
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigations into mechanism and origin of regioselectivity in the metallaphotoredox-catalyzed α-arylation of N-alkylbenzamides†
Alexander W.
Rand
 *,
Mo
Chen
and
John
Montgomery
*
*,
Mo
Chen
and
John
Montgomery
*
Department of Chemistry, University of Michigan, 930 N. University Ave., Ann Arbor, MI 48109-1055, USA. E-mail: jmontg@umich.edu
First published on 19th August 2022
Abstract
A mechanistic study on the α-arylation of N-alkylbenzamides catalyzed by a dual nickel/photoredox system using aryl bromides is reported herein. This study elucidates the origins of site-selectivity of the transformation, which is controlled by the generation of a hydrogen atom transfer (HAT) agent by a photocatalyst and bromide ions in solution. Tetrabutylammonium bromide was identified as a crucial additive and source of a potent HAT agent, which led to increases in yields and a lowering of the stoichiometries of the aryl bromide coupling partner. NMR titration experiments and Stern–Volmer quenching studies provide evidence for complexation to and oxidation of bromide by the photocatalyst, while elementary steps involving deprotonation of the N-alkylbenzamide or 1,5-HAT were ruled out through mechanistic probes and kinetic isotope effect analysis. This study serves as a valuable tool to better understand the α-arylation of N-alkylbenzamides, and has broader implications in halide-mediated C–H functionalization reactions.
Introduction
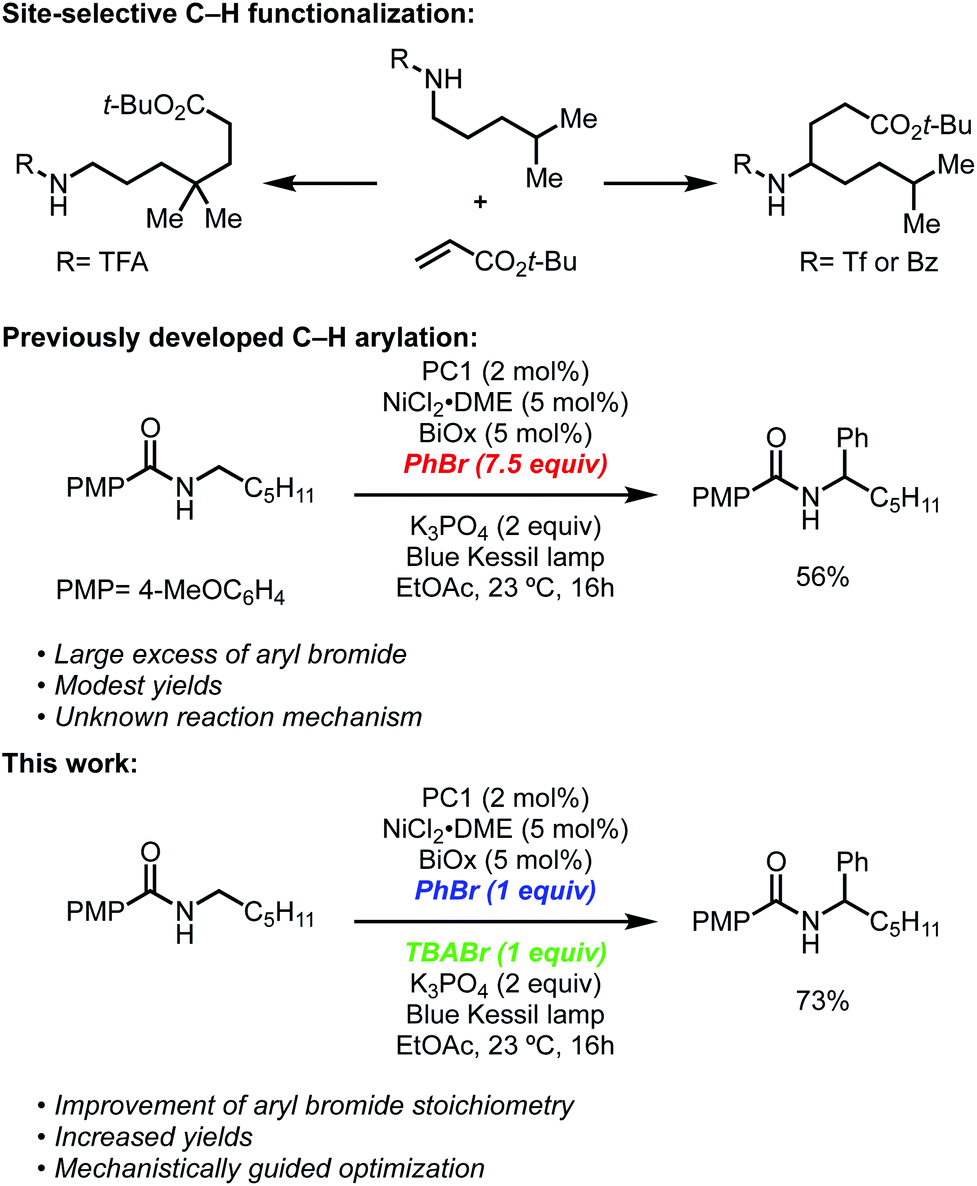
Site-selective C–H functionalization reactions allow streamlined access to valuable products in an efficient manner from commonly available starting materials.1 The ability to differentiate C–H bonds that are in similar steric and electronic environments is a challenge that requires both the development of new reactions and a better understanding of existing mechanistic underpinnings.2 The exploration of strategies in selective functionalization targeted towards specific C–H bonds, however, often rely on directing groups,3 which can sometimes be tedious to install and remove in cases where the directing functionality is not desired in the final structure.Complementary to transition metal-catalyzed approaches, processes involving proton-coupled electron transfer (PCET) and hydrogen atom transfer (HAT) initiated by a photocatalyst have allowed for the selective functionalization of sp3 C–H bonds.4 Notably, independent studies by Rovis5 and Knowles6 showed that PCET could be used to alkylate distal C–H bonds using Michael acceptors by leveraging an amidyl radical followed by a 1,5-HAT. Several other groups, including Rovis,7 Alexanian,8 Tambar,9 Martin,10 Nagib,11 and Roizen12 showed that similar strategies could generate either distal or proximal alkylation/allylation under a variety of conditions. Despite these important developments in the functionalization of unactivated C–H bonds, the origins of site-selectivity have often been elusive.
Recently, our group and the Martin lab collaboratively reported a metallaphotoredox-catalyzed α-arylation and alkylation of N-alkylbenzamides using aryl or alkyl bromides (Scheme 1).13 This methodology was found to preferentially activate aryl bromides in the presence of aryl or alkyl chlorides and could tolerate a number of sensitive functional groups including boronic esters, alkyl amides, and challenging heterocyclic units including pyridines and thiophenes. Furthermore, the unique site-selectivity was highlighted through the orthogonal α- and δ-functionalization of a common starting material using either our method or conditions developed by Knowles.6 Lastly, this method was rendered asymmetric through the use of a chiral bioxazoline (BiOx) ligand, which allowed for high enantioinduction when conducted at low temperatures.
 | ||
| Scheme 1 Site-selective C–H functionalization of protected amines. | ||
Despite the broad scope for both the N-alkylbenzamide and bromide coupling partner, these reactions, in some cases, suffered from low conversions and often required a large excess of either component to attain synthetically useful yields. During the optimization, it was noted that the reactions exhibited unique profiles that led us to believe a different mechanism than that reported by Rovis7a and Tambar9 was responsible for the observed regio- and chemoselectivity. Among these observations, nickel(II) salts vastly outperformed nickel(0) sources such as Ni(COD)2, strong inorganic and amine bases completely inhibited these reactions, and the insolubility of the base (K3PO4) did not have a negative effect on the reaction. Furthermore, the use of solvents that contained weak C–H bonds, such as DMF and THF, consumed the aryl bromide through deleterious solvent functionalization, which led us to use more robust solvents such as EtOAc.
In recent years, there has been a renewed interest in understanding the underpinnings of metallaphotoredox reactions.14 Among these studies, small deviations in procedures have resulted in substantial changes in regio- and chemoselectivity, as well as new modes of reactivity. In our pursuit to improve upon this reaction through a fundamental understanding of its mechanism, we describe a reaction pathway that better explains the exquisite site-selectivity, the requirements for specific nickel pre-catalyst, and the requirement for relatively large excesses of one of the coupling partners. Based on investigations into the reaction mechanism, we describe here that tetrabutylammonium bromide (TBABr) serves as an integral additive, leading to yield improvements while simultaneously allowing each coupling partner to be used in near-stoichiometric ratios. A series of mechanistic experiments, quenching, and titration studies described herein provide a greater understanding of the role of key additives, the nature of the C–H abstraction agent, and the origin of site-selectivity. We anticipate these insights will provide a foundation for other metallaphotoredox systems and will enable the reactivity trends to be applied in new classes of reactions.
Results and discussion
PCET/1,5-HAT
Considering our initial results and evidence in the literature on amide C–H bond functionalization using photoredox catalysis, we set out to develop a working hypothesis to explain the observed regioselectivity for our metallaphotoredox-catalyzed α-arylation of N-alkylbenzamides using aryl bromides. We initially considered the possibility that the reaction was initiated by a PCET between the photocatalyst, N-alkylbenzamides, and K3PO4 to afford an N-centered radical, followed by 1,5-HAT to yield a distal carbon-centered radical (Scheme 2). From this distal radical, capture by a nickel(II)ArBr species, followed by a series of β-hydride eliminations and reinsertions to chain-walk to the proximal α-position, could allow for reductive elimination to afford the observed products. The catalytic cycle could then be closed through electron transfer between the nickel and iridium photocatalyst. While exploring the substrate scope of this reaction, several inconsistencies were noticed. Namely, N-alkylbenzamides that lack distal C–H bond were suitable substrates in this reaction (Scheme 3A). Furthermore, tertiary amides did not provide appreciable yields of the desired product, though when used in solvent quantities, α-arylation could be observed, suggesting that a free N–H was not necessary for reactivity (Scheme 3B). To probe for a PCET/1,5-HAT as observed by Rovis and Knowles, a substrate containing deuterium at the α-position was subjected to the reaction conditions (Scheme 3C). In the initial report, using α,α-dideutero and α,α-diproteo N-alkylbenzamides, a primary kinetic isotope effect (KIE) was observed suggesting that C–H bond cleavage is involved at or before the turnover-limiting step of the catalytic cycle.13 Furthermore, deuterium scrambling along the alkyl chain of this substrate was not observed. If the reaction proceeded through the formation of a distal radical followed by a chain-walking sequence to arrive at an α-nickel species, a KIE would not be expected, and complete retention of deuterium at the α-position would not be observed. Taken together, these results suggest that PCET followed by 1,5-HAT is not operative under these reaction conditions.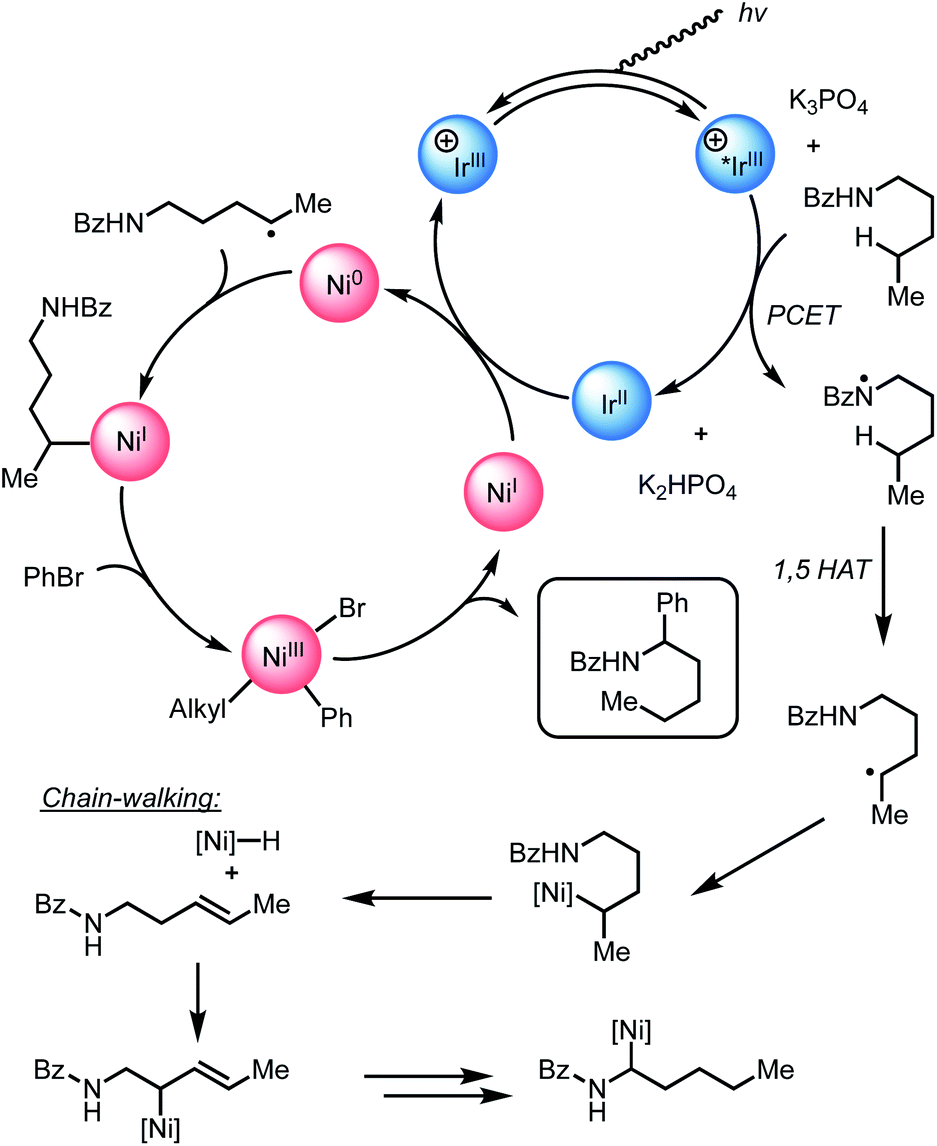 | ||
| Scheme 2 Initial mechanistic hypothesis. | ||
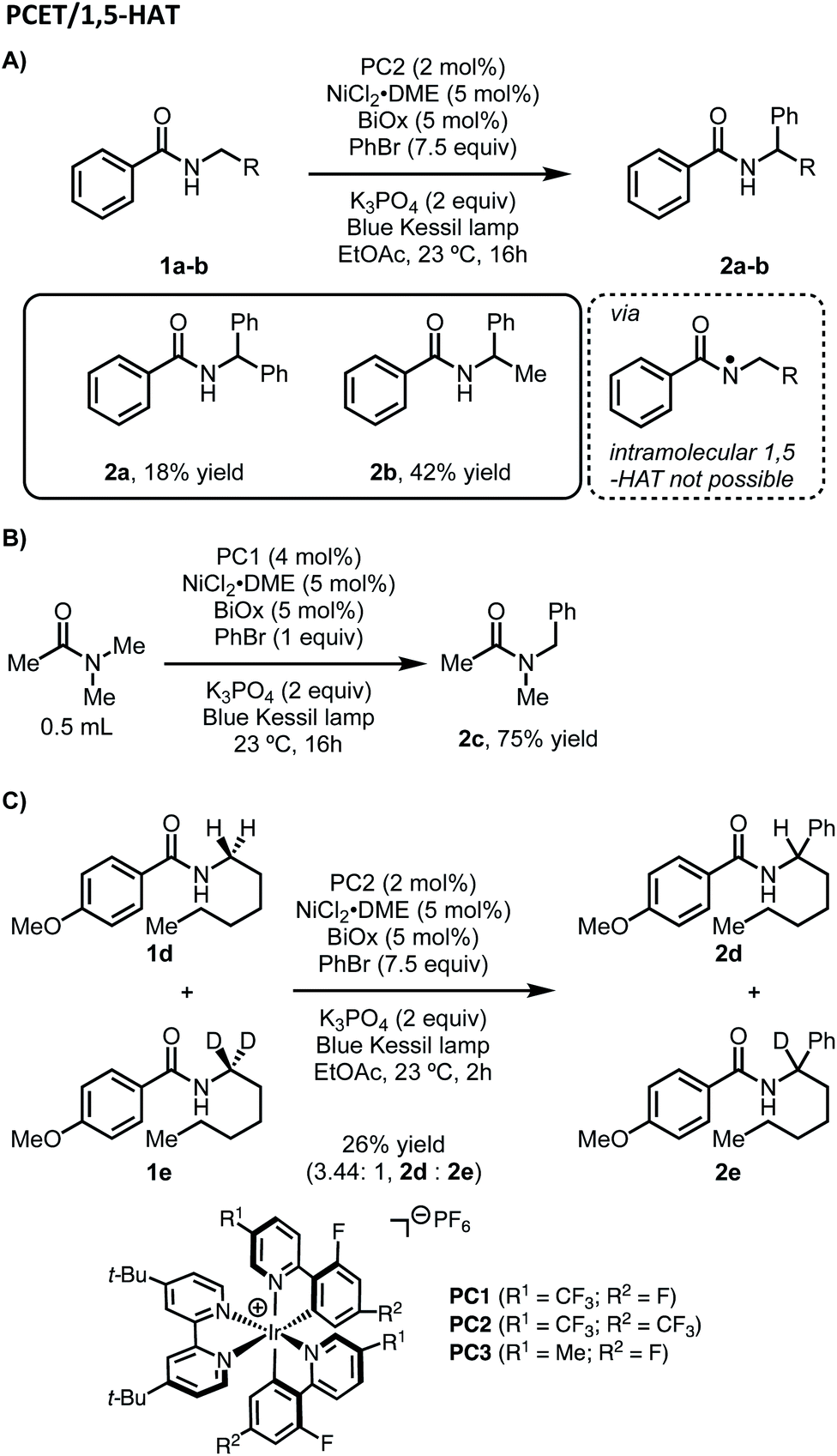 | ||
| Scheme 3 Observed regioselectivity and kinetic isotope studies for the α-arylation of N-alkylbenzamides. | ||
Deprotonation
In addition to their original report of δ-alkylation of trifluoroacetamides with electron deficient olefins,5 the Rovis group, among others, have developed several protocols for the site-selective functionalization of amide derivatives.7c Specifically, by switching from a trifluoroacetamide to a triflamide, α-alkylation became possible under otherwise identical conditions (Scheme 4).7b This change in regioselectivity was attributed to the lower pKa of the triflamide, which allows for deprotonation by K3PO4 followed by oxidation by the photocatalyst to form a triflamidyl radical. The change in site-selectivity was attributed to the stability of the triflamidyl radical, which is persistent enough to undergo intermolecular HAT with a second equivalent of triflamide.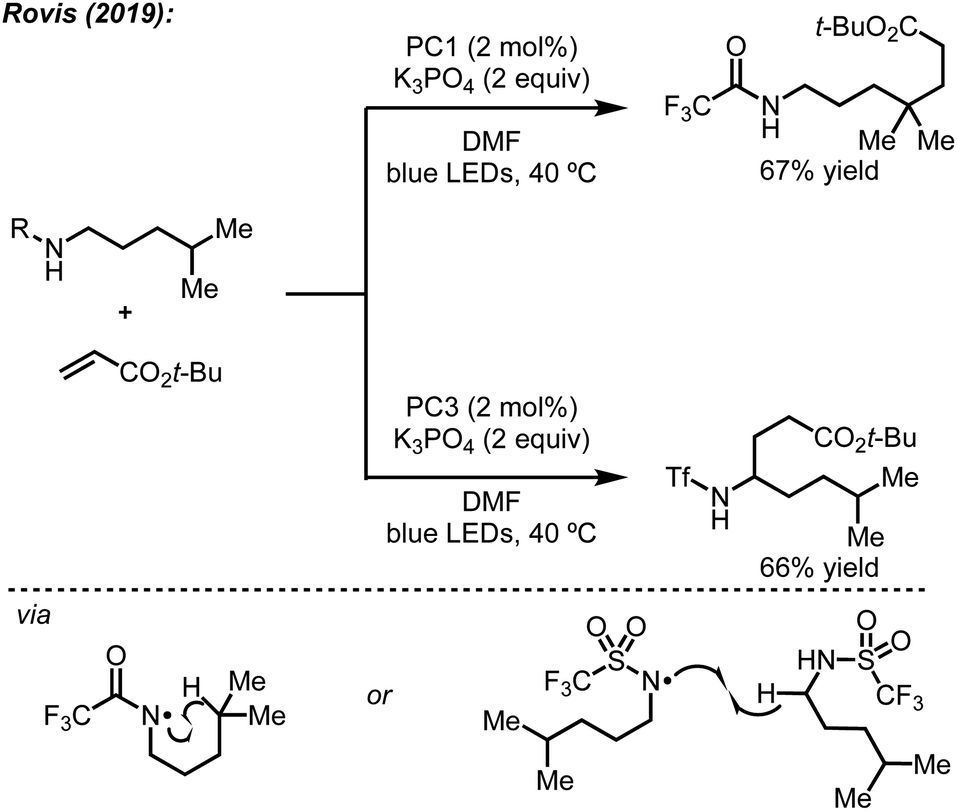 | ||
| Scheme 4 Effects on regioselectivity using different nitrogen protecting groups observed by Rovis. | ||
Based on these past findings, we considered that deprotonation of an N-alkylbenzamide under the reaction conditions might be responsible for the observed regioselectivity. To evaluate this pathway, the potassium salt of N-hexyl-4-methoxybenzamide was synthesized and tested under our reaction conditions for α-arylation. Surprisingly, this substrate did not provide the desired product, and the unreacted N-alkylbenzamide was recovered quantitatively (Scheme 5). This suggests that deprotonation of the N-alkylbenzamides in this reaction is not a productive pathway on the catalytic cycle, and in fact inhibits the reaction. These results could also explain why using K3PO4, which is sparingly soluble in the reaction mixture, still provides the desired product. Having established that the observed regioselectivity is not controlled through deprotonation of the N-alkylbenzamide, we next turned to Stern–Volmer quenching studies to elucidate the origins of radical formation and reaction initiation.
 | ||
| Scheme 5 Unsuccessful α-arylation with deprotonated benzamides. | ||
Stern–Volmer studies
To gain insights into which reaction components were interacting with the photocatalyst for the α-arylation of N-alkylbenzamides, Stern–Volmer quenching studies were next conducted (Scheme 6). Beginning with N-hexyl-4-methoxybenzamide (Ep/2 = +1.78 V vs. SCE), no quenching event was observed, which was expected since its redox potential lies outside the range of PC1 (E1/2red Ir(III*/II) = +1.21 V vs. SCE). Similar results were observed when using PhBr (Ep/2 = −1.76 V vs. SCE) with which reduction is also endergonic using PC1 (E1/2ox Ir(III*/IV) = −0.89 V vs. SCE). While K3PO4 could not be tested in these experiments due to its insolubility in EtOAc and DMF, it was found that i-PrBiOxNiCl2 strongly quenched the photocatalyst. Analysis of the CV of i-PrBiOxNiCl2 revealed a reduction event, Ep/2 Ni(II/I) = −0.83 V vs. SCE, which is thermodynamically feasible with PC1 and also with the reduced form of PC1 (E1/2ox Ir(III/II) = −1.37 V vs. SCE).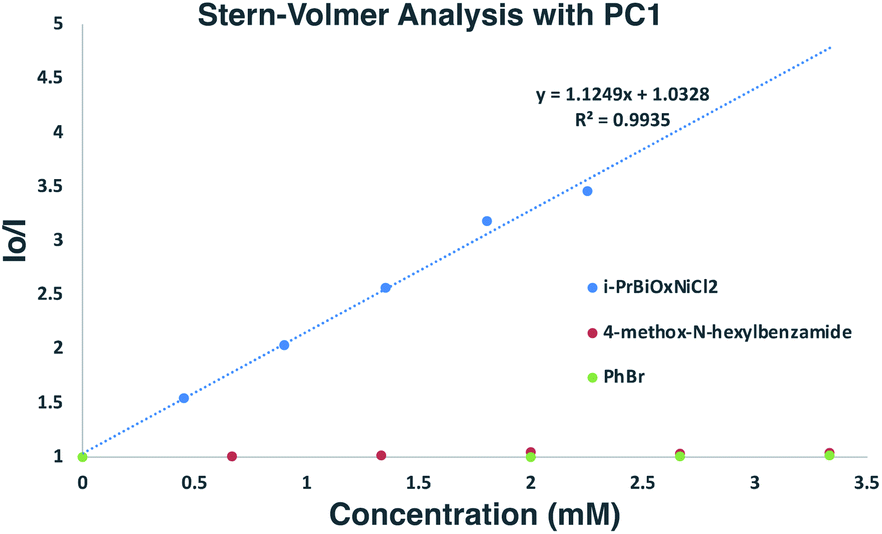 | ||
| Scheme 6 Stern–Volmer analysis of α-arylation reaction components. Linear quenching of PC1 observed with i-PrBiOxNi(II)Cl2. | ||
Given the strong interaction between i-PrBiOxNiCl2 and PC1, we posit that electron transfer from Ir(III*) to Ni(II) would generate Ir(IV), Ni(I), and a halide anion. As halide anions have been shown to undergo oxidation by iridium polypyridyl photocatalysts,15 we envisioned that this newly generated halide anion could be oxidized by Ir(IV) to form a halide radical capable of abstracting weak C–H bonds (TBACl: Ep/2ox = +1.01 vs. SCE in CH3CN; E1/2red Ir(IV/III) = +1.69 V vs. SCE), such as those found in N-hexyl-4-methoxybenzamide.16 We suspected that the lower concentration of halide HAT agents at the beginning of the reaction might lead to sluggish and deleterious off-cycle reactions that necessitate the use of superstoichiometric coupling partners. Since only aryl bromides provided appreciable amounts of product, we envisioned that after initiation of the reaction, Br− derived from PhBr was a likely HAT agent. Further supporting this hypothesis, the Doyle group demonstrated the effectiveness of TBABr as an HAT agent in a nickel/photoredox-catalyzed coupling through C–H abstraction of an acetal.17
Based on our Stern–Volmer quenching studies, we believe that nickel serves to not only activate the aryl bromide for coupling, but also functions as a source of HAT agent for C–H functionalization through the generations of halide anions.15a,16a,b,d,18,19,20,21 Under this premise, we began exploring the effect of adding exogenous halide salts to our model reaction in order to more efficiently generate reactive HAT agents (Table 1). As shown previously, aryl bromides were the only aryl substrates that provided the desired product (Table 1, entries 1–3). However, when adding 1 equivalent of TBABr to a reaction containing PhCl, we were delighted to see 30% yield of the desired product (entry 4).19a,22 While nickel has been shown to activate PhCl, PhBr, and PhI at room temperature, we believe that the addition of TBABr facilitates more facile access to halide radicals through SET with PC1 (TBABr Ep/2ox = +0.71 vs. SCE in CH3CN and TBACl Ep/2ox = +1.01 vs. SCE in CH3CN).23 The addition of TBACl did not have a large effect on the reaction when using PhBr (entry 5). TBAI can also be oxidized by PC1 (TBAI Ep/2ox = +0.26 vs. SCE in CH3CN), but the lower BDE of H–I compared to H–Br (71 kcal mol−1vs. 87 kcal mol−1) might explain why iodide is not an effective HAT agent in this reaction (entry 6). Similarly, examining the thermodynamics for HAT from Br˙ (BDE H–Br = 87 kcal mol−1) could explain the why only α-arylation is observed (H3C(O)NHC–H(CH3)2 BDE = 92 kcal mol−1vs. (H3C)2CH–H BDE = 99 kcal mol−1).24
To probe the feasibility of halide radical generation through the oxidation of a halide anion by a photocatalyst, we conducted Stern–Volmer quenching studies using an exogenous halide salt (Scheme 7). When conducting Stern–Volmer quenching studies using TBABr, we observed strong, static and dynamic quenching of PC1 (Ks = 1.8 × 103 M−1 S−1 and Kd = 2.9 × 104 M−1 S−1), which is indicative of association of Br− to PC1 prior to electron transfer as well as intermolecular electron transfer from PC1 to Br−. Similar observations and magnitudes have been seen in metallaphotoredox systems that employ halide salts as HAT agents.16b Corroborating the observed dynamic quenching, it was also observed through NMR titration experiments that, similar to Knowles and Alexanian,8 a new iridium species was formed when mixing PC1 and TBABr (Keq = 6.3 × 102) (Scheme 8). We believe that counterion exchange between cationic PC1 and TBABr could form an iridium–bromide complex that would serve to bring together these two species in solution for more efficient electron transfer.18,19b,f,25 Taken together, these results support our hypothesis that Br− formed under our reaction conditions could be oxidized to Br˙ by the photocatalyst (TBABr: Ep/2ox = +0.71 vs. SCE), which in turn serves as an HAT agent for the abstraction of C–H bonds from the substrate. This could also explain why neutral photocatalysts with similar excited-state redox potentials, such as 4CzIPN (E1/2red (PC*/PC˙−) = +1.43 vs. SCE, E1/2ox (PC˙+/PC*) = −1.18 vs. SCE),26 provided only trace product in this reaction, as electron transfer would require the photocatalyst and Br− to come into proximity for electron transfer to occur.
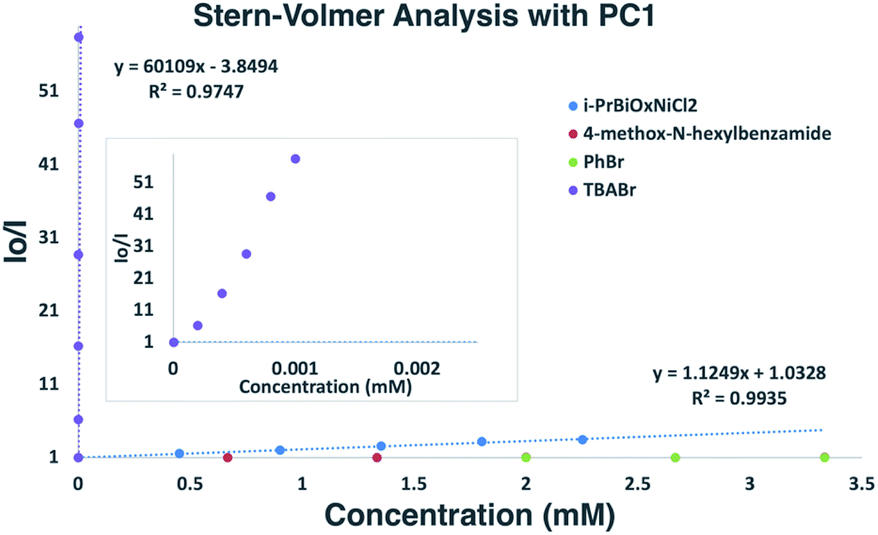 | ||
| Scheme 7 Stern–Volmer plot of reaction components with an expansion for TBABr. Strong, non-linear quenching of PC1 observed with TBABr suggesting static and dynamic quenching. Quenching was also observed with i-PrBiOxNi(II)Cl2, while PhBr and 4-methoxy-N-hexylbenzamide did not quench PC1. | ||
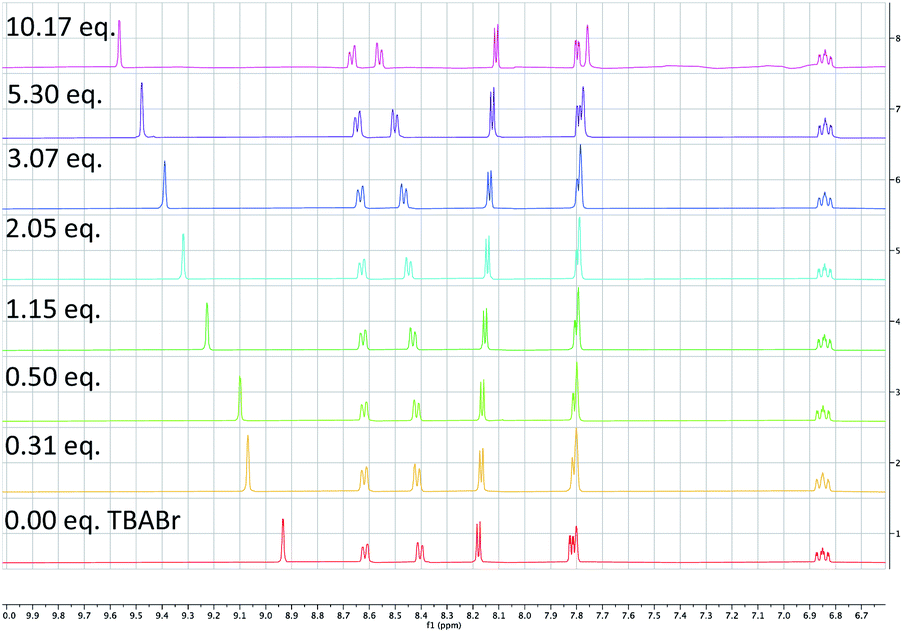 | ||
| Scheme 8 1H NMR of PC1 upon titration with TBABr. A large downfield shift of the C3–H of the bipyridyl ligand was observed at increasing concentrations of TBABr suggesting a complexation of Br− to PC1. | ||
HAT agents
Having identified TBABr as an HAT agent capable of undergoing SET with PC1 to form Br˙, we began reevaluating the parameters of the reaction (Table 2). During our initial optimization, we determined that 7.5 equivalents of PhBr were necessary to achieve high yield of the desired α-arylation product (Table 2, entry 1), and when using an equal ratio of N-alkylbenzamide to PhBr only 23% of the desired product could be attained (entry 2). Through the inclusion of 1 equivalent of TBABr, it was found that using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of N-hexyl-4-methoxybenzamide and PhBr provided 52% yield of the desired product (entry 3). Lastly, when using PhBr as the limiting reagent, 1 equivalent of TBABr significantly improved the yield of α-arylation product from 27% to 73% yield (entries 4 and 5).
:1 ratio of N-hexyl-4-methoxybenzamide and PhBr provided 52% yield of the desired product (entry 3). Lastly, when using PhBr as the limiting reagent, 1 equivalent of TBABr significantly improved the yield of α-arylation product from 27% to 73% yield (entries 4 and 5).
Having observed higher yields when including of TBABr, this method was applied to several previously low-yielding reactions. Aryl bromides with ortho substituents typically lead to lower yields, but when including TBABr, the yield was increased from 33% using 7.5 equivalents of 2-bromotoluene to 41% when using the aryl bromide as the limiting reagent (entry 1, Table 3). Similary, yields when using electron-deficient aryl bromides such as 4-bromobenzotrifluoride and methyl-4-bromobenzoate increased from 53% to 64% and 49% to 67% yield (entries 2 and 3). Lastly, benzamides that previously gave low yields benefitted from the inclusion of TBABr, which resulted in an increase from 15% to 46% yield. These results show that including a HAT agent, such as TBABr, have a beneficial effect by improving reaction efficiency, which allows higher yields of the desired product with much lower excess of any reagent. When LnNiBr2 precatalysts are used, small yield improvements are seen by the addition of TBABr, in contrast to the more substantial yield and stoichiometry improvements seen with LnNiCl2 precatalysts (see ESI†).
|
|
|||
|---|---|---|---|
| Entry | R1 | R2 | % Yielda |
| a Experiment was carried out with 0.2 mmol ArBr and 0.4 mmol benzamide. Yields were determined by 1H NMR using dibromomethane as an internal standard, yields in parentheses were isolated. b Experiment was carried out at 0.1 mmol scale. | |||
| 1 | 4-OMe | 2-Me | 54 (41) |
| 2b | 4-OMe | 4-CF3 | 71 (64) |
| 3b | 4-OMe | 4-CO2Me | 75 (67) |
| 4 | 3-CO2Me | H | 58 (46) |
Based on spectroscopic and experimental evidence, we believe the addition of exogenous Br− promotes facile C–H abstraction through the oxidation of Br− by PC1 to generate Br˙, which is capable of abstracting the α-C–H bonds of N-alkylbenzamides27 and acetals.17 When TBABr is included in this reaction, the formation of higher concentrations of an iridium–bromide complex increases the efficiency of electron transfer between PC1 and Br−, which leads to more productive catalysis despite the lower concentration of PhBr. In the case where Br− is not present at the beginning of the reaction, halide anions must be generated through reduction of i-PrBiOxNiX2 by PC1. When conducting this reaction without TBABr, only small quantities of Br− are available for oxidation by PC1, which results in less productive catalysis.
Proposed mechanism
Guided by the studies presented herein and building upon recent investigations on related systems,13,16a,b,18a,19b,21,27,28 we propose that the α-arylation of N-alkylbenzamides is initiated through the irradiation of PC1 to generate an excited-state Ir(III*) species. Reduction of BiOxNi(II)Cl2 (i-PrBiOxNi(II)Cl2Ep/2 (Ni(II/I)) = −0.83 V vs. SCE) by PC1 (E1/2ox Ir(IV/III*) = −0.89 V vs. SCE) would form Ir(IV), BiOxNi(I)Cl and Cl−. Oxidation of Cl− to Cl˙ or Br− to Br˙ (TBACl Ep/2ox = +1.01 vs. SCE and TBABr: Ep/2ox = +0.71 vs. SCE in CH3CN) by Ir(IV) (E1/2red Ir(IV/III) = +1.69 V vs. SCE) are both thermodynamically favorable, and it has previously been proposed that the binding of halides to the 3,3′-position of bipyridine ligands of the photocatalyst provides a kinetic driving force for this redox event to occur.8,18a,29 Furthermore, we posit a halide radical would then perform an HAT on N-alkylbenzamide (I) to generate radical (II) and HX, which could be sequestered by K3PO4 in the reaction.After reduction of the Ni(II) species by PC1, and HAT by a halide radical, II could then be captured by i-PrBiOxNi(I)Br to form i-PrBiOxNi(II)alkylBr (III) (Scheme 9).28 At this point, reduction of i-PrBiOxNi(II)alkylBr (III) by Ir(II) (E1/2red Ir(III/II) = −1.37 V vs. SCE) could generate a i-PrBiOxNi(I)alkyl species (IV) and Ir(III). Ir(III*) could then go on to oxidize another equivalent of Br− to generate Ir(II) and Br˙, which would then perform an HAT to continue the catalytic cycle. At this point, oxidative addition of PhBr to i-PrBiOxNi(I)alkyl (IV) would form i-PrBiOxNi(I)alkylPhBr (V), which would be poised for reductive elimination to generate i-PrBiOxNi(I)Br and the desired product.30 While the reaction is proposed to be initiated from i-PrBiOxNi(II)Cl2, we envision that i-PrBiOxNi(I)Br generated under after the first catalytic cycle would serve as a suitable catalyst for this transformation.
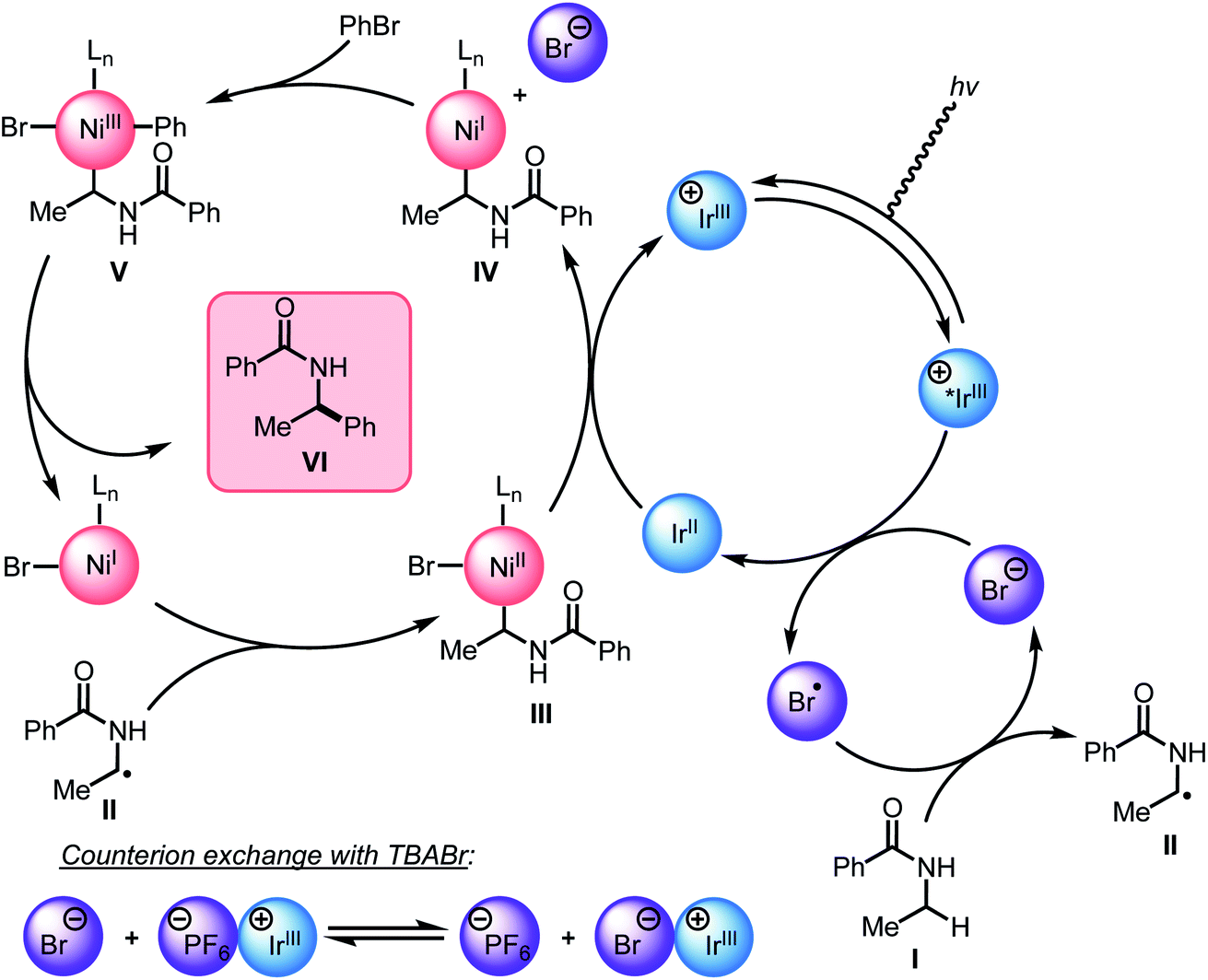 | ||
| Scheme 9 Revised mechanistic hypothesis. | ||
Through these studies, we observed that the inclusion of TBABr leads to higher yields despite using lower concentrations of PhBr. Since exogenous Br− is not necessary to produce the desired product, and the most likely HAT agent in the reaction after the initial catalytic cycle is Br˙, we believe that the primary benefit of TBABr is to more efficiently generate Br˙ through oxidation of Br− by PC1, which serves to abstract the α-C–H bonds of N-alkylbenzamides, during the initiation of the reaction when substrate-derived bromide concentration is low. As evident from NMR titrations with PC1 and TBABr, when including TBABr in this reaction, the exogenous Br− serves to form a higher concentration of Ir(III)–Br complex in solution, which more efficiently promotes oxidation of Br− to Br˙ by Ir(III*). Because the oxidation of Br− to Br˙ is more favorable than the oxidation of the Cl− to Cl˙ (TBABr: Ep/2ox = +0.71 vs. SCE and TBACl Ep/2ox = +1.01 vs. SCE in CH3CN),15a,16b,31 Br˙ is expected the be the predominate HAT agent throughout the initial and subsequent catalytic cycles when TBABr is present at the beginning of the reaction.
Conclusions
This mechanistic study provides evidence that the α-arylation of N-alkylbenzamides using aryl bromides operates through an HAT mechanism that is mediated through the direct generation of a carbon-centered radical by Br˙ and a photocatalyst. The regio- and chemoselectivity observed in the reaction is governed by the BDEs of C–H bonds in the substrate and the ability of radical formation/HAT strength of different halide radicals. Evidence was gathered that refutes a PCET/1,5-HAT/chainwalking sequence or deprotonation/substrate oxidation sequence, and supports the notion that the reaction involves C–H abstraction by a halide radical. Evidence for electron transfer between PC1 and i-PrBiOxNi(II)Cl2 and between PC1 and TBABr were supported by Stern–Volmer quenching studies. NMR titration studies highlight the formation of an iridium bromide species, which may facilitate more efficient oxidation of Br− to Br˙. Based on these studies, improvements in reaction conditions were found using bromide additives that allowed for near equimolar stoichiometries of the N-alkylbenzamides and aryl bromides as well as increases in yield of α-arylation. Overall, this study improves access to valuable α-aryl-N-alkylbenzamides and expands the understanding of the role of additives and the origin of site-selectivity in metallaphotoredox-based C–H functionalization processes.Conflicts of interest
There are no conflicts to declare.Data availability
Supporting data has been provided as ESI† associated with this article.Author contributions
AWR and JM conceived the project and conducted analysis of data. AWR conducted the mechanistic experiments and wrote the first draft of the paper. MC conducted the experimental illustrations of the TBABr additive along with the accompanying characterization. MC and JM revised the manuscript through additions and edits.Acknowledgements
We thank the National Science Foundation under the CCI Center for Selective C–H Functionalization (CHE-1700982) for funding, Cole Cruz for fruitful discussions and assistance with experimental setup, and the Ruben Martin lab for helpful discussions and collaboration during the initial report of this method.References
- B. M. Trost, Science, 1983, 219, 245 CrossRef CAS PubMed.
- H. M. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- (a) D. C. Miller, K. T. Tarantino and R. R. Knowles, Top. Curr. Chem., 2016, 374, 30 CrossRef PubMed; (b) J. M. Mayer and I. J. Rhile, Biochim. Biophys. Acta, 2004, 1655, 51–58 CrossRef CAS PubMed.
- J. C. Chu and T. Rovis, Nature, 2016, 539, 272–275 CrossRef PubMed.
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed.
- (a) S. M. Thullen, S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2019, 141, 14062–14067 CrossRef CAS PubMed; (b) M. A. Ashley, C. Yamauchi, J. C. K. Chu, S. Otsuka, H. Yorimitsu and T. Rovis, Angew. Chem., Int. Ed. Engl., 2019, 58, 4002 CrossRef CAS PubMed; (c) D. F. Chen, J. C. K. Chu and T. Rovis, J. Am. Chem. Soc., 2017, 139, 14897–21490 CrossRef CAS PubMed.
- C. M. Morton, Q. Zhu, H. Ripberger, L. Troian-Gautier, Z. S. D. Toa, R. R. Knowles and E. J. Alexanian, J. Am. Chem. Soc., 2019, 141, 13253–13260 CrossRef CAS PubMed.
- B. Xu and U. K. Tambar, ACS Catal., 2019, 9, 4627–4631 CrossRef CAS PubMed.
- W. J. Yue, C. S. Day and R. Martin, J. Am. Chem. Soc., 2021, 143, 6395–6400 CrossRef CAS PubMed.
- (a) Z. Zhang, X. Zhang and D. A. Nagib, Chem, 2019, 5, 3127–3134 CrossRef CAS PubMed; (b) Z. Zhang, L. M. Stateman and D. A. Nagib, Chem. Sci., 2019, 10, 1207–1211 RSC.
- (a) M. A. Short, J. M. Blackburn and J. L. Roizen, Angew. Chem., Int. Ed. Engl., 2018, 57, 296 CrossRef CAS; (b) M. A. Short, M. F. Shehata, M. A. Sanders and J. L. Roizen, Chem. Sci., 2020, 11, 217–223 RSC; (c) A. L. G. Kanegusuku, T. Castanheiro, S. K. Ayer and J. L. Roizen, Org. Lett., 2019, 21, 6089–6095 CrossRef CAS PubMed.
- A. W. Rand, H. Yin, L. Xu, J. Giacoboni, R. Martin-Montero, C. Romano, J. Montgomery and R. Martin, ACS Catal., 2020, 10, 4671–4676 CrossRef CAS.
- (a) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 52 CrossRef CAS; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (c) C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) G. Li, W. M. Ward and G. J. Meyer, J. Am. Chem. Soc., 2015, 137, 8321–8323 CrossRef CAS PubMed; (b) L. Troian-Gautier, M. D. Turlington, S. A. M. Wehlin, A. B. Maurer, M. D. Brady, W. B. Swords and G. J. Meyer, Chem. Rev., 2019, 119, 4628–4683 CrossRef CAS PubMed.
- (a) D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715–12718 CrossRef CAS PubMed; (b) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS PubMed; (c) S. K. Kariofillis, B. J. Shields, M. A. Tekle-Smith, M. J. Zacuto and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 7683–7689 CrossRef CAS PubMed; (d) L. K. G. Ackerman, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS PubMed; (e) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed. Engl., 2019, 58, 6152–6163 CrossRef CAS; (f) L. Huang and M. Rueping, Angew. Chem., Int. Ed. Engl., 2018, 57, 10333–10337 CrossRef CAS PubMed.
- S. K. Kariofillis, S. Jiang, A. M. Zuranski, S. S. Gandhi, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 1045–1055 CrossRef CAS PubMed.
- (a) L. Barriault, T. McCallum, S. Rohe and A. Morris, Angew. Chem., Int. Ed. Engl., 2018, 57, 15664–15669 CrossRef; (b) Z. Wang, X. Ji, T. Han, G. J. Deng and H. Huang, Adv. Synth. Catal., 2019, 361, 5643–5647 CrossRef CAS.
- (a) L. Huang and M. Rueping, Angew. Chem., Int. Ed. Engl., 2018, 57, 10333–10337 CrossRef CAS PubMed; (b) T. Kawasaki, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2020, 142, 336–3370 CrossRef PubMed; (c) S. J. Hwang, B. L. Anderson, D. C. Powers, A. G. Maher, R. G. Hadt and D. G. Nocera, Organometallics, 2015, 34, 4766–4774 CrossRef CAS; (d) S. J. Hwang, D. C. Powers, A. G. Maher, B. L. Anderson, R. G. Hadt, S. L. Zheng, Y. S. Chen and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 6472–6475 CrossRef CAS PubMed; (e) S. I. Ting, S. Garakyaraghi, C. M. Taliaferro, B. J. Shields, G. D. Scholes, F. N. Castellano and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 5800–5810 CrossRef CAS PubMed; (f) V. Bacauanu, S. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernandez, R. Remy and D. W. C. MacMillan, Angew. Chem., Int. Ed. Engl., 2018, 57, 12543–12548 CrossRef CAS PubMed.
- B. J. Shields, B. Kudisch, G. D. Scholes and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 3035–3039 CrossRef CAS PubMed.
- M. K. Nielsen, B. J. Shields, J. Liu, M. J. Williams, M. J. Zacuto and A. G. Doyle, Angew. Chem., Int. Ed. Engl., 2017, 56, 7191–7194 CrossRef CAS PubMed.
- T. T. Tsou and J. K. Kochi, J. Org. Chem., 1980, 45, 1930–1937 CrossRef CAS.
- (a) D. Nicewicz, H. Roth and N. Romero, Synlett, 2015, 27, 714–723 CrossRef; (b) G. Li, M. D. Brady and G. J. Meyer, J. Am. Chem. Soc., 2018, 140, 5447–5456 CrossRef CAS PubMed.
- R. K. Quinn, Z. A. Konst, S. E. Michalak, Y. Schmidt, A. R. Szklarski, A. R. Flores, S. Nam, D. A. Horne, C. D. Vanderwal and E. J. Alexanian, J. Am. Chem. Soc., 2016, 138, 696–702 CrossRef CAS PubMed.
- P. Zhang, C. C. Le and D. W. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed.
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- X. Shu, L. Huan, Q. Huang and H. Huo, J. Am. Chem. Soc., 2020, 142, 19058–19064 CrossRef CAS PubMed.
- O. Gutierrez, J. C. Tellis, D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS PubMed.
- S. Y. Hsieh and J. W. Bode, ACS Cent. Sci., 2017, 3, 66–72 CrossRef CAS PubMed.
- M. Yuan, Z. Song, S. O. Badir, G. A. Molander and O. Gutierrez, J. Am. Chem. Soc., 2020, 142, 7225–7234 CrossRef CAS PubMed.
- J. M. Saveant, J. Phys. Chem. B, 1994, 98, 3716–3724 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and copies of spectra. See https://doi.org/10.1039/d2sc01962k |
| This journal is © The Royal Society of Chemistry 2022 |