
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe curious case of a sterically crowded Stenhouse salt†
Valentin
Théry
a,
Florian
Molton
a,
Selim
Sirach
a,
Neven
Tillet
a,
Jacques
Pécaut
b,
Eder
Tomás-Mendivil
*a and
David
Martin
 *a
*a
aUniv. Grenoble Alpes, CNRS, DCM, Grenoble 38000, France
bUniv. Grenoble Alpes, CEA, CNRS, INAC-SyMMES, UMR 5819, Grenoble 38000, France
First published on 27th July 2022
Abstract
We report a peculiar Stenhouse salt. It does not evolve into cyclopentenones upon basification, due to the steric hindrance of its bulky stable carbene patterns. This allowed for the observation and characterization of the transient open-chain neutral derivative, which was isolated as its cyclized form. The latter features an unusually long reactive C–O bond (150 pm) and a rich electrochemistry, including oxidation into an air-persistent radical cation.
Introduction
In 1850, John Stenhouse observed that furfural-containing oils deeply stain various organic tissues.1 He later succeeded in crystallising the salt resulting from the addition of aniline to crude furfural in the presence of hydrochloric acid, obtaining “a mass of beautiful iridescent fur”.2 Despite this simple synthesis, the chemistry of so-called Stenhouse salts A·H+ (Scheme 1) has remained fairly unexplored. Sporadic interest revivals have been limited for a long time by a lack of selectivity and reproducibility.3 In recent literature, Stenhouse salts are mostly invoked either as (i) formal parent structures for derivative “donor–acceptor Stenhouse adducts” with photo-switching properties,4 (ii) coloured Maillard-type products in food browning reactions5 or (iii) elusive intermediates in the formation of cyclopentenones.6,7 In fact, only a few conditions and substitution patterns (aryl and H, Me or indole as N,N′-substituents)3h,j,k,6,7a have allowed the isolation of defined salts A·H+. They easily evolve into cyclopentenones B, which can be substituted with a variety of nucleophiles or rearrange into a more stable isomer C. | ||
| Scheme 1 a) Stenhouse salts A·H+ and their conversion into cyclopentenones. (b) 1,3-Di(amino)oxyallyls D, stabilized radical cations D˙+ and putative vinylogous Stenhouse-type A˙+. | ||
The neutral deprotonated forms A have eluded spectroscopic observation to date. Li and Batey proposed that these intermediates exist in solution as cyclic c-A, whose ring opening affords less stable o-A, undergoing fast conrotatory electrocyclization to give cyclopentenones B.6,7a Note that the formation of B from A·H+ is reversible. In some cases, the addition of acids to B gives back A·H+. For this reason, B has been sometimes misattributed to A, causing confusion in the research community.3,7
Open-chain o-A are reminiscent of 1,3-di(amino)oxyallyls D. These diradicaloids have lifetimes from seconds to years in solution at room temperature, depending on substitution patterns.8,9 We recently reported air-stable versions.8g Compounds D undergo reversible electron transfers. They afford radical cations D˙+, which are usually air-persistent. Naturally, we wondered whether vinylogous open-chain o-A could feature similar redox behaviours. In principle the design of sterically hindered models may prevent the electrocyclization of A·H+ into B.
Herein, we describe the synthesis of such a Stenhouse salt featuring the extreme bulky environment provided by a cyclic (alkyl)(amino)carbene (CAAC).10–12 This allowed for the unprecedented isolation of the cyclic deprotonated derivative c-A and the spectroscopic observation of its open-chain counterpart o-A. These peculiar Stenhouse-type compounds feature rich redox chemistry, including the oxidation into open-chain A˙+, a persistent paramagnetic cyanine dye.
Results and discussion
Our approach towards Stenhouse derivatives from a stable carbene involves (i) the formation of an acylium from the addition of a CAAC to an acryloyl chloride,11l followed by (ii) the 1,4-addition of a second carbene to the resulting Michael acceptor, yielding a Stenhouse salt after proton migration (Scheme 2a). As aminocarbenes are strong bases, it seemed likely that deprotonation to afford A would immediately follow. Therefore, we added three equivalents of CAAC to a solution of cinnamoyl chloride in THF. After work-up to remove iminium salts stemming from the protonation of one equivalent of CAAC, a pale-yellow solid was isolated in 44% yield (Scheme 2b). A single-crystal X-ray diffraction study13 showed the formation of a neutral close-chain Stenhouse derivative c-1. Data from 13C and 1H NMR spectra are fully consistent. As expected from the low-symmetry of the molecule, all carbons and protons of the 2,6-di(isopropyl)phenyl group (Dipp) and the gem-dimethyl and CH2 protons of the five-membered ring of both CAAC patterns are diastereotopic. Some aromatic protons and isopropyl signals feature unusual up-field 1H chemical shifts because the rigid structure of c-1 forces the respective shielding cones of the phenyl and Dipp groups into close proximity.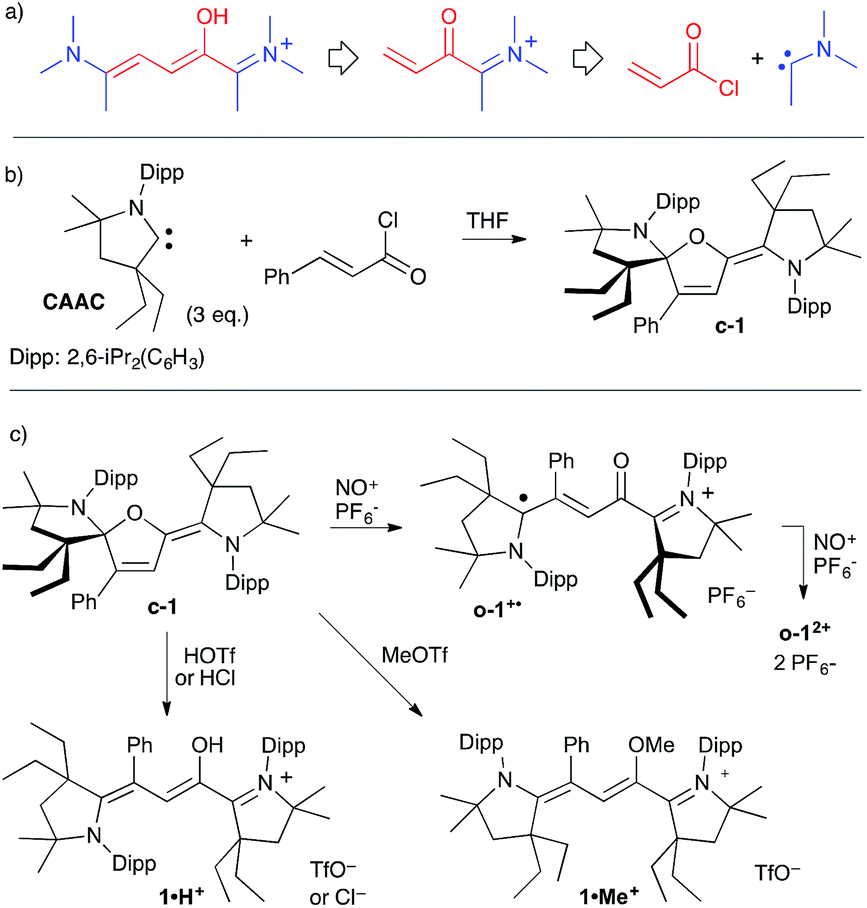 | ||
| Scheme 2 a) Retrosynthetic approach for the synthesis of Stenhouse salt derivatives from stable aminocarbenes; synthesis (b) and reactivity (c) of neutral Stenhouse derivative c-1. | ||
No trace impurity in isotropic NMR spectra could be attributed to open-chain o-1. However, the solid-state structure of c-1 (Fig. 1) features a remarkably long C1–O1 bond length (150.7 pm). Significant deviation from the standard value (143 pm14) is not common for Csp3–O bond lengths, even in strained epoxides,15 and “extreme” lengths above 150 pm are mostly limited to some reactive oxoniums.16,17 We ruled out a possible artefact due to interactions or packing effects in the crystal. Indeed, DFT-optimized structures18,19 corroborated the experimental solid-state geometry (see the ESI†), including the shorter C4–O1 distance (139.1 pm, a typical value for single Csp2–O bonds14). We found that NBO formalism can account for the unusual elongation of C1–O1. Second-order perturbative energy analysis revealed a strong negative hyperconjugation of the lone pair of N1 into the low-lying σ*(C1–O1) bond (E(2) = 21 kcal mol−1). As the cleavage of the weak C1–O1 bond led to o-1, this suggested that the reactivity of c-1 could mimic that of its open-chain counterpart.
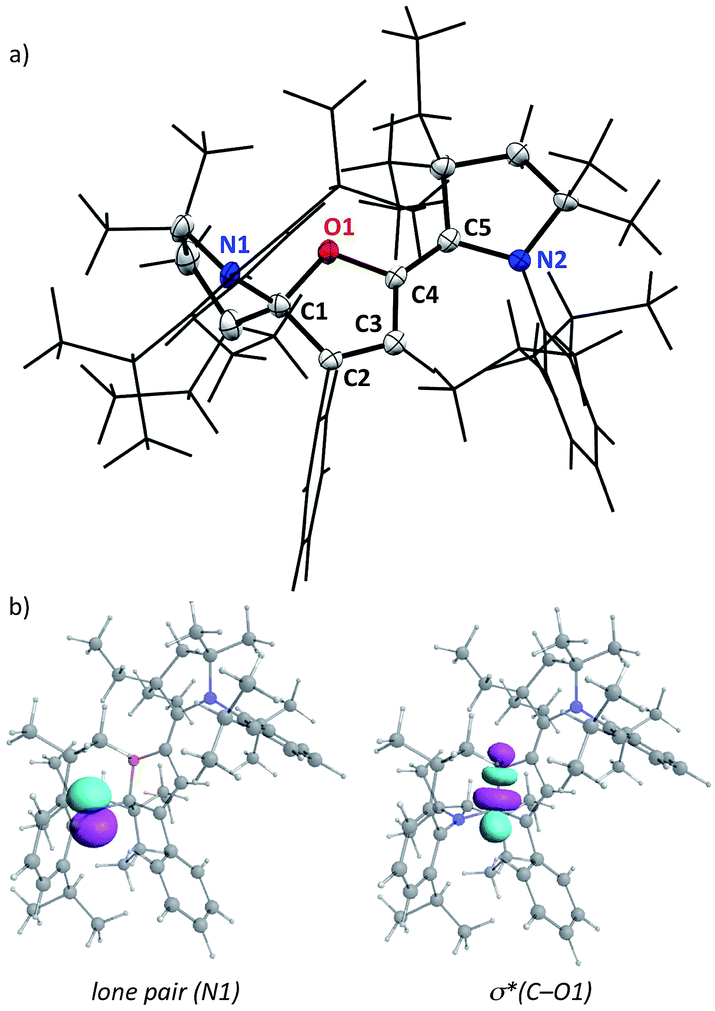 | ||
| Fig. 1 a) Representation of the single crystal X-ray structure of c-1. A wireframe display was used for clarity, with thermal ellipsoids (50% probability) shown only for selected atoms. (b) Representation of relevant natural bond orbitals for c-1. | ||
Compound c-1 is sensitive and affords highly coloured compounds when exposed to air or moisture. The hydrolysis product features a strong UV-vis absorption at 526 nm and was identified as the open-chain protonated product 1·H+. It was synthesized by the addition of trifluoromethanesulfonic acid to c-1. Stenhouse salt [1·H+][OTf−] is a deep pink to violet iridescent solid with a golden shine (Scheme 2c). Initial crystallization attempts afforded nearly-bidimensional plates (monoclinic, I2/a), which could only provide a poorly resolved crystallographic structure determination. Thus, we synthesized [1·H+][Cl−] from the reaction of c-1 with a solution of HCl in diethylether. Satisfyingly, this chloride salt crystalized as well-defined needles (monoclinic, P21/c).
Single-crystal X-ray crystallography confirmed the open-chain structure of 1·H+. In the solid state, the chain is twisted and not fully conjugated, with one dominating resonance structure, as represented in Scheme 2. In solution, the 1H and 13C spectra showed broad signals at room temperature, which sharpened at lower temperature, suggesting a significant conformational flexibility for the π-system. As expected, salts of 1·H+ yielded back c-1 upon addition of triethylamine. Similarly, addition of methyltriflate (MeOTf) to c-1 afforded the O-methylated Stenhouse salt [1·Me+][OTf−], which was fully characterized, including a single-crystal X-ray crystallography study.
EPR of aerated solutions evidenced the presence of a persistent radical, which was attributed to o-1˙+. This was confirmed by stoichiometric preparations, either by electrolysis (see further below) or by the reaction of c-1 with one equivalent of nitrosonium hexafluorophosphate (NOPF6) as the oxidant. The radical cation was isolated as a dark red powder (43% yield). It persists in solution for hours. Note that it is rather insensitive to oxygen, with identical half-lives either in air or under an argon atmosphere (about 15 h in acetonitrile at room temperature). Regardless of conditions, it ultimately yielded 1·H+, likely acting as a H˙ scavenger from impurities or solvents. Isotropic EPR hyperfine spectra (Fig. 2) indicated a large hyperfine constant with one hydrogen atom (10 MHz) and only one nitrogen atom (11 MHz).20 These values were fairly reproduced with DFT calculations (10 and 6 MHz respectively), which also showed that one CAAC moiety is orthogonal to the rest of the π-system, with no significant contribution to the singly occupied molecular orbital (SOMO). As a result, less than 5% Mulliken spin density was found on N2 and C5 (against 20% on O1, 13% on C4, 20% on C3, 13% on C2, 17% on C1 and 17% on N1).
 | ||
| Fig. 2 Left, experimental X-band EPR spectra of 1˙+ in acetonitrile at room temperature (black); the corresponding simulated spectra (blue) with the following set of parameters: line-broadening parameter lw = 0.288 and hyperfine coupling constants a(14N) = 11.4 MHz and a(1H) = 10.1 MHz. Right, representation of computed SOMO. | ||
Further oxidation of [1˙+][PF6−] with a second equivalent of NO(PF6) led to dication salt [12+][PF6−]2 in 60% yield. It was fully characterized, including a single-crystal X-ray crystallography study.
Cyclic voltammetry experiments unveiled a rich electrochemistry (Fig. 3). Closed-chain c-1 undergoes two successive oxidations into c-1˙+ (E0 = −0.5 V vs. Fc/Fc+) and c-12+ (E0 = −0.16 V), respectively (processes 1 and 2 in Fig. 3a). A loss of reversibility is observed at a lower scan rate, which was interpreted as the result of ring opening of the oxidized species at room temperature (Fig. 3b). This hypothesis was confirmed by cyclic voltammetry studies of [o-1˙+][PF6−]. Indeed, the radical cation undergoes a reversible oxidation at E0 = −0.27 V to afford o-12+ (process 3). The reduction wave of o-1˙+ is irreversible at room temperature, but becomes reversible at −30 °C (process 4, E1/2 = −0.84 V), indicating that the fast ring-closure into c-1 follows a stepwise EC process with the transient formation of neutral open-chain o-1. Note that after a first reducing cycle in voltammograms of [1˙+][PF6−], as c-1 is generated, the oxidation waves of c-1 into c-1˙+ (process 1) and c-1˙+ into c-12+ (process 2) appear. Conversely, this also accounts for cyclic voltammograms of c-1 at a low scan-rate, which feature the reduction waves of o-12+ into o-1˙+ (process 3) and of o-1˙+ into c-1 (process 4).
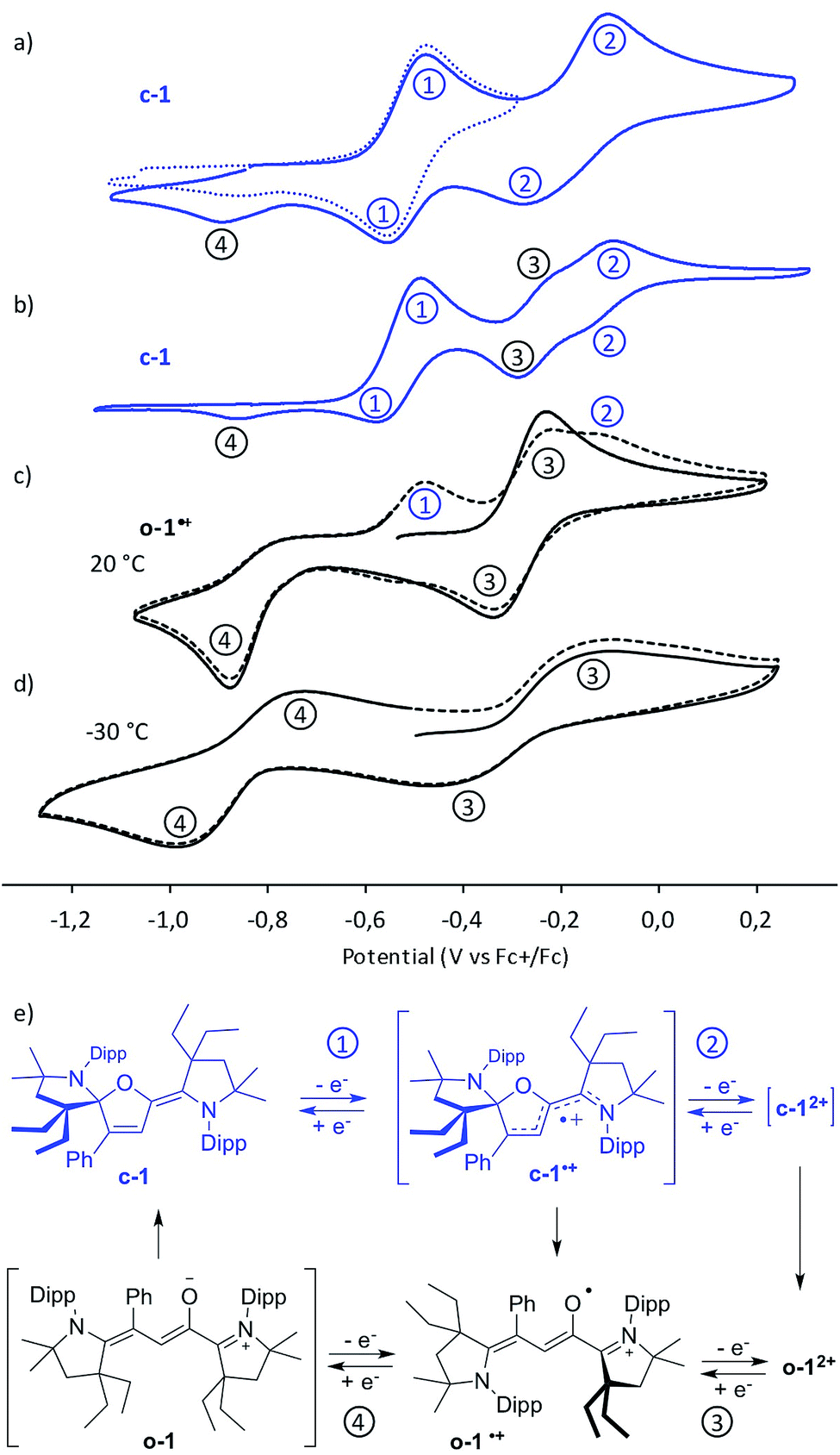 | ||
| Fig. 3 Cyclic voltammetry at 100 mV s−1 rate (if not otherwise stated) with 0.1 M nBu4NPF6 electrolyte of acetonitrile solution (1 mM) of: (a) c-1, (b) c-1 at a rate of 10 mV s−1, (c) o-1˙+ at 20 °C, and (d) o-1˙+ at −30 °C; (e) corresponding electrochemical processes. | ||
Cyclic voltammetry experiments indicated that the ring opening of c-1˙+ was slow enough to allow for its spectroscopic observation. However, UV-monitoring of the electrolysis of c-1 couldn't clearly evidence the transient formation of this radical. We realized that the characteristic signals for c-1˙+ must be hidden by the spectra of o-1˙+. Indeed TD-DFT calculations predicted very similar absorptions for both radicals (c-1˙+, 560 and 450 nm; o-1˙+, 630 and 430 nm). Thus, we turned to the in situ EPR-monitoring of the electrolysis of c-1 at −0.4 V with an in-house device (see the ESI†). We observed the formation of a transient radical with o-1˙+ (Fig. 4). Note that DFT calculations predicted a simple hyperfine structure for the isotropic EPR spectra of c-1˙+ (with only a significant coupling constant of 13 MHz with N2). Accordingly, the EPR spectra of the mixtures could be fitted with a close value (15.5 MHz).
 | ||
| Fig. 4 Left, EPR monitoring of the electrolysis at −0.4 V of c-1 in acetonitrile; middle, the corresponding simulated spectra; right, deconvolution and respective contribution of c-1˙+ (red) and o-1˙+ (black) radicals to the spectra. | ||
Importantly, electrochemical data also showed that the spectroscopic observation of neutral open-chain o-1, an elusive derivative of Stenhouse salts to date, could be possible at low temperature. Thus, we performed the addition of a strong base (potassium hexamethyldisilazide, KHMDS) to a THF solution of 1·H+ at −78 °C. UV-monitoring indicated an instantaneous reaction with the formation of a deep blue compound (Fig. 5). The observation of a strong absorption at 610 nm matched the TD-DFT predictions for o-1 (608 nm; πHOMO → π*LUMO). This band gradually disappeared upon warming to room temperature, as the characteristic band for c-1 at 358 nm grew.
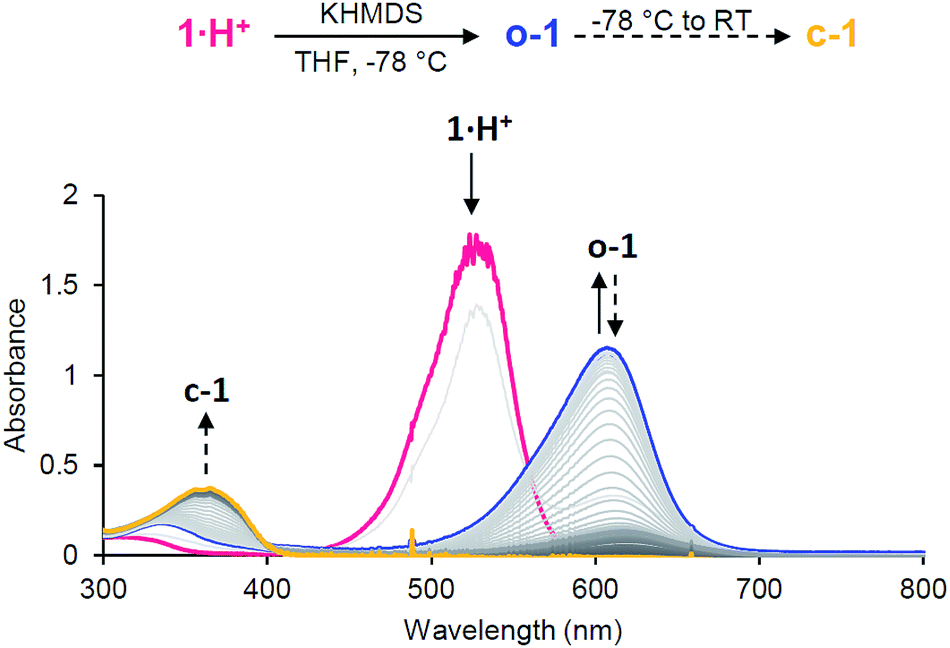 | ||
| Fig. 5 UV-monitoring of the addition of KHMDS to 1·H+ in THF. | ||
Conclusions
The basification of 1·H+ yields neutral spirocyclic c-1. Such hydrofurans have been previously only considered as elusive intermediates on the route from Stenhouse salts to cyclopentenones. Molecule c-1 features a remarkably long C–O bond (150.7 pm), typical of those in reactive oxoniums. NBO analysis indicates a strong π-back donation of the lone pair of an amino group into the σXO* orbital, thus accounting for the unusual elongation and easy cleavage of this bond. Neutral c-1 can undergo two reversible oxidations affording open-chain radical cation o-1˙+ and dication o-12+. All transient forms of c-1 (c-1˙+ and c-12+, as well as zwitterionic o-1) could be characterized prior to their respective ring opening or closure.Open-chain derivatives of 1 stand out among the usual cyanine polymethines.21 Indeed, the electrochemistry of typical representatives of this large family of organic dyes is mostly limited to one electron-oxidation, yielding short-lived radical dications followed by their fast dimerization or decay.22 This is in marked contrast with the rich redox behaviour of derivatives 1. For instance cyanine-based radical o-1˙+ is air-persistent and its oxidation affords stable dication o-12+. Upon one-electron reduction, the deep-red radical o-1˙+ is reversibly switched to yellow hydrofuran c-1. In turn, reversible protonation can switch c-1 to deep-pink cyanine 1·H+.
The steric hindrance of the CAAC pattern of the novel Stenhouse derivatives has manifold implications. As already mentioned, it forbids the cyclisation into the corresponding cyclopentenone. It also considerably slows down conformational evolution of the derivatives, as demonstrated by fluxional 1H NMR spectroscopy of 1·H+ and 1·Me+ at room temperature. It is likely for the same reason that the ring-opening of c-1˙+ and c-12+ and ring-closure of o-1 are remarkably slow intramolecular processes (at least a few seconds at room temperature). Furthermore, steric hindrance prevents optimal conjugation of open-chain derivatives of 1, which all feature twisted systems. Thus, it is clear that fine-tuning of sterics will significantly impact the electronic, spectroscopic and electrochemical properties of the compounds. We envision that the variety of available stable N-heterocyclic carbenes will allow for the development of a large and diverse family of novel switchable redox-active cyanine dyes.
Author contributions
D. M. and E. T. M. co-wrote the paper and conceived and designed the study. V. T., F. M., N. T., S. S., D. M. and E. T. M. performed the chemical experiments and analysed the data. D. M. performed the DFT studies. J. P. performed the crystallographic studies. All authors discussed the results and commented on the manuscript.Data availability
The experimental procedures, analytical data and computational details supporting the findings of this study are available within the manuscript and its ESI file.† Raw and unprocessed NMR data are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no 892162. We acknowledge the CNRS and the University of Grenoble Alpes for a frictionless environment in the context of the Labex Arcane and CBH-EUR-GS (ANR-17-EURE-0003). The authors thank the CECCIC centre of Grenoble for computer resources and the ICMG analytic platform, especially P. Girard, D. Gatineau, R. Sanahuges and N. Altounian for their outstanding service.Notes and references
- J. Stenhouse, Liebigs Ann., 1850, 74, 278–297 CrossRef.
- J. Stenhouse, Liebigs Ann., 1870, 156, 197–205 CrossRef.
- (a) W. König, K. Hey, F. Schulze, E. Silberkweit and K. Trautmann, Ber. Dtsch. Chem. Ges., 1934, 67, 1274–1296 CrossRef; (b) G. Williams and C. L. Wilson, J. Chem. Soc., 1942, 506–507 RSC; (c) A. P. Dillon and K. G. Lewis, Tetrahedron, 1969, 25, 2035–2039 CrossRef CAS; (d) K. G. Lewis and C. E. Mulquiney, Aust. J. Chem., 1970, 23, 2315–2323 CrossRef CAS; (e) K. G. Lewis, Aust. J. Chem., 1973, 26, 893–897 CrossRef CAS; (f) K. G. Lewis and C. E. Mulquiney, Tetrahedron, 1977, 33, 463–475 CrossRef CAS; (g) K. G. Lewis and C. E. Mulquiney, Aust. J. Chem., 1979, 32, 1079–1092 CrossRef CAS; (h) K. Honda, H. Komizu and M. Kawasaki, J. Chem. Soc., Chem. Commun., 1982, 253–254 RSC; (i) B. R. d'Arcy, K. G. Lewis and C. E. Mulquiney, Aust. J. Chem., 1985, 38, 953–965 CrossRef; (j) P. Šafář and J. Kováč, Collect. Czech. Chem. Commun., 1989, 54, 2425–2432 CrossRef; (k) H. S. Patel and G. H. Majmudar, Eur. Polym. J., 1991, 27, 89–92 CrossRef CAS.
- (a) M. M. Lerch, W. Szymański and B. L. Feringa, Chem. Soc. Rev., 2018, 47, 1910–1937 RSC; (b) S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2014, 136, 8169–8172 CrossRef CAS PubMed; (c) S. Helmy, S. Oh, F. A. Leibfarth, C. J. Hawker and J. Read de Alaniz, J. Org. Chem., 2014, 79, 11316–11329 CrossRef CAS PubMed.
- T. Hofmann, J. Agric. Food Chem., 1998, 46, 932–940 CrossRef CAS.
- R. F. A. Gomes, J. A. S. Coelho and C. A. M. Afonso, Chem. – Eur. J., 2018, 24, 9170–9186 CrossRef CAS PubMed.
- (a) S.-W. Li and R. A. Batey, Chem. Commun., 2007, 43, 3759–3761 RSC; (b) A. Procopio, P. Costanzo, M. Curini, M. Nardi, M. Oliverio and G. Sindona, ACS Sustainable Chem. Eng., 2013, 1, 541–544 CrossRef CAS; (c) K. Griffiths, C. W. D. Gallop, A. Abdul-Sada, A. Vargas, O. Navarro and G. E. Kostakis, Chem. – Eur. J., 2015, 21, 6358–6361 CrossRef CAS PubMed; (d) K. Griffiths, P. Kumar, J. D. Mattock, A. Abdul-Sada, M. B. Pitak, S. J. Coles, O. Navarro, A. Vargas and G. E. Kostakis, Inorg. Chem., 2016, 55, 6988–6994 CrossRef CAS PubMed; (e) M. S. Estevão, R. J. V. Martins and C. A. M. Afonso, J. Chem. Educ., 2017, 94, 1587–1589 CrossRef; (f) R. F. A. Gomes, N. R. Esteves, J. A. S. Coelho and C. A. M. Afonso, J. Org. Chem., 2018, 83, 7509–7513 CrossRef CAS PubMed; (g) M. L. Di Gioia, M. Nardi, P. Costanzo, A. De Nino, L. Maiuolo, M. Oliverio and A. Procopio, Molecules, 2018, 23, 1891–1904 CrossRef PubMed; (h) S. I. Sampani, A. McGown, A. Vargas, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, J. Spencer and G. E. Kostakis, J. Org. Chem., 2019, 84, 6858–6867 CrossRef CAS PubMed; (i) J. G. Pereira, J. P. M. António, R. Mendonça, R. F. A. Gomes and C. A. M. Afonso, Green Chem., 2020, 22, 7484–7490 RSC; (j) K. Peewasan, M. P. Merkel, O. Fuhr and A. K. Powell, Dalton Trans., 2020, 49, 2331–2336 RSC; (k) T. B. Shah, R. S. Shiny, R. B. Dixit and B. C. Dixit, J. Saudi Chem. Soc., 2014, 18, 985–992 CrossRef.
- (a) D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7014–7017 CrossRef CAS PubMed; (b) T. Schulz, C. Farber, M. Leibold, C. Bruhn, W. Baumann, D. Selent, T. Porsch, M. C. Holthausen and U. Siemeling, Chem. Commun., 2013, 49, 6834 RSC; (c) V. Regnier and D. Martin, Org. Chem. Front., 2015, 2, 1536–1545 RSC; (d) V. Regnier, F. Molton, C. Philouze and D. Martin, Chem. Commun., 2016, 52, 11422–11425 RSC; (e) M. Devillard, V. Regnier, M. Tripathi and D. Martin, J. Mol. Struct., 2018, 1172, 3–7 CrossRef CAS; (f) M. Tripathi, V. Regnier, Z. Ziani, M. Devillard, C. Philouze and D. Martin, RSC Adv., 2018, 8, 38346–38350 RSC; (g) E. Tomás-Mendivil, M. Devillard, V. Regnier, J. Pecaut and D. Martin, Angew. Chem., Int. Ed., 2020, 59, 11516–11520 CrossRef PubMed.
- See also: (a) U. Siemeling, C. Farber, C. Bruhn, M. Leibold, D. Selent, W. Baumann, M. von Hopffgarten, C. Goedecke and G. Frenking, Chem. Sci., 2010, 1, 697 RSC; (b) S. D. Ursula and U. Radius, Organometallics, 2017, 36, 1398 CrossRef; (c) G. Kuzmanich, F. Spänig, C. K. Tsai, J. M. Um, R. M. Hoekstra, K. N. Houk, D. M. Guldi and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2011, 133, 2342 CrossRef CAS PubMed.
- (a) V. Lavallo, Y. Canac, C. Prasang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed; (b) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (c) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; (d) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed; (e) U. S. D. Paul and U. Radius, Eur. J. Inorg. Chem., 2017, 3362–3375 CrossRef CAS; (f) M. Melaimi, R. Jazzar, M. Soleihavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed.
- CAAC-based organic radicals: (a) J. K. Mahoney, D. Martin, C. Moore, A. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 18766–18769 CrossRef CAS PubMed; (b) L. Jin, M. Melaimi, L. L. Liu and G. Bertrand, Org. Chem. Front., 2014, 1, 351–354 RSC; (c) Y. Li, K. C. Mondal, P. P. Samuel, H. Zhu, C. M. Orben, S. Panneerselvam, B. Dittrich, B. Schwederski, W. Kaim, T. Mondal, D. Koley and H. W. Roesky, Angew. Chem., Int. Ed., 2014, 53, 4168–4172 CrossRef CAS PubMed; (d) J. K. Mahoney, D. Martin, F. Thomas, C. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519–7525 CrossRef CAS PubMed; (e) S. Styra, M. Melaimi, C. E. Moore, A. L. Rheingold, T. Augenstein, F. Breher and G. Bertrand, Chem. – Eur. J., 2015, 21, 8441–8446 CrossRef CAS PubMed; (f) D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886–12890 CrossRef CAS PubMed; (g) J. K. Mahoney, R. Jazzar, G. Royal, D. Martin and G. Bertrand, Chem. – Eur. J., 2017, 23, 6206–6212 CrossRef CAS PubMed; (h) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 15620–15623 CrossRef CAS PubMed; (i) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2206–2213 CrossRef CAS PubMed; (j) P. W. Antoni and M. M. Hansmann, J. Am. Chem. Soc., 2018, 140, 14823–14835 CrossRef PubMed; (k) P. W. Antoni, T. Bruckhoff and M. M. Hansmann, J. Am. Chem. Soc., 2019, 141, 9701–9711 CrossRef CAS PubMed; (l) V. Regnier, E. A. Romero, F. Molton, R. Jazzar, G. Bertrand and D. Martin, J. Am. Chem. Soc., 2019, 141, 1109–1117 CrossRef CAS PubMed; (m) J. Messelberger, A. Grünwald, S. J. Goodner, F. Zeilinger, P. Pinter, M. E. Miehlich, F. W. Heinemann, M. M. Hansmann and D. Munz, Chem. Sci., 2020, 11, 4138–4149 RSC.
- For stable organic radicals based on other types of N-heterocyclic carbenes, see also: (a) J. Back, J. Park, Y. Kim, H. Kang, Y. Kim, M. J. Park, K. Kim and E. Lee, J. Am. Chem. Soc., 2017, 139, 15300–15303 CrossRef CAS PubMed; (b) C. L. Deardorff, R. E. Sikma, C. P. Rhodes and T. W. Hudnall, Chem. Commun., 2016, 52, 9024–9027 RSC; (c) L. Y. M. Eymann, A. G. Tskhovrebov, A. Sienkiewicz, J. L. Bila, I. Zikhovic, H. M. Ronnow, M. D. Wodrich, L. Vannay, C. Corminboeuf, P. Pattison, E. Solari, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2016, 138, 15126–15129 CrossRef CAS PubMed; (d) D. Rottschafer, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 4765–4768 CrossRef PubMed; (e) N. M. Gallagher, H.-Z. Ye, S. Feng, J. Lopez, Y. G. Zhu, T. Van Voorhis, Y. Shao-Horn and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 3952–3955 CrossRef CAS PubMed; (f) Y. Kim, J. E. Byeon, G. Y. Jeong, S. S. Kim, H. Song and E. Lee, J. Am. Chem. Soc., 2021, 143(23), 8527–8532 CrossRef CAS PubMed; (g) J. Zhao, X. Li and Y.-F. Han, J. Am. Chem. Soc., 2021, 143, 14428–14432 CrossRef CAS PubMed; (h) L. Delfau, S. Nichilo, F. Molton, J. Broggi, E. Tomás-Mendivil and D. Martin, Angew. Chem., Int. Ed., 2021, 60, DOI:10.1002/anie.202111988.
- CCDC 2163356–2163359 contains the crystallographic data for this paper. These data are provided free of charge by the Cambridge crystallographic data centre.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin trans. II, 1987, S1 RSC.
- See for example: A. J. Leyhane and M. L. Snapper, Org. Lett., 2006, 8, 5183–5186 CrossRef CAS PubMed.
- G. Gunbas, N. Hafezi, W. L. Sheppard, M. M. Olmstead, I. V. Stoyanova, F. S. Tham, M. P. Meyer and M. Mascal, Nat. Chem., 2012, 4, 1018–1023 CrossRef CAS PubMed.
- A long C–O bond was also reported in strained acenaphthofurans: Y. Uchimura, T. Shimajiri, Y. Ishigaki, R. Katoono and T. Suzuki, Chem. Commun., 2018, 54, 10300–10303 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, et al., Calculations were performed with the Gaussian suite of programs: Gaussian09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009. See the ESI† for complete citation.
- If not otherwise stated, calculations were carried out at the B3LYP/6-311g(d,p) level of theory with the PCM model for acetonitrile and Grimme's D3 empirical dispersion: S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Experimental values for isotropic hyperfine constants were extracted from EPR spectra by fitting with the EasySpin simulation package: S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- For general reviews on cyanine dyes: (a) A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973–2012 CrossRef CAS PubMed; (b) J. L. Bredas, C. Adant, P. Tackx, A. Persoons and B. M. Pierce, Chem. Rev., 1994, 94, 243–278 CrossRef CAS.
- (a) R. Lenhard and A. D. Cameron, J. Phys. Chem., 1993, 97, 4916–4925 CrossRef; (b) J. E. H. Buston, F. Marken and H. L. Anderson, Chem. Commun., 2001, 37, 1046–1047 RSC; (c) J. L. Lyon, D. M. Eisele, S. Kirstein, J. P. Rabe, D. A. Vanden Bout and K. J. Stevenson, J. Phys. Chem. C, 2008, 112, 1260–1268 CrossRef CAS; (d) J. Lenhard and R. Parton, J. Am. Chem. Soc., 1987, 109, 5808–5813 CrossRef CAS; (e) R. Parton and J. Lenhard, J. Org. Chem., 1990, 55, 49–57 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. Experimental procedures, characterization data and DFT studies. CCDC 2163356–2163359. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01895k |
| This journal is © The Royal Society of Chemistry 2022 |