
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular copolymerization through self-correction of non-polymerizable transient intermediates†
Ganyu
Chen
a,
Peichen
Shi
a,
Longhui
Zeng
a,
Liubin
Feng
a,
Xiuxiu
Wang
a,
Xujing
Lin
a,
Yibin
Sun
a,
Hongxun
Fang
a,
Xiaoyu
Cao
 a,
Xinchang
Wang
*b,
Liulin
Yang
*a and
Zhongqun
Tian
a
a,
Xinchang
Wang
*b,
Liulin
Yang
*a and
Zhongqun
Tian
a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Key Laboratory of Chemical Biology of Fujian Province, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: llyang@xmu.edu.cn
bSchool of Electronic Science and Engineering (National Model Microelectronics College), Xiamen University, Xiamen 361005, P. R. China
First published on 17th June 2022
Abstract
Kinetic control over structures and functions of complex assembly systems has aroused widespread interest. Understanding the complex pathway and transient intermediates is helpful to decipher how multiple components evolve into complex assemblies. However, for supramolecular polymerizations, thorough and quantitative kinetic analysis is often overlooked. Challenges remain in collecting the information of structure and content of transient intermediates in situ with high temporal and spatial resolution. Here, the unsolved evolution mechanism of a classical self-sorting supramolecular copolymerization system was addressed by employing multidimensional NMR techniques coupled with a microfluidic technique. Unexpected complex pathways were revealed and quantitatively analyzed. A counterintuitive pathway involving polymerization through the ‘error-correction’ of non-polymerizable transient intermediates was identified. Moreover, a ‘non-classical’ step-growth polymerization process controlled by the self-sorting mechanism was unraveled based on the kinetic study. Realizing the existence of transient intermediates during self-sorting can encourage the exploitation of this strategy to construct kinetic steady state assembly systems. Moreover, the strategy of coupling a microfluidic technique with various characterization techniques can provide a kinetic analysis toolkit for versatile assembly systems. The combined approach of coupling thermodynamic and kinetic analyses is indispensable for understanding the assembly mechanisms, the rules of emergence, and the engineering of complex assembly systems.
Introduction
Complex assembly systems are characterized by multi-component, complex pathways towards hierarchical assemblies.1–3 In recent years, kinetic control over structures and functions of complex assembly systems has aroused widespread interest. Kinetic products that are more diverse than thermodynamic products can be obtained by external stimuli.4–7 Fine regulation of the kinetics process is essential for engineering complex assembly systems, such as non-equilibrium assemblies that require constant influx of energy or matter,8–14 living supramolecular polymerizations,15–21 and supramolecular copolymerizations.21–23Kinetic studies for revealing the detailed assembly pathways and corresponding intermediates are helpful to decipher how multiple components evolve into complex assemblies.24 However, the assembly process for supramolecular polymerizations is typically interpreted based on thermodynamic data, while quantitative kinetic analysis for detailed assembly pathways and possible intermediates is often overlooked. Previous studies have elegantly employed photophysical spectroscopy (e.g. stopped-flow circular dichroism and temperature-jump fluorescence) to reveal the complex pathways of supramolecular polymerizations.24,25 Challenges remain in collecting the information of structure and content of transient intermediates in situ with high temporal and spatial resolution.
Here, the unsolved evolution mechanism of a classical self-sorting supramolecular copolymerization system26 (Scheme 1) was revealed by employing a quantitative kinetic analysis approach. This approach included multidimensional NMR coupled with a microfluidic technique (μF-NMR)27–29 for high structural and temporal resolution, combined with mathematical modeling. Self-sorting is an exquisite strategy to construct sequence-controlled supramolecular copolymers.26,30–45 The successful construction of such systems follows a simple thermodynamic rule which requires a large difference in binding constants among interaction pairs. However, the detailed self-sorting pathways, and the evolution of possible transient intermediates are relatively unexplored and thereby are still elusive. In this case study, unexpected complex pathways were revealed and quantitatively analyzed. A counterintuitive pathway of polymerization through the ‘error-correction’ of non-polymerizable transient intermediates was identified. Furthermore, a ‘non-classical’ step-growth polymerization process controlled by the self-sorting mechanism was unraveled through the kinetic study. Realizing the existence of transient intermediates during self-sorting can inspire researchers to rediscover the value of this classical assembly strategy in constructing kinetically controlled steady state assembly systems.
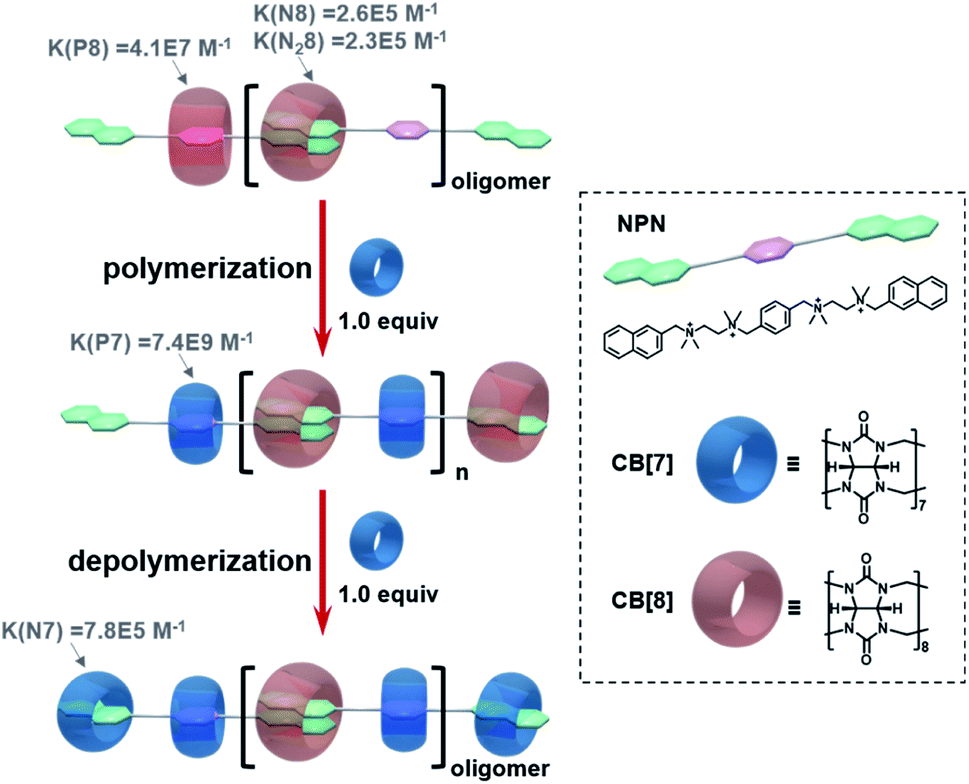 | ||
| Scheme 1 Schematic representation of cucurbituril-based self-sorting supramolecular polymerization and depolymerization. The binding constants refer to ref. 26. | ||
Results and discussion
Thermodynamic interpretation
As shown in Scheme 1, supramolecular alternating copolymers have been constructed by the self-sorting recognition of cucurbit[8]uril (CB[8]), cucurbit[7]uril (CB[7]), and a bifunctional guest molecule NPN containing two naphthalene end groups and a p-phenylene group. The driving force for self-sorting can be understood through thermodynamic study (Scheme 1 and Fig. S1–S15†). By mixing equimolar CB[8] and NPN, only oligomers can be obtained, since the competitive binding of the p-phenylene group with CB[8] hinders the polymerization. The oligomers can be efficiently converted to polymers after adding one equivalent of CB[7]. This is because CB[7] has more than two orders of magnitude higher binding constants with p-phenylene than CB[8], which ensures the specific combination of CB[8] with naphthalene instead of p-phenylene. Moreover, supramolecular polymers can be depolymerized by adding another equimolar amount of CB [7], since CB[7] has a higher binding constant with naphthalene than CB[8].Based on the thermodynamic data, it can be inferred that the pathway for this self-sorting supramolecular polymerization follows three steps: (1) the instant dissociation of CB[8] from naphthalene groups, (2) CB[7] selectively combines with p-phenylene groups, and (3) CB[8] recombines with two naphthalene end groups to realize the polymerization. However, whether the system follows this intuitive shortcut or other pathways with any transient intermediates involved requires an in-depth kinetic investigation.
Identifying the evolution of transient intermediates
The system was first monitored by conventional NMR to identify potential intermediates. Indeed, multiple dynamic peaks in 1H NMR spectra were observed during the polymerization (Fig. S16 and 17†). Three representative peaks at 5.35, 3.20, and 2.79 ppm not found in raw components were observed and gradually attenuated during the polymerization (Fig. 1), suggesting the existence of intermediates. Further NMR studies, including HSQC, HMBC, NOESY, HSQC-NOESY, DOSY, and 1H experiments, were conducted to identify the structures of these intermediates (Fig. S1–S32†). The single peak at 5.35 ppm was ascribed to a structural fragment formed when a CB[7] molecule combines with a naphthalene group, which represents a common structural fragment of various intermediates (abbreviated as N7, including two possible structures N7P7 and N7P8 in Fig. 1a); the peak at 3.20 ppm was identified as the complex formed by both a naphthalene group and the adjacent p-phenylene complexing with CB[7] (abbreviated as N7P7, as shown in Fig. 1a); the peak at 2.79 ppm was ascribed to a structural fragment in which two naphthalene groups and an adjacent p-phenylene group are complexed with CB[8] in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mode and 1:1 mode, respectively (abbreviated as N28P8, as shown in Fig. 1a). Compared with the structure of final supramolecular polymers that require the correct association of naphthalene with CB[8] and p-phenylene with CB[7], these intermediates are off-pathway kinetic products that are non-polymerizable by themselves (N7) or thermodynamically unfavored (N28P8). This suggests a counterintuitive complex pathway for this three-component assembly system.
:1 mode and 1:1 mode, respectively (abbreviated as N28P8, as shown in Fig. 1a). Compared with the structure of final supramolecular polymers that require the correct association of naphthalene with CB[8] and p-phenylene with CB[7], these intermediates are off-pathway kinetic products that are non-polymerizable by themselves (N7) or thermodynamically unfavored (N28P8). This suggests a counterintuitive complex pathway for this three-component assembly system.
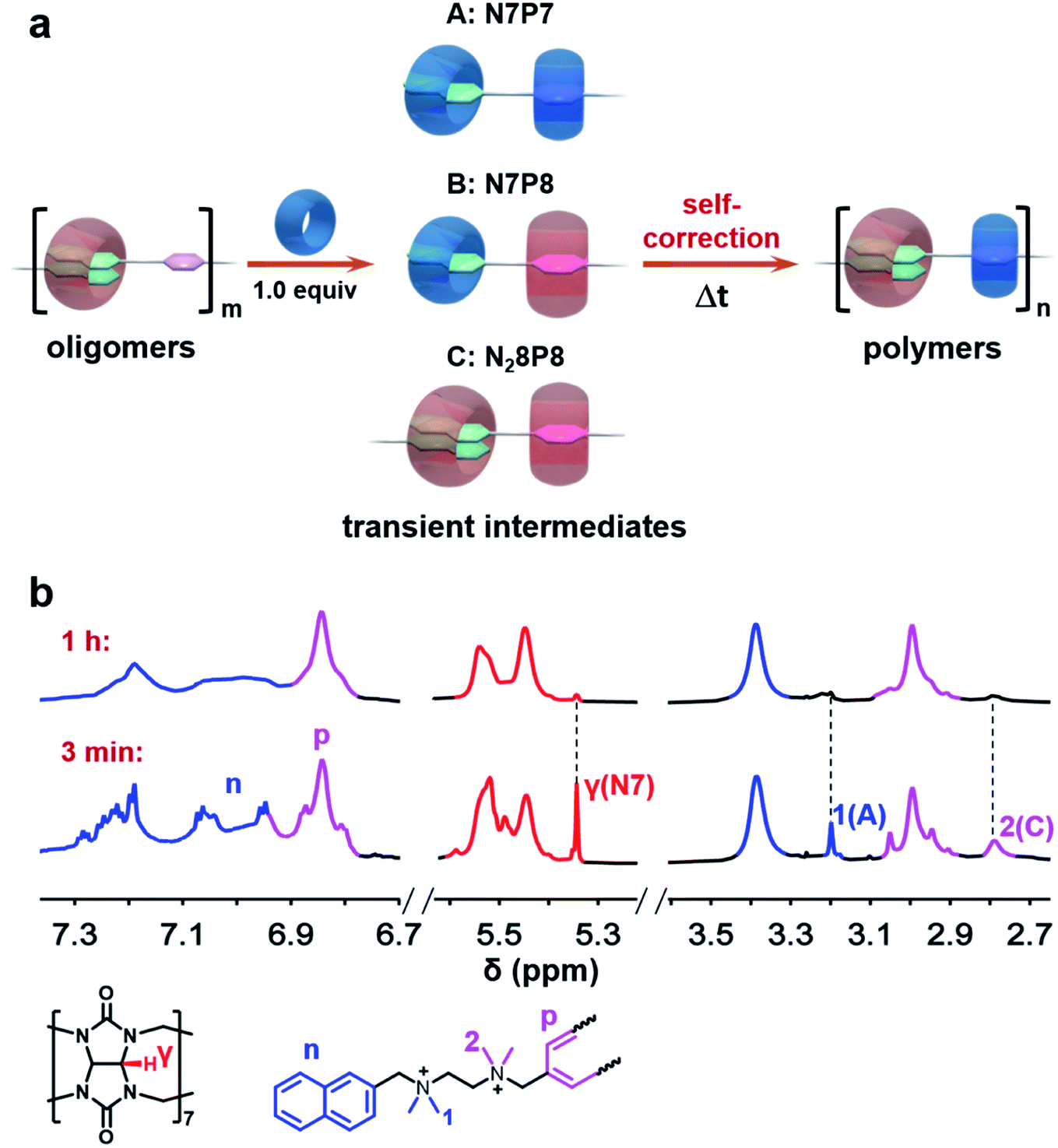 | ||
| Fig. 1 (a) Schematic representations of intermediates observed in the self-sorting supramolecular polymerization. (b) 1H NMR (850 MHz, 298 K, D2O) spectra of the polymerization at about 3 min and 1 h. ‘γ(N7)’ indicates the proton γ of CB[7] which complexed with naphthalene, and ‘1(A)’ and ‘2(C)’ indicate the methyl protons 1 and 2 of NPN in the intermediates A and C, respectively. It took about 3 minutes to collect the first 1H NMR spectrum, and the polymerization took about 1 hour to complete. Initial concentration of each component in the oligomer: [NPN]0 = [CB[8]]0 = 2 mM; [CB[7]]0 = 2 mM. | ||
To understand the generation and transition of these intermediates, a quantitative kinetic analysis is indispensable. For conventional NMR, it took at least 3 minutes to collect the first 1H NMR spectrum. That means it is difficult to monitor the earlier period of supramolecular polymerization, especially the generation process of intermediates by conventional NMR. In contrast, μF-NMR marries the merits of the microfluidic technique and NMR spectroscopy. Therefore, it is capable of revealing more details of the polymerization period as early as 1.5 s from the onset. Specifically, reactants are pumped constantly into the microfluidic chip (Scheme S1†) at the same volumetric rate, which can then be transferred to the NMR sampling cell. Due to the fast mixing (millisecond time scale) in the microfluidic chip as well as online monitoring, the NMR spectra of the early assembly period (typically starting from 1.5 s) can be obtained. By changing the flow rate, the concentration of intermediates at different points in time can be readily determined by the integration of NMR peaks (Fig. S27 and S28†). As shown in Fig. 2a and b, the concentration of N7 rapidly increased within 12 s, and then slowly decreased in the following 360 s. The period of polymerization was divided into stage I and stage II according to the maximum concentration of N7. It is noteworthy that the maximum concentration of N7 almost reached 1.3 mM, accounting for almost two-thirds of the total concentration of CB[7], suggesting that they were dominant intermediates during polymerization.
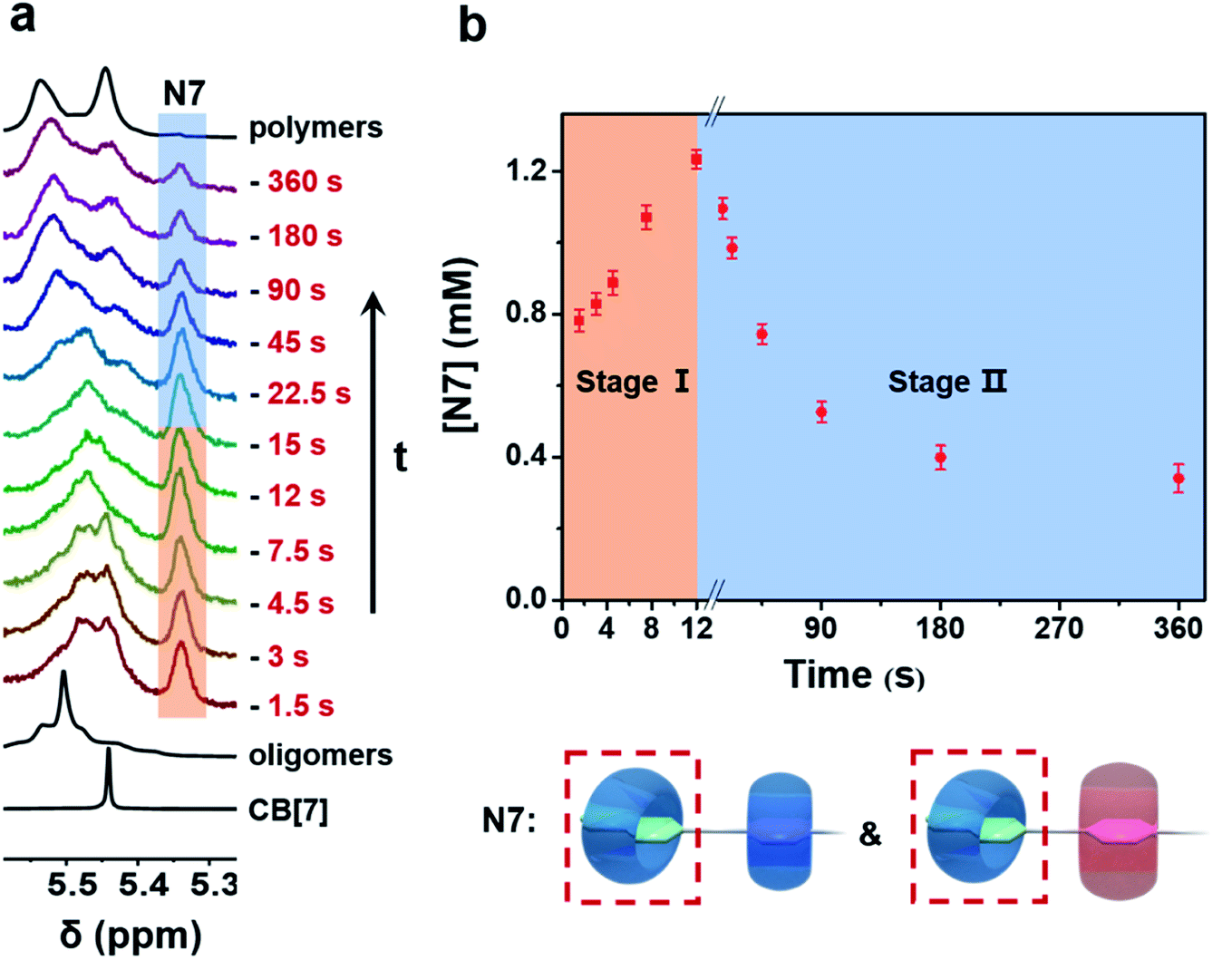 | ||
| Fig. 2 (a) 1H μF-NMR (500 MHz, 298 K, D2O) spectra of the supramolecular polymerization system recorded at various points in time. (b) Time-dependent concentration of N7 intermediates from 1.5 s to 360 s (extended period can be observed by the conventional NMR technique). Note that the error bars were obtained from three parallel μF-NMR experiments. Initial concentration of each component in oligomer solution: [NPN]0 = [CB[8]]0 = 2 mM; [CB[7]]0 = 2 mM. | ||
The formation of the final supramolecular polymers requires the threading of CB[7] from naphthalene to the p-phenylene group. However, the corresponding intermediate N7P was not observed, suggesting that the threading process was too fast to be detected by μF-NMR. Considering the structures of N7 intermediates, including N7P7 and N7P8 but not N7P, reasons for the long lifetime of N7 may be the following two conjectures: (1) when p-phenylene is complexed with CB[7] or CB[8], the adjacent naphthalene group can still combine with CB[7], resulting in the formation of these intermediates. In this case, the direct threading of CB[7] from naphthalene to the p-phenylene group is blocked. (2) The dissociation rate of CB[7] from the naphthalene group into the solvent is much slower than the association rate considering the high association constant (K = 7.8 × 105 M−1).46
To validate the above conjectures, control experiments were carried out by monitoring the self-sorting process of equimolar NPN and CB[7] without CB[8]. The thermodynamic equilibrium product was NP7N, of which CB[7] combines with the p-phenylene group rather than the naphthalene group. However, the kinetic monitoring also found the metastable intermediate N7P7 in this system (Fig. S33–S36†), which verified the first conjecture. In addition, neither free CB[7] nor intermediate N7P (in which only the naphthalene group is complexed with CB[7]) could be observed, indicating that the complexation and threading of CB[7] from naphthalene to p-phenylene were too fast to be detected by μF-NMR. Despite the fast association and shuttling, the conversion of N7P7 into the final product NP7 was slower and could be observed, and the whole self-sorting process took about 45 s to finish (Fig. S36b†). This indicates that the disassociation of CB[7] from the naphthalene group, i.e. the error-correction process, is the rate-determining step in the whole self-sorting process. A corresponding kinetic model based on this mechanism was then established (see Section 2.9.1 in the ESI†), which successfully described the experimental data (Fig. S36b†). These results further confirm the second conjecture.
Kinetics of the supramolecular polymerization
Although N7 intermediates were observed in both the above systems, the kinetic profile was remarkably different for the supramolecular polymerization system. In the presence of CB[8], the full generation of N7 intermediates at stage I was slower than the one without CB[8] which was not observed at all. At stage II, the error-correction of N7 intermediates took about 1 h to finish, which stands in sharp contrast to the ones without CB[8] of ∼45 s. Therefore, it is reasonable to infer that CB[8] also plays a key role in rate-determining steps both at stage I and stage II.To confirm this hypothesis, the starting species for the supramolecular polymerization, i.e. the thermodynamic products of the equimolar mixture of NPN and CB[8], were first identified. According to the association constants of CB[8] with two groups (Table S1†),26i.e. the naphthalene and p-phenylene groups of the NPN monomer, it can be calculated that about 75 percent of CB[8] should combine with two naphthalene end groups, and the remaining CB[8] should complex with p-phenylene groups, resulting in oligomers with an average degree of polymerization of about 4.47 The quantitative calculation was further confirmed by 1H NMR (Fig. S37†) and DOSY (Fig. S38†) measurements. After adding CB[7], the generation of N7 intermediates at stage I may proceed through two steps as shown in Fig. 3a: (1) the disassociation of one naphthalene group from the ternary complex of naphthalene groups and CB[8] (N28P), resulting in the NP fragment with a free naphthalene group and the N8P fragment with one naphthalene group complexed with CB[8]. Only the dissociation of one naphthalene from CB[8] is considered since the threading of CB[8] on N8P from the residual naphthalene to p-phenylene is faster as well as more thermodynamically favored than the dissociation of CB[8] into the solvent. (2) The fast association of CB[7] with NP and NP8 results in N7 intermediates (including N7P7 and N7P8). Regarding the proposed pathway at stage I, the dissociation of one naphthalene group from CB[8] is likely the rate-determining step. The corresponding kinetic model (see Section 2.9.2 in the ESI†) is in good agreement with the experimental data (Fig. 3b), which confirms the above hypothesis.
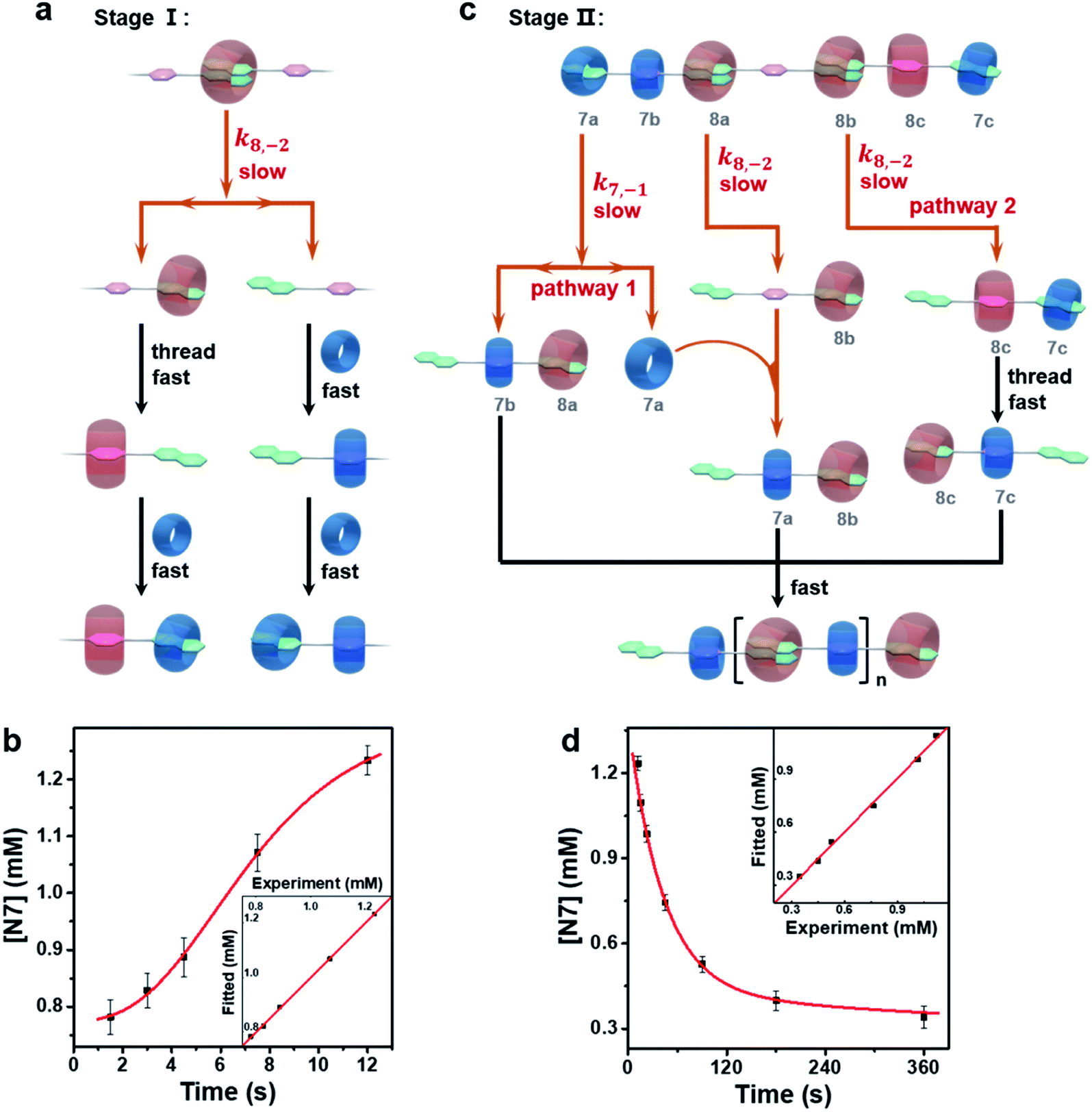 | ||
| Fig. 3 (a) Pathways for the formation of N7 intermediates at stage I. The red arrow lines indicate the rate-determining step. (b) Time-dependent concentration of N7 intermediates at stage I and the simulated kinetic trace with R2 = 0.999. (c) Pathways for the error-correction of the averaged intermediate at stage II. For clarity, the three CB[7] molecules and the three CB[8] molecules are marked as 7a, 7b, 7c, and 8a, 8b, 8c, respectively. (d) Time-dependent concentration of N7 intermediates at stage II and the simulated kinetic traces with R2 = 0.999. Initial concentration of each component in oligomer solution: [NPN]0 = [CB[8]]0 = 2 mM; [CB[7]]0 = 2 mM. | ||
The N7 intermediates can be viewed as kinetic off-pathway products which retard the supramolecular polymerization. However, these kinetic intermediates are metastable and follow an error-correction process at stage II to promote polymerization. For the sake of simplicity, all the intermediates were treated as an equivalent intermediate (Fig. 3c), according to the kinetic analysis of stage I as well as the initial contents of N7 intermediates at stage II. In this equivalent intermediate, two-thirds of CB[7] combine with naphthalene groups, which is consistent with the maximum content of N7 detected, and the equimolar monomers, CB[7] and CB[8], also meet the initial stoichiometric setting. Last but not least, most of the structural fragments of this equivalent intermediate were evidenced by NMR spectra (Fig. S16–S32†). According to the equivalent intermediate, two parallel pathways are proposed for the error-correction of N7 intermediates resulting in the correct polymerizable monomer N8P7N: (1) the disassociation of N7P7 fragments into NP7 and CB[7], meanwhile, the disassociation of N28P fragments into NP, and then the released CB[7] threading through NP to associate with p-phenylene; (2) the disassociation of N28P8N7 fragments into NP8N7 following fast sequential threading into N8P7N. It is worth noting that, for the first pathway, the complete dissociation of N7 must be coordinated by the dissociation of N28. This is the major reason for the much slower error-correction during polymerization. According to the analysis of the above pathways, a corresponding kinetic model was constructed (see Section 2.9.3 in the ESI†), and the curve fits well with the experimental data (Fig. 3d).
To further verify the transition of intermediates corresponding to the supramolecular polymerization process, real-time molecular weights of polymers were monitored by conventional diffusion-ordered spectroscopy (DOSY). Due to the limitation of DOSY, only the data from ∼150 s after the onset of the process were obtained (Fig. S39†). The results showed that the growth of the polymer chain was governed by the transition of intermediates (Fig. 4a). Interestingly, it was found that as the conversion rate of N7 intermediates increased from 82% to 90%, the average molecular weight of polymers gradually increased from ∼200 K to ∼550 K (Fig. 4b). However, the growth rate slowed down rapidly, and the molecular weight approached the final value. This is different from the law of classical step-growth polymerization,47 in which the molecular weight increases more and more rapidly when the reaction degree exceeds 90%. Two reasons are proposed for the abnormal phenomenon in this self-sorting supramolecular polymerization system: (1) the growth of the polymer chain is mainly determined by the complexation of CB[8] and two naphthalene groups. Restricted by thermodynamic equilibrium, the conversion rate cannot reach 100%, resulting in the limited molecular weight of the polymers. (2) CB[7] promotes polymerization by inhibiting the ‘error’ binding of CB[8] to p-phenylene groups, but this effect gradually diminishes at the end of polymerization. The reason is that when CB[8] molecules are fully bound to naphthalene groups, complexation of the residual CB[7] with p-phenylene groups would not promote further polymerization, and thereby would not lead to drastic changes in molecular weight. From another point of view, it means that this self-sorting supramolecular polymerization system is less sensitive to monomer ratio than conventional polycondensations.47
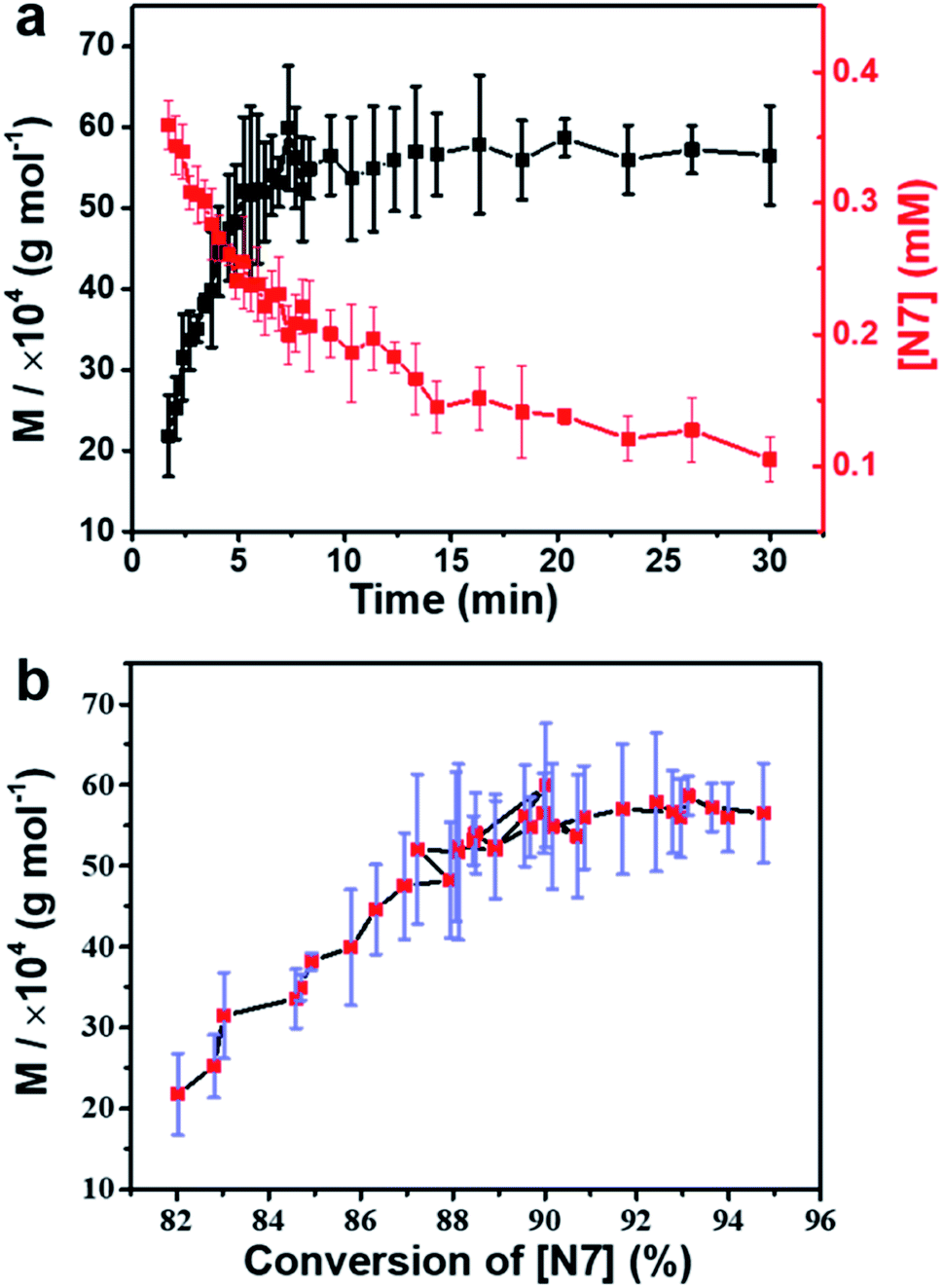 | ||
| Fig. 4 (a) Plots of the changes in the molecular weight of polymers and the concentration of intermediates N7 with time. (b) Plots of the molecular weight of polymers as a function of the conversion rate of N7. Note that the error bars were obtained from three parallel DOSY as well as 1H NMR monitoring experiments. | ||
Kinetics of the supramolecular depolymerization
The depolymerization process induced by additional equimolar CB[7] took only about 25 s to complete (Fig. 5), which was much faster than the polymerization. The degradation products were various oligomers containing N7P7 fragments (Fig. S5–S13†), indicating that the depolymerization mainly proceeded through the competitive binding between CB[7] and CB[8] to the naphthalene group. The prerequisite for CB[7] binding to the naphthalene group is the dissociation of ternary CB[8] complex N28. According to the above results, it can be inferred that the dissociation of N28 should be the rate-determining step during depolymerization (Fig. 5a). As shown in Fig. 5c, the corresponding kinetic model (see Section 2.9.4 in the ESI†) showed a good fitting to the experimental data. Considering that depolymerization involves only one kind of dissociation process, i.e. the dissociation of the N28 complex, it is reasonable that the depolymerization is much faster than self-sorting supramolecular polymerization.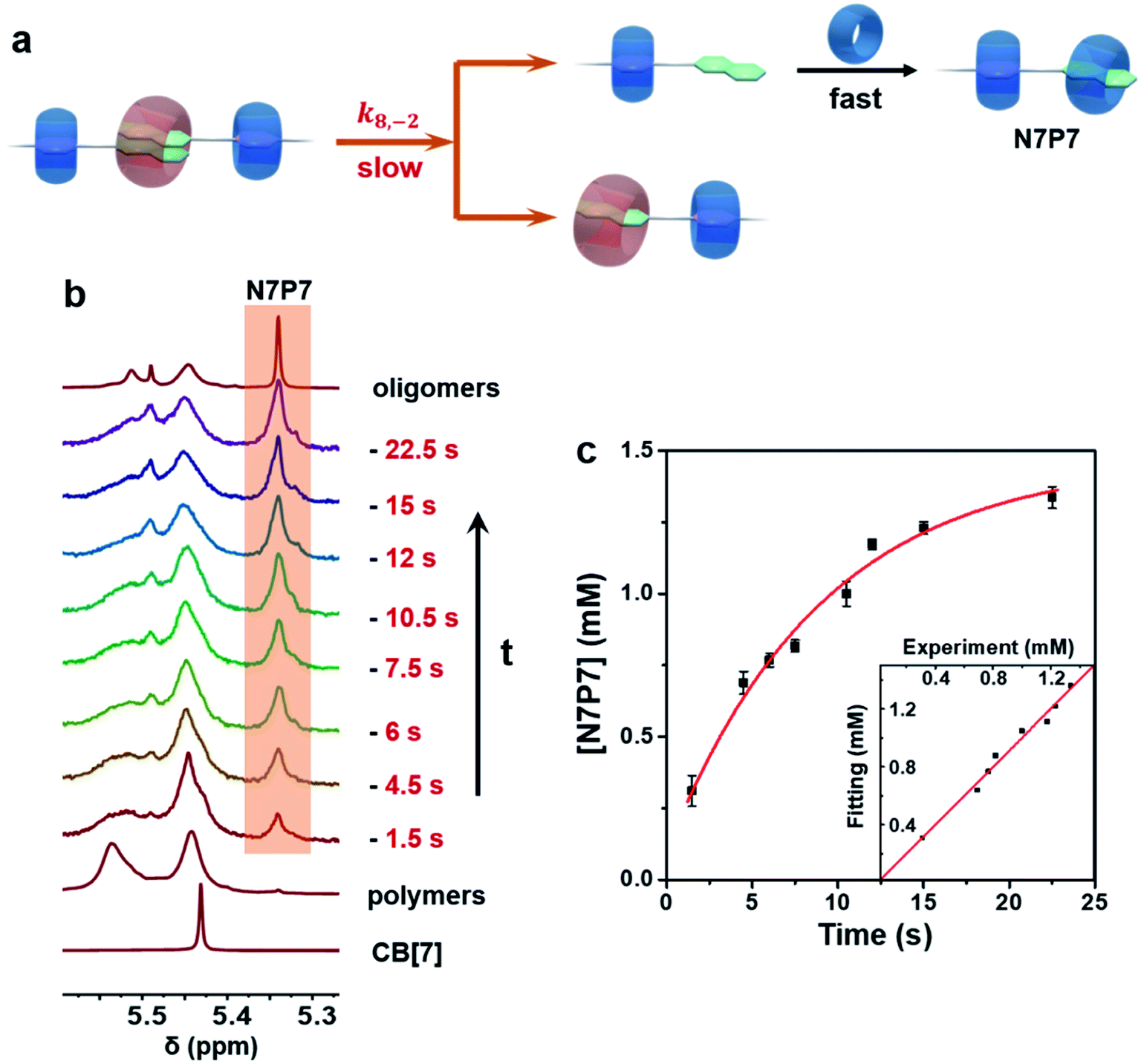 | ||
| Fig. 5 (a) Pathways of depolymerization. The red arrow lines represent the rate-determining step. (b) 1H μF-NMR (500 MHz, 298 K, D2O) spectra of the supramolecular depolymerization induced by additional equimolar CB[7] recorded at various points in time. (c) Time-dependent concentration of product N7P7 and the simulated kinetic traces with R2 = 0.983. Initial concentration of each component in polymer solution: [NPN]0 = [CB[8]]0 = [CB[7]]0 = 1.5 mM; initial concentration of additional CB[7]0 = 1.5 mM. | ||
Conclusion
The evolution process of a classical self-sorting supramolecular polymerization system was successfully revealed herein. Representative intermediates were identified and quantified, and unexpected complex self-sorting pathways in this three-component assembly system were revealed. The self-sorting process involved the fast generation of various off-pathway intermediates dominated by kinetics and then followed an ‘error-correction’ process to promote the supramolecular polymerization. Moreover, a ‘non-classical’ step-growth polymerization process controlled by the self-sorting mechanism was revealed through the kinetic study.Identifying the transient intermediates formed during self-sorting is helpful to exploit the merits of such a classical assembly strategy. For instance, new strategies to construct kinetically controlled assemblies might be inspired. Although traditional studies of self-sorting focus on the final equilibrium products, the kinetically controlled steady state intermediates with tunable structure and life-time could also play leading roles, if a meticulous design is considered. Moreover, endeavors should be made to couple various strategies including self-sorting, chemical fuel-driven assembly,8–14 and catassembly,48–51 together with other constitution elements especially the feedback loops,52–58 to construct interaction networks that finally evolve into complex assembly systems, as shown by living beings. With the continuous influx of energy and matter that regulates the evolution process, new structures, patterns, functions, and even ‘new chemistry’ could emerge from sufficiently complex assembly systems.
Complex assembly systems require versatile kinetic analysis techniques with high spatial and temporal resolutions. This work has demonstrated the power of multidimensional NMR techniques coupled with the microfluidic technique for acquiring abundant information from complex assembly processes. It is worth noting that a kinetic analysis toolkit which couples the microfluidic technique with various characterization techniques would be necessary for versatile assembly systems. For the interpretation of kinetic data, mathematical modeling was employed in this study. However, to tackle the possible massive data issue from highly complex systems, machine learning will undoubtedly show great potential and advantages. Therefore, it is necessary to establish general methodologies for the study of complex assembly systems. With increasing interest in the study of system chemistry,2,59 the combined approach of integrating thermodynamic and kinetic analyses as well as big data processing is expected to be critical in understanding the assembly mechanisms, the rules of emergence, and the engineering of complex assembly systems.
Data availability
All data supporting the findings of this study are available within the article and its ESI† files, and from the corresponding author upon reasonable request.Author contributions
Yang, L. L. conceptualized the project. Chen, G. Y., Zeng, L. H., and Feng, L. B. have performed all the conventional NMR experiments described in the manuscript. Chen, G. Y. analyzed the NMR data and prepared the ESI.† Shi, P. C. deduced the mathematical models and performed the kinetic curve fitting, and wrote the “kinetic models” part of the ESI.† Wang, X. X., Chen, G. Y., and Wang, X. C. performed the μF-NMR experiments. Lin, X. J. synthesized all the molecules. Chen, G. Y. and Yang, L. L. wrote the overall manuscript. Chen, G. Y. and Shi, P. C. worked on the figures. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Zehuan Huang for the helpful discussion. This work was supported by the National Natural Science Foundation of China (NSFC) (No. 21971216, 21971217, 21991130, and 21991131), the Fundamental Research Funds for the Central Universities (No. 20720210007), and the Top-Notch Young Talents Program of China.References
- J. M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed.
- E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111–119 CrossRef CAS PubMed.
- C. Rest, R. Kandanelli and G. Fernández, Chem. Soc. Rev., 2015, 44, 2543–2572 RSC.
- M. Wehner and F. Würthner, Nat. Rev. Chem., 2019, 4, 38–53 CrossRef.
- M. Wehner, M. I. S. Rohr, M. Buhler, V. Stepanenko, W. Wagner and F. Wurthner, J. Am. Chem. Soc., 2019, 141, 6092–6107 CrossRef CAS PubMed.
- P. A. Korevaar, C. J. Newcomb, E. W. Meijer and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 8540–8543 CrossRef CAS PubMed.
- A. T. Haedler, S. C. Meskers, R. H. Zha, M. Kivala, H. W. Schmidt and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 10539–10545 CrossRef CAS PubMed.
- J. Boekhoven, A. M. Brizard, K. N. Kowlgi, G. J. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- S. Dhiman and S. J. George, Bull. Chem. Soc. Jpn., 2018, 91, 687–699 CrossRef CAS.
- S. De and R. Klajn, Adv. Mater., 2018, 30, 1706750 CrossRef PubMed.
- Z. Yin, G. Song, Y. Jiao, P. Zheng, J.-F. Xu and X. Zhang, CCS Chem., 2019, 1, 335–342 CrossRef CAS.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188–195 CrossRef CAS PubMed.
- J. Kang, D. Miyajima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646–651 CrossRef CAS PubMed.
- R. D. Mukhopadhyay and A. Ajayaghosh, Science, 2015, 349, 241–242 CrossRef CAS PubMed.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Wurthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef CAS PubMed.
- M. Endo, T. Fukui, S. H. Jung, S. Yagai, M. Takeuchi and K. Sugiyasu, J. Am. Chem. Soc., 2016, 138, 14347–14353 CrossRef CAS PubMed.
- S. Ogi, K. Matsumoto and S. Yamaguchi, Angew. Chem., Int. Ed., 2018, 57, 2339–2343 CrossRef CAS PubMed.
- L. MacFarlane, C. Zhao, J. Cai, H. Qiu and I. Manners, Chem. Sci., 2021, 12, 4661–4682 RSC.
- B. Adelizzi, N. J. Van Zee, L. N. J. de Windt, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6110–6121 CrossRef CAS PubMed.
- W. Wagner, M. Wehner, V. Stepanenko and F. Wurthner, J. Am. Chem. Soc., 2019, 141, 12044–12054 CrossRef CAS PubMed.
- D. van der Zwaag, P. A. Pieters, P. A. Korevaar, A. J. Markvoort, A. J. Spiering, T. F. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 12677–12688 CrossRef CAS PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. Smulders, P. A. Hilbers, A. P. Schenning, T. F. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed.
- Z. Huang, L. Yang, Y. Liu, Z. Wang, O. A. Scherman and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 5351–5355 CrossRef CAS PubMed.
- G. Finch, A. Yilmaz and M. Utz, J. Magn. Reson., 2016, 262, 73–80 CrossRef CAS PubMed.
- H. Fang, Y. Sun, X. Wang, M. Sharma, Z. Chen, X. Cao, M. Utz and Z. Tian, Sci. China: Chem., 2018, 61, 1460–1464 CrossRef CAS.
- Y. Sun, H. Fang, X. Lin, X. Wang, G. Chen, X. Wang, Z. Tian, L. Yang, M. Utz and X. Cao, CCS Chem., 2022, 4, 557–565 CrossRef CAS.
- F. Wang, C. Han, C. He, Q. Zhou, J. Zhang, C. Wang, N. Li and F. Huang, J. Am. Chem. Soc., 2008, 130, 11254–11255 CrossRef CAS PubMed.
- F. Wang, B. Zheng, K. Zhu, Q. Zhou, C. Zhai, S. Li, N. Li and F. Huang, Chem. Commun., 2009, 4375–4377 RSC.
- Y. Liu, Y. Yu, J. Gao, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6576–6579 CrossRef CAS PubMed.
- Y. Liu, K. Liu, Z. Wang and X. Zhang, Chem.–Eur. J., 2011, 17, 9930–9935 CrossRef CAS PubMed.
- S. Dong, X. Yan, B. Zheng, J. Chen, X. Ding, Y. Yu, D. Xu, M. Zhang and F. Huang, Chem.–Eur. J., 2012, 18, 4195–4199 CrossRef CAS PubMed.
- L. Li, H. Y. Zhang, J. Zhao, N. Li and Y. Liu, Chem.–Eur. J., 2013, 19, 6498–6506 CrossRef CAS PubMed.
- J. del Barrio, P. N. Horton, D. Lairez, G. O. Lloyd, C. Toprakcioglu and O. A. Scherman, J. Am. Chem. Soc., 2013, 135, 11760–11763 CrossRef CAS PubMed.
- C. Li, Chem. Commun., 2014, 50, 12420–12433 RSC.
- S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS PubMed.
- F. Zeng, Y. Han and C. F. Chen, Chem. Commun., 2015, 51, 3593–3595 RSC.
- X. Wang, W. Wang, Y. Wang and H. Yang, Chem. Lett., 2015, 44, 1040–1046 CrossRef CAS.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- N. Nayak and K. R. Gopidas, ChemistrySelect, 2016, 1, 1028–1032 CrossRef CAS.
- T. Hirao, H. Kudo, T. Amimoto and T. Haino, Nat. Commun., 2017, 8, 634 CrossRef PubMed.
- Z. Zhang, Y. Liu, J. Zhao and X. Yan, Polym. Chem., 2020, 11, 367–374 RSC.
- H. Li, Y. Yang, F. Xu, Z. Duan, R. Li, H. Wen and W. Tian, Org. Chem. Front., 2021, 8, 1117–1124 RSC.
- W. L. Mock and N. Y. Shih, J. Am. Chem. Soc., 1989, 111, 2697–2699 CrossRef CAS.
- G. Odian, Principles of Polymerization, Wiley, New York, 4th edn, 2004 Search PubMed.
- Y. Wang, H. X. Lin, L. Chen, S. Y. Ding, Z. C. Lei, D. Y. Liu, X. Y. Cao, H. J. Liang, Y. B. Jiang and Z. Q. Tian, Chem. Soc. Rev., 2014, 43, 399–411 RSC.
- H. Zhang, Y. Wang, H. Zhang, X. Liu, A. Lee, Q. Huang, F. Wang, J. Chao, H. Liu, J. Li, J. Shi, X. Zuo, L. Wang, L. Wang, X. Cao, C. Bustamante, Z. Tian and C. Fan, Nat. Commun., 2019, 10, 1006 CrossRef CAS PubMed.
- Y. Wang, Y. Sun, P. Shi, M. M. Sartin, X. Lin, P. Zhang, H. Fang, P. Peng, Z. Tian and X. Cao, Chem. Sci., 2019, 10, 8076–8082 RSC.
- Y. Jiao, Y. Qiu, L. Zhang, W.-G. Liu, H. Mao, H. Chen, Y. Feng, K. Cai, D. Shen, B. Song, X.-Y. Chen, X. Li, X. Zhao, R. M. Young, C. L. Stern, M. R. Wasielewski, R. D. Astumian, W. A. Goddard and J. F. Stoddart, Nature, 2022, 603, 265–270 CrossRef CAS PubMed.
- T. Fujii and Y. Rondelez, ACS Nano, 2013, 7, 27–34 CrossRef CAS PubMed.
- F. Schaufelberger and O. Ramstrom, J. Am. Chem. Soc., 2016, 138, 7836–7839 CrossRef CAS PubMed.
- S. N. Semenov, L. J. Kraft, A. Ainla, M. Zhao, M. Baghbanzadeh, V. E. Campbell, K. Kang, J. M. Fox and G. M. Whitesides, Nature, 2016, 537, 656–660 CrossRef CAS PubMed.
- B. A. Grzybowski and W. T. Huck, Nat. Nanotechnol., 2016, 11, 585–592 CrossRef CAS PubMed.
- A. J. P. Teunissen, T. F. E. Paffen, I. A. W. Filot, M. D. Lanting, R. J. C. van der Haas, T. F. A. de Greef and E. W. Meijer, Chem. Sci., 2019, 10, 9115–9124 RSC.
- S. Panja and D. J. Adams, Giant, 2021, 5, 100041 CrossRef CAS.
- M. Jain and B. J. Ravoo, Angew. Chem., Int. Ed., 2021, 60, 21062–21068 CrossRef CAS PubMed.
- J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01930b |
| This journal is © The Royal Society of Chemistry 2022 |