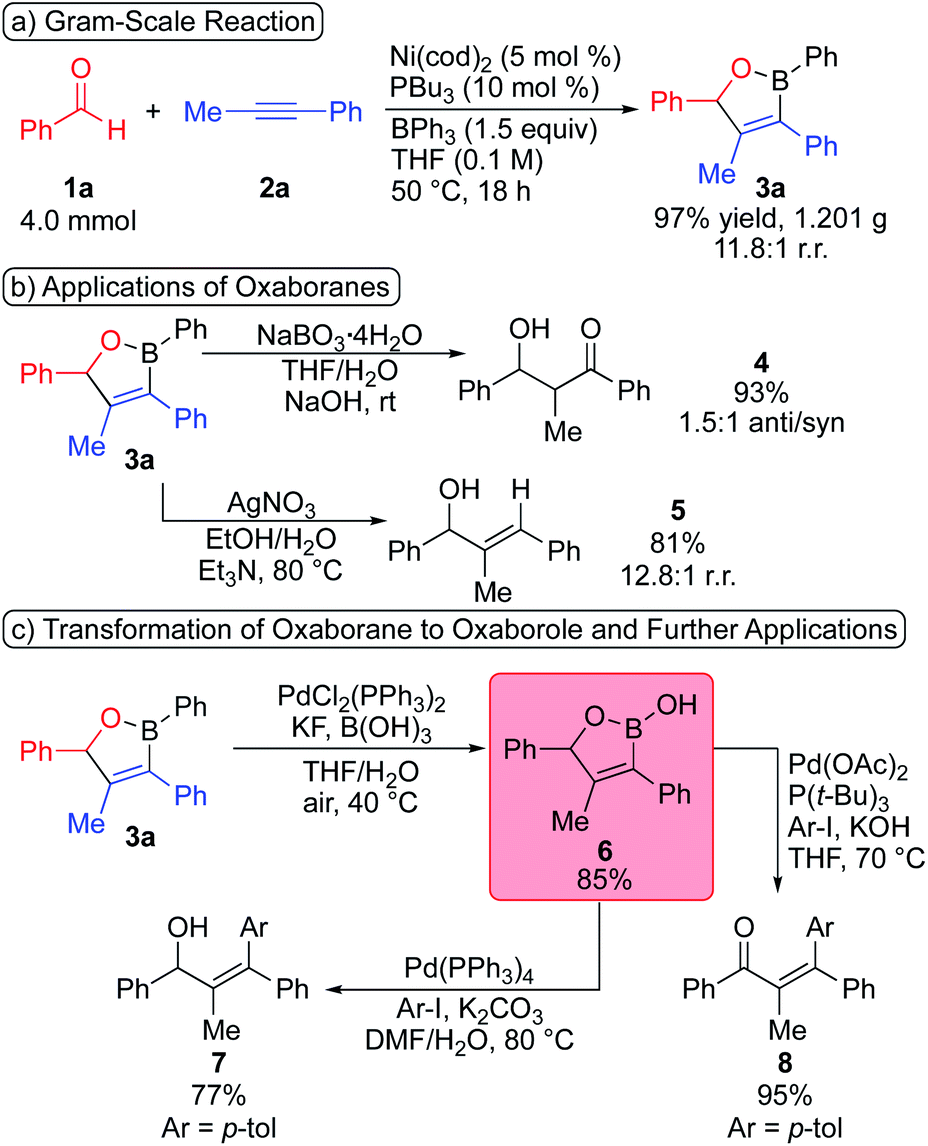 | ||
| Scheme 4 Synthetic transformations of oxaboranes. | ||
We then attempted to transform our oxaborane product to an oxaborole. Dong and coworkers previously reported transformation of oxaborinanes to cyclic hydroxyl boronates via hydrolysis in the presence of H2O/MeOH.11d However, oxaborane 3a is unreactive under analogous hydrolysis conditions. We were pleased to observe that oxaborole 6 can be generated in 85% yield by selective homocoupling of oxaborane 3a in the presence of PdCl2(PPh3)2 (Scheme 4c).21 The modularity of our oxaborane synthesis and ability to transform oxaboranes to oxaboroles provide access to the biologically active oxaborole motif with high levels of customizability. Additionally, these oxaboroles can also be utilized as versatile synthetic intermediates. Oxaborole 6 undergoes Suzuki–Miyaura coupling with 4-iodotoluene to form highly substituted allylic alcohol 7 in 77% yield when Pd(PPh3)4 is used as a precatalyst.22 α,β-Unsaturated ketone 8 is formed in 95% yield through a sequence of Suzuki–Miyaura coupling and oxidation when the catalyst is generated from Pd(OAc)2 and P(t-Bu)3.23
Based on mechanisms proposed by Montgomery and Ogoshi for nickel-catalyzed dehydrogenative7 and dealkylative8,24 cyclocondensation reactions, we propose the following plausible catalytic cycles for this dearylative cyclocondensation reaction (Scheme 5). Coordination of the Ni(0) catalyst to the alkyne and aldehyde substrates forms intermediate I. Oxidative cyclization generates oxanickelacyclopentene intermediate II. Transmetalation with triphenylborane forms vinyl-Ni(II)-aryl intermediate III. We envision two potential pathways to generate oxaborane 3 from intermediate III. One potential pathway involves a second intramolecular transmetalation to form diaryl-Ni(II) intermediate IV with release of the oxaborane product 3. Subsequent reductive elimination from IV generates biphenyl and reforms the Ni(0) catalyst. Alternatively, sigma-bond metathesis of the Ni–Ph and B–Ph bonds would lead to direct extrusion of biphenyl from intermediate III to form six-membered nickelacycle V. Reductive elimination of the vinyl and boryl ligands generates oxaborane product 3 and regenerates the Ni(0) catalyst.
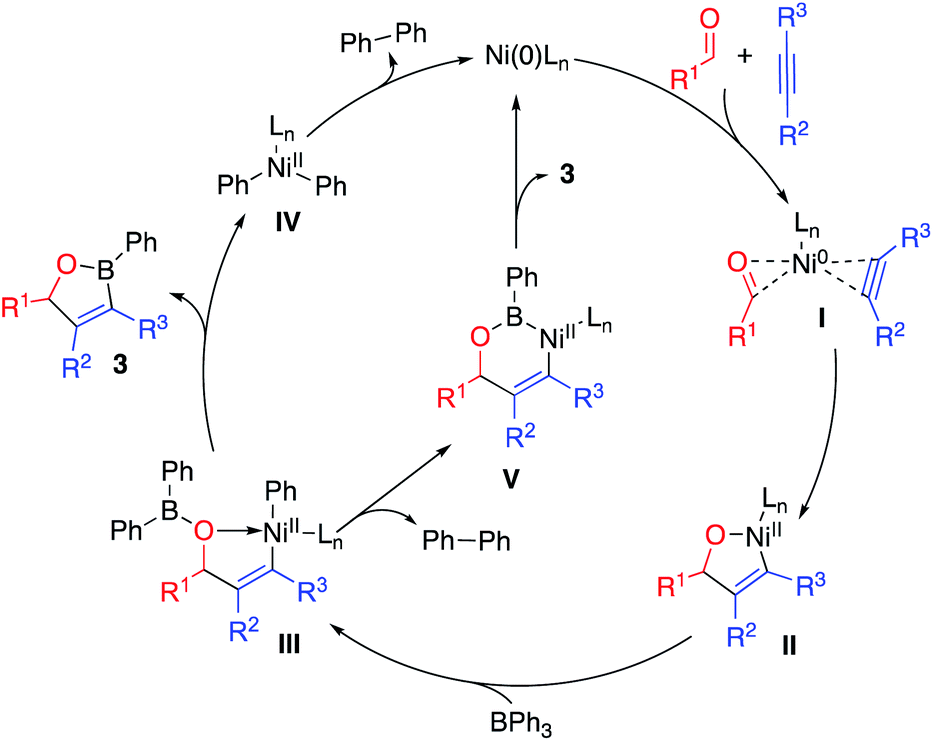 | ||
| Scheme 5 Proposed catalytic cycle. | ||
Conclusions
In summary, we have developed a Ni-catalyzed dearylative cyclocondensation of aldehydes, alkynes, and triphenylborane. This reaction forms C–C and C–B bonds to generate a variety of oxaborane products in up to 99% yield under mild reaction conditions. These oxaborane products are readily transformed into oxaboroles, β-hydroxy ketones, a variety of substituted allylic alcohols, and highly substituted α,β-unsaturated ketones. Further studies to determine the mechanism and expand the scope of dearylative cyclocondensation reactions are ongoing in our laboratory.Data availability
The data for this work, including optimization tables, experimental procedures, and characterization data for all compounds are provided in the ESI.†Author contributions
M. T. K. and H. K. B. contributed equally to this work. M. T. K. and L. M. S. conceived the project. M. T. K. optimized the reaction conditions and performed design of experiment studies. M. T. K., H. K. B., and S. A. P. synthesized starting materials, evaluated the scope of the reaction, and completed synthetic applications. M. T. K., H. K. B., and L. M. S. wrote the manuscript with input from S. A. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Foundation-CHE (grant no. 1955529) for supporting this work. We gratefully acknowledge Prof. Junqi Li (Iowa State University) for her expertise and advice regarding organoboron compounds.Notes and references
- For review of nickel-catalyzed oxidative cyclization reactions, see: (a) J. Montgomery, Angew. Chem., Int. Ed., 2004, 43, 3890–3908 CrossRef CAS PubMed; (b) Y. Hoshimoto, M. Ohashi and S. Ogoshi, Acc. Chem. Res., 2015, 48, 1746–1755 CrossRef CAS PubMed; (c) S. Ogoshi, Bull. Chem. Soc. Jpn., 2017, 90, 1401–1406 CrossRef CAS; (d) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed.
- For select examples of nickel-catalyzed oxidative cyclization reactions involving aldehydes and alkynes, see: (a) E. Oblinger and J. Montgomery, J. Am. Chem. Soc., 1997, 119, 9065–9066 CrossRef CAS; (b) W.-S. Huang, J. Chan and T. F. Jamison, Org. Lett., 2000, 2, 4221–4223 CrossRef CAS PubMed; (c) E. A. Colby and T. F. Jamison, J. Org. Chem., 2003, 68, 156–166 CrossRef CAS PubMed; (d) K. M. Miller, W.-S. Huang and T. F. Jamison, J. Am. Chem. Soc., 2003, 125, 3442–3443 CrossRef CAS PubMed; (e) G. M. Mahandru, G. Liu and J. Montgomery, J. Am. Chem. Soc., 2004, 126, 3698–3699 CrossRef CAS PubMed; (f) S. Ogoshi, T. Arai, M. Ohashi and H. Kurosawa, Chem. Commun., 2008, 1347–1349, 10.1039/B717261C; (g) P. R. McCarren, P. Liu, P. H.-Y. Cheong, T. F. Jamison and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 6654–6655 CrossRef CAS PubMed; (h) H. Wang, G. Lu, G. J. Sormunen, H. A. Malik, P. Liu and J. Montgomery, J. Am. Chem. Soc., 2017, 139, 9317–9324 CrossRef CAS PubMed.
- For select examples of nickel-catalyzed oxidative cyclization reactions involving alkynes and other π-components, see: (a) J. Montgomery and A. V. Savchenko, J. Am. Chem. Soc., 1996, 118, 2099–2100 CrossRef CAS; (b) C. Molinaro and T. F. Jamison, J. Am. Chem. Soc., 2003, 125, 8076–8077 CrossRef CAS PubMed; (c) S. J. Patel and T. F. Jamison, Angew. Chem., Int. Ed., 2003, 42, 1364–1367 CrossRef CAS PubMed; (d) S. Ogoshi, H. Ikeda and H. Kurosawa, Angew. Chem., Int. Ed., 2007, 46, 4930–4932 CrossRef PubMed; (e) X. You, X. Xie, G. Wang, M. Xiong, R. Sun, H. Chen and Y. Liu, Chem.–Eur. J., 2016, 22, 16765–16769 CrossRef CAS PubMed.
- For select examples of nickel-catalyzed oxidative cyclization reactions involving alkenes, see: (a) S. Ogoshi, M. Ueta, T. Arai and H. Kurosawa, J. Am. Chem. Soc., 2005, 127, 12810–12811 CrossRef CAS PubMed; (b) M. Ohashi, M. Ikawa and S. Ogoshi, Organometallics, 2011, 30, 2765–2774 CrossRef CAS; (c) Y. Hoshimoto, Y. Hayashi, H. Suzuki, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2012, 51, 10812–10815 CrossRef CAS PubMed; (d) Y. Hayashi, Y. Hoshimoto, R. Kumar, M. Ohashi and S. Ogoshi, Chem. Commun., 2016, 52, 6237–6240 RSC; (e) H. Shirataki, M. Ohashi and S. Ogoshi, Eur. J. Org. Chem., 2019, 2019, 1883–1887 CrossRef CAS; (f) W.-M. Feng, T.-Y. Li, L.-J. Xiao and Q.-L. Zhou, Org. Lett., 2021, 23, 7900–7904 CrossRef CAS PubMed.
- For select examples of nickel-catalyzed oxidative cyclization reactions involving dienes, see: (a) S. Ogoshi, K.-i. Tonomori, M.-a. Oka and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 7077–7086 CrossRef CAS PubMed; (b) M. Morimoto, Y. Nishida, T. Miura and M. Murakami, Chem. Lett., 2013, 42, 550–552 CrossRef CAS; (c) Y.-Q. Li, G. Chen and S.-L. Shi, Org. Lett., 2021, 23, 2571–2577 CrossRef PubMed.
- For an example of nickel-catalyzed oxidative cyclization reaction involving allenes, see: W. Li, N. Chen and J. Montgomery, Angew. Chem., Int. Ed., 2010, 49, 8712–8716 CrossRef CAS PubMed.
- R. D. Baxter and J. Montgomery, J. Am. Chem. Soc., 2008, 130, 9662–9663 CrossRef CAS PubMed.
- M. Ohashi, H. Saijo, T. Arai and S. Ogoshi, Organometallics, 2010, 29, 6534–6540 CrossRef CAS.
- J. Chen, X. Fang, Z. Bajko, J. W. Kampf and A. J. Ashe, Organometallics, 2004, 23, 5088–5091 CrossRef CAS.
- (a) G.-H. Fang, Z.-J. Yan, J. Yang and M.-Z. Deng, Synthesis, 2006, 2006, 1148–1154 Search PubMed; (b) Y. Nagashima, K. Hirano, R. Takita and M. Uchiyama, J. Am. Chem. Soc., 2014, 136, 8532–8535 CrossRef CAS PubMed; (c) G. Lafitte, K. Kunihiro, C. Bonneaud, B. Dréan, F. Gaigne, V. Parnet, R. Pierre, C. Raffin, R. Vatinel, J.-F. Fournier, B. Musicki, G. Ouvry, C. Bouix-Peter, L. Tomas and C. S. Harris, Tetrahedron Lett., 2017, 58, 3757–3759 CrossRef CAS; (d) M. Nogami, K. Hirano, M. Kanai, C. Wang, T. Saito, K. Miyamoto, A. Muranaka and M. Uchiyama, J. Am. Chem. Soc., 2017, 139, 12358–12361 CrossRef CAS PubMed; (e) L.-J. Cheng and N. P. Mankad, Angew. Chem., Int. Ed., 2018, 57, 10328–10332 CrossRef CAS PubMed; (f) R. Fritzemeier, A. Gates, X. Guo, Z. Lin and W. L. Santos, J. Org. Chem., 2018, 83, 10436–10444 CrossRef CAS PubMed; (g) M.-B. Li, D. Posevins, A. Geoffroy, C. Zhu and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2020, 59, 1992–1996 CrossRef CAS PubMed.
- (a) H. Saito, S. Otsuka, K. Nogi and H. Yorimitsu, J. Am. Chem. Soc., 2016, 138, 15315–15318 CrossRef CAS PubMed; (b) F. Yang, M. Zhu, J. Zhang and H. Zhou, Med. Chem. Commun., 2018, 9, 201–211 RSC; (c) S. Tsuchiya, H. Saito, K. Nogi and H. Yorimitsu, Org. Lett., 2019, 21, 3855–3860 CrossRef CAS PubMed; (d) H. Lyu, I. Kevlishvili, X. Yu, P. Liu and G. Dong, Science, 2021, 372, 175–182 CrossRef CAS PubMed; (e) H. E. Hackney and D. G. Hall, ChemPhotoChem, 2022, e202100219, DOI:10.1002/cptc.202100219.
- (a) F. L. Rock, W. Mao, A. Yaremchuk, M. Tukalo, T. Crépin, H. Zhou, Y.-K. Zhang, V. Hernandez, T. Akama, S. J. Baker, J. J. Plattner, L. Shapiro, S. A. Martinis, S. J. Benkovic, S. Cusack and M. R. K. Alley, Science, 2007, 316, 1759–1761 CrossRef CAS PubMed; (b) S. J. Baker, C. Z. Ding, T. Akama, Y.-K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275–1288 CrossRef CAS PubMed; (c) S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279–4285 RSC; (d) D. D. Dixon, J. W. Lockner, Q. Zhou and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 8432–8435 CrossRef CAS PubMed; (e) X. Li, Y.-K. Zhang, J. J. Plattner, W. Mao, M. R. K. Alley, Y. Xia, V. Hernandez, Y. Zhou, C. Z. Ding, J. Li, Z. Shao, H. Zhang and M. Xu, Bioorg. Med. Chem. Lett., 2013, 23, 963–966 CrossRef CAS PubMed.
- (a) M. Nogami, K. Hirano, K. Morimoto, M. Tanioka, K. Miyamoto, A. Muranaka and M. Uchiyama, Org. Lett., 2019, 21, 3392–3395 CrossRef CAS PubMed; (b) M. Nakano, R. Nakamura, Y. Sumida, K. Nagao, T. Furuyama, F. Inagaki and H. Ohmiya, Chem. Pharm. Bull., 2021, 69, 526–528 CrossRef CAS PubMed.
- (a) G. Zweifel, G. M. Clark and N. L. Polston, J. Am. Chem. Soc., 1971, 93, 3395–3399 CrossRef CAS; (b) N. W. J. Ang, C. S. Buettner, S. Docherty, A. Bismuto, J. R. Carney, J. H. Docherty, M. J. Cowley and S. P. Thomas, Synthesis, 2018, 50, 803–808 CrossRef CAS.
- For select examples of design of experiment in organic synthesis, see: (a) Design and Optimization in Organic Synthesis, ed. R. Carlson and J. E. Carlson, Elsevier B. V., Amsterdam, Netherlands, 2005 Search PubMed; (b) S. E. Denmark and C. R. Butler, J. Am. Chem. Soc., 2008, 130, 3690–3704 CrossRef CAS PubMed; (c) S. Stone, T. Wang, J. Liang, J. Cochran, J. Green and W. Gu, Org. Biomol. Chem., 2015, 13, 10471–10476 RSC; (d) S. A. Weissman and N. G. Anderson, Org. Process Res. Dev., 2015, 19, 1605–1633 CrossRef CAS; (e) P. M. Murray, F. Bellany, L. Benhamou, D.-K. Bučar, A. B. Tabor and T. D. Sheppard, Org. Biomol. Chem., 2016, 14, 2373–2384 RSC; (f) G. D. Bowden, B. J. Pichler and A. Maurer, Sci. Rep., 2019, 9, 11370 CrossRef PubMed; (g) M. T. Koeritz, R. W. Burgett, A. A. Kadam and L. M. Stanley, Org. Lett., 2020, 22, 5731–5736 CrossRef CAS PubMed.
- (a) K. M. Miller, T. Luanphaisarnnont, C. Molinaro and T. F. Jamison, J. Am. Chem. Soc., 2004, 126, 4130–4131 CrossRef CAS PubMed; (b) P. Liu, P. McCarren, P. H.-Y. Cheong, T. F. Jamison and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2050–2057 CrossRef CAS PubMed.
- (a) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901–2916 CrossRef CAS PubMed; (b) A. F. Orsino, M. Gutiérrez del Campo, M. Lutz and M.-E. Moret, ACS Catal., 2019, 9, 2458–2481 CrossRef CAS PubMed; (c) A. F. Orsino and M.-E. Moret, Organometallics, 2020, 39, 1998–2010 CrossRef CAS.
- (a) O. Vogl, J. Macromol. Sci., Part A: Pure Appl.Chem., 1967, 1, 243–266 CrossRef; (b) O. Vogl, Makromol. Chem., 1974, 175, 1281–1308 CrossRef CAS.
- Y. S. Zhao, X. Q. Tang, J. C. Tao, P. Tian and G. Q. Lin, Org. Biomol. Chem., 2016, 14, 4400–4404 RSC.
- C. Liu, X. Li and Y. Wu, RSC Adv., 2015, 5, 15354–15358 RSC.
- E. R. Darzi, B. M. White, L. K. Loventhal, L. N. Zakharov and R. Jasti, J. Am. Chem. Soc., 2017, 139, 3106–3114 CrossRef CAS PubMed.
- S. Roscales and A. G. Csákÿ, Org. Lett., 2015, 17, 1605–1608 CrossRef CAS PubMed.
- N. Rodríguez, M. Medio-Simón and G. Asensio, Adv. Synth. Catal., 2007, 349, 987–991 CrossRef.
- M. Ohashi, O. Kishizaki, H. Ikeda and S. Ogoshi, J. Am. Chem. Soc., 2009, 131, 9160–9161 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all new compounds. See https://doi.org/10.1039/d2sc01840c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |