
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAzodioxy compounds as precursors for C-radicals and their application in thermal styrene difunctionalization†
Stefanie
Plöger
,
Christian
Mück-Lichtenfeld
 ,
Constantin G.
Daniliuc
and
Armido
Studer
*
,
Constantin G.
Daniliuc
and
Armido
Studer
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
First published on 5th August 2022
Abstract
An atom-economic thermal α,β-difunctionalization of various styrenes with readily prepared azodioxy compounds is reported. Mechanistic studies reveal that the starting azodioxy compounds can thermally be cleaved to the corresponding C-nitroso compounds, which under these thermal conditions further homolyze to generate reactive C-radicals along with the persistent NO radical. In the presence of a styrene, C-radical addition with subsequent nitrosylation followed by tautomerization is occurring, resulting in an overall styrene β-alkylation-α-oximation reaction.
Introduction
For decades, carbon-centered radicals have found extensive use as valuable reactive intermediates in organic synthesis.1 In particular, radical difunctionalizations of simple alkenes have emerged as powerful tools for preparing functionalized skeletons.2 Various alkyl radical precursors have been successfully used for alkene functionalization, such as cyclic ethers,3 amides,4 α-bromo esters,5 diazo compounds,6 alkoxyamines,7 unactivated ketones8 or aliphatic alcohols9 – just to mention a few. In most of these alkene difunctionalizations, an additional reagent to trap the transient adduct C-radical has to be added, decreasing atom economy. In that regard, bifunctional reagents providing the C-radical and also the trapping moiety would be beneficial.10 For example, this was realized for thermal alkoxyamine addition reactions7 and also in atom as well as group transfer additions.2a,7,11,12Recently, we developed a mild photomediated radical 1,2-difunctionalization of various electron-poor alkenes with α-acetoxy nitrosoalkanes as bifunctional reagents (Scheme 1a).13 In analogy to the Barton nitrite ester reaction14 that involves the homolytic O–NO bond cleavage of a nitrite ester to generate an alkoxy radical and the persistent nitric oxide radical (NO), we used the visible light induced homolytic C–NO bond cleavage of acyloxy nitroso compounds to generate a transient C radical and the persistent NO radical. The electron-rich α-oxy-C radical undergoes radical addition to an electrophilic alkene to give a transient adduct C-radical.13 Persistent radical effect (PRE)15 mediated highly selective cross-coupling of the NO radical with the adduct C-radical eventually provides the corresponding nitrosoalkane, which upon tautomerization leads to the final oxime product.
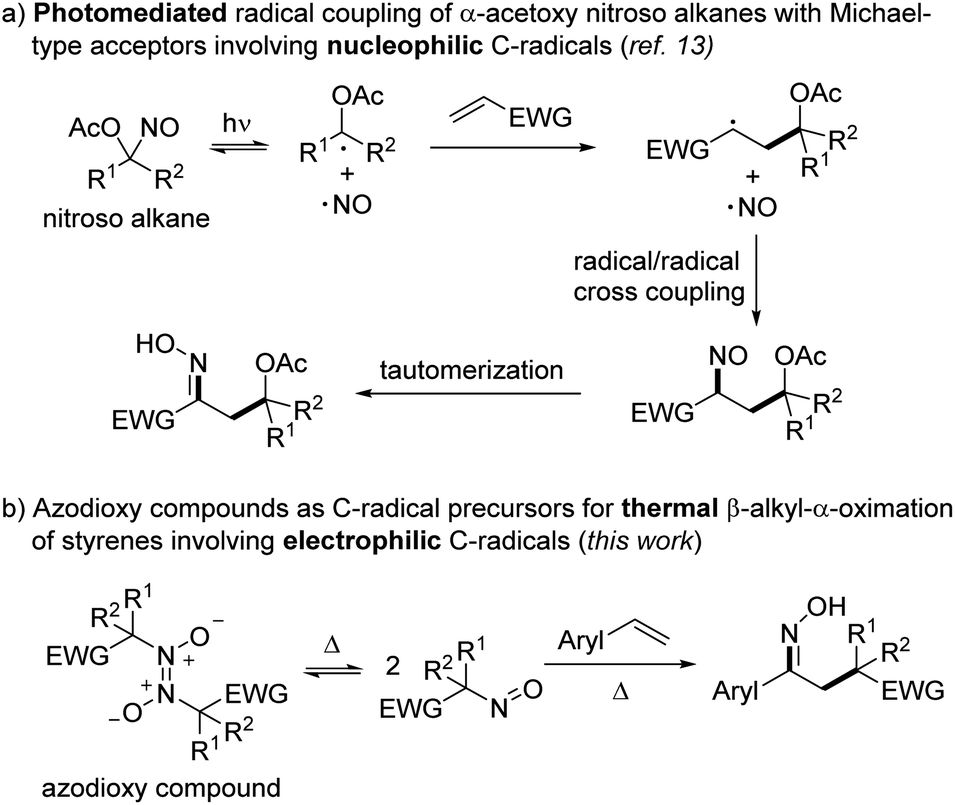 | ||
| Scheme 1 Use of C-nitroso compounds as bifunctional reagents for radical alkene 1,2-difunctionalization. | ||
Notably, C-nitroso compounds were intensively studied due to their interesting reactivity and important bioactivity.16 However, their synthetic utility in chemistry has so far mostly been limited to the role as C radical acceptors.17 Nonetheless, we demonstrated in this initial study their promising potential as C-radical precursors.13 However, it is well established that the majority of the C-nitroso compounds are not stable in solution and dimerize to the corresponding azodioxy compounds.18 Unfortunately, in contrast to the monomeric blue nitroso compounds, the dimers are generally transparent and show no photoactivity upon irradiation with visible light. We therefore decided to develop an alternative entry into C-nitroso radical chemistry that uses the azodioxy dimers as the C-radical precursors applying thermal conditions. Moreover, the reported photochemical approach allowed to generate exclusively nucleophilic α-acetoxy alkyl radicals, while electrophilic C-radicals are not accessible using that strategy. The concept is presented in Scheme 1b.
The azodioxy dimer should be thermally reversibly cleaved to the corresponding monomeric nitroso compound which then engages in a thermal homolytic cleavage of the C–NO bond to generate the corresponding electron-deficient C-radical along with the NO radical. As in the photochemical process, C-radical addition to an alkene, subsequent trapping of the adduct radical with NO and tautomerization should eventually provide the corresponding 1,2-difunctionalized products. Herein, we report the realization of that strategy and show that azodioxy compounds react with styrenes to the targeted oximes. DFT calculations provide further insights into the process.
Results and discussion
Initial experiments were conducted with the azodioxy compound 1a and styrene 2a in DMF. For details on the preparation of all azodioxy compounds used in this study, see ESI.† While no product formation could be observed at room temperature (Table 1, entry 1), we were delighted to see that at 100 °C for 3.5 h, 1,2-difunctionalization proceeded smoothly. We found that the targeted oxime further cyclizes to the dihydro-1,2-oxazine 3a which was isolated in 53% yield as a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-mixture of diastereoisomers (Table 1, entry 2). Of note, the temperature necessary for successful transformation is readily monitored by the appearance of the typical blue color of the corresponding monomeric C-nitroso compound and a slightly better yield (60%) was obtained upon running the reaction at 60 °C (Table 1, entry 3). Solvent screening revealed that in DMSO the yield further increased to 66%, while worse results were achieved in acetone and MeOH (Table 1, entries 4–6).
:1-mixture of diastereoisomers (Table 1, entry 2). Of note, the temperature necessary for successful transformation is readily monitored by the appearance of the typical blue color of the corresponding monomeric C-nitroso compound and a slightly better yield (60%) was obtained upon running the reaction at 60 °C (Table 1, entry 3). Solvent screening revealed that in DMSO the yield further increased to 66%, while worse results were achieved in acetone and MeOH (Table 1, entries 4–6).
|
|
|||
|---|---|---|---|
| Entrya | Solvent | Temp. [°C] | Yieldb [%] |
| a Reaction conditions: 1a (0.1 mmol), 2b (0.6 mmol), solvent (2 mL), Ar, 1a was added in three portions every 30 min. b Yield of isolated product. c 1a was added in six portions every 30 min. d Reaction was conducted open to air. | |||
| 1 | DMF | r.t. | n.d. |
| 2 | DMF | 100 | 53 |
| 3 | DMF | 60 | 60 |
| 4 | DMSO | 60 | 66 |
| 5 | Acetone | 60 | n.d. |
| 6 | MeOH | 60 | 32 |
| 7 | DMSO | 60 | 73 |
| 8c,d | DMSO | 60 | 44 |
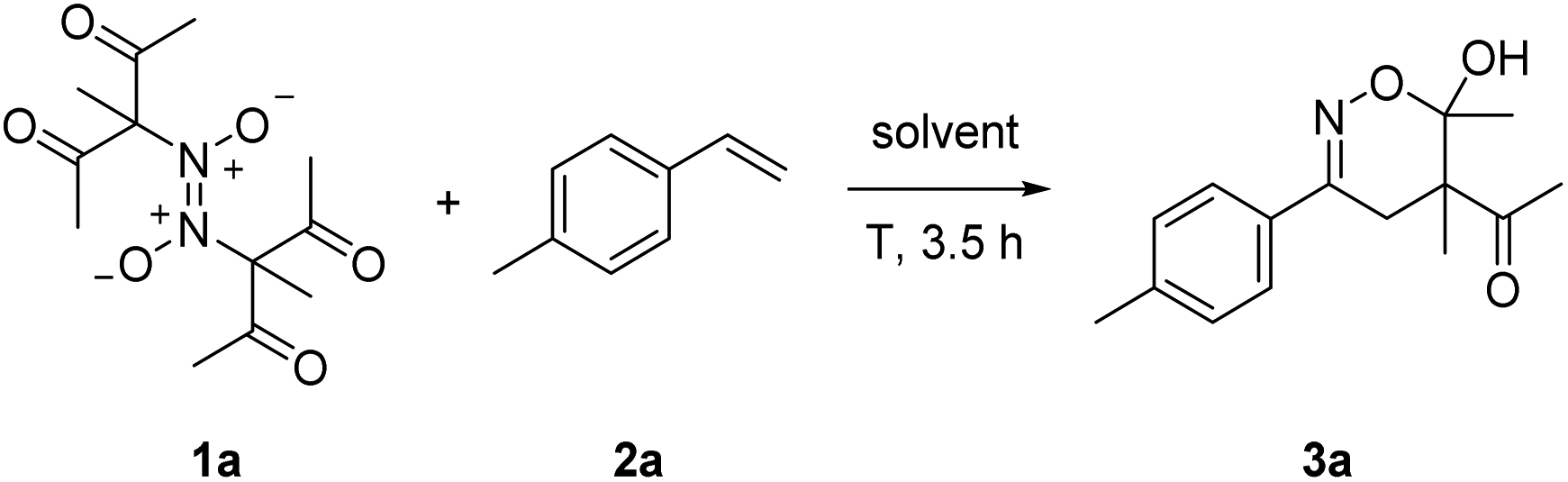
The yield could be further increased (73%) by keeping the concentration of the azodioxy compound and accordingly also the nitrosoalkane concentration low upon adding 1a in six portions every 30 min (Table 1, entry 7). Unfortunately, the use of syringe pump technique to keep the nitroso-compound concentration even lower was not feasible, since 1a is insoluble in DMSO at room temperature. Exclusion of air is beneficial, as running the reaction under air atmosphere led to a decreased yield of 44% (Table 1, entry 8).
With the optimized reaction conditions in hand, we varied the radical acceptor keeping azodioxy 1a as the bifunctional reagent (Scheme 2). We were pleased to see that along with styrene (3b) various para-substituted styrenes engaged in the 1,2-difunctionalization. Not only electron-donating substituents (3c, 3d, 3g, 3h) but also halogen atoms (3e, 3f) and electron-withdrawing functional groups (3k) were tolerated as para-substituents providing the corresponding products in 60–78% yields. Notably, a free carboxylic acid functionality (3j) and the synthetically valuable Bpin-moiety (3i) were also tolerated. As expected, with the most electron-deficient 2,3,4,5,6-pentafluorostyrene the lowest yield was obtained (3n, 23%), indicating the relevance of polar effects considering the electrophilicity of the C-radical derived from 1a. Steric effects at the aryl moiety are not of importance, as the ortho-tolyl derivative provided a good yield (3l, 66%). The meta-congener afforded a similar result (3m, 70%). Naphthalene and heteroarene based systems could also be used as radical acceptors and the desired products 3o–3r were obtained in moderate to good yields (38–84%). Interestingly, conjugated dienes 2s and 2t also serve as coupling partners. In the case of 2s, a 3:1 mixture of the two regioisomers resulting from the 1,4-difunctionalization (3s′, 3 isomers, see ESI†) and the 1,2-difunctionalization (3s) were formed in 81% overall yield. In contrast, the diene 2s with a methyl group at the 2-position provided exclusively the 1,4-difunctionalization product 3t′ in 86% yield with complete E-selectivity. For non-activated aliphatic alkenes, the C-radical addition is slow so that direct trapping with NO competes. Indeed, reaction of 1a with 1-octene (2u) under the optimized condition did not work. However, C-radical precursor 1ae with 2u provided the desired compound, albeit in low yield (3u′, 18%). In this case, 1ae was added via syringe pump to keep the NO concentration low.
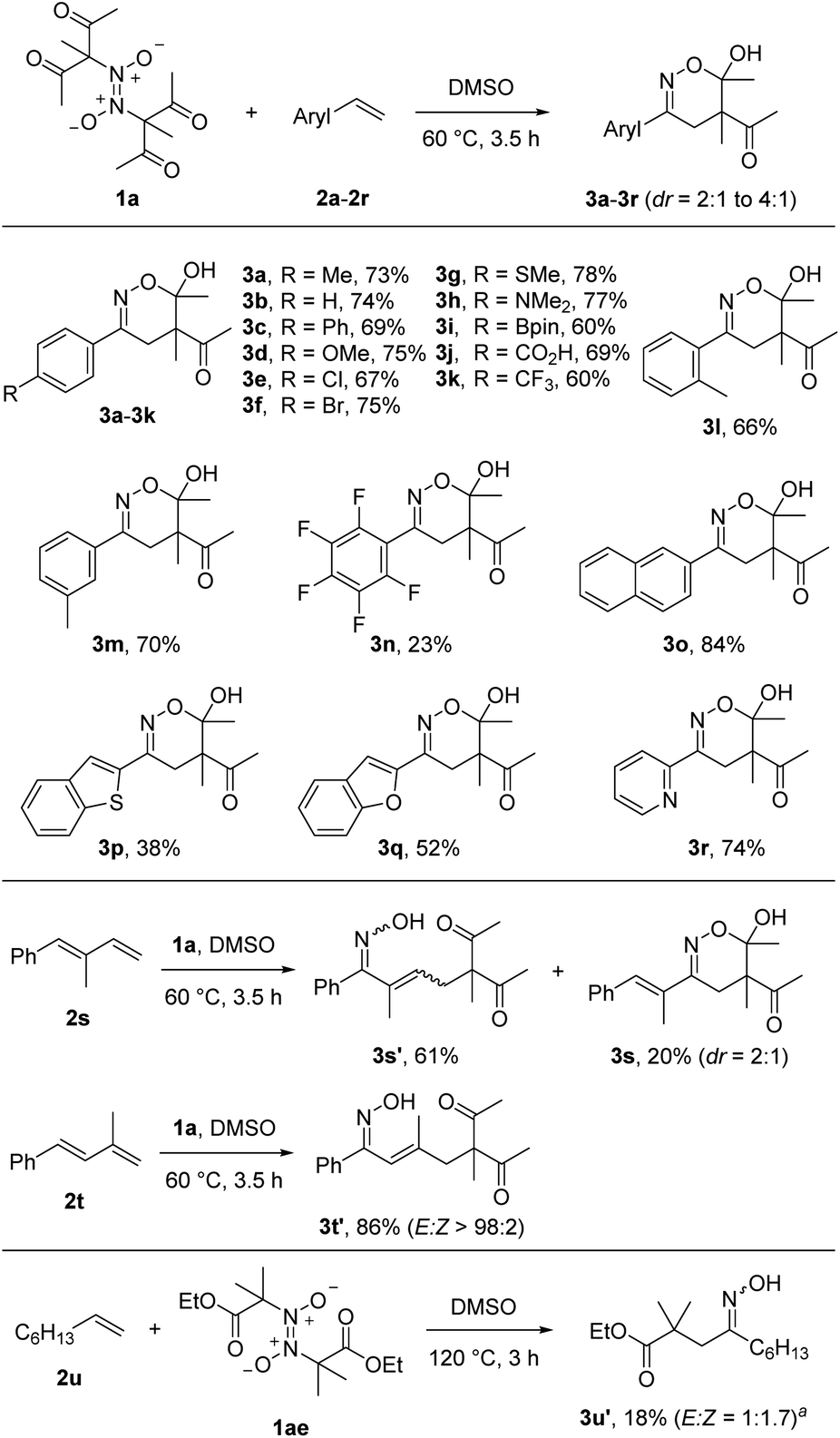 | ||
| Scheme 2 Variation of the radical acceptor 2. a40 equiv. of the alkene 2u were used. | ||
Switching back to styrene 2a as radical acceptor, we next tested various azodioxy compounds 1 as radical precursors. The methyl group in 1a could be replaced by a benzyl (3v) and allyl (3w) substituent and the targeted products were isolated in 56% and 57% yield with moderate diastereoselectivities (3:1). Along with the acetylacetone derivatives other 1,3-diketones engaged in the 1,2-difunctionalization. As an example, 3x was obtained in 67% yield with complete diastereoselectivity.
The doubly activating 1,3-diketo moiety in the C-radical is not required and azodioxy compounds leading to α-monoketo-C-radicals also worked, as documented by the successful preparation of the dihydro-1,2-oxazines 3y–3aa (40–59%). In this series, the lowest yield was noted for the azodioxy compound leading to the more electron-rich para-MeO-phenyl-keto C-radical. The structure of 3y was confirmed by single-crystal X-ray diffraction analysis (see Scheme 3).19 Similar results were noted for other “mono ketones” (see 3ab, 3ac′ and 3al, 3al′). Interestingly, the keto functionality in 3ac′ is obviously sterically too shielded and cyclization to the corresponding dihydro-1,2-oxazine did not occur, while for 3al a mixture of the cyclized/non-cyclized forms was isolated. The azodioxy compound derived from 2-methylcyclohexane-1,3-dione participated in the reaction to give the annulated oxazine 3ad in 68% yield.
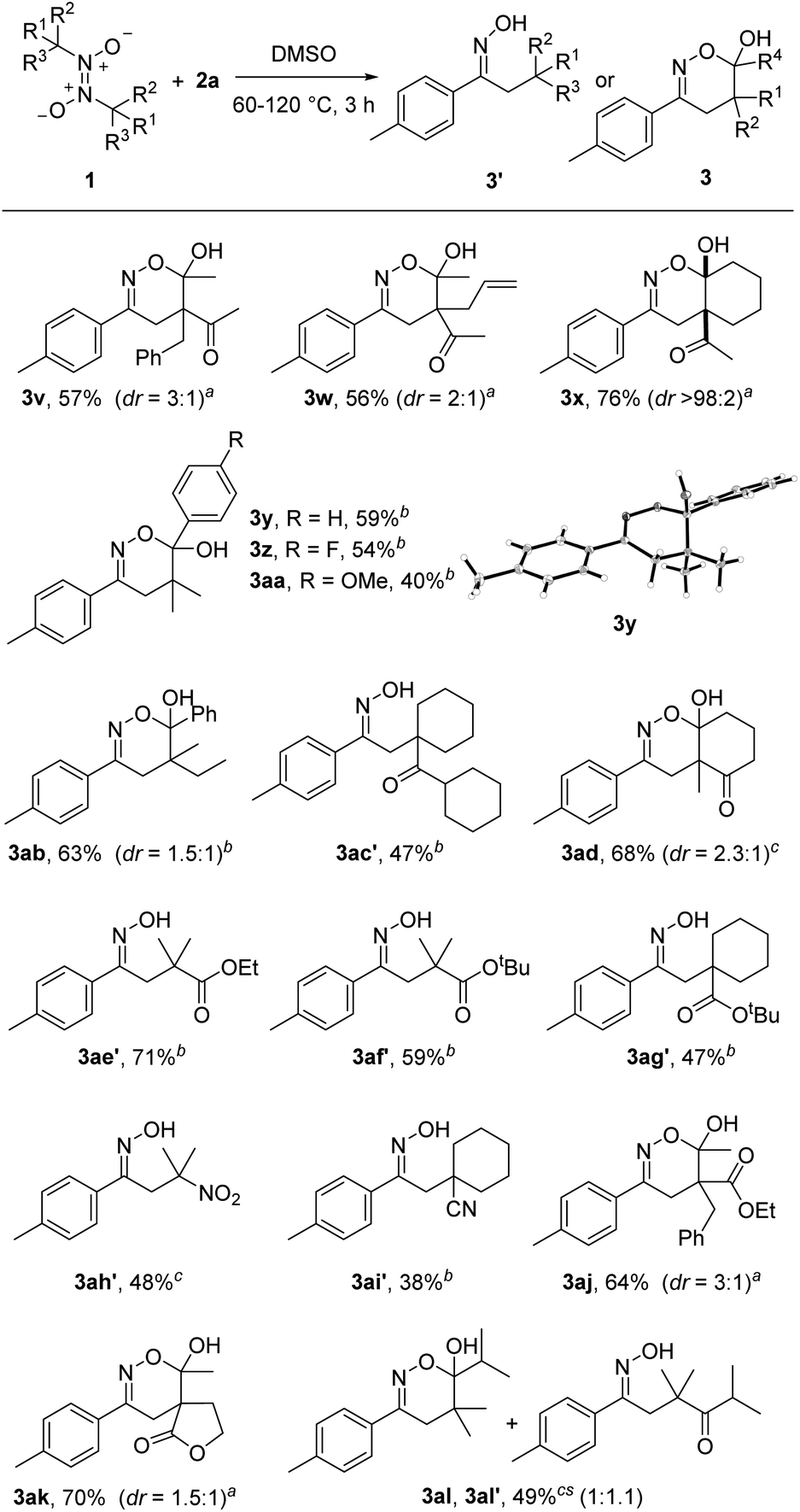 | ||
| Scheme 3 Variation of the azodioxy compound. aThe reaction was conducted at 60 °C. bThe reaction was conducted at 120 °C. cThe reaction was conducted at 80 °C. | ||
Pleasingly, along with the α-keto C-radicals, other electrophilic radicals could be generated using this novel strategy. Thus, α-ester radicals are accessible and the reaction with 2a provided the N-hydroximinoesters 3ae′ (71%), 3af′ (59%) and 3ag′ (47%). For the esters, cyclization did not occur and oximes were isolated as products. The scope could be further expanded by the successful implementation of α-nitroalkyl and α-cyanoalkyl radicals. As examples, β-nitrooxime 3ah′ (48%) and nitrileoxime 3ai′ (38%) were prepared, albeit in slightly lower yields. Moreover, β-ketoester α-C-radicals (3aj, 64%) and β-ketolactone radicals (3ak, 70%) could be thermally generated from the corresponding azodioxy compounds, further broaden the versatility of the herein presented method.
To testify the practicality of the method, the dihydro-1,2-oxazine 3c and the oxime 3ae′ were prepared on a 7 mmol scale without compromising the yield (Scheme 4a). We also investigated the follow-up chemistry using 3c and 3ae′ as model substrates to document the synthetic value of our products (Scheme 4b). The oxime functionality of 3ae′ could be reduced to a primary amine with zinc in acetic acid. Subsequent N-acetylation gave the N-protected γ-amino acid ester 4 in 58% overall yield. Since N-heterocycles are important structures for pharmaceuticals,20 the 5,6-dihydro-1,2-oxazine 3c was reduced with NaCNBH3 to obtain the N-hydroxy pyrroline 5 in 49% yield as a mixture of diastereoisomers (see ESI†). This reaction proceeds via oxime reduction to the corresponding hydroxylamine, that cyclizes to give the intermediate nitrone which eventually gets reduced to the hydroxyl amine. The remaining keto functionality gets also reduced. Hydrolysis of the oxime functionality in 3c afforded the triketone 6 in 68% yield.
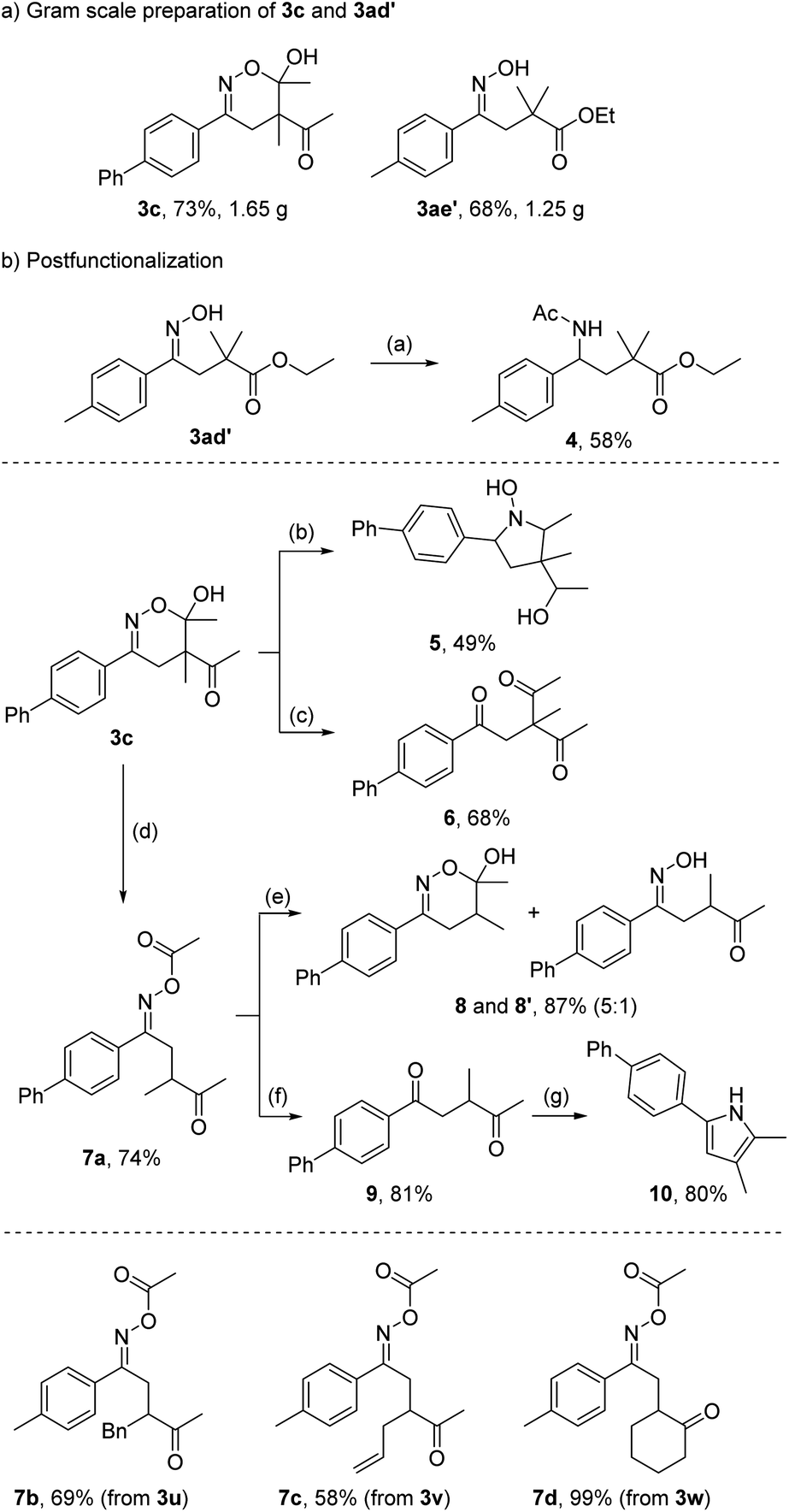 | ||
| Scheme 4 Gram scale preparation of 3c and 3ad′ and follow-up chemistry. Reaction conditions: (a) Zn (5.0 equiv.), AcOH (10 equiv.), Ac2O, 68 °C, 18 h; (b) NaCNBH3 (3.2 equiv.), AcOH, 18 h; (c) CH2O, HCl (3 mol L−1), EtOH, reflux, 3 h; (d) DMF, 150 °C, 1.5 h; (e) Co2(CO)8 (1.0 equiv.), NEt3 (1.0 equiv.), Et2O, 15 min; (f) Co2(CO)8 (1.0 equiv.), NEt3 (1.0 equiv.), Et2O, 15 min, then CH2O, HCl (3 mol L−1), EtOH, reflux, 3 h; (g) NH4OAc (7.6 equiv.), EtOH, reflux, 4 h. | ||
We observed a clean migration of one acetyl group to the oxime group upon heating of the hemiacetal 3c in DMF to 150 °C for 90 min. The O-acetyl oxime 7a was isolated in 74% yield. In analogy, oxime esters 7b–7d were obtained in good to excellent yields from the corresponding dihydro-1,2-oxazines. Importantly, the acetyl migration leading to compounds 7a–7d represents a valuable transformation since it formally allows to add secondary C-radicals onto radical acceptors via the C-nitroso approach. The direct implementation of secondary alkyl-nitroso compounds is impossible, because immediate tautomerization to the corresponding oximes would take place and the oximes do not show any activity as C-radical precursors. The equilibrium lies entirely – or at least almost exclusively – on the side of the oxime.18b,21 Accordingly, the installation of the acetyl group not only activates the nitroso compound but also prevents as a temporal protecting group its unwanted tautomerization.
Cleavage of the acetyl group in 7a led to a mixture of 5,6-dihydro-1,2-oxazine 8 and the corresponding uncyclized oxime 8′ in high combined yield (87%). Acetyl removal and subsequent hydrolysis of the oxime functionality afforded the 1,4-diketone 9 (81%) which could be further converted in a Paal–Knorr reaction with ammonium acetate to pyrrole 10 (80%).
To show that azodioxy compounds of type 1 serve as thermal C-radical precursors, a probe experiment was conducted. Reaction of 1a with vinyl cyclopropane 2v provided the ring-opened β,γ-unsaturated oxime 11 in 49% yield, supporting the presence of radical intermediates (Scheme 5). We also tested whether the azodioxy 1a can be photochemically activated. However, irradiation of 1a in the presence of 2a at room temperature with white LEDs did not lead to any 1,2-difunctionalization product 3a. Moreover, at 60 °C white LEDs did not show a measurable effect on the rate and yield of the transformation.
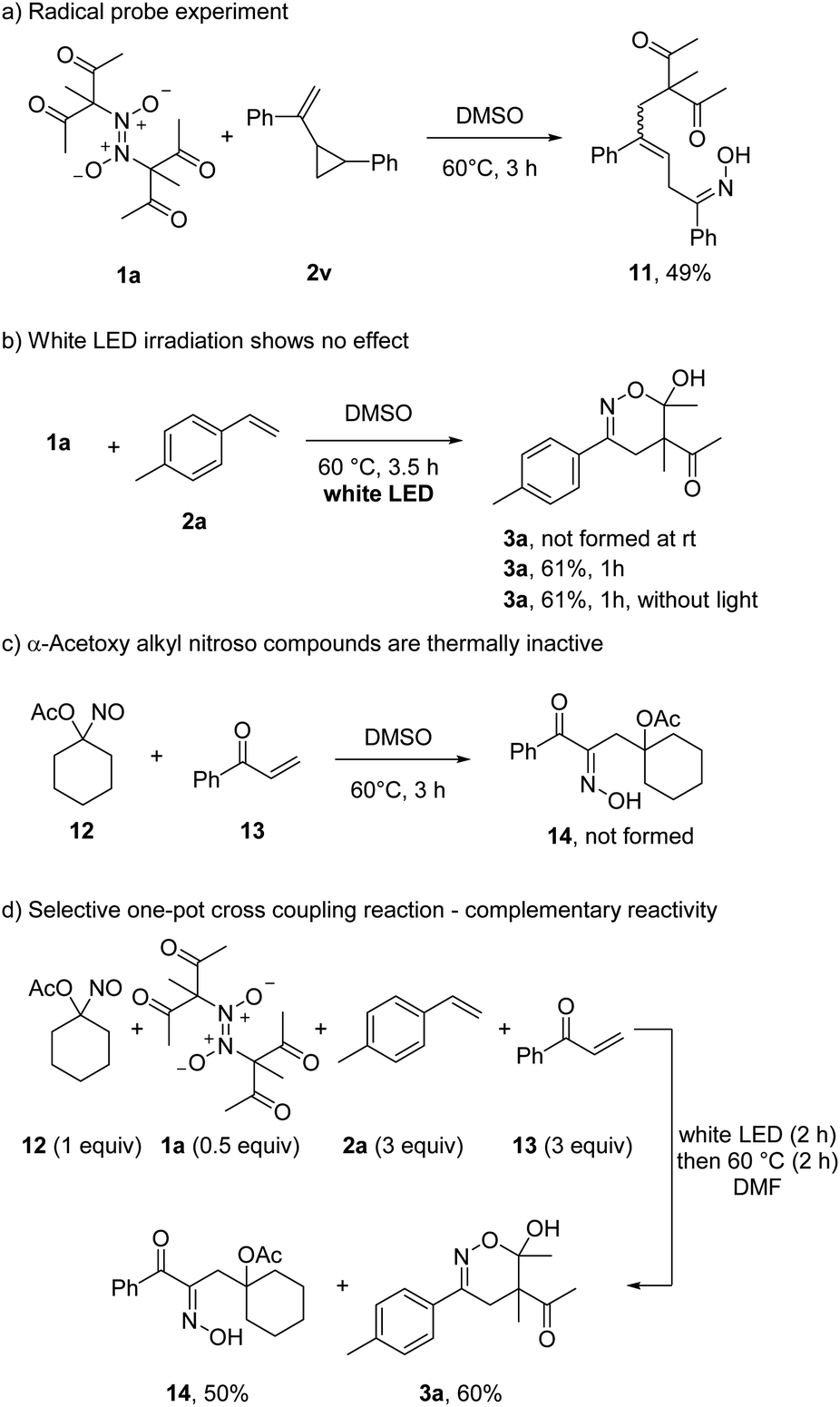 | ||
| Scheme 5 Mechanistic experiments and complementary reactivity. | ||
Comparing the herein presented reaction with the visible light induced radical coupling of α-acyloxy alkyl nitroso compounds,13 the homolytic C–NO cleavage of electron-deficient C-nitroso compounds occurs thermally. Surprisingly, the acetoxy C-nitroso compound 12 did not react thermally with phenyl vinyl ketone (13). Even at 120 °C, the targeted oxime 14 was not formed (not shown). Thus, while electrophilic C-radicals are accessible upon thermal treatment of in situ generated C-nitroso compounds, the α-acetoxy alkyl nitroso congeners (stable in the monomeric form) leading to nucleophilic C-radicals do not engage in thermal C–NO bond homolysis. This surprising observation allowed us to run a self-sorting process where 4 different reaction components are mixed and pairwise selectively addressed depending on the reaction conditions chosen. Hence, a mixture of 12, 1a, 2a and 13 in DMF at room temperature was first irradiated for 2 h with white LEDs. Then, the mixture was heated to 60 °C without irradiation for additional 2 h resulting in the selective formation of 3a (60%) and 14 (50%). The other potential coupling products (1a with 13 and 12 with 2a) were not identified.
DFT calculations show that dimer 1a is formed exothermically from 1,1-diacetyl-nitrosomethane (ΔG = −7.8 kcal mol−1) while the formation of the dimer of 12 is slightly endothermic (ΔG = +0.2 kcal mol−1). The C-nitroso bond dissociation energy of 12 (40.1 kcal mol−1) is significantly higher than the bond energy of the nitroso monomer of 1a (22.2 kcal mol−1), proving the higher thermal stability of the former. Two model nitroso compounds with 1-acetoxy and 1-methoxycarbonyl substituents show the same trends (see ESI for details†).
Conclusions
In summary, we have shown that azodioxy compounds are general precursors for electrophilic C-radicals that engage in thermal styrene 1,2-difunctionalization reactions. The method shows a broad functional group tolerance and the reactions leading to interesting products are experimentally easy to conduct (just mixing and heating). Different tertiary C-radicals can be thermally generated via this strategy. We have noted that C-nitroso compounds leading to electrophilic C-radicals exist at room temperature mainly in the dimeric azodioxy form, whereas the previously studied α-acetoxy alkyl nitroso compounds mainly exist in their monomeric form, as supported by DFT calculations. Further, the monomeric α-acetoxy alkyl nitroso systems are photoactive and white LED irradiation leads to clean C-radical generation. However, these monomeric nitroso compounds are thermally not active. The complementary photo/thermal reactivity of α-acetoxy alkyl nitroso compounds with respect to the azodioxy systems allows for highly chemoselective cross coupling chemistry.Data availability
The data that support the findings of this study are available in the ESI.†Author contributions
S. P. conducted all experiments and characterized the novel compounds. C. M.-L. conducted the DFT calculations. C. G. D. conducted the single-crystal X-ray diffraction analysis. S. P. and A. S. designed the experiments and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG) and the Fonds der Chemischen Industrie for supporting this work.Notes and references
- For selected reviews: (a) S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790 CrossRef CAS PubMed; (b) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef CAS PubMed; (c) C. Chatgililaloglu and A. Studer, Encyclopedia of Radicals in Chemistry, Biology and Materials, Wiley, Chichester, 2012 CrossRef; (d) P. Renaud and M. P. Sibi, Radicals in Organic Synthesis, Wiley-VCH, Weinheim, 2001 CrossRef.
- (a) T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087 RSC; (b) M.-Y. Cao, X. Ren and Z. Lu, Tetrahedron Lett., 2015, 56, 3732 CrossRef CAS; (c) X.-W. Lan, N.-X. Wang and Y. Xing, Eur. J. Chem., 2017, 5821 CrossRef CAS; (d) G. Sauer and S. Lin, ACS Catal., 2018, 8, 5175 CrossRef CAS; (e) H. Yao, W. Hu and W. Zhang, Molecules, 2021, 26, 105 CrossRef CAS PubMed.
- (a) K. Cheng, L. Huang and Y. Zhang, Org. Lett., 2009, 11, 2908 CrossRef CAS PubMed; (b) H. Sun, Y. Zhang, F. Guo, Z. Zha and Z. Wang, J. Org. Chem., 2012, 77, 3563 CrossRef CAS PubMed.
- X.-H. Yang, W.-T. Wei, H.-B. Li, R.-J. Song and J.-H. Li, Chem. Commun., 2014, 50, 12867 RSC.
- (a) F. Zhang, P. Du, J. Chen, H. Wang, Q. Luo and X. Wan, Org. Lett., 2014, 16, 1932 CrossRef CAS PubMed; (b) X.-H. Ouyang, R.-J. Song, M. Hu, Y. Yang and J.-H. Li, Angew. Chem., Int. Ed., 2016, 55, 3187 CrossRef CAS PubMed.
- (a) J. Zhang, J. Jiang, D. Xu, Q. Luo, H. Wang, J. Chen, H. Li, Y. Wang and X. Wan, Angew. Chem., Int. Ed., 2015, 54, 1231 CrossRef CAS PubMed; (b) J. Jiang, J. Liu, L. Yang, Y. Shao, J. Cheng, X. Bao and X. Wan, Chem. Commun., 2015, 51, 14728 RSC.
- C. Wetter and A. Studer, Chem. Commun., 2004, 174 RSC.
- (a) B. Schweitzer-Chaput, J. Demaerel, H. Engler and M. Klussmann, Angew. Chem., Int. Ed., 2014, 53, 8737 CrossRef CAS PubMed; (b) X.-W. Lan, N.-X. Wang, W. Zhang, J.-L. Wen, C.-B. Bai, Y. Xing and Y.-H. Li, Org. Lett., 2015, 17, 4460 CrossRef CAS PubMed.
- J.-K. Cheng and T.-P. Loh, J. Am. Chem. Soc., 2015, 137, 42 CrossRef CAS PubMed.
- H.-M. Huang, P. Bellotti, J. Ma, T. Dalton and F. Glorius, Nat. Rev. Chem., 2021, 5, 301 CrossRef CAS.
- (a) T. Pintauer, Eur. J. Inorg. Chem., 2010, 2449 CrossRef CAS; (b) K. Weidner, A. Giroult, P. Panchaud and P. Renaud, J. Am. Chem. Soc., 2010, 132, 17511 CrossRef CAS PubMed; (c) J. M. Muñoz-Molina, T. R. Belderrain and P. J. Pérez, Eur. J. Inorg. Chem., 2011, 3155 CrossRef; (d) J. D. Nguyen, J. W. Tucker, M. D. Konieczynaska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160 CrossRef CAS PubMed; (e) T. Pintauer and K. Matyjaszewski in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgrilialoglu and A. Studer, Wiley, Chichester, 2012, p. 1851 Search PubMed; (f) Y. Li, Y. Han, H. Xiong, N. Zhu, B. Qian, C. Ye, E. A. B. Kantchev and H. Bao, Org. Lett., 2016, 18, 392 CrossRef CAS PubMed; (g) L.-L. Liao, G.-M. Cao, Y.-X. Jiang, X.-H. Jin, X.-L. Hu, J. J. Chruma, G.-Q. Sun, Y.-Y. Gui and D.-G. Yu, J. Am. Chem. Soc., 2021, 143, 2812 CrossRef CAS PubMed.
- (a) S. Fujiwara, Y. Shimizu, T. Shin-ike and N. Kambe, Org. Lett., 2001, 3, 2085 CrossRef CAS PubMed; (b) I. Fernández, F. M. Bickelhaupt and F. P. Cossío, Chem.–Eur. J., 2009, 15, 13022 CrossRef PubMed; (c) B. Dinda, Essentials of Pericyclic and Photochemical Reactions, Springer, Cham, 2017, pp. 161–177 CrossRef; (d) D.-L. Wang, N.-Q. Jiang, Z.-J. Cai and S.-J. Ji, Chem.–Eur. J., 2021, 27, 17765 CrossRef CAS PubMed.
- D. Zheng, S. Plöger, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 8547 CrossRef PubMed.
- (a) D. H. R. Barton, J. M. Beaton, L. E. Geller and M. M. Pechet, J. Am. Chem. Soc., 1960, 82, 2640 CrossRef CAS; (b) D. H. R. Barton and J. M. Beaton, J. Am. Chem. Soc., 1960, 82, 2641 CrossRef CAS; (c) D. H. R. Barton, J. M. Beaton, L. E. Geller and M. M. Pechet, J. Am. Chem. Soc., 1961, 83, 4076 CrossRef CAS; for a review, see: (d) G. Majetich and K. Wheless, Tetrahedron, 1995, 51, 7095 CrossRef CAS.
- (a) H. Fischer, Chem. Rev., 2001, 101, 3581 CrossRef CAS PubMed; (b) A. Studer, Chem.–Eur. J., 2001, 7, 1159 CrossRef CAS PubMed; (c) D. Leifert and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 74 CrossRef CAS PubMed.
- (a) P. Zuman and B. Shah, Chem. Rev., 1994, 94, 1621 CrossRef CAS; (b) D. M. Gooden, H. Chakrapani and E. J. Toone, Curr. Top. Med. Chem., 2005, 5, 687 CrossRef CAS PubMed; (c) H. Yamamoto and N. Momiyama, Chem. Commun., 2005, 3514 RSC; (d) H. Yamamoto and M. Kawasaki, Bull. Chem. Soc. Jpn., 2007, 80, 595 CrossRef CAS; (e) H. Vančik, Aromatic C-Nitroso Compound, Springer, New York, 2013 CrossRef.
- Y. Gao, S. Yang, W. Xiao, J. Nie and X.-Q. Hu, Chem. Commun., 2020, 56, 13719 RSC.
- (a) R. Hoffmann, R. Gleiter and F. B. Mallory, J. Am. Chem. Soc., 1970, 92, 1460 CrossRef CAS; (b) B. G. Gowenock and G. B. Richter-Addo, Chem. Rev., 2004, 104, 3315 CrossRef PubMed; (c) D. Beaudoin and J. D. Wuest, Chem. Rev., 2016, 116, 258 CrossRef CAS PubMed.
- Deposition numbers 2178219 (for 3y) contain the ESI† crystallographic data for this paper.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- J. Long, N. Harris and K. Lammertsma, J. Org. Chem., 2001, 66, 6762 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2178219. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03860a |
| This journal is © The Royal Society of Chemistry 2022 |