
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic structure analysis of electrochemical CO2 reduction by iron-porphyrins reveals basic requirements for design of catalysts bearing non-innocent ligands†
Maxime
Tarrago
a,
Shengfa
Ye
 *ab and
Frank
Neese
*a
*ab and
Frank
Neese
*a
aMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, D-45470 Mülheim an der Ruhr, Germany. E-mail: shengfa.ye@dicp.ac.cn; frank.neese@kofo.mpg.de
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
First published on 29th June 2022
Abstract
Electrocatalytic CO2 reduction is a possible solution to the increasing CO2 concentration in the earth’s atmosphere, because it enables storage of energy while using the harmful CO2 feedstock as a starting material. Notably, iron(II) tetraphenylporphyrin, [FeII(TPP)]0 (TPP2− = tetraphenylporphyrin tetra-anion diradical), and its derivatives have been established as one of the most promising families of homogeneous catalysts for CO2 reduction into CO. Our earlier work has demonstrated that [Fe(TPP)]2−, a catalytically active species, is best described as an Fe(II) center antiferromagnetically coupled with a TPP4− diradical. In fact, [Fe(TPP)]2− represents a prototypical example of a diverse array of highly efficient molecular catalysts that feature non-innocent ligands. To obtain valuable insights for future catalyst design, their outstanding catalytic performance warrants an investigation aimed at elucidating the role played by the ligand non-innocence in the reaction. To this end, the reactivity of [Fe(TPP)]2− was first investigated in detail by using density functional theory calculations, and the theoretical results were then validated by reproducing available experimental kinetic and thermodynamic data. Further in-depth analyses pinpointed the electronic-structure feature of the non-innocent TPP ligand that is responsible for the high efficiency of the reaction. Finally, we analyzed the electronic-structure evolution found for the reactions catalyzed by ten related representative non-innocent systems. Our results revealed that for the reactions under consideration, the reducing equivalents are stored on the non-innocent ligand, while CO2 functionalization takes place at the metal center. Therefore, all of the transformations invariably entail two synchronized electron-transfer events: (1) a metal-to-CO2 transfer and (2) a ligand-to-metal electron transfer. The former is affected by σ-donation from the metal dz2 orbital to the CO2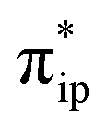 orbital, and the latter is facilitated by orbital coupling between the ligand and the metal center. Our results suggested that ligand non-innocence plays a fundamental role in stabilizing highly active intermediates while realizing high product selectivity for CO2 reduction and that the metal–ligand cooperativity is essential to the high reaction kinetics. On the basis of these findings, we proposed fundamental requirements for design of catalysts with non-innocent ligands.
orbital, and the latter is facilitated by orbital coupling between the ligand and the metal center. Our results suggested that ligand non-innocence plays a fundamental role in stabilizing highly active intermediates while realizing high product selectivity for CO2 reduction and that the metal–ligand cooperativity is essential to the high reaction kinetics. On the basis of these findings, we proposed fundamental requirements for design of catalysts with non-innocent ligands.
Introduction
Over the course of the last 30 years, the mean atmospheric CO2 concentration has increased by 20% and reached a new record of 411 ppm in 2020.1 On one hand, CO2 is a major greenhouse gas and largely contributes to global warming. On the other hand, CO2 is a ubiquitous C1 feedstock that can be used to produce value-added chemicals and biofuels, thereby closing the hydrocarbon cycle.2 However, due to its high thermodynamic stability and kinetic inertness, CO2 functionalization typically requires not only an external energy input, but also, more importantly, appropriate catalysts.Recently, tremendous effort has been devoted to developing efficient catalysts for photo-3 and electro-chemical4 CO2 reduction generating CO, formic acid, oxalate, methanol and other hydrocarbon compounds.2b,5 In particular, recent research effort has been directed towards catalysts containing earth-abundant base metals for economic reasons. Besides heterogeneous catalysts, homogeneous molecular systems also attract much attention,6 because such systems often can provide considerable mechanistic insights on reactions. Typically, mechanistic understanding can be used to direct systematic tuning of available systems and, more importantly, rational design of new catalysts with higher efficiency and product selectivity. To date, a plethora of homogeneous catalysts have been reported in the literature. Among them, [Fe(TPP)] (1, TPP2− = tetraphenylporphyrinate dianion, Scheme 1) and its derivatives exhibit one of the highest catalytic performances in dimethylformamide (DMF), i.e. relatively low overpotentials (−1.64 V vs. the standard calomel electrode, SCE, for [Fe(TPP)]), high turnover frequencies and a faradaic efficiency for CO generation close to 100% in the presence of sufficiently weak acid.7,8
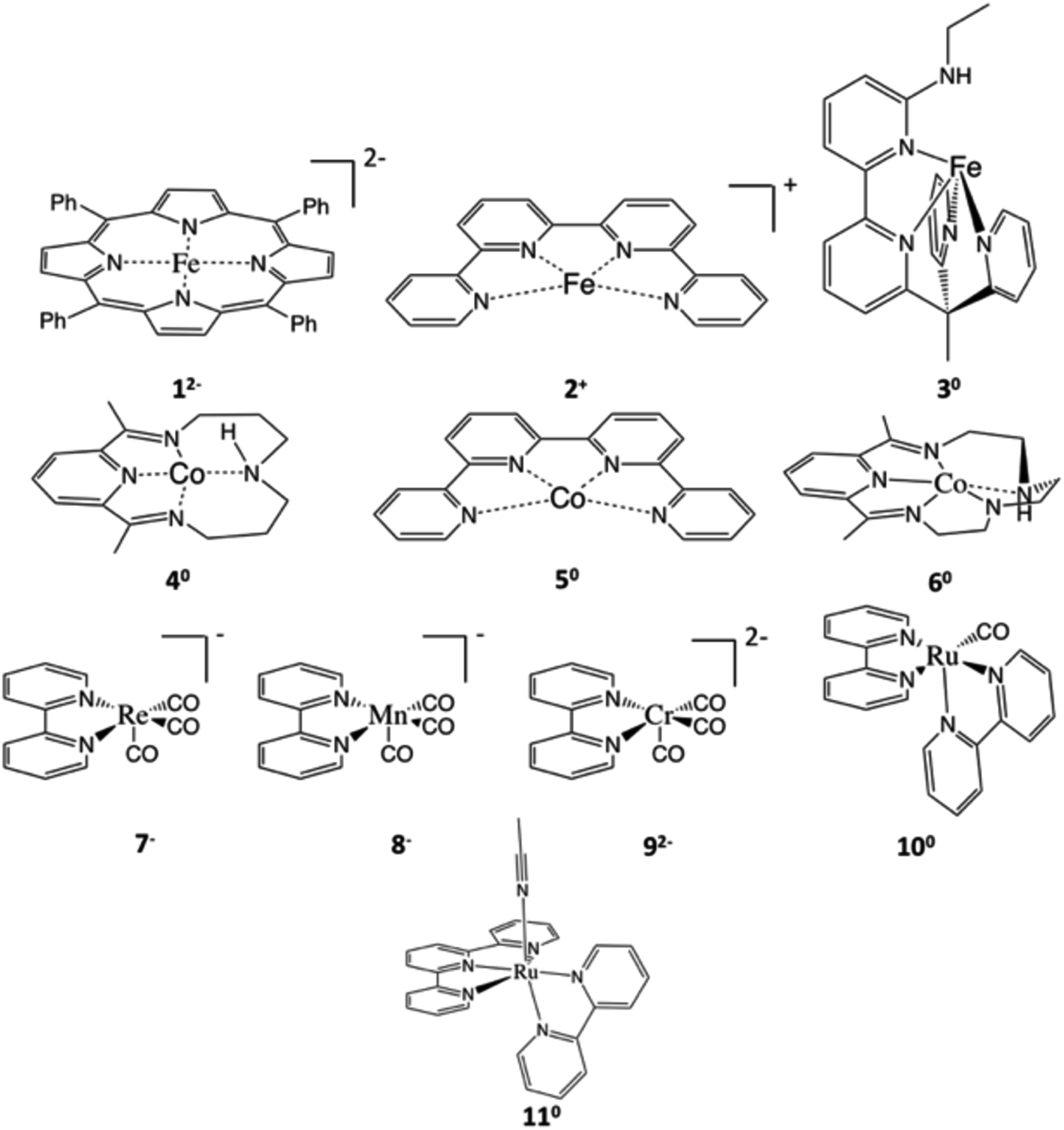 | ||
| Scheme 1 Representative examples of CO2 reduction catalysts studied in this work. Systems supported by non-innocent ligands include 12−,8,16 [M(qpy)]n+ (M = Fe (2+), Co (50), qpy = quaterpyridine),12 [Fe(bpyNHEtPY2Me)]0 (30, bpyNHEtPY2Me = 6′-(1,1-di(pyridin-2-yl)ethyl)-N-ethyl-[2,2′-bipyridin]-6-amine),19 [Co(N4H)]0 (40, N4H = 2,12-dimethyl-3,7,11,17-tetraazabicyclo[11.3.1]-heptadeca-1(7),2,11,13,15-pentane),14 [Co(L)]0 (60, L = 2,13-dimethyl-3,6,9,12,18-pentaazabicyclo-[12.3.1]octadeca-1(18),2,12,14,16-pentaene),13 [M(bpy)(CO)3]n− (M = Re (7−), Mn (8−), Cr (92−); bpy = bipyridine),15,26,27 [Ru(bpy)2(CO)]0 (100),28 and [Ru(tpy)(bpy)]0 (110, tpy = terpyridine).20 All species are labelled as Yz, where z corresponds to the charge of the active species prior to CO2 binding in the catalytic cycle. | ||
Experimental findings suggested that the active species responsible for CO2 conversion is [Fe(TPP)]2− (12−), a formal Fe0 complex, generated by two-electron reduction of 1.8 It is generally accepted that the reaction is initiated by CO2 binding to 12− to yield an η1-CO2 adduct (Scheme 2); however, the following transformation is rather controversial. An earlier experimental study8b reported by Costentin and coworkers suggested that formation of two hydrogen bonds between the leaving O atom of the CO2 complex and two Brønsted acids activates the C–O bond being cleaved (pathway I in Scheme 2). Subsequently, the C–O bond scission is accompanied by a single proton transfer, thereby yielding an FeII-carbonyl species and releasing a hydroxide anion. Although this mechanistic hypothesis likely accounted for the influence of the pKa value of the Brønsted acid on the reaction rate,8b the postulated tri-molecular reaction is expected to suffer from a prohibitively large positive entropic term.9 Theoretical calculations10 instead pointed out that the η1-CO2 adduct first gets protonated to afford a metallacarboxylic acid, which then undergoes C–O bond breaking concerted with second protonation, ultimately furnishing an FeII-carbonyl adduct and H2O (pathway B in Scheme 2). Similar mechanisms have been proposed in the literature for CO2 reduction mediated by related systems.11–15
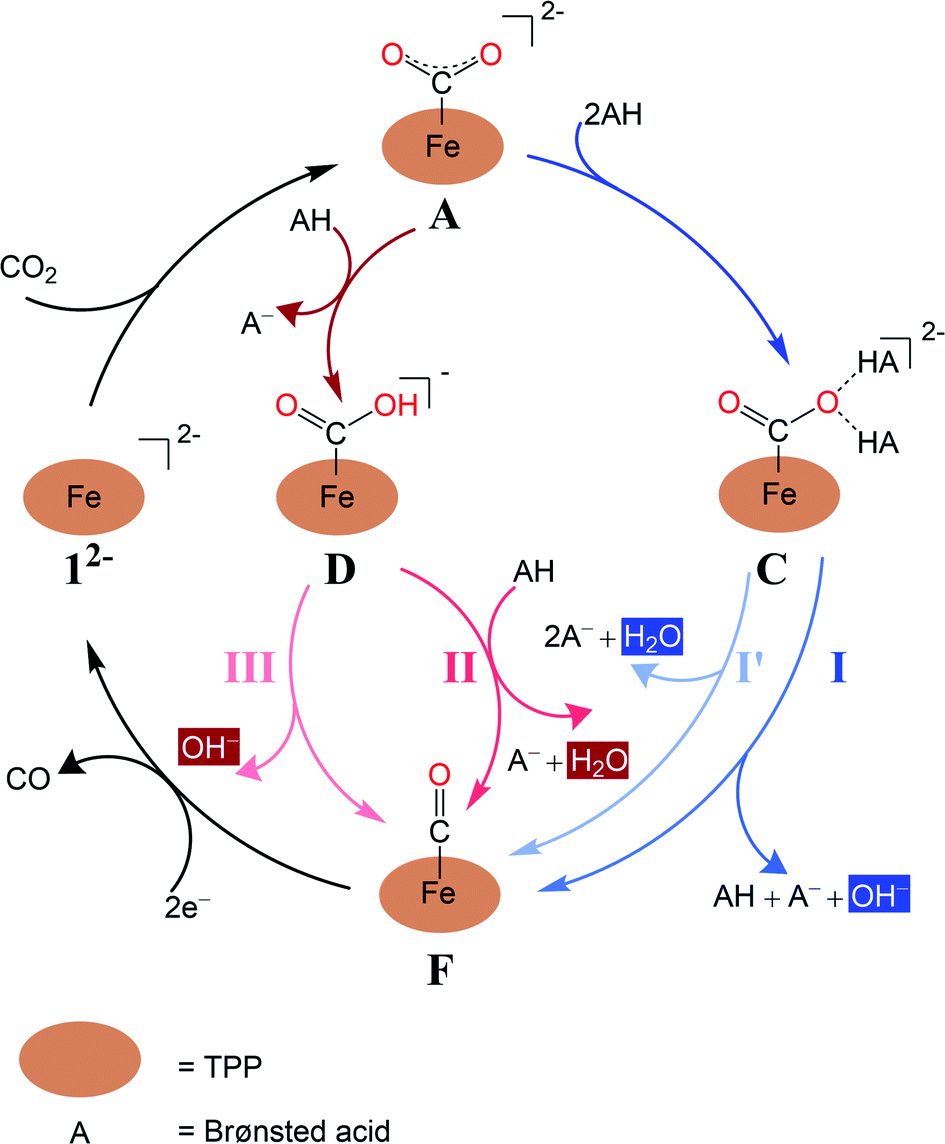 | ||
| Scheme 2 Four mechanisms of CO2-to-CO conversion catalyzed by Fe(TPP) investigated in the present work. | ||
To understand reaction mechanisms, thoroughly elucidating the electronic structures of key intermediates is typically a prerequisite. Our earlier work using a combined spectroscopic and computational approach unequivocally revealed that 12− in fact contains an intermediate spin ferrous center (SFe = 1) that is antiferromagnetically coupled with a TPP4− diradical (STPP = 1), thereby yielding an overall singlet ground state, viz. [FeII(TPP˙˙4−)]2−.16 As such, the two electrons used to reduce CO2 are stored in the non-innocent TPP ligand rather than the Fe center. However, the earlier mechanistic investigations10 did not clarify the role of TPP in the CO2 transformation by 12−. Furthermore, a range of homogeneous CO2 activation catalysts reported thus far are supported by well-known redox active non-innocent ligands, such as porphyrins,8 corroles,17 pyridine-diimines13,14,18 polypyridines,4b,15,19,20 and N-heterocyclic carbenes21 to name a few. The reactions with such a diverse array of systems thus give rise to an intriguing question about how the non-innocent ligand interacts with the metal center to trigger the two-electron CO2 reduction to CO, because apparently not all coordination-unsaturated transition metal complexes bearing non-innocent ligands are capable of activating CO2. In fact, although ligand non-innocence has been intensely discussed in general catalysis,22–25 its implication with respect to CO2 functionalization has received relatively little attention.
The present work serves a dual purpose. We first present a detailed analysis of the electronic-structure evolution in the course of CO2-to-CO conversion catalyzed by 12−, which shows that the non-innocent nature of TPP is at the core of the high catalytic activity. Then, we analyze the electronic structure changes of the same reaction mediated by ten related catalysts, which also exhibit excellent catalytic performances (Table S1†). They include eight metal–polypyridine systems and two metal–pyridine-diimine derivatives, all containing well-known non-innocent ligands (Scheme 1). Comparison of all systems under consideration enables us to identify the crucial role of non-innocent ligands in CO2 activation. On the basis of this, we propose fundamental electronic structure requirements for CO2 reduction with non-innocent systems, which can be used as a guideline for future design of similar catalytic systems.
Computational details
All calculations were performed using the ORCA 4.2 program package.29 For geometry optimizations and frequency calculations, the hybrid B3LYP density functional30 was used in combination with the Def2-TZVP basis set for iron and the first coordination sphere, and the Def2-SVP basis set for all remaining atoms,31 referred to as the B3LYP/Def2-TZVP/Def2-SVP level of theory hereafter. Tight geometry convergence settings and default SCF convergence settings were used for all geometry optimizations. The final electronic energies were computed with the B3LYP functional in combination with the Def2-TZVPP basis set for all atoms (referred to as the B3LYP/Def2-TZVPP level of theory hereafter). Default SCF convergence settings were employed. To account for solvation effects and non-covalent dispersion interactions, the solvation model C-PCM32 for DMF and the Grimme’s D3BJ dispersion corrections were employed, respectively, for all calculations.33,34 Grid level 5 was used for all the calculations. All calculations were accelerated by using the RIJCOSX approximation.35The initial guesses of transition state geometries were obtained at the B3LYP/Def2-TZVP/Def2-SVP level of theory by running relaxed surface scans in the sensible normal modes and/or by calculating using the nudged elastic band method,36 particularly for delicate transition states. The approximate transition state geometries were then optimized by maximizing the energy in a given normal mode and minimizing the energy in all other normal modes. Subsequent frequency analyses showed that local minima have no imaginary frequency, and transition states have only one imaginary frequency, occasionally besides an additional imaginary frequency of less than 16 cm−1 (see the ESI†). Despite our repeated attempts, this residual frequency could not be removed but is attributed to numerical noise owing to its magnitude.
Enthalpies were calculated by adding the zero-point energy (EZPE) and the thermal energy at 298 K (Eth) calculated at the aforementioned level of theory to the electronic energy (Eel) calculated at the B3LYP/Def2-TZVPP level of theory. A kBT term was also added to account for the PV term in an ideal gas approximation. To estimate more accurate energies of intermediates featuring antiferromagnetic spin coupling between the ligand and the metal center, an electronic energy correction (ΔEel) calculated by using the method proposed by Malrieu and Trinquier37 was added to the energy of the broken-symmetry solution (eqn (1a)). Free energies at 298 K were calculated by adding the electronic, translational, vibrational and rotational entropy contributions multiplied by the temperature to the enthalpy.38 In the case of bimolecular reactions in the gas phase, translational entropic contributions typically account for +10 to +15 kcal mol−1 to Gibbs free energy changes,9 as suggested by our earlier work on O2 and CO2 association with transition metal centers.39d,40 Apparently, the gas-phase approximation overestimates the condensed-phase free energy significantly for two reasons. First, translational freedom is largely quenched in the condensed phase, because the volume is occupied by the solvent.41 Consequently, the translation entropy, which is directly dependent on the volume accessible to the solute, is affected by the passage from the gas to condensed phase. The subsequent loss of entropy can be estimated by calculating the loss of accessible volume (ΔSAV).41 Second, gas-phase free energy does not account for the cavitation free energy (ΔGcav-disp). The latter corresponds to the free energy affording the formation of the solvent cage around the solutes. Typically, both effects induce negative Gibbs free energy variations for bimolecular reactions, which partially compensate the large entropic gains calculated in the gas phase. To account for them, we added two correction terms −TΔSAV and ΔGcav-disp to the free energy computed in the gas phase (eqn (1b)). The former was calculated following a procedure described elsewhere with the van der Waals radii of the solutes and solvent.41 The latter was estimated using a linear regression of the cavity surface using the van der Waals radii of the solute.42
| H = Eel + ΔEel + EZPE + Eth + kBT | (1a) |
| G = H − SgT − TΔSAV + ΔGcav-disp | (1b) |
All redox potentials against the SCE electrode in DMF were calculated from the Gibbs free energies of the species of the redox couple, according to the formula:
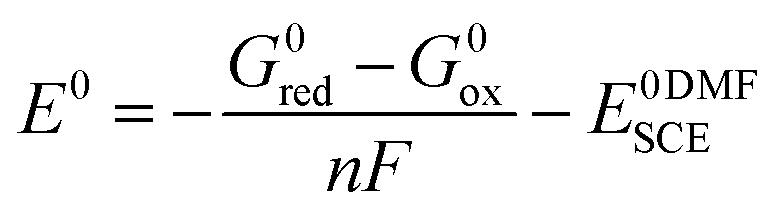 | (2) |
For orbital visualization analysis, the unrestricted corresponding orbitals (UCO)48 with an overlap between the alpha and beta ones greater than 0.95 were localized using the Pipek–Mezey algorithm.49 The alpha and beta sets in this subspace were approximated to be identical, and the orbitals thus obtained from the localized subspace were considered as doubly occupied. The singly-occupied UCO and the magnetic orbitals (overlap ranging from 0 to 0.95) were not localized. Among the resulting orbitals, the d orbitals were identified by their predominant Fe character (>70%) according to the molecular orbital (MO) Löwdin population analysis.
Results and discussion
Potential energy surfaces
Experimental evidence points out that CO2 reduction to CO mediated by 12− is initiated by CO2 association with catalytically active 12−, leading to an η1-CO2 adduct (A in Fig. 1). Accordingly, in the following section, only pathways involving 12− as the catalytically active species are considered.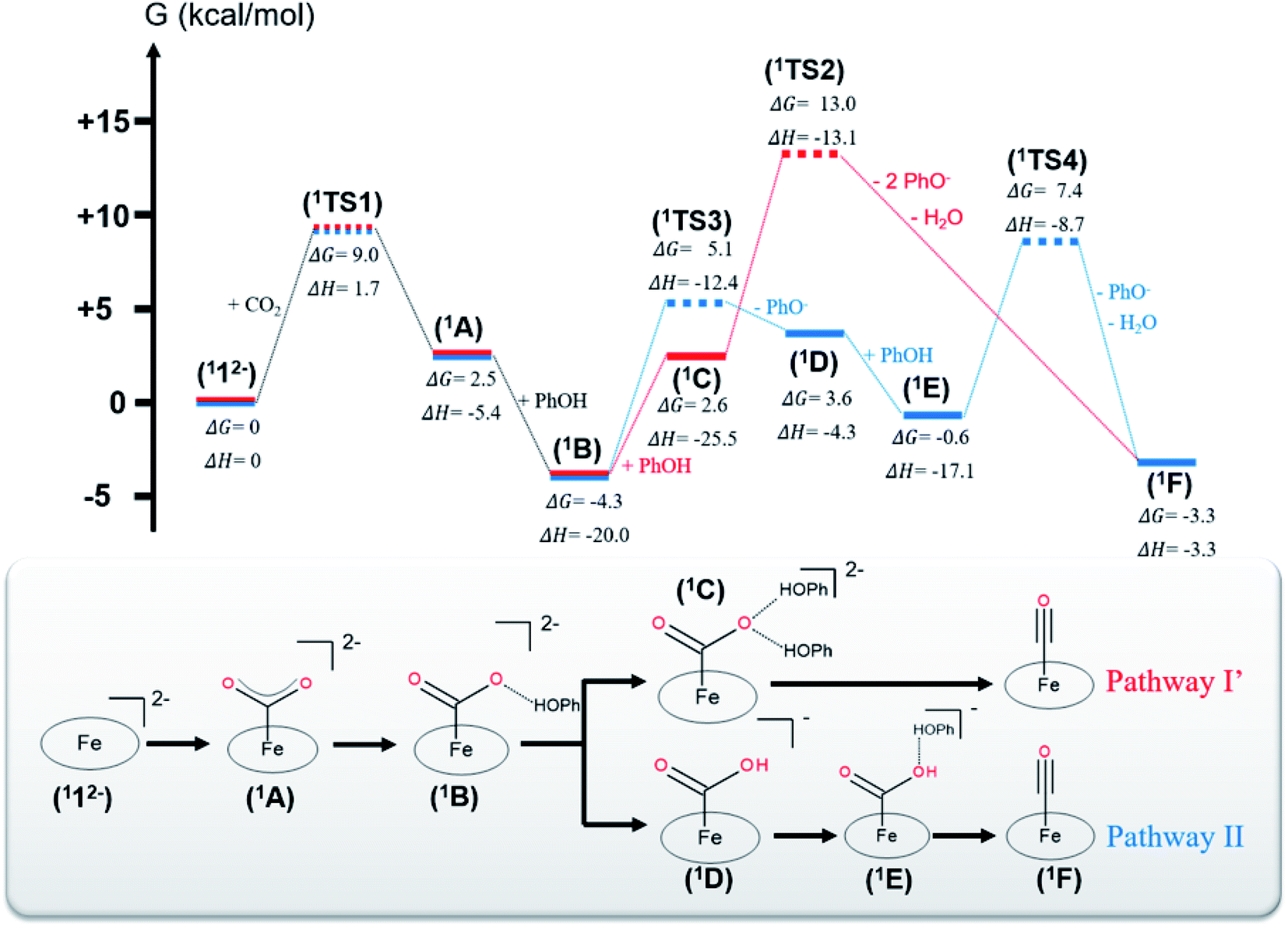 | ||
| Fig. 1 (Top) Energy landscape of the intermediates and transition states involved in the CO2-to-CO reduction. Intermediates are displayed in solid lines, and transition states are displayed in dashed lines. Intermediates belonging to pathway I′ are represented by red lines and those belonging to pathway II by blue lines. The Gibbs free energy changes and enthalpy changes were computed with respect to intermediate 112− and all reactants infinitely separated in DMF. (Bottom) Schematic representations of the intermediates in the order of appearance in pathways I′ (red) and II (blue). Hydrogen bonds are displayed by dashed lines. | ||
The CO2 adduct A then undergoes proton-assisted C–O bond cleavage to produce CO and H2O in acidic media. As discussed above, the mechanistic details of the C–O bond breaking have not reached a consensus yet.8b,10 To address this question, we initially tested the mechanistic proposition due to Costentin and coworkers, pathway I schematized in Scheme 2.8b In this mechanistic scenario, the interactions of the leaving O atom of A with two phenol (PhOH) molecules (the proton donor employed in the experiments8b) furnish complex C that features two hydrogen bonds between the CO2 moiety and PhOH. The following C–O bond cleavage is concomitant with the transfer of only one proton, thereby generating an OH− and the product metal-carbonyl complex [Fe(CO)(TPP)] (F) in the end. The relaxed surface scans (Fig. S2†) in which the C–O distance was systematically varied showed that such a process cannot occur for reasons discussed below. We thus envisioned two more feasible mechanistic scenarios referred to as pathways I′ and II in Fig. 1. In pathway I′, the C–O bond rupture is realized by simultaneous two proton transfer from each PhOH to CO2 in C, leading to F and H2O. In pathway II, A first reacts with one PhOH molecule (rather than two in pathway I′) to form complex B with only one hydrogen bond. After releasing a phenoxide, B transforms into metallacarboxylic acid [Fe(COOH)(TPP)]− (D). Subsequently, D forms a hydrogen bond of its COOH group with an incoming PhOH to convert into complex E, and this intermediate undergoes C–O bond scission assisted by a second proton transfer from PhOH, thereby resulting in F and H2O.
Although 12− was irrefutably identified to feature a singlet ground state,16 we investigated the reaction mechanisms on singlet, triplet and quintet potential energy surfaces in order to explore the possibility of the multi-state reactivity.8g However, our theoretical results predicted that all aforementioned intermediates in the S = 1 and 2 states lie at substantially higher energies than the corresponding diamagnetic ones (Table S2†). Hence, they are very unlikely to participate in the actual reaction (for details, see the ESI†). Therefore, in the present section, we focus on the singlet reaction mechanism. To avoid confusion, hereafter we add a superscript to each species to denote its spin multiplicity.
The two steps leading to the formation of 1B are the same for both pathways I′ and II. For the CO2 adduct 1A, we tested two different CO2 binding modes with the Fe center, namely, η1-CO2 and η1-OCO. However, during the geometry optimizations, the η1-OCO complex spontaneously decomposes into 112− and CO2. Therefore, the η1-CO2 adduct is the most appropriate model for 1A. DFT calculations predicted that formation of 1A traverses a transition state (1TS1) with a moderate barrier of ΔG‡ = +9.0 kcal mol−1 and is slightly endergonic with ΔG = 2.5 kcal mol−1. This positive ΔG value originates from the unfavorable entropic term and the limited exothermicity of the CO2 association (ΔH = −5.4 kcal mol−1). The latter finding can be readily traced back to the fact that typically CO2 is a weak ligand and the metal–CO2 interaction is quite weak.14c,15d,39d In line with this reasoning, the entropy contribution accounts for more than 80% of the barrier. Formation of 1B from 1A is appreciably exergonic (ΔG = −6.8 kcal mol−1) and has no detectable barrier (Fig. S2†). In fact, the step is strongly exothermic due to the formation of a hydrogen bond between the CO2 molecule and the incoming PhOH (ΔH = −14.6 kcal mol−1), but this exothermicity is partially balanced by the entropic cost of associating two fragments (1A and a PhOH).
In pathway I′, formation of 1C from 1B was computed to be significantly endergonic (ΔG = +6.9 kcal mol−1) and barrierless (Fig. S2†). Unlike for the formation of 1B, the enthalpy gain relative to the formation of an additional hydrogen bond between the oxygen of the CO2 and a PhOH is only moderate (ΔH = −4.5 kcal mol−1) and does not compensate for the unfavorable entropic cost relative to the association of 1B and PhOH. The subsequent step that consists of the C–O bond cleavage in 1C followed by the dissociation of the weakly bound H2O and PhO− yielding 1F was estimated to be moderately exergonic (ΔG = −5.9 kcal mol−1). The exergonicity arises from the tremendous entropic contribution due to the dissociation of 1C into four fragments, i.e.1F, H2O and two phenolates (PhO−), even if this step involves a large positive enthalpy change (ΔH = +22.2 kcal mol−1). The conversion of 1C to 1F has to overcome a high barrier of ΔG‡ = +10.4 kcal mol−1 (1TS2), which can be attributed to lack of enough driving force for the C–O bond cleavage in 1C as suggested by the estimated enthalpy change (ΔH = +9.4 kcal mol−1, Table S10†). Interestingly, the free energy of the final product 1F is on par with that of intermediate 1B. Despite this, it should be noted that the reaction still proceeds, because the reduction of 1F to regenerate the catalyst is driven by the potential applied at the electrode.
In pathway II, the transformation of 1B into 1D consists of a proton transfer in 1B followed by the dissociation of the PhO− from the metallacarboxylic acid. The step is moderately endergonic (ΔG = +7.9 kcal mol−1), which originates from an unfavorable enthalpy contribution, although 1D is stabilized by an intramolecular hydrogen bond between the COOH group and one nitrogen of the porphyrin ligand as indicated by a short N–H interatomic distance of 2.02 Å. This step needs to overcome a sizeable barrier (1TS3, ΔG‡ = +9.4 kcal mol−1) that mostly arises from the need of sufficient driving force associated with the proton transfer in 1B (ΔH = +7.3 kcal mol−1, Table S10†). Subsequently, the formation of complex 1E from 1D is exergonic (ΔG = −4.2 kcal mol−1), wherein a strong enthalpic contribution is balanced by an unfavorable entropic term, owing to the association of two fragments (1D and PhOH). No detectable kinetic barrier could be found for this step (Fig. S2†). The next step that is composed of the C–O bond scission in 1E and dissociation of the weakly bound H2O and PhO− is slightly exergonic by ΔG = −2.7 kcal mol−1. The tremendous entropy contribution arising from the dissociation of 1E into three fragments (1F, PhO− and H2O) offsets the positive enthalpy change, ΔH = +13.8 kcal mol−1, similar to the formation of 1F in pathway I′. This transformation needs to pass through a barrier of +8.0 kcal mol−1 (1TS4) that is on par with that of 1TS2 in pathway I′. In analogy to the formation of 1F in pathway I′, this barrier can be attributed to the significant endothermicity of the cleavage of the C–O bond in 1E (ΔH = +6.5 kcal mol−1, Table S10†). However, although for both pathways, the barriers leading to 1F are commensurate, the energy of 1TS2 is +5.6 kcal mol−1 above that of 1TS4. This difference largely arises from the prohibitive entropic term resulting from the association of 1A with two PhOH molecules, because the enthalpy term of 1TS2 is 4.4 kcal mol−1 lower than that of 1TS4.
To investigate whether the second proton transfer is essential to the reaction, we examined another reaction channel, in which the C–O bond cleavage takes place at intermediate 1D to release 1F and a hydroxide ion (pathway III, Fig. S3†) rather than a water molecule. The C–O bond cleavage is significantly uphill (ΔG = +15.2 kcal mol−1) due to a prohibitive positive enthalpic change (ΔH = +23.6 kcal mol−1) and, more importantly, has to cross an unconquerable barrier of ΔG‡ = +43.3 kcal mol−1 (1TS5 in Fig. S3†). In comparison with pathways I′ and II, the high enthalpic cost of the bond cleavage in the present case apparently stems from the absence of protons to trap the resulting OH− to produce H2O, a thermodynamic sink. As a consequence, the C–O bond breaking involves an exceedingly high kinetic barrier. Our finding thus revealed that the C–O bond cleavage is necessarily concurrent with the formation of a water molecule. Following this reasoning, pathway I advocated by Costentin and co-workers8b in which 1C directly dissociates into 1F, PhO−, PhOH and OH− should involve a much higher barrier than that of 1TS2 in pathway I′ and can be safely ruled out as being a plausible mechanism.
As shown in Fig. 1, our theoretical results showed that the highest barrier in pathway I (1TS2) is 7.9 kcal mol−1 higher than that in pathway II (1TS3). Hence, the CO2 reduction process is most likely to proceed along pathway II. To further strengthen this conclusion, we carried out calculations using the double-hybrid w-B2PLYP functional in conjunction with the Def2-QZVPP basis set (Table S10†). The results indicate that 1TS2 is 12.0 kcal mol−1 lower in energy than 1TS3. The energy differences estimated by both density functionals considerably exceeds the typical error range of density functional theory computations.50 This confirms that pathway II is indeed energetically more favorable, congruent with an earlier computational study.10 However, the cited work did not investigate the singlet reaction, but only considered the reaction occurring on the triplet and quintet surfaces. Because the estimated barrier differences between the formation of the CO2 adduct 1A (ΔG‡ = +9.0 kcal mol−1), the first proton transfer (ΔG‡ = +9.4 kcal mol−1) and the C–O bond cleavage in pathway II (ΔG‡ = +7.9 kcal mol−1) fall within the typical uncertainty range of computations with hybrid DFT functionals,51 we cannot unequivocally determine which one is the rate-determining step of the entire catalytic process. Despite this uncertainty, our calculations strongly suggest that the C–O bond breaking involves a sizeable barrier and has to be concerted with a proton transfer, both notions consistent with earlier experimental studies.7,8
To investigate whether our computational model is consistent with the observed kinetics of the reaction, we computed the turnover frequency of the reaction that requires a complete and precise energy landscape of all intermediates and transition states connecting them.52 For pathways I′ and II, the maximum turnover frequency estimated for three different phenol concentrations, [PhOH] = 0.1, 0.75 and 3 M using the method of Costentin and co-workers53 is summarized in Table 1 (see the ESI† for details). The turnover frequencies calculated for pathway II are in reasonable agreement with the experimental values, while those for pathway I′ show substantial deviations. More importantly, pathway II is a first-order reaction with respect to [PhOH], consistent with the experiment, whereas pathway I′ is a second-order reaction. Therefore, pathway II qualitatively and quantitatively reproduces the experimental findings, which thus further lends credence to the reliability of our theoretical results, a necessary premise for the following analyses aiming at obtaining qualitative insights into the reaction mechanism.
| [PhOH] = 0.1 M | [PhOH] = 0.75 M | [PhOH] = 3 M | |
|---|---|---|---|
| Pathway I′ | 4.3 s−1 | 2.4 × 102 s−1 | 3.9 × 103 s−1 |
| Pathway II | 3.6 × 104 s−1 | 2.8 × 104 s−1 | 1.1 × 106 s−1 |
| Experiment | 1.8 × 103 s−1 | 1.5 × 104 s−1 | 1 × 105 s−1 |
We also computationally investigated side reactions that lead to H2 and formic acid (for details, see the ESI†). Typically, these reactions start with formation of a metal-hydride species rather than a CO2 adduct.54 However, the former transformation was observed to suffer from a prohibitive kinetic barrier, compared to the generation of 1A. Furthermore, a previous study has pointed out that η1-OCO adducts can also be the precursors for producing formic acid.39d As specified above, our calculations suggested that such an η1-OCO adduct probably cannot exist in the present case. Both findings likely account for the observed high product selectivity of the CO2 reduction catalyzed by 12−.
Electronic structure analysis of the reaction
In the following, the electronic-structure evolution of pathway II was scrutinized. The purpose is to correlate the electronic structure of 112− with its exceedingly high activity toward CO2 functionalization, in particular, to pinpoint the role played by the non-innocent TPP ligand.As elaborated in our previous work,16 the bonding of 112− is best described as an intermediate spin FeII center (SFe = 1) antiferromagnetically coupled with a triplet TPP˙˙4− diradical (STPP = 1), thereby yielding an overall singlet ground state (Fig. 2b). Specifically, the Fe center features an electronic configuration of (dxy)2(dz2)2(dxz)1(dyz)1, and there are two electrons occupying the low lying TPP centered π* eg orbitals labelled as 1eg(x) and 1eg(y) (in the D4h point group representation). The Fe dxz/yz and TPP 1eg magnetic orbitals form two spin-coupled pairs that represent two antiferromagnetic exchange coupling pathways. It should be noted that the fragment orbitals of Fe dxz/yz and TPP eg belong to the same representation (eg) of the effective D4h point group of 112−; therefore, their interactions are symmetry-allowed as indicated by the computed considerable overlap of the two spin coupled pairs (0.33). If both fragment orbital sets transformed as different irreducible representations of the effective D4h point group, their exchange interaction would feature ferromagnetic coupling on the grounds of the Goodenough–Kanamori rule55 rather than antiferromagnetic coupling as determined experimentally.16,47 In line with this reasoning, the corresponding quintet state (512−), which features ferromagnetic coupling of these two fragments, was estimated to lie 12.1 kcal mol−1 higher in energy than the singlet state.
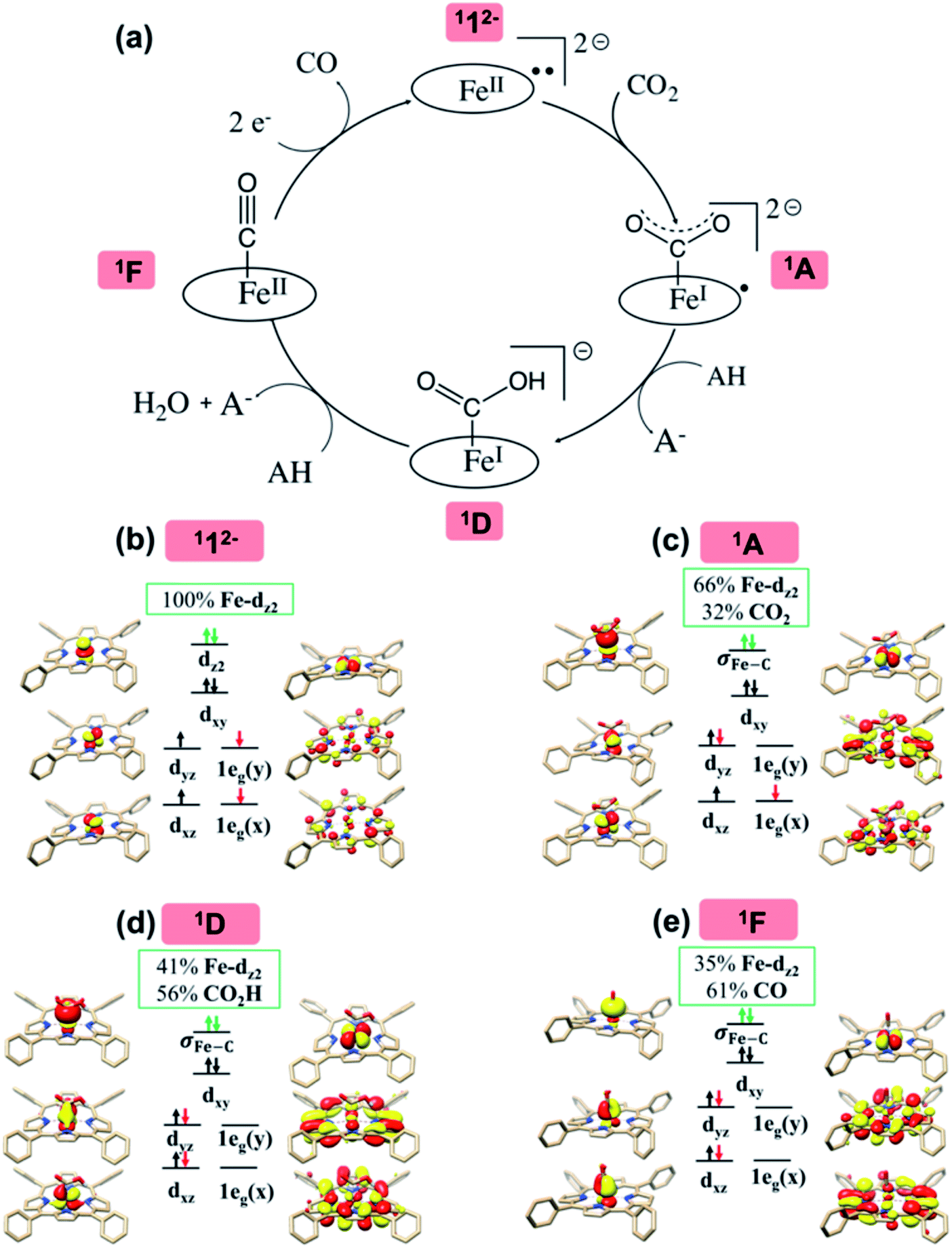 | ||
| Fig. 2 The proposed catalytic cycle of CO2-to-CO conversion catalyzed by 112− (a), and the electronic structures of intermediates 112− (b), 1A (c), 1D (d) and 1F (e). The electrons involved in the TPP-to-Fe intramolecular electron transfer are shown by red arrows, and those in the Fe-to-CO2 electron transfer by green arrows. The weight of Fe and CO2/CO2H/CO in the σFe–C bonding MO is displayed in green boxes. For clarity, all hydrogens are omitted except those in the CO2H moiety. | ||
As depicted in Fig. 2c, the driving force to generate 1A largely stems from the σ donation from the doubly occupied Fe dz2 orbital to the vacant CO2 in-plane π*  orbital. The electron acceptor is one of the lowest unoccupied molecular orbitals (LUMOs) of free CO2 formed by an out-of-phase combination of the 2pz orbitals of the central C atom and the two terminal O atoms and has a larger lobe at the central C atom than those at the two terminal O atoms. Upon CO2 binding, it gets considerably bent with an O–C–O angle of 130° compared to 180° for uncoordinated CO2 molecules. As elaborated earlier,39d such a geometric distortion not only significantly decreases the energy of the CO2
orbital. The electron acceptor is one of the lowest unoccupied molecular orbitals (LUMOs) of free CO2 formed by an out-of-phase combination of the 2pz orbitals of the central C atom and the two terminal O atoms and has a larger lobe at the central C atom than those at the two terminal O atoms. Upon CO2 binding, it gets considerably bent with an O–C–O angle of 130° compared to 180° for uncoordinated CO2 molecules. As elaborated earlier,39d such a geometric distortion not only significantly decreases the energy of the CO2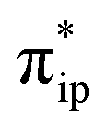 orbital, but also increases the C-p character in it. Thus, the energy difference between the CO2
orbital, but also increases the C-p character in it. Thus, the energy difference between the CO2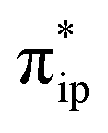 and Fe dz2 orbitals drops, and their overlap becomes more favored. Consequently, both factors work in synergy to enhance the Fe–CO2 interactions. Despite this, the resulting bonding MO, hereafter referred to as σFe–C, contains only 32% CO2 π* parentage; hence, the bonding remains quite weak, consistent with a moderate enthalpy change of −5.4 kcal mol−1 estimated for the formation of 1A (vide supra). Nevertheless, this essentially dative interaction can be viewed as partial electron transfer from the Fe center to the CO2 moiety. Interestingly, the MO analyses revealed that the formation of 1A is accompanied by an intramolecular β electron transfer from TPP 1eg(y) to Fe dyz, whereas the other spin-coupled pair consisting of the Fe dxz and TPP 1eg(x) MOs remains intact. Different from 112−, the Fe dxz and dyz orbitals in 1A are not energetically degenerate anymore, because the former is destabilized by the repulsion with the doubly occupied σ orbitals of the two C–O bonds; while the latter is stabilized by its back-donation to the CO2 out-of-plane π* orbital, the other LUMO of free CO2. Consequently, the electronic structure of 1A is best formulated as having a low spin FeI center (SFe = 1/2) that is bound to an approximately charge-neutral CO2 and is antiferromagnetically coupled with a TPP˙3− radical (STPP = 1/2), thus giving an overall singlet ground state. More importantly, the TPP-to-Fe electron transfer mitigates the depletion of the electron density of the Fe center resulting from the Fe-to-CO2 σ donation. Therefore, the CO2 association process does not engender substantial variation of the electron density of the Fe center.
and Fe dz2 orbitals drops, and their overlap becomes more favored. Consequently, both factors work in synergy to enhance the Fe–CO2 interactions. Despite this, the resulting bonding MO, hereafter referred to as σFe–C, contains only 32% CO2 π* parentage; hence, the bonding remains quite weak, consistent with a moderate enthalpy change of −5.4 kcal mol−1 estimated for the formation of 1A (vide supra). Nevertheless, this essentially dative interaction can be viewed as partial electron transfer from the Fe center to the CO2 moiety. Interestingly, the MO analyses revealed that the formation of 1A is accompanied by an intramolecular β electron transfer from TPP 1eg(y) to Fe dyz, whereas the other spin-coupled pair consisting of the Fe dxz and TPP 1eg(x) MOs remains intact. Different from 112−, the Fe dxz and dyz orbitals in 1A are not energetically degenerate anymore, because the former is destabilized by the repulsion with the doubly occupied σ orbitals of the two C–O bonds; while the latter is stabilized by its back-donation to the CO2 out-of-plane π* orbital, the other LUMO of free CO2. Consequently, the electronic structure of 1A is best formulated as having a low spin FeI center (SFe = 1/2) that is bound to an approximately charge-neutral CO2 and is antiferromagnetically coupled with a TPP˙3− radical (STPP = 1/2), thus giving an overall singlet ground state. More importantly, the TPP-to-Fe electron transfer mitigates the depletion of the electron density of the Fe center resulting from the Fe-to-CO2 σ donation. Therefore, the CO2 association process does not engender substantial variation of the electron density of the Fe center.
Compared to uncoordinated CO2, the bent CO2 moiety is primed for protonation to afford 1D. Besides the influence on its LUMO discussed above, the CO2 bending also increases the energy of its highest occupied molecular orbital (HOMO), an antisymmetric combination of the two O lone pairs.39d The interaction of the O lone pairs with a proton is therefore favored by the geometric distortion of the CO2 moiety. Our theoretical results revealed that the protonation causes an increase of the CO2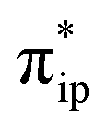 parentage in the doubly occupied σFe–C MO from 32% in 1A to 56% in 1D (Fig. 2d), thereby indicating substantial covalent character for the Fe–CO2H interaction. Consequently, upon going from 1A to 1D, the calculated Fe–CCO2 bond distance shortens from 2.02 Å to 1.89 Å. This bonding description suggests that, on average, the CO2 ligand is reduced by one electron at the stage of 1D. As such, the conversion of 1A to 1D is best described as a concerted proton-electron transfer (CPET) to the CO2 ligand.56 Moreover, the TPP ligand in 1D returns to its usual state of a closed-shell dianion, because the protonation is companied by a β electron transfer from TPP 1eg(x) to Fe dxz. Due to the high covalency of the Fe–CO2H bond, 1D has to be described as a resonance hybrid of a low spin FeII ion (SFe = 0) bound to a −C(O)OH ligand and a low spin Fe0 center (SFe = 0) coordinated by a +C(O)OH ligand. In analogy to the preceding step, the TPP-to-Fe electron transfer balances the Fe-to-CO2 electron transfer, and the electron density of the Fe center remains largely unchanged.
parentage in the doubly occupied σFe–C MO from 32% in 1A to 56% in 1D (Fig. 2d), thereby indicating substantial covalent character for the Fe–CO2H interaction. Consequently, upon going from 1A to 1D, the calculated Fe–CCO2 bond distance shortens from 2.02 Å to 1.89 Å. This bonding description suggests that, on average, the CO2 ligand is reduced by one electron at the stage of 1D. As such, the conversion of 1A to 1D is best described as a concerted proton-electron transfer (CPET) to the CO2 ligand.56 Moreover, the TPP ligand in 1D returns to its usual state of a closed-shell dianion, because the protonation is companied by a β electron transfer from TPP 1eg(x) to Fe dxz. Due to the high covalency of the Fe–CO2H bond, 1D has to be described as a resonance hybrid of a low spin FeII ion (SFe = 0) bound to a −C(O)OH ligand and a low spin Fe0 center (SFe = 0) coordinated by a +C(O)OH ligand. In analogy to the preceding step, the TPP-to-Fe electron transfer balances the Fe-to-CO2 electron transfer, and the electron density of the Fe center remains largely unchanged.
The MO diagram of 1TS4 (Fig. S8†) suggests that, over the course of the heterolytic C–O bond breaking, the C–O σ bonding orbital evolves into a lone pair of the O atom in the H2O product. Accordingly, the resulting C atom formally becomes a carbocation center, and due to its exceedingly high electron-accepting capability, the σFe–C MO of 1F acquires more C character (61%) as the expense of the weight of the Fe dz2 atomic orbital dropping to 35% (Fig. 2e). Hence, at this stage, the two-electron transfer from the Fe dz2 orbital to the CO2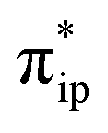 orbital is largely completed. Of note, 1F also features strong back-donation from the doubly-occupied Fe dxz and dyz orbitals to the C–O π* orbitals. Thus, 1F is best described as containing a low spin ferrous center bound to a CO ligand and a closed-shell porphyrin dianion.
orbital is largely completed. Of note, 1F also features strong back-donation from the doubly-occupied Fe dxz and dyz orbitals to the C–O π* orbitals. Thus, 1F is best described as containing a low spin ferrous center bound to a CO ligand and a closed-shell porphyrin dianion.
The role of the non-innocent TPP ligand in the reaction
In the present case, the active species 112− is formed by a TPP centered two-electron reduction of 31 at a mild potential. Because the electron acceptors are the highly delocalized TPP centered 1eg orbitals, the additional electron density is distributed to the twenty-four atoms of the porphyrin ligand. Therefore, the reduction does not significantly escalate the interelectronic repulsion of all delocalized π electrons of the reduced TPP4− ligand. For the same reason, this electron transfer is subjected to a low degree of geometric distortions and hence a reduced reorganization energy. This contrasts with metal centered redox processes for which much more compact and localized d orbitals function as redox active orbitals; consequently, reduction typically causes a much higher gain in interelectronic repulsion. Likewise, a greater reorganization energy is anticipated because of the more significant distortions of the first coordination sphere of the metal center. Typically, two-electron reduction processes of 3d transition metal ions cannot readily occur, because the required reduction potentials are often exceedingly negative. The resulting complexes are likely to be highly reactive and may involve various facile side-reactions or deactivation pathways for electron-richer metal centers, such as H2 or HCOOH generation (see the ESI†), instead of participating in the target reaction. Therefore, the non-innocent nature of TPP explains why 112− can be generated at a mild potential and its catalyzed reaction exhibits high faradaic efficiency for CO production.Although the reducing equivalents are stored on the TPP ligand, CO2 must associate with the metal center for further activation. This is due to the completely delocalized nature of the π-electrons; because none of the C atoms of TPP possesses sufficient electron density allowing for facile CO2 binding. In this regard, the Fe center is superior, because its high-lying doubly populated dz2 orbital has an appropriate shape and can efficiently overlap with the CO2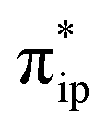 orbital.39d Hence, the Fe center should be the active site for CO2 functionalization. However, the required reducing equivalents are stored at the ligand.
orbital.39d Hence, the Fe center should be the active site for CO2 functionalization. However, the required reducing equivalents are stored at the ligand.
The above described situation gives rise to an intriguing question of how the metal center communicates with the ligand in order to efficiently transfer the reducing equivalents to the CO2 moiety. As elaborated above, the TPP to CO2 two-electron transfer required for conversion of CO2 to CO is achieved by two simultaneous electron transfer events. One is the two-electron transfer from Fe dz2 to CO2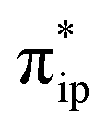 , and the other is the two-electron transfer from TPP 1eg to Fe dxz/yz. Both electron-transfer events are coupled in such a way that the electron density of the Fe center does not vary significantly throughout the reaction. This reduces the energy resulting from the adjustments in the first coordination sphere of the Fe center, while maintaining the electron donating abilities of the Fe center, thereby preventing the formation of highly energetic intermediates or transition states. This analysis underscores the fundamental importance of the non-innocence of the TPP ligand and its cooperativity with the Fe center to the reactivity, which, at least in part, rationalizes why 112− exhibits the exceedingly high catalytic activity towards CO2 reduction.
, and the other is the two-electron transfer from TPP 1eg to Fe dxz/yz. Both electron-transfer events are coupled in such a way that the electron density of the Fe center does not vary significantly throughout the reaction. This reduces the energy resulting from the adjustments in the first coordination sphere of the Fe center, while maintaining the electron donating abilities of the Fe center, thereby preventing the formation of highly energetic intermediates or transition states. This analysis underscores the fundamental importance of the non-innocence of the TPP ligand and its cooperativity with the Fe center to the reactivity, which, at least in part, rationalizes why 112− exhibits the exceedingly high catalytic activity towards CO2 reduction.
What happens if the TPP-to-Fe electron transfer cannot take place? As detailed in the ESI,† the triplet state of [Fe(TPP)]2− (312−) was computed to lie 5.7 kcal mol−1 above 112−. It also contains an intermediate spin ferrous center but interacts with an open-shell singlet TPP˙˙4− diradical with one α electron and one β electron occupying its 1eg(x) and 1eg(y) orbitals, respectively (Fig. S5†). In analogy to the electronic structure changes found for the singlet reaction, CO2 binding to 312− is also driven by a weak σ donation from Fe dz2 to CO2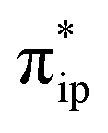 , concomitant with a spin-allowed electron transfer of a β electron from TPP 1eg(y) to Fe dyz. Consequently, the triplet Fe–CO2 adduct (3A) is best described as having a low spin FeI center ferromagnetically coupled with a TPP˙3− radical, and its formation was estimated to be thermodynamically equally probable (ΔG = −0.2 kcal mol−1) with that of 1A (ΔG = +2.5 kcal mol−1) within the error range of DFT computations. Subsequently, protonation of 3A to yield 3D indeed induces further electron transfer from Fe to CO2, but unlike on the singlet surface, the electron transfer from TPP 1eg(x) to Fe dxz cannot occur because this process is spin-forbidden. Hence, upon going from 3A to 3D, the electron density at the Fe center is significantly depleted. Congruent with this reasoning, generation of 3D was calculated to be uphill to ΔG = +10.0 kcal mol−1, whereas that of 1D is only ΔG = +1.1 kcal mol−1. In fact, the cost for the formation of 3D is even greater than the highest barrier in the singlet pathway. As such, these findings demonstrate that synchronization of the TPP-to-Fe electron transfer with the Fe-to-CO2 electron transfer is fundamentally crucial to the reaction kinetics.
, concomitant with a spin-allowed electron transfer of a β electron from TPP 1eg(y) to Fe dyz. Consequently, the triplet Fe–CO2 adduct (3A) is best described as having a low spin FeI center ferromagnetically coupled with a TPP˙3− radical, and its formation was estimated to be thermodynamically equally probable (ΔG = −0.2 kcal mol−1) with that of 1A (ΔG = +2.5 kcal mol−1) within the error range of DFT computations. Subsequently, protonation of 3A to yield 3D indeed induces further electron transfer from Fe to CO2, but unlike on the singlet surface, the electron transfer from TPP 1eg(x) to Fe dxz cannot occur because this process is spin-forbidden. Hence, upon going from 3A to 3D, the electron density at the Fe center is significantly depleted. Congruent with this reasoning, generation of 3D was calculated to be uphill to ΔG = +10.0 kcal mol−1, whereas that of 1D is only ΔG = +1.1 kcal mol−1. In fact, the cost for the formation of 3D is even greater than the highest barrier in the singlet pathway. As such, these findings demonstrate that synchronization of the TPP-to-Fe electron transfer with the Fe-to-CO2 electron transfer is fundamentally crucial to the reaction kinetics.
How does the system synchronize the TPP-to-Fe electron transfer with the Fe-to-CO2 electron transfer? First, the TPP eg and Fe dxz,yz fragment orbitals have comparable energy, otherwise the electron transfer would not be thermodynamically feasible. Most importantly, both sets transform as the same irreducible representation of the effective D4h point group of 112−. Consequently, they form two spin-coupled pairs with considerable overlaps. The antiferromagnetic coupling between the ligand and the metal center lowers the energy of the singlet energy surface compared to that of higher spin multiplicities and ensures that the electron transfer does not incur an energetically unfavorable spin crossover.57 As seen in Fig. 3, along the reaction coordinates of the formation of 1A, the sum of Fe and CO2 populations of the β-1eg(x) magnetic orbital slowly increases from about 6% in 112− to 20% in 1TS1 and then increases drastically and finally reaches 98% in 1A. Such a continuous electron transfer minimizes the variation of electron density of the metal center along the reaction coordinates. Consequently, the loss of Fe electron density due to the Fe to CO2 transfer is immediately compensated by an increasing TPP to Fe electron delocalization in the magnetic orbitals, which likely lowers the activation barrier by increasing the donating abilities of the metal center, even when the complete ligand-to-metal electron transfer lacks significant thermodynamic driving forces. Hence, the antiferromagnetic coupling is pivotal in synchronizing the two electron-transfer events.
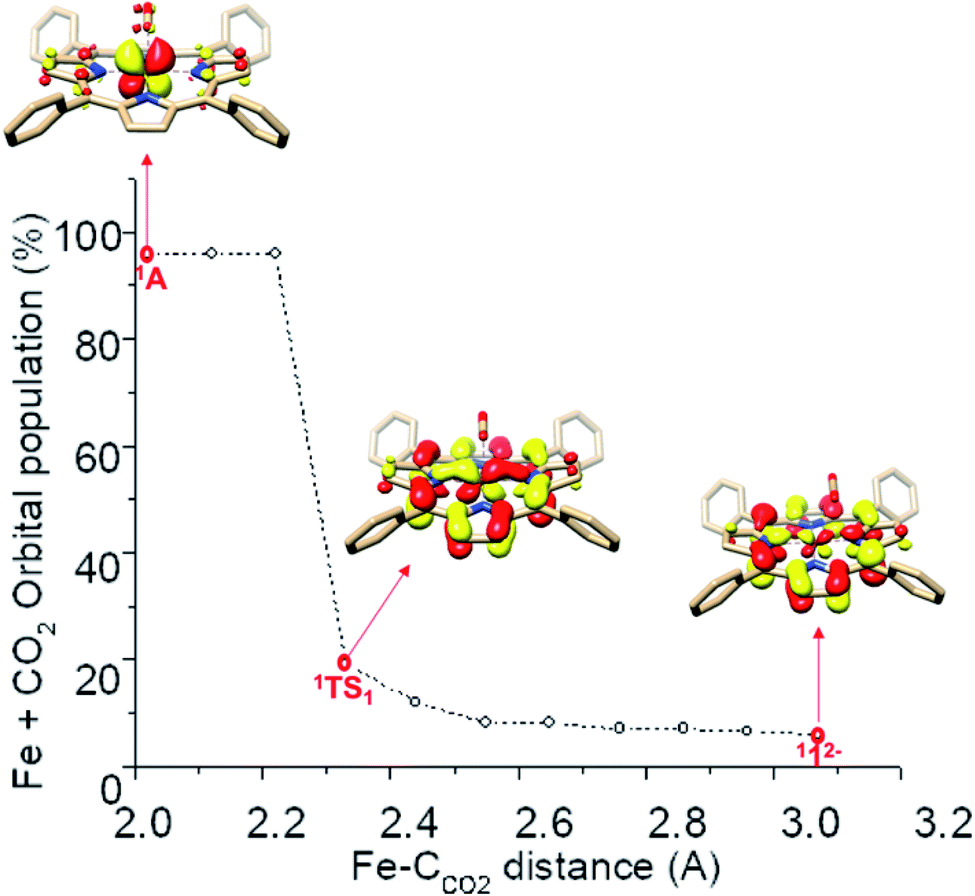 | ||
| Fig. 3 Evolution of the sum of Fe and CO2 Löwdin population of the β (spin-down) magnetic orbital 1eg(y) as a function of the Fe–CCO2 distance during the formation of intermediate 1A. The dots correspond to the structures calculated along the relaxed surface scan. The red dot corresponds to 1A, 1TS1 and 112− (from left to right). | ||
The above analyses correlate the electronic structure of 112− with its high activity. In fact, non-innocent ligands were found for a range of CO2 reduction catalysts. Besides 112−, we have analyzed the electronic-structure evolution in the course of the CO2-to-CO reactions mediated by at least ten related catalysts (Scheme 2) that are also supported by non-innocent ligand platforms (for details, see the ESI†). For all these systems, the metal center acts as the active site for CO2 functionalization, but the non-innocent ligand acts as the electron reservoir. Like 112−, this requires the metal-to-CO2 electron transfer to be coupled with the ligand-to-metal electron transfer. Our results demonstrated that the main differences among them lie in the nature of the chemical steps accomplishing the ligand-to-metal electron transfer, according to which these catalysts are divided into three categories.
Category I catalysts include 112− (Fig. 2), 22+ (Fig. S9†)12 and 330 (Fig. S10†).19 The active species consists of a metal center in its usual oxidation state that is antiferromagnetically coupled with a one- (22+) or two-electron reduced ligand diradical (112− and 330). This category is distinguished by the ligand-to-metal electron transfer being achieved by two separate one-electron transfer events; one takes place during the CO2 adduct formation step, and the other during the first protonation step.
Category II catalysts consist of 240, 250 and 260,12–14 which either feature a metal center antiferromagnetically coupled with a two-electron reduced ligand in a triplet state (240, 250), or a metal center coupled with a doubly reduced ligand in a singlet state (260). Irrespective of the electronic structures, the electron transfer pathways are identical for these three catalysts. The characteristics of category II catalysts are that, for two separate ligand-to-metal electron transfer events, one occurs during the CO2 adduct formation step, and the other during the final C–O bond cleavage step. Here we take the reaction with 240 as an example to discuss the electronic-structure evolution along the reaction coordinate, and summarize those for 250 and 260 in the ESI (Fig. S11 and S12†).
The reaction mechanism of selective CO2 to CO conversion catalyzed by 240 under wet conditions has been subjected to extensive computational and experimental studies.14 It was found that 240 first binds CO2 to form a η1-CO2 adduct 2G, which then undergoes protonation to yield metallacarboxylic acid 2H, and the C–O bond is cleaved concomitant with the second proton transfer, furnishing metal-carbonyl intermediate 2I and releasing a water molecule.
According to the reported reaction pathway14 shown in Fig. 4a, we analyzed the electronic structure changes during the reaction. Our present investigation supports the notion that the pyridine-diimine ligand in 240 is non-innocent, consistent with earlier studies.14c,18,58,59 As shown in Fig. 4b, 240 was found to be composed of a low spin CoII (SCo = 1/2) antiferromagnetically coupled with a N4H˙˙2− diradical (SN4H = 1). This bonding description indicates that, similar to TPP2−, the N4H0 ligand can harbor additional two electrons in its conjugated π system. Our MO analysis (Fig. 4b) suggests that 240 contains a CoII center featuring an electron configuration of (dxy)2(dz2)2(dxz)2(dyz)1 and two delocalized unpaired electrons populating the N4H π* based 1a′ and 1a′′ MOs, labelled according to their symmetry representation of Cs point group. Of note, besides the N4H 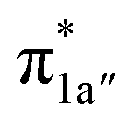 fragment orbital (71%), the Co dyz atomic orbital (29%) makes a sizeable contribution to the 1a′′ MO, because both transform as the A′′ representation of Cs point group. Consequently, the magnetic orbitals of N4H
fragment orbital (71%), the Co dyz atomic orbital (29%) makes a sizeable contribution to the 1a′′ MO, because both transform as the A′′ representation of Cs point group. Consequently, the magnetic orbitals of N4H 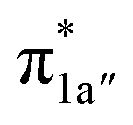 and Co dyz overlap significantly, hence leading to strong antiferromagnetic coupling between the ligand and the metal center.
and Co dyz overlap significantly, hence leading to strong antiferromagnetic coupling between the ligand and the metal center.
 | ||
| Fig. 4 The proposed catalytic cycle of CO2-to-CO conversion catalyzed by 240 (a),14e and the electronic structures of the intermediates 240 (b), 2G (c), 2H (d) and 2I (e). Dashed straight lines between atoms represent hydrogen bonds. Electrons involved in the ligand-to-metal intramolecular transfer are shown by red arrows. Electrons involved in the metal-to-CO2 electron transfer are shown by green arrows. The weight of Co and CO2/CO2H/CO in the σCo–C molecular orbital is displayed in green boxes. In 2H, the weight of Co and N4H in the singly-occupied 1a′ molecular orbital is displayed in red boxes. For clarity, all hydrogens are omitted except the hydrogen of the CO2H moiety and that involved in the intramolecular hydrogen bond between the CO2H moiety and the amine of the N4H ligand. | ||
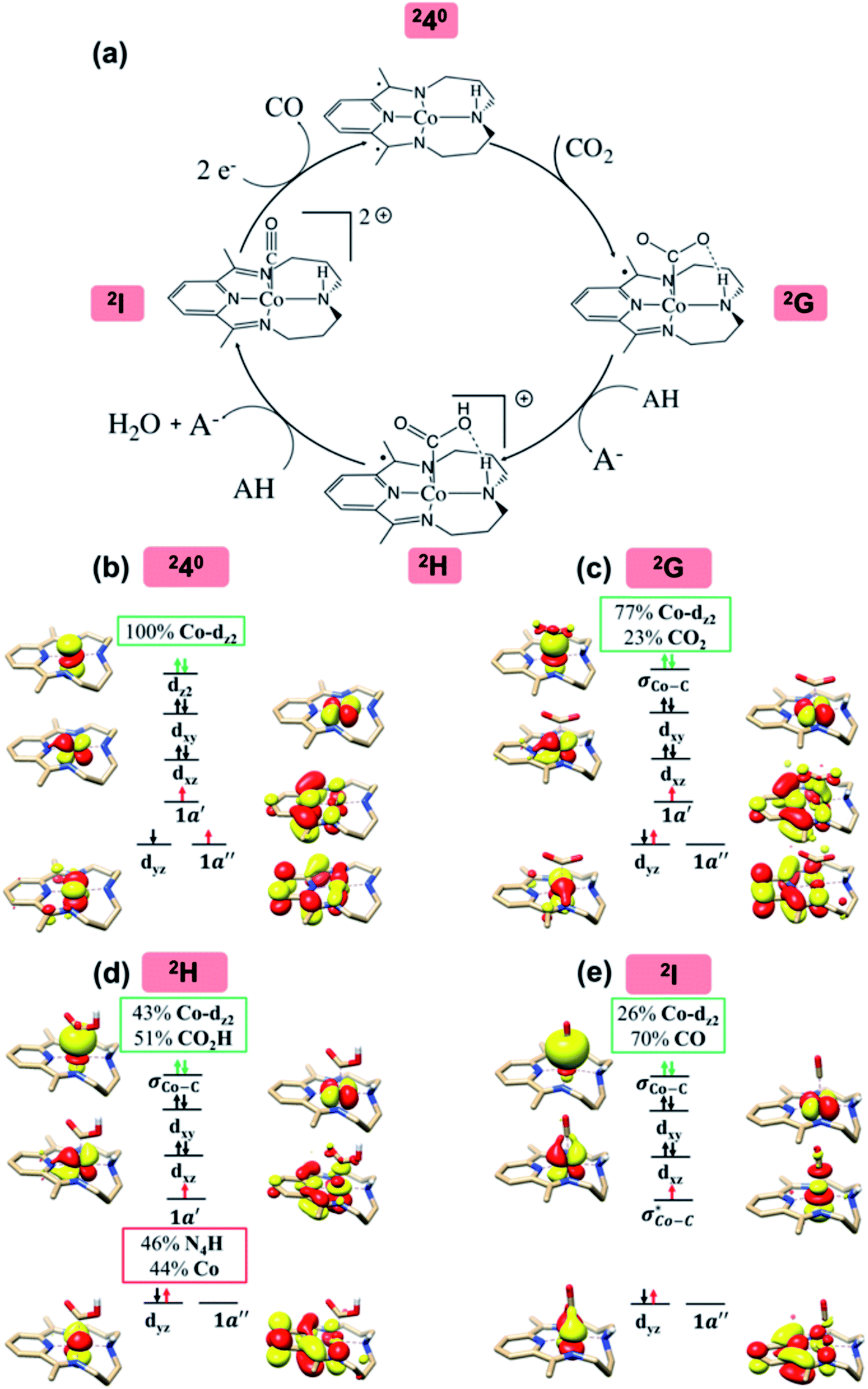
Upon formation of 2G from 240, the doubly-occupied Co dz2 orbital slightly mixes with the unoccupied CO2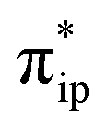 orbital. The resulting Co–CO2 bond is thus essentially dative, as suggested by the estimated dominant Co percentage in the σCo–C MO (77% Co, 23% CO2, Fig. 4c). This step is coupled with an electron transfer from N4H
orbital. The resulting Co–CO2 bond is thus essentially dative, as suggested by the estimated dominant Co percentage in the σCo–C MO (77% Co, 23% CO2, Fig. 4c). This step is coupled with an electron transfer from N4H 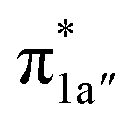 to Co dyz such that the resulting intermediate (2G) is best described as a low-spin (SCo = 0) CoI center weakly bound to a CO20 ligand and to an N4H˙− radical (SN4H = 1/2). The subsequent conversion of 2G into 2H substantially increases the covalency of the Co–CCO2H interaction, as suggested by σCo–C containing nearly identical percentages of Co and CO2H (43% Co and 51% CO2H, Fig. 4d). Hence, this step can be viewed as the first Co-to-CO2 electron transfer. Because
to Co dyz such that the resulting intermediate (2G) is best described as a low-spin (SCo = 0) CoI center weakly bound to a CO20 ligand and to an N4H˙− radical (SN4H = 1/2). The subsequent conversion of 2G into 2H substantially increases the covalency of the Co–CCO2H interaction, as suggested by σCo–C containing nearly identical percentages of Co and CO2H (43% Co and 51% CO2H, Fig. 4d). Hence, this step can be viewed as the first Co-to-CO2 electron transfer. Because 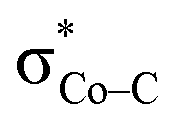 and N4H
and N4H 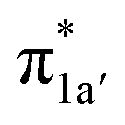 belong to the A′ representation of Cs point group, both fragment orbitals can interact with each other, and the resulting 1a′ MO acquires sizeable character of
belong to the A′ representation of Cs point group, both fragment orbitals can interact with each other, and the resulting 1a′ MO acquires sizeable character of 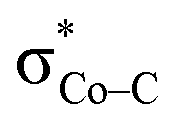 (Co dz2 44% and CO2H 10%). This orbital mixing can be interpreted as a partial electron transfer from the N4H ligand to the Co center. The final C–O bond heterolytic cleavage to afford 2I and H2O further increases the CO character in σCo–C (26% Co and 70% CO, Fig. 4e). Therefore, it is reasonable to consider this step as the second Co-to-CO2 electron transfer, accompanied by completion of the N4H
(Co dz2 44% and CO2H 10%). This orbital mixing can be interpreted as a partial electron transfer from the N4H ligand to the Co center. The final C–O bond heterolytic cleavage to afford 2I and H2O further increases the CO character in σCo–C (26% Co and 70% CO, Fig. 4e). Therefore, it is reasonable to consider this step as the second Co-to-CO2 electron transfer, accompanied by completion of the N4H 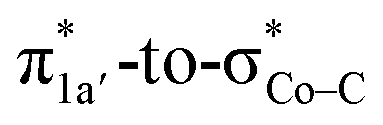 electron transfer. Consequently, this results in a CoII center bound to a carbonyl ligand in 2I.
electron transfer. Consequently, this results in a CoII center bound to a carbonyl ligand in 2I.
The reaction entails two intramolecular electron transfer events: a metal-to-CO2 transfer that is affected by σ donation from the doubly-occupied Co dz2 to the unoccupied CO2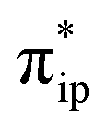 orbital, and a N4H-to-Co electron transfer that makes up for the loss of electron density on the metal center. Both events are coupled efficiently via the mixing of the ligand-centered electron donating orbitals (N4H
orbital, and a N4H-to-Co electron transfer that makes up for the loss of electron density on the metal center. Both events are coupled efficiently via the mixing of the ligand-centered electron donating orbitals (N4H 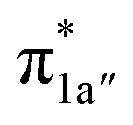 and
and 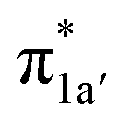 ) and the metal-based electron accepting orbitals (Co dyz and dz2) of the appropriate symmetry and comparable energies. This orbital mixing gives rise to a spin-coupled orbital pair involving Co dyz and N4H
) and the metal-based electron accepting orbitals (Co dyz and dz2) of the appropriate symmetry and comparable energies. This orbital mixing gives rise to a spin-coupled orbital pair involving Co dyz and N4H 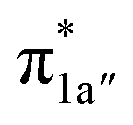 and a singly-occupied delocalized MO composed of N4H
and a singly-occupied delocalized MO composed of N4H 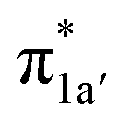 and
and 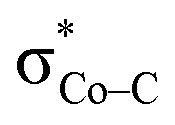 . The first electron transfer route involving the spin-coupled pair is the same as that found for the reaction with 112−. On the other hand, the second pathway chooses
. The first electron transfer route involving the spin-coupled pair is the same as that found for the reaction with 112−. On the other hand, the second pathway chooses 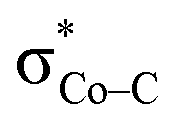 instead of Co dxz, because Co dxz is doubly populated in 240, and
instead of Co dxz, because Co dxz is doubly populated in 240, and 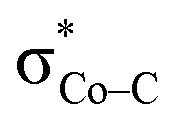 is the lowest-lying vacant Co d orbital besides Co dyz. However, due to the high energy of
is the lowest-lying vacant Co d orbital besides Co dyz. However, due to the high energy of 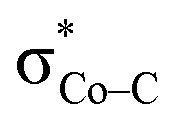 , a significant depletion of the electron density on the metal center is required to give this electron transfer a sufficient thermodynamic driving force. Hence, unlike in 112−, the transfer is not yet complete in 2H.
, a significant depletion of the electron density on the metal center is required to give this electron transfer a sufficient thermodynamic driving force. Hence, unlike in 112−, the transfer is not yet complete in 2H.
Category III catalysts include 17−,1518−,26192−,271100,28 and 1110,20 all of which contain metal centers coupled with closed-shell, doubly-reduced ligands. For category III catalysts, the ligand-to-metal transfer is entirely completed during the formation of the metal–CO2 adduct. Here we take the reaction with 17− as an example to discuss the electronic structure evolution along the reaction coordinate. The reactions with 192−, 18−, 1100 and 1110 are discussed in the ESI (Fig. S13–S16†).
It was found in the 1980s that 17− and its derivatives can catalyze selective CO2-to-CO reduction, and the reaction exhibits one of the highest turnover frequencies reported for homogeneous catalysis (Table S1†).15h The mechanism of CO2 reduction by 17− has been extensively studied, particularly in the work published by Keith and co-workers.15d In their original study, it was shown that the inclusion of a K+ counterion in the vicinity of 17− drastically improved the description of the redox potentials associated with the formation of the active species.15d Hence, the active species was proposed to be the ion pair 17−–K+ (Fig. 5a), rather than anionic complex 17−.15d The reduction of CO2 is initiated by binding of CO2 to 17−–K+ to form an η1-CO2 adduct 1J, which is then protonated to afford carboxylic acid 1K. Subsequently, 1K undergoes a second reduction at the electrode, and the C–O bond breaking is concomitant with a second proton transfer, ultimately yielding 2L and releasing a water molecule and the K+ counterion.
 | ||
| Fig. 5 The proposed catalytic cycle of CO2-to-CO conversion catalyzed by 17− (a),15d and the electronic structure of the intermediates 17−–K+ (b), 1J (c), 1K (d) and 2L (e). Electrons involved in the ligand-to-metal and metal-to-CO2 intramolecular transfer are shown by green arrows. The weight of Re and bpy in the 1π* molecular orbital (in 17−–K+) and the weight of Re, bpy and CO2/CO2H/CO in the σRe–C bonding orbital (in all other intermediates) are displayed in green boxes. For clarity, all hydrogens are omitted except the hydrogen of the CO2H moiety. | ||
As the electronic structure changes of the proposed reaction pathway have been elaborated previously,15d here we recapitulate only important features in order to compare them with the aforementioned cases. Earlier experimental and computational studies15e,60 revealed that the diamagnetic ground state of 17− is best formulated as a low spin ReI center (SRe = 0) ligated by a singlet bpy2− ligand. Binding of K+ to 17− does not discernibly change the electronic structure. Specifically, the Re center of 17−–K+ features an electronic configuration of (dxy)2(dxz)2(dyz)2, and its HOMO (1π*), albeit with dominant bpy π* parentage (74%), contains nonnegligible Re dz2 character (16%) (Fig. 5b). The mixing of these two fragment orbitals is symmetry-allowed, because both belong to the 1A′ representation of Cs point group. Furthermore, the Re center moves out of the bipyridine plane, allowing a stronger overlap between the two fragments.
As CO2 approaches the Re center of 17−–K+ to form 1J, two-electron transfer from bpy π* to the formally empty Re dz2 orbital takes place, and simultaneously the latter orbital donates the electron density to the CO2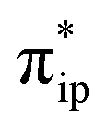 orbital (Fig. 5c). The resulting bonding σRe–C MO of 1J has a 59% contribution from the CO2
orbital (Fig. 5c). The resulting bonding σRe–C MO of 1J has a 59% contribution from the CO2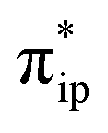 orbital. Thus, during this elementary step, on average, approximately one electron is transferred from the ligand to CO2via the metal center. Due to the substantial covalent character of the Re–CO2 interaction, the electronic structure of 1J is thus best described as a resonance hybrid between a ReI center (SRe = 0) bound to a CO22− ligand and a Re−I ion (SRe = 0) interacting with a CO20 moiety. As shown in Fig. 5d, the subsequent protonation further polarizes the Re–C bond in 1K as indicated by the increased –CO2H weight in the σRe–C orbital (Re dz2 26% and CO2H 66%). Finally, an outer-sphere electron transfer from the electrode to the bpy π* orbital occurs, along with the C–O bond cleavage, thereby yielding intermediate 2L and releasing a water molecule.
orbital. Thus, during this elementary step, on average, approximately one electron is transferred from the ligand to CO2via the metal center. Due to the substantial covalent character of the Re–CO2 interaction, the electronic structure of 1J is thus best described as a resonance hybrid between a ReI center (SRe = 0) bound to a CO22− ligand and a Re−I ion (SRe = 0) interacting with a CO20 moiety. As shown in Fig. 5d, the subsequent protonation further polarizes the Re–C bond in 1K as indicated by the increased –CO2H weight in the σRe–C orbital (Re dz2 26% and CO2H 66%). Finally, an outer-sphere electron transfer from the electrode to the bpy π* orbital occurs, along with the C–O bond cleavage, thereby yielding intermediate 2L and releasing a water molecule.
Again, the reaction with 17− also involves two coupled metal-to-CO2 and ligand-to-metal electron transfer events, which shift the two electrons stored on bpy directly to the CO2 ligand while leaving the Re oxidation state essentially unchanged. The coupling of the two electron transfer events is accomplished by mixing of the bpy π* (electron donor) and Re dz2 (electron acceptor) fragment orbitals, because of their appropriate symmetry and considerable overlap. Different from 112−, in the present case, the orbital mixing leads to a doubly occupied delocalized MO formed by Re dz2 and bpy π* rather than two spin-coupled pairs. Because the Re–CO2 interaction lowers the energy of the Re dz2 orbital, CO2 association triggers the direct bpy-to-Re two-electron transfer. This contrasts with the two categories discussed above, whose ligand-to-metal electron transfer proceeds via two separate events.
Basic electronic structure requirements for catalysts with non-innocent ligands
All the CO2-to-CO catalytic reactions studied above involve two synchronized electron transfer events: (1) a metal-to-CO2 two-electron transfer, and (2) a ligand-to-metal one- or two-electron transfer. The former is always effected mainly via σ-donation of two electrons from the doubly-occupied metal-centered dz2 orbital to the empty CO2 π* orbital. The latter transfer connects highly delocalized occupied ligand π* orbitals with formally unoccupied metal d orbitals. The ligand–metal cooperativity is at the core of the reactivity for all these complexes, thereby ensuring that the electrons stored on the ligand are transferred to CO2via the metal center.How does the system ensure the synchronization of the two electron transfer events? All of the investigated catalysts feature strong interactions between the ligand-based electron-donating π* orbital and the metal-based electron-accepting d-orbital, which give rise to molecular orbitals delocalized over the ligand platform and the metal center. In 112−, the strong antiferromagnetic coupling of the TPP eg orbitals with the dxz,yz atomic orbitals yields the delocalized 1eg MOs. In 240, the N4H 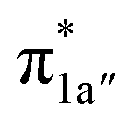 fragment orbital mixes with the Co dyz atomic orbital, and at the later stage, the N4H
fragment orbital mixes with the Co dyz atomic orbital, and at the later stage, the N4H 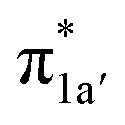 fragment orbital interacts with the
fragment orbital interacts with the 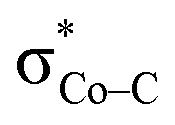 fragment orbital to give the orbitals 1a′′ and 1a′, respectively. In 17−, the bpy π* fragment orbital mixes with the Re dz2 orbital to generate the 1π* molecular orbital. Irrespective of different interaction modes, the coupling of ligand- and metal-based orbitals is pivotal in ensuring that even marginal losses of the electron density on the metal center is compensated by a continuous ligand-to-metal electron delocalization, as found for the case of 12−. This is because the aforementioned orbitals have the same symmetries and similar energies; otherwise, such coupling could not happen.
fragment orbital to give the orbitals 1a′′ and 1a′, respectively. In 17−, the bpy π* fragment orbital mixes with the Re dz2 orbital to generate the 1π* molecular orbital. Irrespective of different interaction modes, the coupling of ligand- and metal-based orbitals is pivotal in ensuring that even marginal losses of the electron density on the metal center is compensated by a continuous ligand-to-metal electron delocalization, as found for the case of 12−. This is because the aforementioned orbitals have the same symmetries and similar energies; otherwise, such coupling could not happen.
This work is perfectly in line with the earlier studies on the role of ligand non-innocence in general catalysis.22–25 In terms of reactivity, although ligand-centered mechanisms have been occasionally reported,61,62 in the vast majority of cases, the reactions completely occur at the metal center. In the latter situations, the ligands serve as an “electron reservoir”, thus enabling the metal center to maintain its common oxidation state as the reaction progresses,63,64,68 and the electrons stored on the ligand are transferred to the metal center during the reaction.22,63–65 Again, such catalysts also feature significant mixing between the ligand-based redox-active orbitals and unoccupied metal d-orbitals,66 in analogy to our present analysis.
Taken together, for all systems under investigation, non-innocent ligands endorse the following properties to CO2-to-CO electrocatalysts. First and foremost, ligand-centered reductions enhance the stability of the active species by providing low-lying redox-active orbitals. By contrast, metal-centered reduction would involve the formation of highly reactive species which are likely deactivated by side reactions or initiate unwanted reactions at electron-rich metal centers, like H2 or HCOOH production. Furthermore, the ligand–metal cooperativity is essential to the kinetics of the reaction. Owing to the synchronization of both electron transfer events, the metal oxidation state remains unchanged throughout the reaction. In the absence of such synchronization, the reaction would necessarily generate high-energy intermediates as exemplified by the triplet reaction of 312−.
General molecular structure requirements for catalysts with non-innocent ligands
So, how can one design catalytic systems that take the best advantage of the aforementioned properties? First of all, systems with open coordination sites are preferred, which facilitates the σ-bonding with CO2. This step often lacks suitable driving force,10,15d,20 and, in particular, ligand substitution by CO2 can hardly occur. In this regard, four-coordinated planar or five-coordinate square-pyramidal complexes are excellent candidates.The synchronization of the ligand-to-metal and metal-to-CO2 electronic transfer is also dependent on the coupling between the ligand and the metal center, which requires orbitals of similar energies and the same symmetry. Typically, highly conjugated π-systems have a wide range of available MOs, and their varying energies are more likely to match those of the metal d-orbitals. These set orbitals also feature various symmetries, which ensures that such coupling is symmetry allowed. For these two reasons, highly conjugated ligand platforms are clearly excellent candidates. Furthermore, in order to maximize overlap between the lobes of the interacting ligand- and metal-based fragment orbitals, the atoms of the ligand interacting with the metal center should employ their pz orbitals in the π-system. For instance, conjugated sp2 nitrogen donors or carbenes are excellent candidates.
Lastly, the choice of the metal center influences the metal–ligand cooperativity. The second or third row transition metals have larger, less compact d orbitals than first-row congeners and are likely to delocalize more electron density to the ligand. For instance, the molecular orbital of [Mn(bpy)(CO)3]− is much more centered at the metal (24%, Fig. S13†) than its rhenium counterpart, [Re(bpy)(CO)3]− (16%, Fig. 5). For the same reason, 4d and 5d metals are also expected to donate more electron density to the CO2 ligand, thus facilitating CO2 reduction. This, at least in part, explains why rhenium-bipyridine catalysts typically exhibit higher catalytic performances than their manganese counterparts.
In a given row of the periodic table, metals on the left tend to be less electronegative than those on the right. Consequently, the latter are expected to donate less electron density to the CO2 molecules than the former. Besides this, the number of valence d-electrons also plays a significant role in the reactivity. The number differs among metals of the same oxidation state; therefore, the available intramolecular electron transfer pathways also change, leading to drastic differences in mechanisms and catalytic performances. For instance, [Fe(qpy)] belongs to category I, while [Co(qpy)] belongs to category II. Because the dxz orbital is doubly-occupied in the latter, one of the electron transfer pathways available in the former case is inactivated in the latter, which results in the shift from category I to category II. Furthermore, relative to [Fe(qpy)], [Co(qpy)] exhibits a better catalytic performance (Table S1†). In sharp contrast, [Fe(TPP)] by far outperforms [Co(TPP)], although both Fe compounds feature an analogous electronic configuration and so do the Co compounds.67 Clearly, more detailed investigations are required to pinpoint the exact relationship between the type of metal–ligand cooperativity and the catalytic performance.
Conclusion
Our computational investigation of the CO2-to-CO reduction reaction catalyzed by 112− proposes the following mechanism: (1) Formation of the adduct, (2) protonation of the adduct, (3) cleavage of the C–O bond, and (4) reduction of the metal-carbonyl and release of a CO molecule. We found a satisfying agreement between the predicted mechanism and the available thermodynamic and kinetic data. In particular, we found that the highest barrier was indeed compatible with the observed turnover frequency of this catalytic reaction.Most importantly, our present investigation provides a profound understanding of the structure–activity relationship of 112−. Indeed, the reaction involves two electron transfer events: (1) a metal-to-CO2 transfer and (2) a ligand-to-metal electron transfer. These two electron transfer events are synchronized in such a way that the electron density of the metal center hardly varies along the reaction coordinates. It was shown that the synchronization of the two electron transfer events is fundamental to the reactivity, because it circumvents the formation of intermediates having highly electron-rich or -deficient metal centers. Optimal synchronization is realized by the antiferromagnetic spin coupling between the porphyrin ligand and the metal center, which serves as a conduit between them; consequently, the variation of electron density on the metal center is minimized.
Comparison of the reactivity of 12− with those of ten related catalysts with non-innocent ligand platforms enables the generalization of the feature of ligand non-innocence in CO2 reduction observed for 12−. On the basis of these findings, we propose that ligand non-innocence in CO2 reduction plays a central role in ensuring a high selectivity and stability, while maintaining fast kinetics through ligand–metal cooperativity. Finally, fundamental requirements to design catalysts with non-innocent ligands are proposed.
Author contributions
S. Y. and F. N. conceived and supervised the project; M. T. carried out theoretical computations. All authors wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge generous financial support from the European Union Marie Sklodowska Curie Initial Training NoNoMeCat (Grant 675020-MSCA-ITN-2015-ETN) and the Max-Planck Society, in particular, the joint workspace of MPI Chemische Energiekonversion and MPI Kohlenforschung.References
- P. Tans, and R. Keeling, NOAA/ESRL (https://www.esrl.noaa.gov/gmd/ccgg/trends/) and Scripps Institution of Oceanography (https://scrippsco2.ucsd.edu/), accessed January 14, 2019.
- (a) B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Advances in the Use of CO2 as a Renewable Feedstock for the Synthesis of Polymers, Chem. Soc. Rev., 2019, 48(16), 4466–4514 RSC; (b) E. E. Benson, C. P. Kubiac, A. J. Sathrum and J. M. Smieja, Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels, Chem. Soc. Rev., 2009, 38(1), 89–99 RSC; (c) M. E. Dry, The Fischer-Tropsch process: 1950-2000, Catal. Today, 2002, 71(3–4), 227–241 CrossRef CAS.
- (a) J. M. Lehn and R. Ziessel, Photochemical reduction of carbon dioxide to formate catalyzed by 2,2′t-bipyridine or 1,10-phenanthroline-ruthenium(II) complexes, J. Org. Chem., 1990, 382(1–2), 157–173 CrossRef CAS; (b) J. Grodkowski, D. Behar, P. Neta and P. Hambright, Iron-porphyrin-catalyzed reduction of CO2. Photochemical and radiation chemical studies, J. Phys. Chem. A, 1997, 101(3), 248–254 CrossRef CAS; (c) D. Behar, T. Dhanasekaran, P. Neta, C. M. Hosten, D. Ejeh, P. Hambright and E. Fujita, Cobalt-porphyrin catalyzed reduction of CO2. Radiation chemical, photochemical and electrochemical studies, J. Phys. Chem. A, 1998, 102(17), 2870–2877 CrossRef CAS.
- (a) B. Mondal, J. Song, F. Neese and S. Ye, Bio-Inspired Mechanistic Insights into CO2 Reduction, Curr. Opin. Chem. Biol., 2015, 25, 103–109 CrossRef CAS PubMed; (b) N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Molecular Polypyridine-Based Metal Complexes as Catalysts for the Reduction of CO2, Chem. Soc. Rev., 2017, 46(3), 761–796 RSC; (c) H. Takeda, C. Cometto, O. Ishitani and M. Robert, Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction, ACS Catal., 2017, 7(1), 70–88 CrossRef CAS; (d) R. Francke, B. Schille and M. Roemelt, Homogeneously Catalyzed Electroreduction of Carbon Dioxide—Methods, Mechanisms, and Catalysts, Chem. Rev., 2018, 118(9), 4631–4701 CrossRef CAS PubMed; (e) T. N. Huan, E. S. Andreiadis, J. Heodkamp, P. Simon, E. Derat, S. Cobo, G. Royal, A. Bergmann, P. Strasser, H. Dau, V. Artero and M. Fontecave, From molecular copper complexes to composite electrocatalytic materials for selective reduction of CO2 to formic acid, J. Mater. Chem. A, 2015, 3(7), 3901–3907 RSC; (f) F. H. Haghighi, H. Hadadzadeh, H. Farrokhpour, N. Serri, K. Abdi and H. A. Rudbari, Computational and experimental study on the electrocatalytic reduction of CO2 to CO by a mononuclear ruthenium complex, Dalton Trans., 2014, 43(29), 11317–11332 RSC.
- (a) R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Electrocatalytic CO2 reduction to oxalate by a copper complex, Science, 2010, 327(5963), 313–315 CrossRef CAS PubMed; (b) J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2, Science, 2014, 345(6196), 546–550 CrossRef CAS.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, Y. Wang, X. Xue, R. Chen, S. Yang and Z. Jin, Progress and perspective of electrocatalytic CO2 reduction for renewable Carbonaceous fuels and chemicals, Adv. Sci., 2018, 5(1), 1700275 CrossRef.
- G. F. Manbeck and E. Fujita, A Review of Iron and Cobalt Porphyrins, Phthalocyanines and Related Complexes for Electrochemical and Photochemical Reduction of Carbon Dioxide, J. Porphyrins Phthalocyanines, 2015, 19(01–03), 45–64 CrossRef CAS.
- (a) C. Costentin, S. Drouet, M. Robert and J. M. Saveant, A local proton source enhances CO2 electroreduction to CO by a molecular Fe catalyst, Science, 2012, 338(6103), 90–94 CrossRef CAS PubMed; (b) C. Costentin, S. Drouet, G. Passard, M. Robert and J. M. Saveant, Proton-coupled electron transfer cleavage of heavy-atom bonds in electrocatalytic processes. Cleavage of a C–O bond in the catalyzed electrochemical reduction of CO2, J. Am. Chem. Soc., 2013, 135(24), 9023–9031 CrossRef CAS PubMed; (c) C. Costentin, G. Passard, M. Robert and J. M. Saveant, Ultraefficient homogeneous catalyst for the CO2-to-CO electrochemical conversion, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(42), 14990–14994 CrossRef CAS PubMed; (d) C. Costentin, G. Passard, M. Robert and J. M. Saveant, Pendant acid–base groups in molecular catalysis: H-bond promoters or proton relays? Mechanism of the conversion of CO2 to CO by electrogenerated iron(0) Porphyrins bearing prepositioned phenol functionalities, J. Am. Chem. Soc., 2014, 136(33), 11821–11829 CrossRef CAS PubMed; (e) I. Azcarate, C. Costentin, M. Robert and J. M. Savéant, Through-space charge interaction substituent effects in molecular catalysis leading to the design of the most efficient catalyst of CO2-to-CO electrochemical conversion, J. Am. Chem. Soc., 2016, 138(51), 16639–16644 CrossRef CAS PubMed; (f) A. Khadhraoui, P. Gotico, B. Boitrel, W. Leibl, Z. Halime and A. Aukauloo, Local Ionic Liquid Environment at a Modified Iron Porphyrin Catalyst Enhances the Electrocatalytic Performance of CO2 to CO Reduction in Water, Chem. Commun., 2018, 54(82), 11630–11633 RSC; (g) Y. Zhang, J. Chen, P. E. Siegbahn and R. Z. Liao, Harnessing Non-Innocent Porphyrin Ligand to Circumvent Fe-Hydride Formation in the Selective Fe-Catalyzed CO2 Reduction in Aqueous Solution, ACS Catal., 2020, 10(11), 6332–6345 CrossRef CAS.
- J. N. Harvey, F. Himo, F. Maseras and L. Perrin, Scope and Challenge of Computational Methods for Studying Mechanism and Reactivity in Homogeneous Catalysis, ACS Catal., 2019, 9(8), 6803–6813 CrossRef CAS.
- P. Davethu and S. P. de Visser, CO2 Reduction on an Iron-Porphyrin Center: A Computational Study, J. Phys. Chem. A, 2019, 123(30), 6527–6535 CrossRef CAS PubMed.
- A. Chapovetsky, T. H. Do, R. Haiges, M. K. Takase and S. C. Marinescu, Proton-assisted reduction of CO2 by cobalt aminopyridine macrocycles, J. Am. Chem. Soc., 2016, 138(18), 5765–5768 CrossRef CAS PubMed.
- C. Cometto, L. Chen, P. K. Lo, Z. Guo, K. C. Lau, E. Anxolabéhère-Mallart, C. Fave, T. C. Lau and M. Robert, Highly Selective Molecular Catalysts for the CO2-to-CO Electrochemical Conversion at Very Low Overpotential. Contrasting Fe vs. Co Quaterpyridine Complexes upon Mechanistic Studies, ACS Catal., 2018, 8(4), 3411–3417 CrossRef CAS.
- L. Chen, Z. Guo, X. G. Wie, C. Gallenkamp, J. Bonin and E. Anxolabéhère-Mallart, Molecular catalysis of the electrochemical and photochemical reduction of CO2 with earth-abundant metal complexes. Selective production of CO vs. HCOOH by switching of the metal center, J. Am. Chem. Soc., 2015, 137(34), 10918–10921 CrossRef CAS.
- (a) A. Tinnemans, T. Koster, D. Thewissen and A. Mackor, Tetraaza-macrocyclic Cobalt(II) and Nickel(II) Complexes as Electron-transfer Agents in the Photo (Electro) Chemical and Electrochemical Reduction of Carbon Dioxide, Recl. Trav. Chim. Pays-Bas, 1984, 103(10), 288–295 CrossRef CAS; (b) D. C. Lacy, C. C. L. McCrory and J. C. Peters, Studies of cobalt-mediated electrocatalytic CO2 reduction using a redox-active ligand, Inorg. Chem., 2014, 53(10), 4980–4988 CrossRef CAS PubMed; (c) M. Zhang, M. El-Roz, H. Frei, J. Mendoza-Cortes, M. Head-Gordon, D. C. Lacy and J. C. Peters, Visible Light Sensitized CO2 Activation by the Tetraaza [CoIIN4H(MeCN)]2+ Complex Investigated by FT-IR Spectroscopy and DFT Calculations, J. Phys. Chem. C, 2015, 119(9), 4645–4654 CrossRef CAS; (d) H. Sheng and H. Frei, Direct Observation by Rapid-Scan FT-IR Spectroscopy of Two-Electron-Reduced Intermediate of Tetraaza Catalyst [CoIIN4H(MeCN)]2+ Converting CO2 to CO, J. Am. Chem. Soc., 2016, 138(31), 9959–9967 CrossRef CAS PubMed; (e) A. J. Garza, S. Pakhira, A. T. Bell, J. L. Mendoza-Cortes and M. Head-Gordon, Reaction Mechanism of the Selective Reduction of CO2 to CO by a Tetraaza [CoIIN4H]2+ Complex in the Presence of Protons, Phys. Chem. Chem. Phys., 2018, 20(37), 24058–24064 RSC.
- (a) J. Hawecker, J. M. Lehn and R. Ziessel, Electrocatalytic Reduction of Carbon Dioxide Mediated by Re(Bipy)(CO)3Cl (Bipy = 2,2′-Bipyridine), J. Chem. Soc., Chem. Commun., 1984, 6, 328–330 RSC; (b) T. Scheiring, A. Klein and W. Kaim, EPR Study of Paramagnetic Rhenium(I) Complexes (Bpy˙−)Re(CO)3X Relevant to the Mechanism of Electrocatalytic CO2 Reduction, J. Chem. Soc., Perkin Trans. 2, 1997,(12), 2569–2572 RSC; (c) J. M. Smieja and C. P. Kubiac, Re(bipy-tBu)(CO)3Cl− improved catalytic activity for reduction of carbon dioxide: IR-spectroelectrochemical and mechanistic studies, Inorg. Chem., 2010, 49(20), 9283–9289 CrossRef CAS PubMed; (d) J. A. Keith, K. A. Grice and C. P. Kubiak, Elucidation of the Selectivity of Proton- Dependent Electrocatalytic CO2 Reduction by a fac-Re(bpy)(CO)3Cl, J. Am. Chem. Soc., 2013, 135(42), 15823–15829 CrossRef CAS PubMed; (e) E. E. Benson, M. D. Sampson, K. A. Grice, J. M. Smieja, J. D. Froehlich, D. Friebel, J. A. Keith, E. A. Carter, A. Nilsson and C. P. Kubiak, The Electronic States of Rhenium Bipyridyl Electrocatalysts for CO2 Reduction as Revealed by X-ray Absorption Spectroscopy and Computational Quantum Chemistry, Angew. Chem., Int. Ed., 2013, 52(18), 4841–4844 CrossRef CAS PubMed; (f) C. Riplinger, M. D. Sampson, A. M. Ritzmann, C. P. Kubiak and E. A. Carter, Mechanistic Contrasts between Manganese and Rhenium Bipyridine Electrocatalysts for the Reduction of Carbon Dioxide, J. Am. Chem. Soc., 2014, 136(46), 16285–16298 CrossRef CAS PubMed; (g) E. Haviv, D. Azaiza-Dabbah, R. Carmieli, L. Avram, J. M. Martin and R. A. Neumann, Thiourea Tether in the Second Coordination Sphere as a Binding Site for CO2 and a Proton Donor Promotes the Electrochemical Reduction of CO2 to CO Catalyzed by a Rhenium Bipyridine-Type Complex, J. Am. Chem. Soc., 2018, 140(39), 12451–12456 CrossRef CAS PubMed; (h) M. L. Clark, P. L. Cheung, M. Lessio, E. A. Carter and C. P. Kubiak, Kinetic and Mechanistic Effects of Bipyridine (Bpy) Substituent, Labile Ligand, and Brønsted Acid on Electrocatalytic CO2 Reduction by Re (Bpy) Complexes, ACS Catal., 2018, 8(3), 2021–2029 CrossRef CAS.
- (a) C. Römelt, J. Song, M. Tarrago, J. A. Rees, M. Van Gastel, T. Weyhermüller, S. Debeer, E. Bill, F. Neese and S. Ye, Electronic Structure of a Formal Iron(0) Porphyrin Complex Relevant to CO2 Reduction, Inorg. Chem., 2017, 56(8), 4745–4750 CrossRef PubMed; (b) C. Römelt, S. Ye, E. Bill, T. Weyhermüller, M. van Gastel and F. Neese, Electronic Structure and Spin Multiplicity of Iron Tetraphenylporphyrins in Their Reduced States as Determined by a Combination of Resonance Raman Spectroscopy and Quantum Chemistry, Inorg. Chem., 2018, 57(4), 2141–2148 CrossRef PubMed.
- (a) J. Grodkowski, P. Neta, E. Fujita, A. Mahammed, L. Simkhovich and Z. Gross, Reduction of Cobalt and Iron Corroles and Catalyzed Reduction of CO2, J. Phys. Chem. A, 2002, 106(18), 4772–4778 CrossRef CAS; (b) S. Gonglach, S. Paul, M. Haas, F. Pillwein, S. S. Sreejith, S. Barman, R. De, S. Müllegger, P. Gerschel and U. P. Apfel, Molecular Cobalt Corrole Complex for the Heterogeneous Electrocatalytic Reduction of Carbon Dioxide, Nat. Commun., 2019, 10(1), 1–10 CrossRef CAS PubMed.
- (a) B. J. Fisher and R. Eisenberg, Electrocatalytic Reduction of Carbon Dioxide by Using Macrocycles of Nickel and Cobalt, J. Am. Chem. Soc., 1980, 102(24), 7361–7363 CrossRef CAS; (b) J. Lewis and M. Schröder, Reduction of Schiff-Base Macrocyclic Complexes. Stabilisation of Nickel(I) Conjugated Macrocyclic Complexes via a Reversible Ligand-to-Metal Electron-Transfer Process, J. Chem. Soc., Dalton Trans., 1982,(6), 1085–1089 RSC; (c) M. Ghosh, T. Weyhermüller and K. Wieghardt, Electronic Structure of the Members of the Electron Transfer Series [NiL]z (Z = 3+, 2+, 1+, 0) and [NiL(X)]n (X = Cl, CO, P(OCH3)3) Species Containing a Tetradentate, Redox-Noninnocent, Schiff Base Macrocyclic Ligand L: An Experimental and Density Functional Theoretical Study, Dalton Trans., 2010, 39(8), 1996–2007 RSC.
- (a) D. Z. Zee, M. Nippe, A. E. King, C. J. Chang and J. R. Long, Tuning Second Coordination Sphere Interactions in Polypyridyl–Iron Complexes to Achieve Selective Electrocatalytic Reduction of Carbon Dioxide to Carbon Monoxide, Inorg. Chem., 2020, 59(7), 5206–5217 CrossRef CAS PubMed; (b) M. Loipersberger, D. Z. Zee, J. A. Panetier, C. J. Chang, J. R. Long and M. Head-Gordon, Computational Study of an Iron(II) Polypyridine Electrocatalyst for CO2 Reduction: Key Roles for Intramolecular Interactions in CO2 Binding and Proton Transfer, Inorg. Chem., 2020, 59(12), 8146–8160 CrossRef CAS.
- Z. Chen, C. Chen, D. R. Weinberg, P. Kang, J. J. Concepcion, D. P. Harrison, M. S. Brookhart and T. J. Meyer, Electrocatalytic Reduction of CO2 to CO by Polypyridyl Ruthenium Complexes, Chem. Commun., 2011, 47(47), 12607–12609 RSC.
- (a) J. Agarwal, T. W. Shaw, C. J. Stanton III, G. F. Majetich, A. B. Bocarsly and H. F. Schaefer III, NHC-Containing Manganese(I) Electrocatalysts for the Two-Electron Reduction of CO2, Angew. Chem., 2014, 126(20), 5252–5255 CrossRef; (b) M. Sheng, N. Jiang, S. Gustafson, B. You, D. H. Ess and Y. A. Sun, Nickel Complex with a Biscarbene Pincer-Type Ligand Shows High Electrocatalytic Reduction of CO2 over H2O, Dalton Trans., 2015, 44(37), 16247–16250 RSC; (c) N. P. Liyanage, H. A. Dulaney, A. J. Huckaba, J. W. Jurss and J. H. Delcamp, Electrocatalytic Reduction of CO2 to CO with Re-Pyridyl-NHCs: Proton Source Influence on Rates and Product Selectivities, Inorg. Chem., 2016, 55(12), 6085–6094 CrossRef CAS PubMed; (d) X. Su, K. M. McCardle, J. A. Panetier and J. W. Jurss, Electrocatalytic CO2 Reduction with Nickel Complexes Supported by Tunable Bipyridyl-N-Heterocyclic Carbene Donors: Understanding Redox-Active Macrocycles, Chem. Commun., 2018, 54(27), 3351–3354 RSC.
- V. K. Praneeth, M. R. Ringenberg and T. R. Ward, Redox-Active Ligands in Catalysis, Angew. Chem., Int. Ed., 2012, 51(41), 10228–10234 CrossRef CAS PubMed.
- J. L. Boyer, T. R. Cundari, N. J. DeYonker, T. B. Rauchfuss and S. R. Wilson, Redox Activation of Alkene Ligands in Platinum Complexes with Non-Innocent Ligands, Inorg. Chem., 2009, 48(2), 638–645 CrossRef CAS.
- W. Kaim and B. Schwederski, Non-Innocent Ligands in Bioinorganic Chemistry—An Overview, Coord. Chem. Rev., 2010, 254(13–14), 1580–1588 CrossRef CAS.
- D. L. Broere, B. De Bruin, J. N. Reek, M. Lutz, S. Dechert and J. I. Van Der Vlugt, Intramolecular Redox-Active Ligand-to-Substrate Single-Electron Transfer: Radical Reactivity with a Palladium(II) Complex, J. Am. Chem. Soc., 2014, 136(33), 11574–11577 CrossRef CAS PubMed.
- (a) M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, [Mn(Bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction, Angew. Chem., Int. Ed., 2011, 50(42), 9903–9906 CrossRef CAS; (b) M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, Manganese Catalysts with Bulky Bipyridine Ligands for the Electrocatalytic Reduction of Carbon Dioxide: Eliminating Dimerization and Altering Catalysis, J. Am. Chem. Soc., 2014, 136(14), 5460–5471 CrossRef CAS PubMed; (c) K. T. Ngo, M. McKinnon, B. Mahanti, R. Narayanan, C. C. Grills, M. Z. Ertem and J. Rochford, Turning on the Protonation-First Pathway for Electrocatalytic CO2 Reduction by Manganese Bipyridyl Tricarbonyl Complexes, J. Am. Chem. Soc., 2017, 139(7), 2604–2618 CrossRef CAS PubMed.
- J. Tory, B. Setterfield-Price, R. A. Dryfe and F. Hartl, [M(CO)4(2,2′-Bipyridine)] (M = Cr, Mo, W) as Efficient Catalysts for Electrochemical Reduction of CO2 to CO at a Gold Electrode, ChemElectroChem, 2015, 2(2), 213–217 CrossRef CAS.
- H. Ishida, K. Tanaka and T. Tanaka, Electrochemical CO2 Reduction Catalyzed by Ruthenium Complexes [Ru(Bpy)2(CO)2]2+ and [Ru(Bpy)2(CO)Cl]+. Effect of pH on the Formation of CO and HCOO, Organometallics, 1987, 6(1), 181–186 CrossRef CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA Quantum Chemistry Program Package, J. Chem. Phys., 2020, 152(22), 224108 CrossRef CAS PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38(6), 3098 CrossRef CAS PubMed.
- F. Weigend and R. Alhrichs, Balanced basis sets of split valence, triple zeta valence and quadrupole zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24(6), 669–681 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Efficient, approximate and parallel Hartree-Fock and hybrid DFT calculations. A ‘chain-of-sphere’ algorithm for the Hartree–Fock exchange, Chem. Phys., 2009, 356(1–3), 98–109 CrossRef CAS.
- (a) G. Mills, H. Jónsson and G. K. Schenter, Reversible work transition state theory: application to dissociative adsorption of hydrogen, Surf. Sci., 1994, 324(2–3), 305–337 Search PubMed; (b) G. Henkelman and H. Jónsson, Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points, J. Chem. Phys., 2000, 113(22), 9978–9985 CrossRef CAS; (c) G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113(22), 9901–9904 CrossRef CAS.
- J. P. Malrieu and G. Trinquier, A Recipe for Geometry Optimization of Diradicalar Singlet States from Broken-Symmetry Calculations, J. Phys. Chem. A, 2012, 116(31), 8226–8237 CrossRef CAS PubMed.
- J. W. Ochterski, Thermochemistry in Gaussian, Gaussian Inc., 2000 Search PubMed.
- (a) M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, Nickel(II)-Cyclam: an Extremely Selective Electrocatalyst for Reduction of CO2 in water, J. Chem. Soc., Chem. Commun., 1984, 19, 1315–1316 RSC; (b) M. Beley, J. P. Collin, R. Ruppert and J. P. Sauvage, Electrocatalytic Reduction of Carbone Dioxide by Nickel Cyclam2+ in Water: Study of the Factors Affecting the Efficiency and the Selectivity of the Process, J. Am. Chem. Soc., 1986, 108(24), 7461–7467 CrossRef CAS PubMed; (c) J. D. Froehlich and C. P. Kubiak, Homogeneous Reduction by Ni(cyclam) at a Glassy Carbon Electrode, Inorg. Chem., 2012, 51(7), 3932–3934 CrossRef CAS PubMed; (d) J. Song, E. L. Klein, F. Neese and S. Ye, The mechanism of homogeneous CO2 reduction by Ni(cyclam): product selectivity, concerted proton-electron transfer and C–O bond cleavage, Inorg. Chem., 2014, 53(14), 7500–7507 CrossRef CAS PubMed.
-
(a) G. Xue, C. Geng, S. Ye, A. T. Fiedler, F. Neese and L. Que Jr, Hydrogen-Bonding Effects on the Reactivity of [X–FeIII–O–FeIV
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O](X = OH, F) Complexes toward C–H Bond Cleavage, Inorg. Chem., 2013, 52(7), 3976–3984 CrossRef CAS PubMed;
(b) M. Wirstam, S. J. Lippard and R. A. Friesner, Reversible Dioxygen Binding to Hemerythrin, J. Am. Chem. Soc., 2003, 125(13), 3980–3987 CrossRef CAS PubMed;
(c) S. Ye, C. Riplinger, A. Hansen, C. Krebs, J. M. Bollinger and F. Neese, Electronic Structure Analysis of the Oxygen-Activation Mechanism by FeII-and A-Ketoglutarate (AKG)-Dependent Dioxygenases, Chem.–Eur. J., 2012, 18(21), 6555–6567 CrossRef CAS PubMed.
O](X = OH, F) Complexes toward C–H Bond Cleavage, Inorg. Chem., 2013, 52(7), 3976–3984 CrossRef CAS PubMed;
(b) M. Wirstam, S. J. Lippard and R. A. Friesner, Reversible Dioxygen Binding to Hemerythrin, J. Am. Chem. Soc., 2003, 125(13), 3980–3987 CrossRef CAS PubMed;
(c) S. Ye, C. Riplinger, A. Hansen, C. Krebs, J. M. Bollinger and F. Neese, Electronic Structure Analysis of the Oxygen-Activation Mechanism by FeII-and A-Ketoglutarate (AKG)-Dependent Dioxygenases, Chem.–Eur. J., 2012, 18(21), 6555–6567 CrossRef CAS PubMed. - A. Garza, Solvation entropy made simple, J. Chem. Theory Comput., 2019, 15(5), 3204–3214 CrossRef CAS PubMed.
- (a) T. N. Truong and E. V. Stefanovich, A new method for incorporating solvent effect into the classical, ab initio molecular orbital and density functional theory frameworks for arbitrary shape cavity, Chem. Phys. Lett., 1995, 240(4), 253 CrossRef CAS; (b) D. Sitkoff, K. A. Sharp and B. Konig, Accurate calculation of hydration free energies using macroscopic solvent models, J. Chem. Phys., 1994, 98(7), 1978–1988 CrossRef CAS.
- H. G. Roth, N. A. Romero and D. A. Nicewitcz, Experimental and calculated electrochemical potentials of common organic molecules for applications to single-electron redox chemistry, Synlett, 2016, 27(05), 714–723 CAS.
- A. A. Isse and A. Gennaro, Absolute potential of the standard hydrogen electrode and the problem of interconversion of potentials in different solvents, J. Phys. Chem. B, 2010, 114(23), 7894–7899 CrossRef CAS PubMed.
- R. J. Donohoe, M. Atamian and D. F. Bocian, Characterization of Singly Reduced Iron(II) Porphyrins, J. Am. Chem. Soc., 1987, 109(19), 5593–5599 CrossRef CAS.
- S. J. Konezny, M. D. Doherty, O. R. Luca, R. H. Crabtree, G. L. Soloveichik and V. Batista, Reduction of systematic uncertainty in DFT redox potentials of transition-metal complexes, J. Phys. Chem. C, 2012, 116(10), 6349–6356 CrossRef CAS.
- T. Mashiko, C. A. Reed, K. J. Haller and W. R. Scheidt, Nature of iron(I) and iron(0) tetraphenylporphyrin complexes. Synthesis and molecular structure of (dibenzo-18-crown-6)bis(tetrahydrofuran)sodium (meso-tetraphenylporphinato)ferrate and bis[tris(tetrahydrofuran)sodium](meso-tetraporphinato)ferrate, Inorg. Chem., 1984, 23(20), 3192–3196 CrossRef CAS.
- F. Neese, Definition of corresponding orbitals and the diradical character in broken symmetry DFT calculations on spin coupled system, J. Phys. Chem. Solids, 2004, 65(4), 781–785 CrossRef CAS.
- J. Pipek and P. G. Mezey, A fast intrinsic localization procedure applicable for ab initio and semiempirical linear combination of atomic orbital wave-functions, J. Chem. Phys., 1989, 90(9), 4916–4926 CrossRef CAS.
- E. Bremond, H. Li, A. J. Perez-Jimenez, J. C. Sancho-Garcia and C. Adamao, Tackling an accurate description of molecular reactivity with double-hybrid density functionals, J. Chem. Phys., 2022, 156(16), 161101 CrossRef CAS PubMed.
- M. Korth and S. Grimme, “Mindless” DFT benchmarking, J. Chem. Theory Comput., 2009, 5(4), 993–1003 CrossRef CAS PubMed.
- (a) J. A. Christiansen, The Elucidation of Reaction Mechanisms by the Method of Intermediates in Quasi-Stationary Concentrations, Adv. Catal., 1953, 5, 311–353 CAS; (b) S. Kozuch and S. Shaik, A Combined Kinetic–Quantum Mechanical Model for Assessment of Catalytic Cycles: Application to Cross-Coupling and Heck Reactions, J. Am. Chem. Soc., 2006, 128(10), 3355–3365 CrossRef CAS PubMed; (c) S. Kozuch and S. Shaik, Kinetic-Quantum Chemical Model for Catalytic Cycles: The Haber–Bosch Process and the Effect of Reagent Concentration, J. Phys. Chem. A, 2008, 112(26), 6032–6041 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J. M. Savéant, Turnover Numbers, Turnover Frequencies, and Overpotential in Molecular Catalysis of Electrochemical Reactions. Cyclic Voltammetry and Preparative-Scale Electrolysis, J. Am. Chem. Soc., 2012, 134(27), 11235–11242 CrossRef CAS PubMed.
- J. Schneider, H. Jia, J. T. Muckerman and E. Fujita, Thermodynamics and Kinetics of CO2, CO, and H+ Binding to the Metal Centre of CO2 Reduction Catalysts, Chem. Soc. Rev., 2012, 41(6), 2036–2051 RSC.
- H. Weihe and H. U. Güdel, Quantitative Interpretation of the Goodenough–Kanamori Rules: A Critical Analysis, Inorg. Chem., 1997, 36(17), 3632–3639 CrossRef CAS PubMed.
- (a) S. Hammes-Schiffer, Proton-Coupled Electron Transfer: Classification Scheme and Guide to Theoretical Methods, Energy Environ. Sci., 2012, 5(7), 7696–7703 RSC; (b) M. T. M. Koper, Theory of the transition from sequencial to concerted electrochemical proton-electron transfer, Phys. Chem. Chem. Phys., 2013, 15(5), 1399–1407 RSC; (c) C. Costentin, M. Robert, J. M. Savéant and C. Tard, Breaking Bonds with Electrons and Protons. Models and Examples, Acc. Chem. Res., 2014, 47(1), 271–280 CrossRef CAS PubMed.
- J. N. Harvey, Spin-forbidden Reactions: Computational Insight into Mechanisms and Kinetics, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4(1), 1–14 CAS.
- B. M. Wile, R. J. Trovitch, S. C. Bart, A. M. Tondreau, E. Lobkovsky, C. Milsmann, E. Bill, K. Wieghardt and P. J. Chirik, Reduction Chemistry of Aryl-and Alkyl-Substituted Bis(Imino)Pyridine Iron Dihalide Compounds: Molecular and Electronic Structures of [(PDI)2Fe] Derivatives, Inorg. Chem., 2009, 48(9), 4190–4200 CrossRef CAS PubMed.
- A. C. Bowman, C. Milsmann, E. Bill, E. Lobkovsky, T. Weyhermüller, K. Wieghardt and P. J. Chirik, Reduced N-Alkyl Substituted Bis(Imino)Pyridine Cobalt Complexes: Molecular and Electronic Structures for Compounds Varying by Three Oxidation States, Inorg. Chem., 2010, 49(13), 6110–6123 CrossRef CAS PubMed.
- (a) J. M. Smieja, E. E. Benson, B. Kumar, K. A. Grice, C. S. Seu, A. J. Miller, J. M. Mayer and C. P. Kubiak, Kinetic and Structural Studies, Origins of Selectivity, and Interfacial Charge Transfer in the Artificial Photosynthesis of CO, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(39), 15646–15650 CrossRef CAS PubMed; (b) E. E. Benson and C. P. Kubiak, Structural Investigations into the Deactivation Pathway of the CO2 Reduction Electrocatalyst Re(Bpy)(CO)3Cl, Chem. Commun., 2012, 48(59), 7374–7376 RSC; (c) M. Wang, T. Weyhermüller, E. Bill, S. Ye and K. Wieghardt, Structural and Spectroscopic Characterization of Rhenium Complexes Containing Neutral, Monoanionic, and Dianionic Ligands of 2,2′-Bipyridines and 2,2′:6,2′′-Terpyridines: An Experimental and Density Functional Theory (DFT)-Computational Study, Inorg. Chem., 2016, 55(10), 5019–5036 CrossRef CAS PubMed.
- L. Que and W. B. Tolman, Biologically Inspired Oxidation Catalysis, Nature, 2008, 455(7211), 333–340 CrossRef CAS PubMed.
- P. Chaudhuri, M. Hess, U. Flörke and K. Wieghardt, From Structural Models of Galactose Oxidase to Homogeneous Catalysis: Efficient Aerobic Oxidation of Alcohols, Angew. Chem., Int. Ed., 1998, 37(16), 2217–2220 CrossRef CAS PubMed.
- W. Kaim, Manifestations of Noninnocent Ligand Behavior, Inorg. Chem., 2011, 50(20), 9752–9765 CrossRef CAS PubMed.
- P. J. Chirik and K. Wieghardt, Radical Ligands Confer Nobility on Base-Metal Catalysts, Science, 2010, 327(5967), 794–795 CrossRef CAS PubMed.
- W. I. Dzik, J. I. van der Vlugt, J. N. Reek and B. de Bruin, Ligands That Store and Release Electrons during Catalysis, Angew. Chem., Int. Ed., 2011, 50(15), 3356–3358 CrossRef CAS PubMed.
- (a) F. Mao, D. R. Tyler, M. R. Bruce, A. E. Bruce, A. L. Rieger and P. H. Rieger, Solvent Effects on Electron Delocalization in Paramagnetic Organometallic Complexes: Solvent Manipulation of the Amount of 19-Electron Character in Co(CO)3L2 (L2 = a Chelating Phosphine), J. Am. Chem. Soc., 1992, 114(16), 6418–6424 CrossRef CAS; (b) D. M. Schut, K. J. Keana, D. R. Tyler and P. H. Rieger, Measurement and Manipulation of the Unpaired Electron Density in 18+. Delta. Complexes. Correlation of the Charge Density with Reactivity, J. Am. Chem. Soc., 1995, 117(35), 8939–8946 CrossRef CAS; (c) A. Klein, C. Vogler and W. Kaim, The δ in 18+ δ Electron Complexes: Importance of the Metal/Ligand Interface for the Substitutional Reactivity of “Re (0)” Complexes (α-Diimine-)ReI(CO)3(X), Organometallics, 1996, 15(1), 236–244 CrossRef CAS.
- X. M. Hu, M. H. Rønne, S. U. Pedersen, T. Skrydstrup and K. Daasjberg, Enhanced catalytic activity of cobalt porphyrin in CO2 electroreduction upon immobilization on carbon materials, Angew. Chem., Int. Ed., 2017, 56(23), 6468–6472 CrossRef CAS PubMed.
- B. Mondal and S. Ye, Hidden ligand noninnocence: A combined spectroscopic and computational perspective, Coord. Chem. Rev., 2020, 405, 213115–213119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Catalytic performances of the catalysts mentioned in Scheme 2. Relaxed surface scans confirming the absence of transition states for the formation of some intermediates. Supplementary information on the reactivity of 112−. Detailed calculations of the turnover frequency. The electronic structure of the transition states. Detailed electronic structure evolution of the catalysts not detailed in the main text. Absolute electronic energy, enthalpy and free energy of the intermediates in CO2 reduction catalyzed by 112−. Cartesian coordinates of all the investigated catalysts and intermediates; imaginary frequencies of all calculated intermediates and transition states in CO2 reduction catalyzed by 112−. See https://doi.org/10.1039/d2sc01863b |
| This journal is © The Royal Society of Chemistry 2022 |